
Abstract
This chapter briefly summarizes the most important processes by which hypoxia, lactate accumulation, and acidosis may influence malignant progression and therapeutic resistance of solid malignant tumors. While these phenomena are often elements of an integrated reaction, they may occur independently of each other under certain circumstances. The latter information may be of interest with regard to possible “targeted” therapeutic interventions.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Hypoxia
Evidence supporting the existence of hypoxic tissue areas in solid tumors is derived from data originating from a variety of methods [1]. These include invasive measurements of intratumoral oxygen partial pressures using polarographic needle electrodes (“Eppendorf” microsensor) and histological assays based on the immunodetection of so-called endogenous or exogenous hypoxia markers. In addition, different imaging methods have been developed, which, however, at the present time have not been adopted widely in the clinic. The major cause of tumor hypoxia is an enlargement of the intratumoral diffusion distances of oxygen beyond a critical threshold, which is estimated to be equal to approximately 80 μm at the arterial end of the microvessel. This main origin of continuous or “chronic” hypoxia is modified by other factors, including a reduced oxygen-transport capacity of the blood (anemia) and an increased interstitial fluid pressure, which may lead to a flow stop in microvessels. Besides the phenomenon of continuous tumor hypoxia, one also observes intermittent or “acute” hypoxia, which may be caused by fluctuations in the flux of erythrocytes or by temporary obstructions of tumor capillaries, e.g., by cell aggregates.
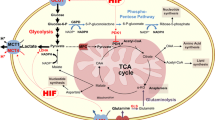
Figure 28.1 shows a synopsis of the various mechanisms by which tumor hypoxia may contribute to a more aggressive phenotype and to an increased resistance to therapy. The discovery that hypoxia is one of the most important factors mediating radioresistance can be traced back to the beginning of the twentieth century. We know today that the mechanism behind this observation is a modification of the free radical chemistry under hypoxic conditions, which has also been shown to be important for some forms of chemotherapy and photodynamic therapy [2]. Since the early 1990s, clinical studies indicated that the pathophysiological significance of hypoxia is clearly not limited to this modification of the radiosensitivity of tumor cells [3]. Hypoxia can lead to an increase of the genetic instability of cancer cells both by inducing mutations and by inhibiting DNA repair [4]. Hypoxia may also act as a selective force favoring the emergence of genetically hypoxia-resistant phenotypes. For example, p53-negative, apoptosis-resistant cell populations may emerge after repeated exposures of cells to hypoxia and reoxygenation [5]. Hypoxia has been shown to be important for the maintenance of the stem cell phenotype, and some types of stem cells have been observed to reside in a “hypoxic niche” in vivo [6]. Furthermore, hypoxia can play an important role in the attenuation of an antitumor immune response. For example, macrophages of the pro-tumorigenic M2 phenotype have been found preferentially in hypoxic tumor areas [7]. Consistent with this finding, other reports have demonstrated that hypoxic tumors contain a higher number of macrophages compared to non-hypoxic tumors. Additionally, higher quantities of intratumoral macrophages have been shown to correlate with a poorer patient prognosis [8]. Under hypoxic conditions, an increased expression of the cytokine CCL28 has been detected, which may lead to intratumoral accumulation of immunosuppressive regulatory T cells which express the cognate receptor CXCR10 [9]. Under hypoxic conditions, adenosine may accumulate in the extracellular space and stimulate adenosine receptors (of the A2A and A2B subtypes) on T cells, thereby leading to an inhibition of antitumor T cell responses [10]. Hypoxia has also been shown to be able to trigger the unfolded protein response and autophagy, which may promote tumor growth and resistance to anticancer therapy [11]. It should be mentioned, however, that both processes can also be antitumorigenic, depending on the specific experimental conditions. Although mTOR inhibition is currently being evaluated as a therapeutic strategy, e.g., in malignant gliomas, hypoxia-mediated suppression of mTOR has recently been shown to prevent irreversible cellular senescence, which may attenuate the efficacy of DNA-damaging agents [12]. Arguably, the overall most significant consequence of hypoxia is a large-scale change of the proteome, which is mediated by the activity of several transcription factors, among which the hypoxia-inducible factor 1 (HIF-1) plays the most important role [13].


Tumor hypoxia is a central driver of malignant progression and resistance to therapy (selection of mechanisms)
More than 800 direct target genes of HIF-1 are known, and a large number of these have been shown to have a direct pathogenic role within the malignant phenotype (see Fig. 28.2, [13]). HIF-1 is a major trigger of proangiogenic cytokines (e.g., VEGF) in tumor cells. Furthermore, HIF-1 can promote vasculogenesis by the recruitment of CXCR4-positive stem cells from the bone marrow via SDF-1. HIF-1 increases the oxygen-transport capacity of the blood by upregulating EPO. Activation of HIF-1 leads to increased cell motility and invasiveness, mediates the ability to remodel the extracellular matrix, and can confer an augmented metastatic potency. These pivotally important processes may be initiated directly by HIF-1, e.g., via the urokinase-type plasminogen activator and matrix metalloproteinases. Additionally, HIF-1 can transactivate transcription factors (e.g., TWIST) which induce the metastasis-promoting cellular program of epithelial-to-mesenchymal transition [14]. HIF-1 may promote radioresistance by allowing cells to survive in hypoxic areas. Moreover, basal HIF-1 expression, but – interestingly – not hypoxia-induced expression of HIF-1, has been demonstrated to play a role for the expression of genes involved in DNA repair [15]. Target genes of HIF-1 can also mediate chemoresistance, e.g., by induction of the MDR-1 gene. There have also been reports describing a role of HIF-1 in mediating increased genetic instability by decreased homologous recombination repair and reduced mismatch repair [16]. HIF-1 can promote the differentiation of TH17 cells [17], which, depending on the experimental paradigm, have been described to both promote and inhibit the growth of tumors. HIF-1-induced SDF-1 may also contribute to the aforementioned accumulation of macrophages in hypoxic tumors [18]. HIF-1 may stimulate proliferation through the induction of autocrine growth factor loops. A number of publications have described an HIF-1-induced upregulation of telomerase and HIF-1 activated genes which are considered to play a role in the stem cell phenotype. Finally, a central mechanism of HIF-1-mediated maladaptation consists of an extensive metabolic reprogramming which leads to a downregulation of mitochondrial oxidative phosphorylation, e.g., via inhibition of the pyruvate dehydrogenase reaction by PDK-1 and promotion of selective autophagy of mitochondria. Simultaneously, HIF-1 mediates the induction of a glycolytic phenotype by increasing glucose influx (e.g., via GLUT-1), upregulation of key enzymes of glycolysis, and by an increase in the efflux of lactate via the monocarboxylate transporter subtype MCT-4 [13, 19].


HIF-1 as the central driver of hypoxia-induced transcriptional “maladaptation” in cancer (selection of mechanisms)
2 Lactate
A substantial part of intratumoral lactate accumulation is the result of HIF-1-mediated metabolic reprogramming. However, comparative analyses of the distribution patterns of hypoxia (as assessed by pimonidazole staining) and locoregional lactate concentrations (analyzed using imaging bioluminescence) have revealed that both parameters are not necessarily co-localized in all cases [20]. Indeed, several HIF-1-independent mechanisms of intratumoral lactate accumulation have been described, e.g., the activation of MYC [21]. Additionally, high lactate levels may also be the consequence of an insufficient waste drainage in poorly vascularized tumor areas. The matter is further complicated by the existence of an intratumoral lactate shuttle between hypoxic (lactate-producing) and normoxic (lactate-consuming) cells [22]. Lactate has been hypothesized to mediate radioresistance by virtue of its antioxidant properties. Lactate also exhibits immunosuppressive properties and promotes cell motility, invasion, and metastasis. Furthermore, lactate may induce angiogenesis, mediate resistance to apoptosis, and may promote a stem cell phenotype. Importantly, lactate can indirectly stabilize HIF-1α and may thus perpetuate the activation of HIF-1 independent of hypoxia [23].
3 Acidosis
HIF-1-induced metabolic reprogramming also contributes to the marked extracellular acidosis often found in malignant tumors by upregulating glycolysis. Nevertheless, direct measurements of intratumoral oxygen and pH levels have revealed unequal distributions of both parameters at the microregional level [24, 25], and glycolysis-deficient cells have been shown to retain the ability to acidify the extracellular environment in vivo [26]. Additional pathogenetic mechanisms yielding an intensified tissue acidosis are based on substantial hydrolysis of ATP (derived from breakdown of substrates other than glucose), glutaminolysis, ketogenesis, and CO2/carbonic acid production [27]. The spectrum of the pathophysiological consequences of intratumoral acidosis includes many processes mentioned for HIF-1 and lactate: acidosis plays a role in mediating radioresistance (e.g., [28]), immune evasion [29], increased cell motility, invasion, metastasis [30, 31], promotion of angiogenesis through VEGF [32], and the stem cell phenotype [33]. Moreover, an acidic extracellular milieu diminishes the effectiveness of basic chemotherapeutic drugs (e.g., doxorubicin, daunorubicin, [34]). Similar to hypoxia and HIF-1, acidosis may contribute to the genetic instability of tumor cells [35] and – similar to hypoxia – is a possible trigger for autophagy [36]. Finally, acidosis has been shown to stabilize HIF-1α independent of hypoxia by nucleolar sequestration of VHL [37].
4 Conclusions
Factors of the microenvironment presented in this report trigger an overlapping range of processes which promote tumor growth and mediate resistance to therapy. The broadest spectrum of these processes is initiated by hypoxia and HIF-1, which are also often at the root of lactate accumulation and intratumoral acidosis. With this in mind, the three factors may be regarded as “siblings.” However, both of the latter factors may also be triggered independently of hypoxia and, importantly, similar pathogenic processes (e.g., radioresistance) may be initiated by all three factors via entirely independent mechanisms (e.g., modification of the spectrum of free radicals generated by radiation vs. scavenging of free radicals). Therefore, the factors discussed here may also act as “accomplices,” depending on the specific triggers for each of them in individual tumors.
References
Vaupel P, Mayer A (2007) Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 26(2):225–239
Vaupel P (2009) Physiological mechanisms of treatment resistance. In: Molls M, Vaupel P, Nieder C, Anscher MS (eds) The impact of tumor biology on cancer treatment and multidisciplinary strategies. Springer, Berlin\Heidelberg, pp 273–290
Höckel M, Schlenger K, Aral B, Mitze M, Schäffer U, Vaupel P (1996) Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res 56(19):4509–4515
Klein TJ, Glazer PM (2010) The tumor microenvironment and DNA repair. Semin Radiat Oncol 20(4):282–287
Graeber TG, Osmanian C, Jacks T et al (1996) Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379(6560):88–91
Suda T, Takubo K, Semenza GL (2011) Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9(4):298–310
Movahedi K, Laoui D, Gysemans C et al (2010) Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 70(14):5728–5739
Lewis C, Murdoch C (2005) Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am J Pathol 167(3):627–635
Facciabene A, Peng X, Hagemann IS et al (2011) Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475(7355):226–230
Sitkovsky M, Lukashev D (2005) Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol 5(9):712–721
Wouters BG, Koritzinsky M (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8(11):851–864
Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV (2012) Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci USA 109(33):13314–13318
Semenza GL (2012) Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci 33(4):207–214
Yang MH, Wu MZ, Chiou SH et al (2008) Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol 10(3):295–305
Wirthner R, Wrann S, Balamurugan K, Wenger RH, Stiehl DP (2008) Impaired DNA double-strand break repair contributes to chemoresistance in HIF-1α-deficient mouse embryonic fibroblasts. Carcinogenesis 29(12):2306–2316
To KK, Sedelnikova OA, Samons M, Bonner WM, Huang LE (2006) The phosphorylation status of PAS-B distinguishes HIF-1α from HIF-2α in NBS1 repression. EMBO J 25(20):4784–4794
Dang EV, Barbi J, Yang HY et al (2011) Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146(5):772–784
Palazón A, Aragonés J, Morales-Kastresana A, de Landázuri MO, Melero I (2012) Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res 18(5):1207–1213
Semenza GL (2011) Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 1813(7):1263–1268
Yaromina A, Quennet V, Zips D et al (2009) Co-localisation of hypoxia and perfusion markers with parameters of glucose metabolism in human squamous cell carcinoma (hSCC) xenografts. Int J Radiat Biol 85(11):972–980
Osthus RC, Shim H, Kim S et al (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275(29):21797–21800
Sonveaux P, Vegran F, Schroeder T et al (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118(12):3930–3942
Hirschhaeuser F, Sattler UG, Mueller-Klieser W (2011) Lactate: a metabolic key player in cancer. Cancer Res 71(22):6921–6925
Vaupel PW, Frinak S, Bicher HI (1981) Heterogeneous oxygen partial pressure and pH distribution in C3H mouse mammary adenocarcinoma. Cancer Res 41(5):2008–2013
Helmlinger G, Yuan F, Dellian M, Jain RK (1997) Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med 3(2):177–182
Newell K, Franchi A, Pouyssegur J, Tannock I (1993) Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proc Natl Acad Sci USA 90(3):1127–1131
Vaupel P (2004) Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol 14(3):198–206
Haveman J (1980) The influence of pH on the survival after X-irradiation of cultured malignant cells. Effects of carbonylcyanide-3-chlorophenylhydrazone. Int J Radiat Biol Relat Stud Phys Chem Med 37(2):201–205
Lardner A (2001) The effects of extracellular pH on immune function. J Leukoc Biol 69(4):522–530
Raghunand N, Gatenby RA, Gillies RJ (2003) Microenvironmental and cellular consequences of altered blood flow in tumours. Br J Radiol 76(1):S11–S22
Calorini L, Peppicelli S, Bianchini F (2012) Extracellular acidity as favouring factor of tumor progression and metastatic dissemination. Exp Oncol 34(2):79–84
Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK (2001) Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res 61(16):6020–6024
Hjelmeland AB, Wu Q, Heddleston JM et al (2011) Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 18(5):829–840
Thews O, Gassner B, Kelleher DK, Schwerdt G, Gekle M (2006) Impact of extracellular acidity on the activity of P-glycoprotein and the cytotoxicity of chemotherapeutic drugs. Neoplasia 8(2):143–152
Morita T, Nagaki T, Fukuda I, Okumura K (1992) Clastogenicity of low pH to various cultured mammalian cells. Mutat Res 268(2):297–305
Wojtkowiak JW, Rothberg JM, Kumar V et al (2012) Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res 72(16):3938–3947
Mekhail K, Gunaratnam L, Bonicalzi ME, Lee S (2004) HIF activation by pH-dependent nucleolar sequestration of VHL. Nat Cell Biol 6(7):642–647
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this paper
Cite this paper
Mayer, A., Vaupel, P. (2013). Hypoxia, Lactate Accumulation, and Acidosis: Siblings or Accomplices Driving Tumor Progression and Resistance to Therapy?. In: Van Huffel, S., Naulaers, G., Caicedo, A., Bruley, D.F., Harrison, D.K. (eds) Oxygen Transport to Tissue XXXV. Advances in Experimental Medicine and Biology, vol 789. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7411-1_28
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7411-1_28
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7256-8
Online ISBN: 978-1-4614-7411-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)