
Abstract
Hypoxia is an important component of the tumor microenvironment and has been the target of drug discovery efforts for almost half a century. These efforts have evolved from offsetting the impact of hypoxia on radiotherapy with oxygen-mimetic radiosensitizers to using hypoxia as a means to selectively target tumors. The more recent description of hypoxia-inducible factors and their role in the hypoxia response network has revealed a host of new drug targets to selectively target tumors. We are developing hypoxia-directed drugs in each of the following areas: novel radiosensitizers for hypofractionated radiotherapy, a second-generation benzotriazine di-N-oxide hypoxia-activated prodrug, and a hypoxia-inducible factor-1–dependent cytotoxin that targets glucose transport. These projects are discussed in the context of hypoxia-directed drug discovery.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
- Hypoxia
- Drug discovery
- Nitroimidazole
- Radiosensitizer
- Hypoxia-activated prodrug
- Biomarker
- Tirapazamine
- SN30000
- HIF-1α
- Glucose transport
6.1 Introduction
6.1.1 Hypoxia as a Therapeutic Target
Hypoxia initially arises as a consequence of oxygen consumption in small tumors or metastases. The cellular response to this hypoxia plays a significant role in the development of the tumor microenvironment and influences the expansion of tumor vasculature, resulting in a disorganized, inefficient tumor microvascular network that has irregular blood flow (Jain 2005; Pries et al. 2009). In turn, this exacerbates existing hypoxia and leads to considerable heterogeneity in oxygen concentrations that may fluctuate spatially and temporally (Dewhirst et al. 2008) (Fig. 6.1). The characterization of hypoxia accordingly depends on the techniques used to measure it. Whereas fine-needle oxygen electrode measurements provide a direct gauge of oxygen tension and have demonstrated a wide range of oxygen concentrations in human tumors (Vaupel et al. 2007), the use of exogenous molecular probes such as 2-nitroimidazoles or endogenous markers such as downstream products of genes regulated by hypoxia-inducible factors (HIFs) report different levels of hypoxia (Fig. 6.2). Nitroimidazole probes are typically activated at levels of less than 1 μM of oxygen, whereas HIF-1 is stabilized at higher oxygen concentrations (Tuttle et al. 2007).

Detection of hypoxic cells in a human colon cancer xenograft HCT116 grown subcutaneously in nude mice. Yellow box shows diffusion-limited hypoxia. White oval shows perfusion-limited hypoxia. Hypoxic marker EF5 (60 mg/kg) was administrated intraperitoneally 1.5 h and blood vessel perfusion marker Hochest33342 (40 mg/kg) was administered intravenously 2 min before the mice were killed. The tumor was removed immediately and frozen in octanol. Frozen sections (8 μm) were immunostained for the hypoxic marker (EF5, red), blood vessels (CD1, green), and perfused blood vessels (Hochest33342, blue). (Jingli Wang, unpublished data)

Illustration of oxygen dependence for cellular response. Radiation sensitivity of clonogenic cell killing increases with increasing oxygen concentration, reaching half-maximal at approximately 4–5 μM (Wouters and Brown 1997), whereas it decreases for hypoxia-activated prodrugs PR-104 (Hicks et al. 2007) and tirapazamine/SN30000 (Hicks et al. 2004, 2010). For nitroimidazoles, oxygen dependence of intracellular binding of EF5 (Tuttle et al. 2007) is similar to the oxygen dependence of the sensitizer enhancement ratio of misonidazole (Carlson et al. 2011). Stabilization of the hypoxia-inducible factor generally occurs under more moderate hypoxic conditions (Tuttle et al. 2007)
The majority of clinical studies have shown that hypoxia results in compromised outcomes across a wide range of diseases and treatment modalities (Horsman et al. 2012; Nordsmark et al. 2005; Vaupel and Mayer 2007). Both chronic hypoxia (Gray et al. 1953; Thomlinson and Gray 1955) and intermittent, or cycling, hypoxia within solid tumors can limit radiotherapy (Brown 1979). Poor perfusion and significant diffusion gradients exist within tumors (Dewhirst et al. 2008) that, along with high interstitial pressures (Heldin et al. 2004), can limit the diffusion of chemotherapeutic agents into hypoxic regions (Minchinton and Tannock 2006). This, when combined with a slowing of proliferation in these areas, can cause resistance to commonly used antiproliferative agents. Identification of the role of HIF-1 in the hypoxia response network (Semenza 2003) has revealed how hypoxia influences survival processes such as increased angiogenesis (Gnarra et al. 1996) and vasculogenesis (Kioi et al. 2010), resistance to cell death (Graeber et al. 1996), aerobic glycolysis (Semenza 2010), and genomic instability (Bindra et al. 2007; Huang et al. 2007). Furthermore, the contribution of tumor hypoxia to invasiveness and metastasis (Chang et al. 2011; Hill et al. 2009) potentially compromises a third treatment modality: surgery. The prevalence of tumor hypoxia, combined with its effect on tumor survival, progression, and resistance to therapy, marks hypoxia as a compelling target for current drug discovery efforts.
6.1.2 Drug Development
In tandem with the growing understanding of the effect of hypoxia, an evolving series of drug discovery efforts have sought to overcome or leverage the effects of hypoxia for therapeutic gain (Denny 2010; Semenza 2007; Wilson and Hay 2011). The major drug discovery effort was centered on chemical radiosensitizers and spanned several decades (Wardman 2007), but minimal clinical success resulted in dwindling efforts in this field (Overgaard 2007).
A paradigm shift in hypoxia targeting occurred in the mid-1980s. Rather than minimizing the impact of hypoxia on radiotherapy, a new strategy sought to use hypoxia as a physiological target that could promote activation of prodrugs to kill tumor cells. Hypoxia-activated prodrugs (HAPs) have been extensively explored over several decades (Brown and Wilson 2004; Chen and Hu 2009; McKeown et al. 2007; Rockwell et al. 2009; Wilson and Hay 2011).
Identification of the HIF as the “master regulator” of hypoxic response and a key drug target (Giaccia et al. 2003; Semenza 2003) resulted in the discovery of a plethora of small molecules that could be potential HIF inhibitors (Xia et al. 2012). However, much of this work has had limited application. Many agents identified as HIF-1α inhibitors actually target upstream or downstream components in the HIF response network or have pleiotropic effects. In addition, these agents are not necessarily cytostatic or cytotoxic, nor are they necessarily selective for hypoxic cells.
Overall, few agents developed to target hypoxia have been registered. Tumor hypoxia has been a niche area, predominantly the preserve of academic groups, and only with the elaboration of the hypoxia response network has hypoxia received mainstream attention as a validated target for drug development.
6.1.3 Defining the Hypoxic “Target”
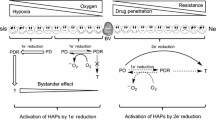
Definition of the “target” depends on the approach directed against the hypoxic cells. For oxygen-mimetic radiosensitizers the target is a DNA radical generated by ionizing radiation (Fig. 6.3a), and selectivity results from competition between oxygen and the electron-affinic nitroimidazole group for this radical. Since oxygen is vastly more efficient at scavenging these radicals, nitroimidazoles effectively sensitize only hypoxic cells.

(a) Mechanism of oxygen-mimetic radiosensitization by electron-affinic nitroaryl radiosensitizers. (b) Mechanism of hypoxia-activated prodrugs
A more complex situation exists for HAPs (Fig. 6.3b). The prodrug is reduced by a one-electron reductase to form a radical anion. This radical anion may be “scavenged” by oxygen to reproduce the prodrug with production of superoxide. In the absence of oxygen the radical anion may undergo a variety of transformations, depending on chemical class, leading to the activated drug. Reduction of the prodrug by two-electron reductases removes the potential for back-oxidation of the radical anion, leading to a loss of hypoxic selectivity. As a consequence, the target is the intersection of three elements: hypoxia, enzymes to activate the prodrug, and intrinsic sensitivity to the activated drug. The initial concept invoked tumor hypoxia as unique to tumor tissue and, essentially, a binary switch, suggesting it is an ideal drug target for HAPs (Denny et al. 1996). However, hypoxia exists in normal tissues, and tumor cells at intermediate oxygen tension are important for tumor progression (Wouters and Brown 1997), making hypoxia a more complex target. The second component requires the location of appropriate enzymes to activate the prodrug within the tumor. NADPH:cytochrome P450 oxidoreductase has been identified as a key one-electron reductase responsible for the activation of many HAPs (Guise et al. 2007; Meng et al. 2012; Patterson et al. 1998; Wang et al. 2012b), but contributions from other one-electron reductases (e.g., aldehyde oxidase, xanthine oxidase, nitric oxide synthases, thioredoxin reductase, NADH-dependent cytochrome b5 reductase, methionine synthase reductase, and NADPH-dependent diflavin oxidoreductase) also have been reported (Adams and Rickert 1995; Ask et al. 2003; Cenas et al. 2006; Chandor et al. 2008; Guise et al. 2012; Papadopoulou et al. 2003; Patterson et al. 1998; Tatsumi et al. 1986; Ueda et al. 2003). The expression of these enzymes and their relative contributions to HAP activity across human tumors is incompletely understood; however, significant variations between cell lines (Guise et al. 2012; Wang et al. 2012b) and individual human tumors (Evans et al. 2000; Patterson et al. 1997) has been demonstrated. The electron affinity of the prodrug seems to be the key determinant of activation (Wardman 2001), indicating little substrate specificity for most of the one-electron reductases. The third constituent of the HAP target is the intrinsic sensitivity of the target cells to the activated drug. Strategies using prodrugs that release cytotoxins that cross-link DNA will be dependent on DNA repair status and may cause normal tissue toxicity if activated inappropriately. Prodrugs delivering inhibitors of specific molecular targets, such as the human epidermal growth factor receptor, have been reported more recently (Patterson et al. 2009). For these agents, the relative expression of the molecular target in hypoxic and normal tissues contributes to the overall target. In the case of HIF inhibitors, the molecular targets are structurally diverse, and hypoxic selectivity is provided by the level of overexpression under hypoxic conditions relative to the levels of the target in normal tissue and the specificity of the inhibitor for the particular molecular target.
6.2 Radiosensitizers
6.2.1 Introduction
Attempts to offset the negative effects of hypoxia on radiation therapy initially focused on manipulation of tumor oxygen status (e.g., fractionated radiation schedules to allow reoxygenation between fractions [Kallman and Dorie 1986]; hyperbaric oxygen treatment [Bennett et al. 2012; Overgaard and Horsman 1996], and nicotinamide in combination carbogen breathing with accelerated radiotherapy [ARCON] [Janssens et al. 2012; Kaanders et al. 2002]). Drug discovery efforts have been centered on the development of chemical radiosensitizers and, in particular, oxygen-mimetic sensitizers.
6.2.2 Nitroimidazole Oxygen Mimetics
The concept of an electron-affinic nitroaryl molecule as a radiosensitizer in hypoxic tumor tissue has a long history (Dische 1985, 1991; Wardman 2007). The 5-nitroimidazole antibiotic metronidazole (Fig. 6.4) was identified as an effective radiosensitizer (Asquith et al. 1974) and displayed clinical benefit (Urtasun et al. 1976). More electron-affinic 2-nitroimidazoles were explored, and misonidazole was identified and advanced to clinical trials (Adams et al. 1976). Misonidazole underwent extensively trials with fractionated radiotherapy; despite indications of clinical benefit (Overgaard 1994), delayed peripheral neuropathy limited treatment (Grigsby et al. 1999; Saunders and Dische 1996). The electron affinity of the nitroimidazole group is the key parameter for radiosensitization and toxicity (Adams et al. 1979a, b). Thus, 5-nitroimidazoles with lower electron affinity had lower toxicity and larger doses could be used to offset their weaker radiosensitization. This led to the identification of nimorazole as a radiosensitizer (Overgaard et al. 1982, 1983) that is well tolerated (Overgaard et al. 1998; Timothy et al. 1984) and used clinically, but only in Denmark. Nimorazole is currently undergoing a phase III clinical trial with accelerated radiotherapy (Overgaard 2012).

(a) Clinically investigated nitroimidazoles. (b) Representative novel 2- and 5-nitromidazole sulfonamides. (c) Clonogenic survival curves of HCT116 colorectal carcinoma cells after increasing dose of radiation in oxia (blue) and anoxia (black) in the presence of equitoxic doses of etanidazole (1 mM) and SN35265 (0.7 mmol)
Attempts to design more polar analogs with reduced lipophilicity and increased systemic clearance to minimize the neurotoxicity observed with misonidazole led to the development of etanidazole (Brown et al. 1981) and doranidazole (Murata et al. 2008; Oya et al. 1995). This approach was only partially successful: Etanidazole had reduced neurotoxicity compared to misonidazole (Coleman et al. 1990) but failed to provide benefit in head and neck cancer (Eschwege et al. 1997; Lee et al. 1995). Doranidazole is currently under investigation for pancreatic cancer (Karasawa et al. 2008) and non-small-cell lung carcinoma (Nishimura et al. 2007).
It is salutary to note that although nitroimidazole radiosensitizers have been extensively investigated clinically and that hypoxic modification was shown to be effective in a meta-analysis (Overgaard 2011), only nimorazole is in clinical use. Two main factors have contributed to the limited clinical success of radiosensitizers. Their use with fractionated radiotherapy – where fractionation of the radiation dose is designed to allow tumor reoxygenation between radiation fractions – reduces the potential for radiosensitization (Hill 1986; Kallman 1972). Fractionated radiotherapy ideally requires a dose of radiosensitizer with each fraction of radiation, a schedule that was unachievable with early 2-nitroimidazoles because of cumulative peripheral neurotoxicity. Perhaps most significant is that many of the trials were small and were conducted without prospectively identifying patients with hypoxic tumors, despite considerable heterogeneity in the level and extent of tumor hypoxia among patients (Hoogsteen et al. 2009).
However, the development of stereotactic body radiotherapy (SBRT) may offer a new opportunity for this class. SBRT uses hypofractionated (one to five doses), high-dose (25–60 Gy in total dose) radiation to treat primary tumors and oligometastases. Initial clinical results of using SBRT to treat a variety of primary tumors suggest locoregional control and toxicity profiles that compare to or improve on those of fractionated radiotherapy (Lo et al. 2010). Prospective, randomized trials to confirm these results compared to standard care will drive increasing use of SBRT. In addition, reduced treatment time and fewer patient visits, combined with emerging potential to replace surgery in patients for whom an outpatient procedure presents risk, indicates potential economic health advantages for SBRT. However, SBRT may accentuate the role of hypoxia in radioresistance because of the reduced opportunity for tumor reoxygenation during therapy (Brown et al. 2010; Carlson et al. 2011). This would offer the possibility of a renaissance for nitroimidazole radiosensitizers in conjunction with SBRT. A recent small, phase III trial of doranidazole in conjunction with intraoperative radiotherapy (25 Gy) for pancreatic tumors demonstrated a survival advantage (Nishimura et al. 2007).
Nevertheless, several barriers exist in the development of radiosensitizers for use with SBRT. Limited (doranidazole) or expired (misonidazole, etanidazole, nimorazole) patent protection for clinically evaluated nitroimidazoles will limit their application, while the wide range of analogs prepared across the field restricts discovery of novel, patentable nitroimidazoles. The other challenge for future development of such radiosensitizers is the use of a biomarker to prospectively identify hypoxia in patients (See Sect. 6.5).
In addressing these challenges, we have recently identified a new class of nitroimidazole with a sulfonamide side chain, providing chemical novelty (Bonnet et al. 2012). A series of 2- and 5-nitroimidazole analogs have been designed and synthesized, and preliminary results show that representative compounds (Fig. 6.4b) produce comparable in vitro radiosensitization to etanidazole at nontoxic concentrations in hypoxic HCT-116 human colorectal carcinoma cells (Fig. 6.4c). The electron affinity of these compounds, as measured by one-electron reduction potential, is higher than corresponding 2- and 5-nitroimidazoles because of the influence of the strong electron-withdrawing side chain and results in increased radiosensitization. Metabolism is also increased in the more electron-affinic examples, resulting in hypoxia-selective cytotoxicity. This novel series provides the opportunity to leverage 30 years of drug development around the class and develop a third-generation radiosensitizer while including extravascular transport (EVT) (See Sect. 6.3.5) and hypoxia biomarker studies (See Sect. 6.5) early in the drug design process.
6.2.3 Molecular Targets in DNA Repair as Radiosensitizers
The targeting of DNA repair for radiosensitization using antimetabolites (Brown et al. 1971) is well established, although these agents work through multiple mechanisms (Shewach and Lawrence 2007). A range of histone deacetylation inhibitors also radiosensitize tumor cells through modulation of the DNA damage response (Camphausen and Tofilon 2007). Specific DNA repair proteins such as poly(ADP-ribose) polymerase (PARP) (Chalmers et al. 2010), ataxia telangiectasia mutated (ATM) pharmacokinetics (Sarkaria and Eshleman 2001), ATM- and Rad3-related (ATR) pharmacokinetics (Wang et al. 2004), and DNA-dependent pharmacokinetics (Blunt et al. 1995) are potential targets for radiosensitization (Helleday et al. 2008; Begg et al. 2011). PARP inhibitors can radiosensitize tumors (Albert et al. 2007; Calabrese et al. 2004), although some of their activity may be due to a vascular effect that results in reduced intermittent hypoxia (Senra et al. 2011). A range of PARP inhibitors are in clinical development and offer potential as radiosensitizers. Novel ATM inhibitors (KU55933 [Hickson et al. 2004] and KU60019 [Golding et al. 2009]) and ATR inhibitors (NU6027 [Peasland et al. 2011] and VE821 [Charrier et al. 2011; Reaper et al. 2011]) display radiosensitization in vitro (Pires et al. 2012). The selective DNA pharmacokinetics inhibitor NU7441 can radiosensitize tumor cells in vitro and in vivo (Zhao et al. 2006), whereas IC87361 (Kashishian et al. 2003) was reported to enhance radiation-induced delay in the growth of Lewis lung carcinomas (Shinohara et al. 2005). One concern about this approach is the potential for these agents to radiosensitize normal tissue within the radiation field. Although particular diseases may be identified to provide synthetically lethal combinations (e.g., BRCA1 loss of function in combination with PARP inhibitors), another approach is to selectively target these agents to hypoxic tissues using a prodrug approach (Parveen et al. 1999; Cazares-Korner et al 2013).
6.3 Hypoxia-Activated Prodrugs
6.3.1 Introduction
HAPs (also called bioreductive prodrugs or hypoxia-selective cytotoxins) can be grouped into six classes based on their activation chemistry (Fig. 6.5). Quinone prodrugs such as EO9, based on the reductive activation of mitomycin C, were the first class to be explored (Lin et al. 1972; Phillips et al. 2013). The observation that redox cycling could provide a basis for hypoxia-selective cytotoxicity of nitroaryl compounds (Mason and Holtzman 1975) was followed by observations that some nitroimidazole radiosensitizers were also selectively toxic to hypoxic tumor cells in culture (Hall and Roizin-Towle 1975; Mohindra and Rauth 1976). This led to extensive studies of nitroheterocycles as hypoxia-activated prodrugs (Jenkins et al. 1990; Naylor et al. 1990; Threadgill et al. 1991), culminating in the bifunctional prodrug RB-6145 (Naylor et al. 1993), in which an alkylating bromoethylamine side chain increased cytotoxic potency (Hill et al. 1986). Clinical development of RB-6145 and its R-enantiomer (CI-1010) (Cole et al. 1992) was halted because of retinal toxicity in preclinical models, providing early evidence that hypoxia in normal tissues could result in dose-limiting toxicities (Breider et al. 1998; Lee and Wilson 2000).

(a) Main chemical classes used for hypoxia-activated prodrugs (HAPs). (b) Examples of HAPs
Description of the principles of bioreductive activation of nitroaryl prodrugs of nitrogen mustard (Denny and Wilson 1986) laid the groundwork for the eventual discovery of PR-104 as a HAP (Patterson et al. 2007). The hypoxic selectivity of aromatic N-oxides based on the 1,2,4-benzotriazine system led to the identification of tirapazamine (TPZ) (Brown 1993). Aliphatic N-oxides were shown to compete with oxygen for reduction by two-electron reductases, providing a mechanism for masking the DNA binding of DNA intercalators (Patterson 1993; Wilson et al. 1992), such as AQ4N (banoxantrone). Stable transition metal complexes (e.g., Co[III] [Milbank et al. 2009; Ware et al. 1993; Yamamoto et al. 2012] and Cu[II] [Parker et al. 2004]) can undergo hypoxia-selective, one-electron reduction to relatively unstable complexes (e.g., Co[II] and Cu[I]), releasing a cytotoxic agent. A vast assortment of compounds from these classes has been explored in the laboratory but only a handful have been evaluated clinically. Several of these provide informative examples of the challenges facing HAP discovery and are briefly discussed below.
6.3.2 PR-104
PR-104 arose from the structural optimization of simple nitroaryl nitrogen mustards (Denny and Wilson 1986) to selectively activated, diffusible mustard cytotoxins (Denny and Wilson 1993) and involved several design challenges. Elevation of the electron affinity of the 5-nitro group into a range suitable for bioreduction required additional electron-withdrawing substituents (e.g., a 3-NO2 group) (Palmer et al. 1992). The relative arrangement of the four substituents provides the best combination of potency and hypoxic selectivity (Palmer et al. 1996). Addition of a carboxamide-linked solubilizing side chain (Palmer et al. 1994), combined with a phosphate prodrug approach, provides sufficient aqueous solubility.
The phosphate group is readily cleaved in plasma (Patel et al. 2007), and the nitro group then undergoes one-electron reduction to a nitro radical anion (Guise et al. 2007) (Fig. 6.6a), which is converted back to the prodrug in the presence of oxygen by redox cycling. Further reduction of the radical anion produces a nitrosobenzene that may undergo subsequent reduction to electron-donating hydroxylamine (PR-104H) and aminobenzene (PR-104M). These activated species cross-link DNA, forming cytotoxic lesions (Gu et al. 2009; Patterson et al. 2007; Singleton et al. 2009). PR-104 is activated under low oxygen concentrations (Hicks et al. 2007) (Fig. 6.2), but reduced species are sufficiently lipophilic and stable to diffuse from the cell of activation to surrounding tumor cells, known as the “bystander effect” (Foehrenbacher et al. 2013; Patterson et al. 2007; Wilson et al. 2007).

Mechanism of activation of leading hypoxia-activated prodrugs. (a) PR-104. One-electron reduction of PR-104A to the nitro radical anion is reversed in the presence of oxygen. Under hypoxia, further reduction of the radical anion leads sequentially to the deactivated nitroso and the activated hydroxylamine (PR-104H) and amine PR-104M. Two-electron reduction of PR-104A bypasses the nitro radical anion and is not hypoxia selective. (b) TH-302. One-electron reduction under hypoxia leads to a radical anion. Radiolytic studies have demonstrated direct fragmentation of the radical anion to release the bromo-phosphoramidate mustard (Br2-IPM). An alternate, stepwise two-electron reduction to the 2-hydroxylamine and subsequent fragmentation has been previously proposed. (c) Tirapazamine. One-electron reduction gives an N-oxide radical that may be reoxidized by oxygen. Under hypoxia, protonation and then rearrangement produces a carbon-centered tirapazamine (TPZ) radical. This TPZ radical may then eliminate water to give a DNA-damaging benzotriazinyl nitrogen-centered radical or release a hydroxyl radical. Further reduction of the TPZ radical, or two-electron reduction of TPZ, leads to the relatively nontoxic 1-oxide and nor-oxide. An analogous activation mechanism has been proposed for the related benzotriazine dioxide SN30000
PR-104 displayed excellent in vitro hypoxic selectivity (6- to 160-fold), with single-agent activity and potentiation of radiation in SiHa, HT29, and H460 tumor xenografts (Patterson et al. 2007). PR-104 advanced to clinical trials (Jameson et al. 2010; McKeage et al. 2011), but normal tissue toxicity in humans prevented trials from reaching an efficacious dose (Patel et al. 2011). Activation of PR-104 by the oxygen-insensitive two-electron reductase aldo-ketoreductase AKR-1C3 (Guise et al. 2010), was subsequently suggested as a factor contributing to this toxicity. A new strategy to leverage the presence of both hypoxia and AKR-1C3 expression in particular tumor types, including advanced leukemia (Houghton et al. 2011; Benito et al. 2011), has led to subsequent clinical trials (www.ClinicalTrials.gov identifier NCT01037556).
6.3.3 TH-302
A versatile prodrug strategy based around the 2-nitroimidazole-5-methanol moiety was able to release enediynes (Hay et al. 1999), aspirin (Everett et al. 1999), and a PARP inhibitor (Parveen et al. 1999) in a hypoxia-selective manner; nitroheterocyclic prodrugs of phosphoramidate mustards were shown to release cytotoxins upon reduction (Borch et al. 2000, 2001). These studies were a precursor to the discovery of TH-302, a 2-nitroimidazole-5-methyl phosphoramidite, as a HAP with excellent hypoxic selectivity (Duan et al. 2008; Meng et al. 2012). Steady-state and pulse radiolysis methods showed that TH-302 undergoes one-electron reduction and fragmentation to release bromo-isophosphoramide mustard (Meng et al. 2012) but did not exclude the initially proposed stepwise reduction of 2-nitroimidazole prodrugs to hydroxylamine or amine and fragmentation via an iminomethide (Borch et al. 2001) (Fig. 6.6b). The increased toxicity observed in cells that overexpress bacterial nitroreductase provides evidence of the potential for oxygen-insensitive, two-electron reduction and release of bromo-isophosphoramide mustard (Meng et al. 2012). The released mustard generates DNA cross-links that are responsible for hypoxic cytotoxicity (Meng et al. 2012). Extensive preclinical studies have shown the antitumor activity of TH-302 – either as a single agent (Sun et al. 2012) or in combination with commonly used chemotherapeutic drugs (Liu et al. 2012) and radiation (Lohse et al. 2012) – in many animal xenograft models. The anticancer efficacy of TH-302 correlated well with the levels of xenograft tumor hypoxia, confirming the hypoxic specificity of drug action in vivo (Lohse et al. 2012; Sun et al. 2012). TH-302 is currently the most advanced HAP in clinical development. Promising outcomes from phase II clinical trials (Borad et al. 2012; Chawla et al. 2011) led to the commencement of two randomized, placebo-controlled, phase III trials: one with TH-302 in combination with doxorubicin for advanced soft tissue sarcoma and the other in combination with gemcitabine for advanced pancreatic cancer.
6.3.4 Tirapazamine
TPZ (tirazone) is the prototypic example of a heterocyclic N-oxide HAP and dominated the field for almost two decades (Brown 1993, 2010; Denny and Wilson 2000). TPZ shows highly selective killing in cell culture under hypoxic compared to aerobic conditions (Zeman et al. 1986) as a result of rapid bioreductive metabolism (Baker et al. 1988; Hicks et al. 2003; Siim et al. 1996). One-electron reduction by, for example, NADPH:cytochrome P450 oxidoreductase (Fitzsimmons et al. 1994; Patterson et al. 1997, 1998), inducible nitric oxide synthase (Chinje et al. 2003), or nuclear localized reductases (Evans et al. 1998) produces a N-centered radical (Baker et al. 1988; Laderoute et al. 1988) that is efficiently back-oxidized to TPZ by oxygen (Fig. 6.6c). In the absence of oxygen, protonation and rearrangement leads to an oxidizing radical (Anderson et al. 2003; Shinde et al. 2009, 2010; Yin et al. 2012) or hydroxyl radical (Chowdhury et al. 2007; Daniels and Gates 1996), both of which have been proposed as the species that damages cytotoxic DNA. DNA damage measured by comet assay (Olive et al. 1996; Siim et al. 1996) or induction of γH2AX (Olive et al. 2004; Wang et al. 2012b) correlates with the rates of bioreduction and reductase expression (Wang et al. 2012b) and is repaired by multiple mechanisms, including homologous recombination repair of double-strand breaks (Evans et al. 2008; Hunter et al. 2012). The radical species are short-lived and do not contribute to the killing of surrounding cells. Despite the lack of the bystander effect, TPZ is able to kill cells at intermediate oxygen concentrations because of activation at relatively high oxygen concentrations, with K-values (oxygen concentration for half-maximal hypoxic potency) in the range 1–3 μM (Hicks et al. 2004, 2007; Koch 1993), resulting in good complementarity with radiation (Hicks et al. 2004, 2007; Koch 1993; Wouters and Brown 1997), (Fig. 6.2). In contrast to PR-104 and TH-302, the two- and four-electron reduction products are markedly less cytotoxic than the parent drug (Baker et al. 1988), but this unproductive metabolism reduces potency.
Xenograft studies demonstrated cell killing complementing that of single-dose (Zeman et al. 1988) and fractionated radiation (Brown and Lemmon 1990, 1991). TPZ also demonstrated synergy with cisplatin in preclinical tumor models (Dorie and Brown 1993, 1994), resulting from hypoxia-dependent inhibition of cisplatin DNA cross-link repair (Kovacs et al. 1999).
TPZ has been intensively studied in clinical trials in combination with radiation and chemotherapy in head and neck (Rischin et al. 2005, 2010b), non-small-cell lung (Sandler et al. 2000; Shepherd et al. 2000; von Pawel et al. 2000; Williamson et al. 2005) and cervical carcinomas (Aghajanian et al. 1997; Covens et al. 2006; Craighead et al. 2000; DiSilvestro et al. 2012; Maluf et al. 2006; Rischin et al. 2010a) and has been extensively reviewed (Ghatage and Sabagh 2012; McKeown et al. 2007; Reddy and Williamson 2009). TPZ was well tolerated in early phase trials at doses resulting in plasma drug concentrations in the therapeutic range (Johnson et al. 1997; Senan et al. 1997). Early trials produced signs of activity with the initial phase III trial of TPZ/cisplatin in advanced non-small-cell lung cancer, demonstrating increased overall survival relative to cisplatin and radiation alone (von Pawel et al. 2000). This indication of activity was not confirmed in larger, randomized phase III trials in head and neck (Rischin et al. 2010b) and cervical carcinomas (DiSilvestro et al. 2012), and further development of TPZ has been halted.
Several issues were identified as affecting the efficacy of TPZ as a HAP. TPZ demonstrated significant toxicities that limited the therapeutic ratio (Ghatage and Sabagh 2012; McKeown et al. 2007; Reddy and Williamson 2009). TPZ also has low solubility, which required long infusion times (Graham et al. 1997; Senan et al. 1997). In addition, preclinical studies demonstrated that TPZ is substantially less selective for hypoxic cells in three-dimensional (3D) culture (Durand and Olive 1992) or xenografts (Durand and Olive 1997) than in monolayer cell culture, a consequence of limited EVT (Hicks et al. 1998).
6.3.5 Discovery of a Second-Generation Benzotriazine Dioxide (SN30000)
With these issues in mind we embarked on the discovery of a second-generation benzotriazine dioxide (BTO) as a HAP. Our aim was to identify TPZ analogs with superior activity against hypoxic cells in tumors by improving the solubility-potency product, hypoxia selectivity, and EVT using the end point of improved therapeutic activity in preclinical xenograft models at equivalent toxicity. It also was necessary to identify chemically novel compounds to secure an intellectual property position to support development.
A limited number of TPZ analogs had been prepared and evaluated (Kelson et al. 1998; Minchinton et al. 1992; Zeman et al. 1989), and little information on structure-activity relationships (SARs) existed. We prepared an initial toolset of 42 compounds with a range of substituents to explore SARs and we confirmed the positive relationship between the one-electron reduction potential, E(1), and anoxic potency in both clonogenic and growth inhibition (IC50) assays (Hay et al. 2003).
EVT was investigated using multicellular layers (MCLs), a model of the tumor extravascular compartment in which cells are grown on porous support membranes in culture inserts submerged in culture medium (Cowan et al. 1996; Minchinton et al. 1997) and form diffusion-limited structures with central hypoxia (Hicks et al. 1998). Anoxia reduced TPZ transport in MCLs (Hicks et al. 1998, 2003; Kyle and Minchinton 1999), and reaction diffusion modeling using measured TPZ diffusion coefficients and rate constants for anoxic metabolism predicted steep gradients of TPZ in hypoxic tumor tissue, resulting in reduced cell killing. A spatially resolved pharmacokinetic/pharmacodynamic model for HT29 MCLs incorporating cytotoxic potency measured in anoxic cell cultures predicted increased resistance to TPZ in anoxic MCLs compared to stirred suspensions (Hicks et al. 2003). This confirmed that multicellular resistance to TPZ in anoxic 3D culture was primarily a result of limited transport and was responsible for the reduced efficacy of TPZ in 3D models (Durand and Olive 1992, 1997). This model was extended to tumors by incorporating the measured oxygen dependence (K-curve) of TPZ metabolism (Hicks et al. 2004) and measured TPZ plasma pharmacokinetics to simulate TPZ transport in a mapped microvascular network (Hicks et al. 2006). The model predicted that cell killing by TPZ in the hypoxic region is reduced relative to that achievable with no EVT limitation. In addition, the model successfully predicted activity of TPZ and 15 analogs from the SAR toolset in HT29 xenografts using measured plasma pharmacokinetics, transport parameters, and anoxic cytotoxicity (Hicks et al. 2006).
We also used the molecular toolset to investigate the SAR for transport, demonstrating that diffusion coefficients in HT29 MCLs increased with increasing logP7.4 and decreased with molecular weight, number of hydrogen bond donors, and acceptors (Pruijn et al. 2005, 2008).
After developing the tools to efficiently evaluate novel BTO analogs, we used the screening method guided by the pharmacokinetic/pharmacodynamic model (Fig. 6.7a) to specifically consider EVT at an early stage in the drug design process and to predict in vivo hypoxia selectivity resulting from changes in EVT. Increased bioreduction produces competing effects of increasing potency and decreasing EVT; thus designing improved analogs requires optimizing potency and EVT rather than simply maximizing any individual parameter. A range of structural variations were explored in an effort to optimize these parameters (Fig. 6.7b) (Hay et al. 2007a, b, 2008). The confidence gained using the spatially resolved pharmacokinetic/pharmacodynamic validation allowed us to screen a large number of analogs in vitro and base our SAR on predicted in vivo hypoxic cell killing rather than conduct extensive in vivo testing. Diffusion coefficients and rates of reductive metabolism in the analog series varied by more than 100-fold (Hicks et al. 2010), and a high correlation between predicted and observed activity was found in initial HT29 xenograft screening. The addition of a third saturated ring to the benzotriazine core provided reduced hypoxic metabolism and increased lipophilicity, which increased EVT and created chemical novelty to substantiate an intellectual property position. The addition of a basic amine side chain increased aqueous solubility but reduced lipophilicity and affected hypoxic metabolism, reducing EVT. While the optimization of two SARs for EVT and metabolism provided analogs with superior in vivo hypoxic selectivity, their in vivo activity was influenced by a third SAR for host toxicity. This is exemplified by the variation in maximum tolerated doses, and consequently AUC, as a function of lipophilicity and amine pKa (Fig. 6.8). Whereas SN29143 was predicted to have substantially improved activity compared to TPZ, poor plasma AUC precluded in vivo activity. Attempts to improve the pharmacokinetics by modulating lipophilicity and amine pKa led to a high AUC and improved EVT but very low hypoxic potency, which compromises the activity of SN29434. Increasing both lipophilicity and pKa increased host toxicity and lowered AUC (SN29467). Replacing the strongly electron-donating 3-amino substituent with a weaker 3-alkyl substituent led to increased EVT from increased lipophilicity and increased hypoxic potency from higher rates of metabolism (SN30000). Substituents resulting in higher lipophilicity (SN30124) and higher pKa (SN30080, SN30081) resulted in a similar trend of increasing toxicity and poorer plasma AUC, as described above. SN30000 was predicted to be substantially more active than TPZ and SN29434 as a result of low toxicity and good plasma AUC, and this was demonstrated in the HT29 xenograft model.

(a) A pharmacokinetic/pharmacodynamic (PKPD)-guided screening algorithm that incorporates drug penetration. After initial screening for physicochemical properties and hypoxia selectivity, parameters governing drug penetration (diffusion coefficient and rate of bioreductive metabolism) were measured in vitro or calculated and used in a spatially resolved PKPD model to calculate the drug exposure (AUC) required for 1 log of cell killing in addition to radiation alone. Compounds that demonstrated in vivo hypoxia selectivity at achievable AUC (Prediction A) were advanced to in vivo screening (MTD, plasma PK). The model was then run with measured plasma pharmacokinetics as input, and compounds predicted to add >0.3 log cell killing in addition to radiation alone (Prediction B) were advanced to in vivo clonogenic assay screens. (b) General structure of tricyclic benzotriazine dioxides, indicating drug design considerations

(a) Structure of an amine series of tricyclic benzotriazine dioxides, indicating the range of physicochemical parameters explored. (b) The maximum tolerated doses (MTDs), drug exposure (AUC), and hypoxic cell killing by compounds in combination with radiation (20 Gy) in HT29 xenografts by in vivo clonogenic survival assay
SN30000 emerged as the lead tricyclic BTO from this program, with broadly improved activity relative to TPZ. Aqueous solubility is improved by almost an order of magnitude (Hicks et al. 2010). SN30000 demonstrates higher potency and hypoxic selectivity than TPZ in IC50 assays across a panel of cell lines and in clonogenic assay in HT29 cells (Hicks et al. 2010). It is important to note that the measured K-value of SN30000 is not significantly different (1.14 ± 0.24 μM oxygen) from TPZ (1.21 ± 0.09 μM oxygen), indicating retention of the desirable property of activation at intermediate oxygen concentrations. Improved EVT for SN30000 was confirmed experimentally, with a threefold higher diffusion coefficient than TPZ in HT29 and SiHa MCLs (Hicks et al. 2010). SN30000 shows increased activity relative to TPZ against hypoxic cells in combination with single-dose or fractionated radiation in several tumor xenografts (HT29, SiHa, H460) by in vivo clonogenic assay and superior activity in SiHa xenografts with fractionated radiation by a delay in tumor regrowth (Hicks et al. 2010). SN30000 is currently in preclinical development with Cancer Research UK.
6.4 Targeting the Hypoxia Response Pathway
6.4.1 Introduction
The HIF family of transcription factors is well established as the key mediator of the adaptive response to hypoxia, and their role in cancer has been extensively described (Poon et al. 2009; Semenza 2003, 2010). These transcription factors are the primary oxygen sensors and use oxygen and 2-ketoglutarate as substrates for the hydroxylation of specific proline residues on HIF-1α or HIF-2α by prolyl hydroxylase domain enzymes. This allows binding by the von Hippel-Lindau (VHL) factor and recruitment of an ubiquitin ligase complex that initiates ubiquitination and proteasomal degradation. In the absence of oxygen, HIF-1α is able to bind the constitutively expressed HIF-1β and coactivation partners, bind to hypoxia response elements (HREs), and activate transcription of a variety of genes involved in angiogenesis, metabolic adaption, cell survival, and metastasis. However, HIF-1α activation may also be induced by other stimuli, including genetic changes to tumor suppressors (e.g., VHL [Kaelin 2008]) or tumor activators (e.g., Ras [Mazure et al. 1996]), growth factor stimulation (e.g., IGF-R [Ren et al. 2010]), and depletion of ascorbate (Kuiper et al. 2010). In addition, the differential expression and roles of HIF-1α and HIF-2α need to be considered (Carroll and Ashcroft 2006).
Inhibition of HIF-1α activity has been shown to slow angiogenesis and tumor growth in xenograft models (Maxwell et al. 1997), whereas inhibition of HIF-1α activity sensitizes hypoxic cells to conventional therapies (Moeller et al. 2004, 2005; Williams et al. 2005). The negative impact of HIF1α overexpression on treatment response and outcomes across a range of human tumors is also well described (Jubb et al. 2010; Semenza 2007). Multiple targets within the HIF-1α signaling pathway have been identified as a candidate drug targets (Giaccia et al. 2003; Semenza 2007). As a consequence, there has been a plethora of HIF-1 inhibitors that have been extensively reviewed (Poon et al. 2009; Semenza 2007; Xia et al. 2012). These inhibitors may be characterized as direct (interference with HIF-1α synthesis, stability, or binding to transcription partners and HREs) or indirect via the myriad of upstream or downstream participants in the hypoxia response network.
6.4.2 Direct HIF-1α Inhibitors
Direct inhibition of HIF1α translation has been demonstrated by a wide range of agents through multiple mechanisms, with the topoisomerase-I inhibitor topotecan the best-described example. Topotecan was identified as an inhibitor of HIF-1α translation (Rapisarda et al. 2002) by a topoisomerase-I-dependent mechanism, but at concentrations below those necessary for DNA damage–mediated cytotoxicity (Rapisarda et al. 2004a). As well as inhibiting HIF1α protein expression and tumor growth in a glioma xenograft model (Rapisarda et al. 2004b), combination of daily low-dose topotecan with bevacizumab provided significantly increased tumor cell killing in U251-HRE xenografts compared to either agent alone (Rapisarda et al. 2009). Topotecan recently completed a phase I clinical trial exploring its effect on HIF-1α, and reduced HIF-1α expression was observed in some patients (Kummar et al. 2011). CPT-11 (EZN-2208), a more potent, soluble prodrug (Sapra et al. 2008), provides improved suppression of HIF-1α and downstream gene targets (Sapra et al. 2011) and is in a phase II trial as both a cytotoxin and an HIF-1α inhibitor in combination with bevacizumab (www.ClinicalTrials.gov identifier NCT01251926).
Many of the compounds reported as HIF-1α inhibitors are not specific for HIF-1α or have multiple mechanism of action. Examples of this are seen with the HSP90 inhibitors geldanamycin and 17AAG (Isaacs et al. 2002; Mabjeesh et al. 2002) and with inhibitors of thioredoxin-1, such as PX12, which inhibits assembly of the transcription complex but has other effects (Welsh et al. 2003). PX12 has completed a phase I clinical trial in which stable disease was seen in patients with elevated levels of thioredoxin-1 (Ramanathan et al. 2007).
6.4.3 Indirect HIF Inhibitors
Indirect approaches take advantage of the network of upstream stimulating factors (e.g., the phosphoinositide 3-kinase/AKT/mammalian target of rapamycin pathway [Zhong et al. 2000] and the Ras/mitogen-activated pharmacokinetics pathway [Berra et al. 2000]) and downstream target genes (Semenza 2010) and may provide HIF-1α inhibition via multiple pathway interactions. The use of the multikinase inhibitor sorafenib in the treatment of advanced renal cell carcinoma (RCC) highlights this approach (Rini 2010). Advanced RCC is driven by HIF stabilization via the loss of functional VHL in a majority of cases and displays a highly angiogenic and invasive phenotype. Although sorafenib is primarily aimed at targeting downstream kinases involved directly in angiogenesis (vascular endothelial growth factor receptor-2 and -3 and platelet-derived growth factor receptor), its inhibition of upstream BRAF can also affect HIF-1 activity (Wilhelm et al. 2008).
6.4.4 Targeting Glucose Metabolism
Many of these downstream HIF targets are associated with the cellular reprogramming of metabolism from oxidative phosphorylation to aerobic glycolysis. This shift supports biosynthesis to maintain expansive tumor growth and presents a wide range of potential targets to disrupt tumor cell metabolism (Jones and Schulze 2012). Although regulated by other signaling factors such as p53 and Myc, HIF-1α plays an important role in the regulation of the glycolytic pathway (Cairns et al. 2011). Hypoxic cells are particularly vulnerable to reductions in the production of adenosine triphosphate, and so inhibition of glycolysis is potentially an effective strategy against hypoxic cells (Kurtoglu et al. 2007). This was first demonstrated for 2-deoxy-D-glucose (Song et al. 1976), which, after phosphorylation, inhibits hexokinases and their association with mitochondria. Although tolerated by patients in phase I/II trials, there is a dearth of published information on the efficacy of 2-deoxy-D-glucose in patients (Jones and Schulze 2012).
The glucose transporter GLUT-1 has been shown to be elevated in many tumor types and is a negative prognostic factor (Macheda et al. 2005). Although a variety of glucose transport inhibitors have been reported, many are not selective for GLUT-1 or have multiple mechanisms of action, making assessment of their value for targeting tumor metabolism difficult. For example, phloretin, a competitive inhibitor of GLUT-1, slows tumor growth (Kobori et al. 1997) and can sensitize tumor cells to chemotherapeutics under hypoxic conditions (Cao et al. 2007). However, it can also interact with the monocarboxylate lactate transporter MCT-4 (Dimmer et al. 2000).
A new strategy to identify agents that are selectively cytotoxic to cells overexpressing HIF-1α used a synthetic lethality approach (Kaelin 2005) based on VHL-deficient RCCs (Chan and Giaccia 2008; Sutphin et al. 2007). In this cell line, loss of functional VHL leads to constitutive expression of HIF-1α and mimics chronic hypoxia. A high-throughput screen of small molecules with paired VHL-proficient/-deficient cell lines was used to identify compounds that selectively kill VHL-deficient cells (Sutphin et al. 2007). This approach furnished a series of compounds with diverse properties (Bonnet et al. 2011; Hay et al. 2010; Turcotte et al. 2008) (see Chap. 9). We conducted an SAR study around one class (3-pyridyl benzamidophenyl sulfonamides) and identified analogs with submicromolar cytotoxic potency and selectivity for von Hippel-Lindau negative (VHL-ve) RCC cells in excess of 100-fold in vitro (e.g., SN30408, also known as STF-31) (Sutphin et al. 2011). The presence of a 3-pyridyl carboxamide was key to this activity. Substituents on this ring or the central phenyl ring reduced activity. The methyl sulfonamide linker was required for activity, whereas a wide range of substituents were tolerated on the terminal ring. This SAR was used to design affinity chromatography reagents that selectively bind to GLUT-1 (Chan et al. 2011). Molecular modeling studies using a homology model of GLUT-1 (Salas-Burgos et al. 2004) predicted that SN30408 and related molecules could bind within the central solute channel and interact with ARG126 and TRP412, both key residues for glucose transport (Brockmann et al. 2001). SN30408 seems to occupy a similar binding location to fasentin (Wood et al. 2008) and a series of recently described thiazolidinedione inhibitors that inhibited glucose transport in LNCaP prostate carcinoma cells (Wang et al. 2012a). SN30408 was shown to bind to GLUT-1 and selectively inhibit glucose uptake into VHL-ve RCC cells that overexpress GLUT-1, resulting in necrotic cell death (Chan et al. 2011). A key concern with targeting glucose transport is the effect on normal tissues, such as in the case of GLUT-1, erythrocytes, and the blood-brain barrier. Although it reduced glucose uptake into erythrocytes, SN30408 did not cause hemolysis. This was further monitored in vivo, where 18F-2-fluorodeoxyglucose positron-emission tomography (PET) demonstrated that VHL-ve tumors had high uptake of glucose and that treatment with nontoxic doses of a more soluble analog (SN31154) consistently reduced this uptake while having a minimal effect on the use of glucose in the brain. Daily treatment with a nontoxic dose of SN31154 over 14 days inhibited tumor growth in vivo (Chan et al. 2011).
6.5 Identifying the Target in Patients
As targeted therapies move into the clinic, it becomes increasingly important to identify patients with susceptible tumor cell populations who may benefit clinically (Basu 2010; Mok 2011). To fully exploit hypoxia with targeted therapy, the use of biomarkers to select suitable patients and assess response to treatment will greatly aid clinical development.
While polarographic electrodes have demonstrated a wide range of oxygen tensions in solid tumors (Nordsmark et al. 2005) and hypoxia status has been related to outcome in a range of tumor types (Vaupel and Mayer 2007), this approach is limited to accessible tumors. Tumor oxygenation may also be evaluated using nuclear magnetic resonance techniques with exogenous fluorocarbon markers for 19F nuclear magnetic resonance or blood oxygen level–dependent magnetic resonance imaging (Tatum et al. 2006) (See Chap. 16).
Recent reports of hypoxic gene signatures in various cancer sites (Buffa et al. 2010; Chi et al. 2006; Jubb et al. 2010; Murat et al. 2009; Winter et al. 2007) have related clinical outcome following standard treatments. A signature of 15 hypoxic genes was developed, validated, and used to retrospectively analyze head and neck squamous cell carcinoma (HNSCC) samples from the DAHANCA5 trial (Toustrup et al. 2011). This analysis demonstrated that only patients with hypoxic tumors defined by the hypoxic gene signature benefited from nimorazole.
Exogenous nitroimidazole hypoxia probes such as pimonizadole or EF5, with immunostaining by antibodies to the reduced adducts (Evans et al. 2000; Raleigh et al. 1998), have been used clinically. In a substudy of the ARCON trial pimonidazole was used to measure tumor hypoxia in patients with laryngeal cancer. For patients with higher pimonidazole labeling, ARCON provided benefit in terms of local control and 5 years of disease-free survival (Janssens et al. 2012).
More convenient approaches using circulating surrogate hypoxic markers in blood, such as osteopontin (Le et al. 2003), hepatocyte growth factor, and interleukin-8 (Le et al. 2012) have provided equivocal results. In the DAHANCA5 trial, patients with high levels of plasma osteopontin were shown to benefit from the addition of nimorazole, while patients with intermediate or low osteopontin showed no benefit (Overgaard et al. 2005). However, osteopontin failed to show any correlation with adverse outcome or benefit from the addition of hypoxia-targeted therapy in the TROG 02.02 phase III trial, in which patients with stage III/IV HNSCC received chemoradiotherapy and TPZ (Lim et al. 2012). In the same trial, two other hypoxic markers – hepatocyte growth factor and interleukin-8 – gave some predictive indication (Le et al. 2012).
PET using 2-nitroimidazole–based markers such as 18F-misonidazole (Lee et al. 2009), 18F-EF5 (Koch et al. 2010; Komar et al. 2008), and 18F-HX4 (Dubois et al. 2011; van Loon et al. 2010) has been explored as a noninvasive method for measuring hypoxia (Horsman et al. 2012). In a phase II trial of patients with HNSCC who were treated with chemoradiotherapy with or without TPZ, patients with hypoxic tumors identified using 18F-fluoromisonidazole PET fared significantly better when treated with TPZ compared to standard chemoradiotherapy (Rischin et al. 2006). Despite this, PET was not used for patient selection in the subsequent phase III trial, which failed to demonstrate a benefit for the addition of TPZ to chemoradiotherapy (Ang 2010; Rischin et al. 2010b).
The clinical development of HAPs would benefit from biomarkers that interrogate multiple elements of their sensitivity. We recently demonstrated that the hypoxic activation of EF5 is highly correlated with activation of SN30000 (and TPZ) across a panel of human tumor cell lines (Wang et al. 2012b). This study suggests that PET imaging with [18F]-EF5 will report on both hypoxia and the activity of the one-electron reductases for SN30000 in hypoxic regions of tumors, without having to identify all the contributors to activation.
6.6 Conclusions
Although there is clear evidence that hypoxia limits the response to therapy, extensive drug discovery efforts have delivered limited success in clinically targeting hypoxia. This failure may be attributed in part to difficulties faced by academic groups and small biotechnology companies advancing novel agents to clinical trial. It is beneficial to develop agents in combination with radiotherapy when hypoxia contributes greatly to resistance to therapy. An important issue is the failure to recognize hypoxia-directed drugs as targeted therapies, develop biomarkers to aid in the selection of patients for treatment, and monitor response. In each of three hypoxia-directed approaches under development in our laboratories, we are identifying appropriate biomarkers, while the radiosensitizer and SN30000 will be developed in conjunction with radiotherapy.
References
Adams PC, Rickert DE (1995) Metabolism of [C-14]1,3-dinitrobenzene by rat small intestinal mucosa in vitro. Drug Metab Dispos 23:982–987
Adams GE, Dische S, Fowler JF et al (1976) Hypoxic cell sensitisers in radiotherapy. Lancet 1:186–188
Adams GE, Clarke ED, Flockhart IR et al (1979a) Structure-activity relationships in the development of hypoxic cell radiosensitizers. I. Sensitization efficiency. Int J Radiat Biol Relat Stud Phys Chem Med 35:133–150
Adams GE, Clarke ED, Gray P et al (1979b) Structure-activity relationships in the development of hypoxic cell radiosensitizers. II. Cytotoxicity and therapeutic ratio. Int J Radiat Biol Relat Stud Phys Chem Med 35:151–160
Aghajanian C, Brown C, O’flaherty C et al (1997) Phase I study of tirapazamine and cisplatin in patients with recurrent cervical cancer. Gynecol Oncol 67:127–130
Albert JM, Cao C, Kim KW et al (2007) Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res 13:3033–3042
Anderson RF, Shinde SS, Hay MP et al (2003) Activation of 3-amino-1,2,4-benzotriazine 1,4-dioxide antitumor agents to oxidizing species following their one-electron reduction. J Am Chem Soc 125:748–756
Ang KK (2010) More lessons learned from the suffocation of hypoxia. J Clin Oncol 28:2941–2943
Ask K, Dijols S, Giroud C et al (2003) Reduction of nilutamide by NO synthases: implications for the adverse effects of this nitroaromatic antiandrogen drug. Chem Res Toxicol 16:1547–1554
Asquith JC, Foster JL, Willson RL et al (1974) Metronidazole (“Flagyl”). A radiosensitizer of hypoxic cells. Br J Radiol 47:474–481
Baker MA, Zeman EM, Hirst VK et al (1988) Metabolism of SR 4233 by Chinese hamster ovary cells: basis of selective hypoxic cytotoxicity. Cancer Res 48:5947–5952
Basu S (2010) Personalized versus evidence-based medicine with PET-based imaging. Nat Rev Clin Oncol 7:665–668
Begg AC, Stewart FA, Vens C (2011) Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer 11:239–253
Benito J, Shi Y, Szymanska B et al (2011) Pronounced hypoxia in models of murine and human leukemia: high efficacy of hypoxia-activated prodrug PR-104. PLoS One 6:e23108
Bennett MH, Feldmeier J, Smee R et al (2012) Hyperbaric oxygenation for tumor sensitisation to radiotherapy. Cochrane Database Syst Rev 4, CD005007. doi:10.1002/14651858.CD005007.pub3
Berra E, Pages G, Pouyssegur J (2000) MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev 19:139–145
Bindra RS, Crosby ME, Glazer PM (2007) Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev 26:249–260
Blunt T, Finnie NJ, Taccioli GE et al (1995) Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 80:813–823
Bonnet M, Flanagan JU, Chan DA et al (2011) SAR studies of 4-pyridyl heterocyclic anilines that selectively induce autophagic cell death in von Hippel-Lindau-deficient renal cell carcinoma cells. Bioorg Med Chem 19:3347–3356
Bonnet M, Hay MP, Hicks KO et al (2012) Nitroimidazole compounds and their use in cancer therapy. New Zealand Patent
Borad MJ, Reddy R, Bahary N et al (2012) TH-302 plus gemcitabine vs. gemcitabine in patients with untreated advanced pancreatic adenocarcinoma. In: 37th European Society for Medical Oncology (ESMO) 2012 Congress, Vienna, 28 Sep–2 Oct 2012
Borch RF, Liu J, Schmidt JP et al (2000) Synthesis and evaluation of nitroheterocyclic phosphoramidates as hypoxia-selective alkylating agents. J Med Chem 43:2258–2265
Borch RF, Liu J, Joswig C et al (2001) Antitumor activity and toxicity of novel nitroheterocyclic phosphoramidates. J Med Chem 44:74–77
Breider MA, Ulloa HM, Pegg DG et al (1998) Nitro-Imidazole radiosensitizer-induced toxicity in cynomolgus monkeys. Toxicol Pathol 26:651–656
Brockmann K, Wang D, Korenke CG et al (2001) Autosomal dominant glut-1 deficiency syndrome and familial epilepsy. Ann Neurol 50:476–485
Brown JM (1979) Evidence for acutely hypoxic cells in mouse tumors, and a possible mechanism of reoxygenation. Br J Radiol 52:650–656
Brown JM (1993) SR 4233 (tirapazamine): a new anticancer drug exploiting hypoxia in solid tumors. Br J Cancer 67:1163–1170
Brown M (2010) Henry S. Kaplan Distinguished Scientist Award Lecture 2007. The remarkable yin and yang of tumor hypoxia. Int J Radiat Biol 86:907–917
Brown JM, Lemmon MJ (1990) Potentiation by the hypoxic cytotoxin SR 4233 of cell killing produced by fractionated irradiation of mouse tumors. Cancer Res 50:7745–7749
Brown JM, Lemmon MJ (1991) Tumor hypoxia can be exploited to preferentially sensitize tumors to fractionated irradiation. Int J Radiat Oncol Biol Phys 20:457–461
Brown JM, Wilson WR (2004) Exploiting tumor hypoxia in cancer treatment. Nat Rev Cancer 4:437–447
Brown JM, Goffinet DR, Cleaver JE et al (1971) Preferential radiosensitization of mouse sarcoma relative to normal skin by chronic intra-arterial infusion of halogenated pyrimidine analogs. J Natl Cancer Inst 47:75–89
Brown JM, Yu NY, Brown DM et al (1981) SR-2508: a 2-nitroimidazole amide which should be superior to misonidazole as a radiosensitizer for clinical use. Int J Radiat Oncol Biol Phys 7:695–703
Brown JM, Diehn M, Loo BW (2010) Stereotactic ablative radiotherapy should be combined with a hypoxic cell radiosensitizer. Int J Radiat Oncol Biol Phys 78:323–327
Buffa FM, Harris AL, West CM et al (2010) Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br J Cancer 102:428–435
Cairns RA, Harris IS, Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11:85–95
Calabrese CR, Almassy R, Barton S et al (2004) Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst 96:56–67
Camphausen K, Tofilon PJ (2007) Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol 25:4051–4056
Cao X, Fang L, Gibbs S et al (2007) Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother Pharmacol 59:495–505
Carlson DJ, Keall PJ, Loo BW Jr et al (2011) Hypofractionation results in reduced tumor cell kill compared to conventional fractionation for tumors with regions of hypoxia. Int J Radiat Oncol Biol Phys 79:1188–1195
Carroll VA, Ashcroft M (2006) Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res 66:6264–6270
Cazares-Korner C, Pires IM, Swallow ID et al (2013) CH-01 is a hypoxia-activated prodrug that sensitizes cells to hypoxia/reoxygenation through inhibition of Chk1 and Aurora A ACS. Med Chem Letts 8:1451−1459
Cenas N, Prast S, Nivinskas H et al (2006) Interactions of nitroaromatic compounds with the mammalian selenoprotein thioredoxin reductase and the relation to induction of apoptosis in human cancer cells. J Biol Chem 281:5593–5603
Chalmers AJ, Lakshman M, Chan N et al (2010) Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin Radiat Oncol 20:274–281
Chan DA, Giaccia AJ (2008) Targeting cancer cells by synthetic lethality: autophagy and VHL in cancer therapeutics. Cell Cycle 7:2987–2990
Chan DA, Sutphin PD, Nguyen P et al (2011) Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med 3:94ra70
Chandor A, Dijols S, Ramassamy B et al (2008) Metabolic activation of the antitumor drug 5-(Aziridin-1-yl)-2,4-dinitrobenzamide (CB1954) by NO synthases. Chem Res Toxicol 21:836–843
Chang Q, Jurisica I, Do T et al (2011) Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res 71:3110–3120
Charrier JD, Durrant SJ, Golec JM et al (2011) Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J Med Chem 54:2320–2330
Chawla SP, Ganjoo KN, Adkins D et al (2011) A phase 2 study of TH-302 in combination with doxorubicin in advanced soft tissue sarcoma. In: Connective Tissue Oncology Society (CTOS) annual meeting, Chicago, 26–29 Oct 2011
Chen Y, Hu L (2009) Design of anticancer prodrugs for reductive activation. Med Res Rev 29:29–64
Chi JT, Wang Z, Nuyten DS et al (2006) Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med 3:e47
Chinje EC, Cowen RL, Feng J et al (2003) Non-nuclear localized human NOSII enhances the bioactivation and toxicity of tirapazamine (SR4233) in vitro. Mol Pharmacol 63:1248–1255
Chowdhury G, Junnotula V, Daniels JS et al (2007) DNA strand damage product analysis provides evidence that the tumor cell-specific cytotoxin tirapazamine produces hydroxyl radical and acts as a surrogate for O(2). J Am Chem Soc 129:12870–12877
Cole S, Stratford IJ, Fielden EM et al (1992) Dual function nitroimidazoles less toxic than RSU 1069: selection of candidate drugs for clinical trial (RB 6145 and/or PD 130908. Int J Radiat Oncol Biol Phys 22:545–548
Coleman CN, Wasserman TH, Urtasun RC et al (1990) Final report of the phase I trial of the hypoxic cell radiosensitizer SR 2508 (etanidazole) radiation therapy oncology group 83–03. Int J Radiat Oncol Biol Phys 18:389–393
Covens A, Blessing J, Bender D et al (2006) A phase II evaluation of tirapazamine plus cisplatin in the treatment of recurrent platinum-sensitive ovarian or primary peritoneal cancer: a Gynecologic Oncology Group study. Gynecol Oncol 100:586–590
Cowan DS, Hicks KO, Wilson WR (1996) Multicellular membranes as an in vitro model for extravascular diffusion in tumors. Br J Cancer Suppl 27:S28–S31
Craighead PS, Pearcey R, Stuart G (2000) A phase I/II evaluation of tirapazamine administered intravenously concurrent with cisplatin and radiotherapy in women with locally advanced cervical cancer. Int J Radiat Oncol Biol Phys 48:791–795
Daniels JS, Gates KS (1996) DNA cleavage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (SR4233): Evidence for involvement of hydroxyl radical. J Am Chem Soc 118:3380–3385
Denny WA (2010) Hypoxia-activated prodrugs in cancer therapy: progress to the clinic. Future Oncol 6:419–428
Denny WA, Wilson WR (1986) Considerations for the design of nitrophenyl mustards as agents with selective toxicity for hypoxic tumor cells. J Med Chem 29:879–887
Denny WA, Wilson WR (1993) Bioreducible mustards: a paradigm for hypoxia-selective prodrugs of diffusible cytotoxins (HPDCs). Cancer Metastasis Rev 12:135–151
Denny WA, Wilson WR (2000) Tirapazamine: a bioreductive anticancer drug that exploits tumor hypoxia. Expert Opin Investig Drugs 9:2889–2901
Denny WA, Wilson WR, Hay MP (1996) Recent developments in the design of bioreductive drugs. Br J Cancer 74(Suppl XXVII):S32–S38
Dewhirst MW, Cao Y, Moeller B (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8:425–437
Dimmer KS, Friedrich B, Lang F et al (2000) The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J 350(Pt 1):219–227
Dische S (1985) Chemical sensitisers for hypoxic cells: a decade of experience in clinical radiotherapy. Radiother Oncol 3:97–115
Dische S (1991) A review of hypoxic cell radiosensitization. Int J Radiat Oncol Biol Phys 20:147–152
DiSilvestro P, Ali S, Peter C et al (2012) A Gynecologic Oncology Group phase III randomized trial of weekly cisplatin and radiation versus cisplatin and tirapazamine and radiation in stage IB2, IIA, IIIB and IVA cervical carcinoma limited to the pelvis. Gynecol Oncol 125(Suppl 1):S3
Dorie MJ, Brown JM (1993) Tumor-specific, schedule-dependent interaction between tirapazamine (SR 4233) and cisplatin. Cancer Res 53:4633–4636
Dorie MJ, Brown JM (1994) Potentiation of the anticancer effect of cisplatin by the hypoxic cytotoxin tirapazamine. In: Vaupel PW, Kelleher DK, Gunderoth M (eds) Tumor oxygenation. Fischer-Verlag, Stuttgart/New York, pp 125–135
Duan JX, Jiao H, Kaizerman J et al (2008) Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs. J Med Chem 51:2412–2420
Dubois LJ, Lieuwes NG, Janssen MH et al (2011) Preclinical evaluation and validation of [18F]HX4, a promising hypoxia marker for PET imaging. Proc Natl Acad Sci U S A 108:14620–14625
Durand RE, Olive PL (1992) Evaluation of bioreductive drugs in multicell spheroids. Int J Radiat Oncol Biol Phys 22:689–692
Durand RE, Olive PL (1997) Physiologic and cytotoxic effects of tirapazamine in tumor-bearing mice. Radiat Oncol Investig 5:213–219
Eschwege F, Sancho-Garnier H, Chassagne D et al (1997) Results of a European randomized trial of etanidazole combined with radiotherapy in head and neck carcinomas. Int J Radiat Oncol Biol Phys 39:275–281
Evans JW, Yudoh K, Delahoussaye YM et al (1998) Tirapazamine is metabolized to its DNA-damaging radical by intranuclear enzymes. Cancer Res 58:2098–2101
Evans SM, Hahn S, Pook DR et al (2000) Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res 60:2018–2024
Evans JW, Chernikova SB, Kachnic LA et al (2008) Homologous recombination is the principal pathway for the repair of DNA damage induced by tirapazamine in mammalian cells. Cancer Res 68:257–265
Everett SA, Naylor MA, Patel KB et al (1999) Bioreductively-activated prodrugs for targetting hypoxic tissues: elimination of aspirin from 2-nitroimidazole derivatives. Bioorg Med Chem Lett 9:1267–1272
Fitzsimmons SA, Lewis AD, Riley RJ et al (1994) Reduction of 3-amino-1,2,4-benzotriazine-1,4-di-N-oxide (tirapazamine, WIN 59075, SR 4233) to a DNA-damaging species: a direct role for NADPH:cytochrome P450 oxidoreductase. Carcinogenesis 15:1503–1510
Foehrenbacher A, Patel K, Abbattista M et al (2013) The role of bystander effects in the anti-tumor activity of the hypoxia-activated prodrug PR-104. Front Oncol 3:263. doi:10.3389/fonc.2013.00263
Ghatage P, Sabagh H (2012) Is there a role for tirapazamine in the treatment of cervical cancer? Expert Opin Drug Metab Toxicol 8:1589–1597
Giaccia A, Siim BG, Johnson RS (2003) HIF-1 as a target for drug development. Nat Rev Drug Discov 2:803–811
Gnarra JR, Zhou S, Merrill MJ et al (1996) Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad Sci U S A 93:10589–10594
Golding SE, Rosenberg E, Valerie N et al (2009) Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther 8:2894–2902
Graeber TG, Osmanian C, Jacks T et al (1996) Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumors. Nature 379:88–91
Graham MA, Senan S, Robin H Jr et al (1997) Pharmacokinetics of the hypoxic cell cytotoxic agent tirapazamine and its major bioreductive metabolites in mice and humans: retrospective analysis of a pharmacokinetically guided dose-escalation strategy in a phase I trial. Cancer Chemother Pharmacol 40:1–10
Gray LH, Conger AD, Ebert M et al (1953) Concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol 26:638–648
Grigsby PW, Winter K, Wasserman TH et al (1999) Irradiation with or without misonidazole for patients with stages IIIB and IVA carcinoma of the cervix: final results of RTOG 80–05. Radiation Therapy Oncology Group. Int J Radiat Oncol Biol Phys 44:513–517
Gu Y, Patterson AV, Atwell GJ et al (2009) Roles of DNA repair and reductase activity in the cytotoxicity of the hypoxia-activated dinitrobenzamide mustard PR-104A. Mol Cancer Ther 8:1714–1723
Guise CP, Wang A, Thiel A et al (2007) Identification of human reductases that activate the dinitrobenzamide mustard prodrug PR-104A: a role for NADPH:cytochrome P450 oxidoreductase under hypoxia. Biochem Pharmacol 74:810–820
Guise CP, Abbattista M, Singleton RS et al (2010) The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res 70:1573–1584
Guise CP, Abbattista MR, Tipparaju SR et al (2012) Diflavin oxidoreductases activate the bioreductive prodrug PR-104A under hypoxia. Mol Pharmacol 81:31–40
Hall EJ, Roizin-Towle L (1975) Hypoxic sensitizers: radiobiological studies at the cellular level. Radiology 117:453–457
Hay MP, Wilson WR, Denny WA (1999) Nitrobenzyl carbamate prodrugs of enediynes for nitroreductase gene- directed enzyme prodrug therapy (GDEPT). Bioorg Med Chem Lett 9:3417–3422
Hay MP, Gamage SA, Kovacs MS et al (2003) Structure-activity relationships of 1,2,4-benzotriazine 1,4-dioxides as hypoxia-selective analogues of tirapazamine. J Med Chem 46:169–182
Hay MP, Hicks KO, Pruijn FB et al (2007a) Pharmacokinetic/pharmacodynamic model-guided identification of hypoxia-selective 1,2,4-benzotriazine 1,4-dioxides with antitumor activity: the role of extravascular transport. J Med Chem 50:6392–6404
Hay MP, Pchalek K, Pruijn FB et al (2007b) Hypoxia-selective 3-alkyl 1,2,4-benzotriazine 1,4-dioxides: the influence of hydrogen bond donors on extravascular transport and antitumor activity. J Med Chem 50:6654–6664
Hay MP, Hicks KO, Pchalek K et al (2008) Tricyclic [1,2,4]triazine 1,4-dioxides as hypoxia selective cytotoxins. J Med Chem 51:6853–6865
Hay MP, Turcotte S, Flanagan JU et al (2010) 4-Pyridylanilinothiazoles that selectively target von Hippel-Lindau deficient renal cell carcinoma cells by inducing autophagic cell death. J Med Chem 53:787–797
Heldin CH, Rubin K, Pietras K et al (2004) High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer 4:806–813
Helleday T, Petermann E, Lundin C et al (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8:193–204
Hicks KO, Fleming Y, Siim BG et al (1998) Extravascular diffusion of tirapazamine: effect of metabolic consumption assessed using the multicellular layer model. Int J Radiat Oncol Biol Phys 42:641–649
Hicks KO, Pruijn FB, Sturman JR et al (2003) Multicellular resistance to tirapazamine is due to restricted extravascular transport: a pharmacokinetic/pharmacodynamic study in HT29 multicellular layer cultures. Cancer Res 63:5970–5977
Hicks KO, Siim BG, Pruijn FB et al (2004) Oxygen dependence of the metabolic activation and cytotoxicity of tirapazamine: implications for extravascular transport and activity in tumors. Radiat Res 161:656–666
Hicks KO, Pruijn FB, Secomb TW et al (2006) Use of three-dimensional tissue cultures to model extravascular transport and predict in vivo activity of hypoxia-targeted anticancer drugs. J Natl Cancer Inst 98:1118–1128
Hicks KO, Myint H, Patterson AV et al (2007) Oxygen dependence and extravascular transport of hypoxia-activated prodrugs: comparison of the dinitrobenzamide mustard PR-104A and tirapazamine. Int J Radiat Oncol Biol Phys 69:560–571
Hicks KO, Siim BG, Jaiswal JK et al (2010) Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clin Cancer Res 16:4946–4957
Hickson I, Zhao Y, Richardson CJ et al (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64:9152–9159
Hill RP (1986) Sensitizers and radiation dose fractionation: results and interpretations. Int J Radiat Oncol Biol Phys 12:1049–1054
Hill RP, Gulyas S, Whitmore GF (1986) Studies of the in vivo and in vitro cytotoxicity of the drug RSU-1069. Br J Cancer 53:743–751
Hill RP, Marie-Egyptienne DT, Hedley DW (2009) Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol 19:106–111
Hoogsteen IJ, Lok J, Marres HA et al (2009) Hypoxia in larynx carcinomas assessed by pimonidazole binding and the value of CA-IX and vascularity as surrogate markers of hypoxia. Eur J Cancer 45:2906–2914
Horsman MR, Mortensen LS, Petersen JB et al (2012) Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol 9:674–687
Houghton PJ, Lock R, Carol H et al (2011) Initial testing of the hypoxia activated prodrug PR-104 by the pediatric preclinical testing program. Pediatr Blood Cancer 57:443–453
Huang LE, Bindra RS, Glazer PM et al (2007) Hypoxia-induced genetic instability – a calculated mechanism underlying tumor progression. J Mol Med 85:139–148
Hunter FW, Wang J, Patel R et al (2012) Homologous recombination repair-dependent cytotoxicity of the benzotriazine di-N-oxide CEN-209: comparison with other hypoxia-activated prodrugs. Biochem Pharmacol 83:574–585
Isaacs JS, Jung YJ, Mimnaugh EG et al (2002) Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem 277:29936–29944
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307:58–62
Jameson MB, Rischin D, Pegram M et al (2010) A phase I trial of PR-104, a nitrogen mustard prodrug activated by both hypoxia and aldo-keto reductase 1C3, in patients with solid tumors. Cancer Chemother Pharmacol 65:791–801
Janssens GO, Rademakers SE, Terhaard CH et al (2012) Accelerated radiotherapy with carbogen and nicotinamide for laryngeal cancer: results of a phase III randomized trial. J Clin Oncol 30:1777–1783
Jenkins TC, Naylor MA, O’Neill P et al (1990) Synthesis and evaluation of alpha-[[(2-haloethyl)amino]methyl]-2- nitro- 1H-imidazole-1-ethanols as prodrugs of alpha-[(1-aziridinyl)methyl]-2- nitro-1H-imidazole-1-ethanol (RSU-1069) and its analogues which are radiosensitizers and bioreductively activated cytotoxins. J Med Chem 33:2603–2610
Johnson CA, Kilpatrick D, von Roemeling R et al (1997) Phase I trial of tirapazamine in combination with cisplatin in a single dose every 3 weeks in patients with solid tumors. J Clin Oncol 15:773–780
Jones NP, Schulze A (2012) Targeting cancer metabolism–aiming at a tumor’s sweet-spot. Drug Discov Today 17:232–241
Jubb AM, Buffa FM, Harris AL (2010) Assessment of tumor hypoxia for prediction of response to therapy and cancer prognosis. J Cell Mol Med 14:18–29
Kaanders JH, Bussink J, van der Kogel AJ (2002) ARCON: a novel biology-based approach in radiotherapy. Lancet Oncol 3:728–737
Kaelin WG Jr (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5:689–698
Kaelin WG Jr (2008) The von Hippel-Lindau tumor suppressor protein: O2 sensing and cancer. Nat Rev Cancer 8:865–873
Kallman RF (1972) The phenomenon of reoxygenation and its implications for fractionated radiotherapy. Radiology 105:135–142
Kallman RF, Dorie MJ (1986) Tumor oxygenation and reoxygenation during radiation therapy: their importance in predicting tumor response. Int J Radiat Oncol Biol Phys 12:681–685
Karasawa K, Sunamura M, Okamoto A et al (2008) Efficacy of novel hypoxic cell sensitiser doranidazole in the treatment of locally advanced pancreatic cancer: long-term results of a placebo-controlled randomised study. Radiother Oncol 87:326–330
Kashishian A, Douangpanya H, Clark D et al (2003) DNA-dependent protein kinase inhibitors as drug candidates for the treatment of cancer. Mol Cancer Ther 2:1257–1264
Kelson AB, McNamara JP, Pandey A et al (1998) 1,2,4-Benzotriazine 1,4-dioxides. An important class of hypoxic cytotoxins with antitumor activity. Anticancer Drug Des 13:575–592
Kioi M, Vogel H, Schultz G et al (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120:694–705
Kobori M, Shinmoto H, Tsushida T et al (1997) Phloretin-induced apoptosis in B16 melanoma 4A5 cells by inhibition of glucose transmembrane transport. Cancer Lett 119:207–212
Koch CJ (1993) Unusual oxygen concentration dependence of toxicity of SR-4233, a hypoxic cell toxin. Cancer Res 53:3992–3997
Koch CJ, Scheuermann JS, Divgi C et al (2010) Biodistribution and dosimetry of (18)F-EF5 in cancer patients with preliminary comparison of (18)F-EF5 uptake versus EF5 binding in human glioblastoma. Eur J Nucl Med Mol Imaging 37:2048–2059
Komar G, Seppanen M, Eskola O et al (2008) 18F-EF5: a new PET tracer for imaging hypoxia in head and neck cancer. J Nucl Med 49:1944–1951
Kovacs MS, Hocking DJ, Evans JW et al (1999) Cisplatin anti-tumor potentiation by tirapazamine results from a hypoxia-dependent cellular sensitization to cisplatin. Br J Cancer 80:1245–1251
Kuiper C, Molenaar IGM, Dachs GU et al (2010) Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res 70:5749–5758
Kummar S, Raffeld M, Juwara L et al (2011) Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor-1alpha in advanced solid tumors. Clin Cancer Res 17:5123–5131
Kurtoglu M, Maher JC, Lampidis TJ (2007) Differential toxic mechanisms of 2-deoxy-D-glucose versus 2-fluorodeoxy-D-glucose in hypoxic and normoxic tumor cells. Antioxid Redox Signal 9:1383–1390
Kyle AH, Minchinton AI (1999) Measurement of delivery and metabolism of tirapazamine to tumor tissue using the multilayered cell culture model. Cancer Chemother Pharmacol 43:213–220
Laderoute K, Wardman P, Rauth AM (1988) Molecular mechanisms for the hypoxia-dependent activation of 3-amino- 1,2,4-benzotriazine-1,4-dioxide (SR 4233). Biochem Pharmacol 37:1487–1495
Le QT, Sutphin PD, Raychaudhuri S et al (2003) Identification of osteopontin as a prognostic plasma marker for head and neck squamous cell carcinomas. Clin Cancer Res 9:59–67
Le QT, Fisher R, Oliner KS et al (2012) Prognostic and predictive significance of plasma HGF and IL-8 in a phase III trial of chemoradiation with or without tirapazamine in locoregionally advanced head and neck cancer. Clin Cancer Res 18:1798–1807
Lee AE, Wilson WR (2000) Hypoxia-dependent retinal toxicity of bioreductive anticancer prodrugs in mice. Toxicol Appl Pharmacol 163:50–59
Lee DJ, Cosmatos D, Marcial VA et al (1995) Results of an RTOG phase III trial (RTOG 85-27) comparing radiotherapy plus etanidazole with radiotherapy alone for locally advanced head and neck carcinomas.[comment]. Int J Radiat Oncol Biol Phys 32:567–576
Lee N, Nehmeh S, Schoder H et al (2009) Prospective trial incorporating pre-/mid-treatment [(18)F]-misonidazole positron emission tomography for head-and-neck cancer patients undergoing concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys 75:101–108
Lim AM, Rischin D, Fisher R et al (2012) Prognostic significance of plasma osteopontin in patients with locoregionally advanced head and neck squamous cell carcinoma treated on TROG 02.02 phase III trial. Clin Cancer Res 18:301–307
Lin AJ, Cosby LA, Shanky CW et al (1972) Potential bioreductive alkylating agents. I. Benzoquinone derivatives. J Med Chem 15:1247–1252
Liu Q, Sun JD, Wang J et al (2012) TH-302, a hypoxia-activated prodrug with broad in vivo preclinical combination therapy efficacy: optimization of dosing regimens and schedules. Cancer Chemother Pharmacol 69:1487–1498
Lo SS, Fakiris AJ, Chang EL et al (2010) Stereotactic body radiation therapy: a novel treatment modality. Nat Rev Clin Oncol 7:44–54
Lohse I, Rasowski J, Cao PJ et al (2012) Targeting tumor hypoxia in patient-derived pancreatic xenografts using TH-302. In: AACR Pancreatic Cancer meeting, Lake Tahoe, 18–20 June 2012
Mabjeesh NJ, Post DE, Willard MT et al (2002) Geldanamycin induces degradation of hypoxia-inducible factor 1alpha protein via the proteosome pathway in prostate cancer cells. Cancer Res 62:2478–2482
Macheda ML, Rogers S, Best JD (2005) Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 202:654–662
Maluf FC, Leiser AL, Aghajanian C et al (2006) Phase II study of tirapazamine plus cisplatin in patients with advanced or recurrent cervical cancer. Int J Gynecol Cancer 16:1165–1171
Mason RP, Holtzman JL (1975) The role of catalytic superoxide formation in the O2 inhibition of nitroreductase. Biochem Biophys Res Commun 67:1267–1274
Maxwell PH, Dachs GU, Gleadle JM et al (1997) Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A 94:8104–8109
Mazure NM, Chen EY, Yeh P et al (1996) Oncogenic transformation and hypoxia synergistically act to modulate vascular endothelial growth factor expression. Cancer Res 56:3436–3440
McKeage MJ, Gu Y, Wilson WR et al (2011) A phase I trial of PR-104, a pre-prodrug of the bioreductive prodrug PR-104A, given weekly to solid tumor patients. BMC Cancer 11:432
McKeown SR, Cowen RL, Williams KJ (2007) Bioreductive drugs: from concept to clinic. Clin Oncol 19:427–442
Meng F, Evans JW, Bhupathi D et al (2012) Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol Cancer Ther 11:740–751
Milbank JB, Stevenson RJ, Ware DC et al (2009) Synthesis and evaluation of stable bidentate transition metal complexes of 1-(chloromethyl)-5-hydroxy-3-(5,6,7-trimethoxyindol-2-ylcarbonyl)-2,3-dihydro-1H-pyrrolo[3,2-f]quinoline (seco-6-azaCBI-TMI) as hypoxia selective cytotoxins. J Med Chem 52:6822–6834
Minchinton AI, Tannock IF (2006) Drug penetration in solid tumors. Nat Rev Cancer 6:583–592
Minchinton AI, Lemmon MJ, Tracy M et al (1992) Second-generation 1,2,4-benzotriazine 1,4-di-N-oxide bioreductive anti- tumor agents: pharmacology and activity in vitro and in vivo. Int J Radiat Oncol Biol Phys 22:701–705
Minchinton AI, Wendt KR, Clow KA et al (1997) Multilayers of cells growing on a permeable support. An in vitro tumor model. Acta Oncol 36:13–16
Moeller BJ, Cao Y, Li CY et al (2004) Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 5:429–441
Moeller BJ, Dreher MR, Rabbani ZN et al (2005) Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell 5:99–110
Mohindra JK, Rauth AM (1976) Increased cell killing by metronidazole and nitrofurazone of hypoxic compared to aerobic mammalian cells. Cancer Res 36:930–936
Mok TS (2011) Personalized medicine in lung cancer: what we need to know. Nat Rev Clin Oncol 8:661–668
Murat A, Migliavacca E, Hussain SF et al (2009) Modulation of angiogenic and inflammatory response in glioblastoma by hypoxia. PLoS One 4:e5947
Murata R, Tsujitani M, Horsman MR (2008) Enhanced local tumor control after single or fractionated radiation treatment using the hypoxic cell radiosensitizer doranidazole. Radiother Oncol 87:331–338
Naylor MA, Stephens MA, Cole S et al (1990) Synthesis and evaluation of novel electrophilic nitrofuran carboxamides and carboxylates as radiosensitizers and bioreductively activated cytotoxins. J Med Chem 33:2508–2513
Naylor MA, Threadgill MD, Showalter HD et al (1993) Synthesis of the enantiomers of the bioreductively-activated cytotoxin RSU-1069 and its prodrug RB6145 and lack of stereoselectivity in their cytotoxicity and radiosensitization in vitro. Drug Des Discov 10:249–255
Nishimura Y, Nakagawa K, Takeda K et al (2007) Phase I/II trial of sequential chemoradiotherapy using a novel hypoxic cell radiosensitizer, doranidazole (PR-350), in patients with locally advanced non-small-cell lung cancer (WJTOG-0002). Int J Radiat Oncol Biol Phys 69:786–792
Nordsmark M, Bentzen SM, Rudat V et al (2005) Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol 77:18–24
Olive PL, Vikse CM, Banath JP (1996) Use of the comet assay to identify cells sensitive to tirapazamine in multicell spheroids and tumors in mice. Cancer Res 56:4460–4463
Olive PL, Banath JP, Sinnott LT (2004) Phosphorylated histone H2AX in spheroids, tumors, and tissues of mice exposed to etoposide and 3-amino-1,2,4-benzotriazine-1,3-dioxide. Cancer Res 64:5363–5369
Overgaard J (1994) Clinical evaluation of nitroimidazoles as modifiers of hypoxia in solid tumors. Oncol Res 6:509–518
Overgaard J (2007) Hypoxic radiosensitization: adored and ignored. J Clin Oncol 25:4066–7440
Overgaard J (2011) Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck–a systematic review and meta-analysis. Radiother Oncol 100:22–32
Overgaard J (2012) IAEA-HypoX. Accelerated radiotherapy with or without Nimorazole in squamous cell carcinoma of the head and neck. http://clinicaltrialsgov/ct2/show/NCT01507467
Overgaard J, Horsman MR (1996) Modification of hypoxia-induced radioresistance in tumors by the use of oxygen and sensitizers. Semin Radiat Oncol 6:10–21
Overgaard J, Overgaard M, Nielsen OS et al (1982) A comparative investigation of nimorazole and misonidazole as hypoxic radiosensitizers in a C3H mammary carcinoma in vivo. Br J Cancer 46:904–911
Overgaard J, Overgaard M, Timothy AR (1983) Studies of the pharmacokinetic properties of nimorazole. Br J Cancer 48:27–34
Overgaard J, Hansen HS, Overgaard M et al (1998) A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5–85. Radiother Oncol 46:135–146
Overgaard J, Eriksen JG, Nordsmark M et al (2005) Plasma osteopontin, hypoxia, and response to the hypoxia sensitiser nimorazole in radiotherapy of head and neck cancer: results from the DAHANCA 5 randomised double-blind placebo-controlled trial. Lancet Oncol 6:757–764
Oya N, Shibamoto Y, Sasai K et al (1995) Optical isomers of a new 2-nitroimidazole nucleoside analog (PR-350 series): radiosensitization efficiency and toxicity. Int J Radiat Oncol Biol Phys 33:119–127
Palmer BD, Wilson WR, Cliffe S et al (1992) Hypoxia-selective antitumor agents. 5. Synthesis of water-soluble nitroaniline mustards with selective cytotoxicity for hypoxic mammalian cells. J Med Chem 35:3214–3222
Palmer BD, Wilson WR, Atwell GJ et al (1994) Hypoxia-selective antitumor agents. 9. Structure-activity relationships for hypoxia-selective cytotoxicity among analogues of 5-[N, N-bis(2- chloroethyl)amino]-2,4-dinitrobenzamide. J Med Chem 37:2175–2184
Palmer BD, Wilson WR, Anderson RF et al (1996) Hypoxia-selective antitumor agents. 14. Synthesis and hypoxic cell cytotoxicity of regioisomers of the hypoxia-selective cytotoxin 5-[N, N- bis(2-chloroethyl)amino]-2,4-dinitrobenzamide. J Med Chem 39:2518–2528
Papadopoulou MV, Ji M, Rao MK et al (2003) Reductive activation of the nitroimidazole-based hypoxia-selective cytotoxin NLCQ-1 (NSC 709257). Oncol Res 14:21–29
Parker LL, Lacy SM, Farrugia LJ et al (2004) A novel design strategy for stable metal complexes of nitrogen mustards as bioreductive prodrugs. J Med Chem 47:5683–5689
Parveen I, Naughton DP, Whish WJ et al (1999) 2-nitroimidazol-5-ylmethyl as a potential bioreductively activated prodrug system: reductively triggered release of the PARP inhibitor 5- bromoisoquinolinone. Bioorg Med Chem Lett 9:2031–2036
Patel K, Lewiston D, Gu Y et al (2007) Analysis of the hypoxia-activated dinitrobenzamide mustard phosphate prodrug PR-104 and its alcohol metabolite PR-104A in plasma and tissues by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 856:302–311
Patel K, Choy SF, Hicks KO et al (2011) A combined pharmacokinetic model for the hypoxia-targeted prodrug PR-104A in humans, dogs, rats and mice predicts species differences in clearance and toxicity. Cancer Chemother Pharmacol 67:1145–1155
Patterson LH (1993) Rationale for the use of aliphatic N-oxides of cytotoxic anthraquinones as prodrug DNA binding agents: a new class of bioreductive agent. Cancer Metastasis Rev 12:119–134
Patterson AV, Saunders MP, Chinje EC et al (1997) Overexpression of human NADPH:cytochrome c (P450) reductase confers enhanced sensitivity to both tirapazamine (SR 4233) and RSU 1069. Br J Cancer 76:1338–1347
Patterson AV, Saunders MP, Chinje EC et al (1998) Enzymology of tirapazamine metabolism: a review. Anticancer Drug Des 13:541–573
Patterson AV, Ferry DM, Edmunds SJ et al (2007) Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA crosslinking agent PR-104. Clin Cancer Res 13:3922–3932
Patterson AV, Jaiswal J, Syddall SP et al (2009) Cellular metabolism, murine pharmacokinetics and preclinical antitumor activity of SN29966, a novel hypoxia-activated irreversible pan-HER inhibitor. Mol Cancer Ther 8:Abstract B76
Peasland A, Wang LZ, Rowling E et al (2011) Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer 105:372–381
Phillips RM, Hendriks HR, Peters GJ (2013) EO9 (Apaziquone): from the clinic to the laboratory and back again. Br J Pharmacol 168:11–18
Pires IM, Olcina MM, Anbalagan S et al (2012) Targeting radiation-resistant hypoxic tumor cells through ATR inhibition. Br J Cancer 107:291–299
Poon E, Harris AL, Ashcroft M (2009) Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev Mol Med 11:e26
Pries AR, Cornelissen AJ, Sloot AA et al (2009) Structural adaptation and heterogeneity of normal and tumor microvascular networks. PLoS Comput Biol 5:e1000394
Pruijn FB, Sturman JR, Liyanage HDS et al (2005) Extravascular transport of drugs in tumor tissue: effect of lipophilicity on diffusion of tirapazamine analogs in multicellular layer cultures. J Med Chem 48:1079–1087
Pruijn FB, Patel K, Hay MP et al (2008) Prediction of tumor tissue diffusion coefficients of hypoxia-activated prodrugs from physicochemical parameters. Aust J Chem 61:687–693
Raleigh JA, Calkins-Adams DP, Rinker LH et al (1998) Hypoxia and vascular endothelial growth factor expression in human squamous cell carcinomas using pimonidazole as a hypoxia marker. Cancer Res 58:3765–3768
Ramanathan RK, Kirkpatrick DL, Belani CP et al (2007) A Phase I pharmacokinetic and pharmacodynamic study of PX-12, a novel inhibitor of thioredoxin-1, in patients with advanced solid tumors. Clin Cancer Res 13:2109–2114
Rapisarda A, Uranchimeg B, Scudiero DA et al (2002) Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res 62:4316–4324
Rapisarda A, Uranchimeg B, Sordet O et al (2004a) Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res 64:1475–1482
Rapisarda A, Zalek J, Hollingshead M et al (2004b) Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res 64:6845–6848
Rapisarda A, Hollingshead M, Uranchimeg B et al (2009) Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol Cancer Ther 8:1867–1877
Reaper PM, Griffiths MR, Long JM et al (2011) Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 7:428–430
Reddy SB, Williamson SK (2009) Tirapazamine: a novel agent targeting hypoxic tumor cells. Expert Opin Investig Drugs 18:77–87
Ren H, Accili D, Duan C (2010) Hypoxia converts the myogenic action of insulin-like growth factors into mitogenic action by differentially regulating multiple signaling pathways. Proc Natl Acad Sci U S A 107:5857–5862
Rini BI (2010) New strategies in kidney cancer: therapeutic advances through understanding the molecular basis of response and resistance. Clin Cancer Res 16:1348–1354
Rischin D, Peters L, Fisher R et al (2005) Tirapazamine, cisplatin, and radiation versus fluorouracil, cisplatin, and radiation in patients with locally advanced head and neck cancer: a randomized phase II trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02). J Clin Oncol 23:79–87
Rischin D, Hicks RJ, Fisher R et al (2006) Prognostic significance of [18F]-misonidazole positron emission tomography-detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group Study 98.02. J Clin Oncol 24:2098–2104
Rischin D, Narayan K, Oza AM et al (2010a) Phase 1 study of tirapazamine in combination with radiation and weekly cisplatin in patients with locally advanced cervical cancer. Int J Gynecol Cancer 20:827–833
Rischin D, Peters LJ, O’Sullivan B et al (2010b) Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): a phase III trial of the Trans-Tasman Radiation Oncology Group. J Clin Oncol 28:2989–2995
Rockwell S, Dobrucki IT, Kim EY et al (2009) Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med 9:442–458
Salas-Burgos A, Iserovich P, Zuniga F et al (2004) Predicting the three-dimensional structure of the human facilitative glucose transporter glut1 by a novel evolutionary homology strategy: insights on the molecular mechanism of substrate migration, and binding sites for glucose and inhibitory molecules. Biophys J 87:2990–2999
Sandler AB, Nemunaitis J, Denham C et al (2000) Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol 18:122–130
Sapra P, Zhao H, Mehlig M et al (2008) Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin Cancer Res 14:1888–1896
Sapra P, Kraft P, Pastorino F et al (2011) Potent and sustained inhibition of HIF-1alpha and downstream genes by a polyethyleneglycol-SN38 conjugate, EZN-2208, results in anti-angiogenic effects. Angiogenesis 14:245–253
Sarkaria JN, Eshleman JS (2001) ATM as a target for novel radiosensitizers. Semin Radiat Oncol 11:316–327
Saunders M, Dische S (1996) Clinical results of hypoxic cell radiosensitisation from hyperbaric oxygen to accelerated radiotherapy, carbogen and nicotinamide. Br J Cancer 27:S271–S278
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732
Semenza GL (2007) Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today 12:853–859
Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20:51–56
Senan S, Rampling R, Graham MA et al (1997) Phase I and pharmacokinetic study of tirapazamine (SR 4233) administered every three weeks. Clin Cancer Res 3:31–38
Senra JM, Telfer BA, Cherry KE et al (2011) Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol Cancer Ther 10:1949–1958
Shepherd F, Koschel G, von Pawel J et al (2000) Comparison of Tirazone (tirapazamine) and cisplatin vs. etoposide and cisplatin in advanced non-small cell lung cancer (NSCLC): final results of the international phase III CATAPULT II trial. Lung Cancer 29(Suppl 1):28, abstract No. 87
Shewach DS, Lawrence TS (2007) Antimetabolite radiosensitizers. J Clin Oncol 25:4043–4050
Shinde SS, Hay MP, Patterson AV et al (2009) Spin trapping of radicals other than the *OH radical upon reduction of the anticancer agent tirapazamine by cytochrome P450 reductase. J Am Chem Soc 131:14220–14221
Shinde SS, Maroz A, Hay MP et al (2010) Characterization of radicals formed following enzymatic reduction of 3-substituted analogues of the hypoxia-selective cytotoxin 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). J Am Chem Soc 132:2591–2599
Shinohara ET, Geng L, Tan J et al (2005) DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res 65:4987–4992
Siim BG, van Zijl PL, Brown JM (1996) Tirapazamine-induced DNA damage measured using the comet assay correlates with cytotoxicity towards hypoxic tumor cells in vitro. Br J Cancer 73:952–960
Singleton RS, Guise CP, Ferry DM et al (2009) DNA crosslinks in human tumor cells exposed to the prodrug PR-104A: relationships to hypoxia, bioreductive metabolism and cytotoxicity. Cancer Res 69:3884–3891
Song CW, Clement JJ, Levitt SH (1976) Preferential cytotoxicity of 5-thio-D-glucose against hypoxic tumor cells. J Natl Cancer Inst 57:603–605
Sun JD, Liu Q, Wang J et al (2012) Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res 18:758–770
Sutphin PD, Chan DA, Li JM et al (2007) Targeting the loss of the von Hippel-Lindau tumor suppressor gene in renal cell carcinoma cells. Cancer Res 67:5896–5905
Sutphin PD, Chan D, Turcotte S et al (2011) Heteroaryl benzamides, compositions and methods of use. Patent WO2011011514 A1
Tatsumi K, Kitamura S, Narai N (1986) Reductive metabolism of aromatic nitro compounds including carcinogens by rabbit liver preparations. Cancer Res 46:1089–1093
Tatum JL, Kelloff GJ, Gillies RJ et al (2006) Hypoxia: importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Int J Radiat Biol 82:699–757
Thomlinson RH, Gray LH (1955) The histological structure of some human lung cancers and possible implications for radiotherapy. Br J Cancer 9:539–549
Threadgill MD, Webb P, O’Neill P et al (1991) Synthesis of a series of nitrothiophenes with basic or electrophilic substituents and evaluation as radiosensitizers and as bioreductively activated cytotoxins. J Med Chem 34:2112–2120
Timothy AR, Overgaard J, Overgaard M (1984) A phase I clinical study of Nimorazole as a hypoxic radiosensitizer. Int J Radiat Oncol Biol Phys 10:1765–1768
Toustrup K, Sorensen BS, Nordsmark M et al (2011) Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res 71:5923–5931
Toustrup K, Sorensen BS, Alsner J et al (2012) Hypoxia gene expression signatures as prognostic and predictive markers in head and neck radiotherapy. Semin Radiat Oncol 22:119–127
Turcotte S, Chan DA, Sutphin PD et al (2008) A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell 14:90–102
Tuttle SW, Maity A, Oprysko PR et al (2007) Detection of reactive oxygen species via endogenous oxidative pentose phosphate cycle activity in response to oxygen concentration. J Biol Chem 282:36790–36796
Ueda O, Kitamura S, Ohashi K et al (2003) Xanthine oxidase-catalysed metabolism of 2-nitrofluorene, a carcinogenic air pollutant, in rat skin. Drug Metab Dispos 31:367–372
Urtasun RC, Band P, Chapman JD et al (1976) Radiation and high dose metronidazole in supratentorial glioblastomas. N Engl J Med 294:1364–1367
van Loon J, Janssen MHM, Ollers M et al (2010) PET imaging of hypoxia using [18F]HX4: a phase I trial. Eur J Nucl Med Mol Imaging 37:1663–1668
Vaupel P, Mayer A (2007) Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 26:225–239
Vaupel P, Hockel M, Mayer A (2007) Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal 9:1221–1235
von Pawel J, von Roemeling R, Gatzemeier U et al (2000) Tirapazamine plus cisplatin versus cisplatin in advanced non-small-cell lung cancer: a report of the international CATAPULT I study group. Cisplatin and tirapazamine in subjects with advanced previously untreated non-small-cell lung tumors. J Clin Oncol 18:1351–1359
Wang H, Wang H, Powell SN et al (2004) ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res 64:7139–7143
Wang D, Chu PC, Yang CN et al (2012a) Development of a novel class of glucose transporter inhibitors. J Med Chem 55:3827–3836
Wang J, Foehrenbacher A, Su J et al (2012b) The 2-nitroimidazole EF5 is a biomarker for oxidoreductases that activate bioreductive prodrug CEN-209 under hypoxia. Clin Cancer Res 18:1684–1695
Wardman P (2001) Electron transfer and oxidative stress as key factors in the design of drugs selectively active in hypoxia. Curr Med Chem 8:739–761
Wardman P (2007) Chemical radiosensitizers for use in radiotherapy. Clin Oncol 19:397–417
Ware DC, Palmer BD, Wilson WR et al (1993) Hypoxia-selective antitumor agents. 7. Metal complexes of aliphatic mustards as a new class of hypoxia-selective cytotoxins. Synthesis and evaluation of cobalt(III) complexes of bidentate mustards. J Med Chem 36:1839–1846
Welsh SJ, Williams RR, Birmingham A et al (2003) The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther 2:235–243
Wilhelm SM, Adnane L, Newell P et al (2008) Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 7:3129–3140
Williams KJ, Telfer BA, Xenaki D et al (2005) Enhanced response to radiotherapy in tumors deficient in the function of hypoxia-inducible factor-1. Radiother Oncol 75:89–98
Williamson SK, Crowley JJ, Lara PN Jr et al (2005) Phase III trial of paclitaxel plus carboplatin with or without tirapazamine in advanced non-small-cell lung cancer: Southwest Oncology Group Trial S0003. J Clin Oncol 23:9097–9104
Wilson WR, Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11:393–410
Wilson WR, van Zijl P, Denny WA (1992) Bis-bioreductive agents as hypoxia-selective cytotoxins: nitracrine N-oxide. Int J Radiat Oncol Biol Phys 22:693–696
Wilson WR, Hicks KO, Pullen SM et al (2007) Bystander effects of bioreductive drugs: potential for exploiting pathological tumor hypoxia with dinitrobenzamide mustards. Radiat Res 167:625–636
Winter SC, Buffa FM, Silva P et al (2007) Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res 67:3441–3449
Wood TE, Dalili S, Simpson CD et al (2008) A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol Cancer Ther 7:3546–3555
Wouters BG, Brown JM (1997) Cells at intermediate oxygen levels can be more important than the “hypoxic fraction” in determining tumor response to fractionated radiotherapy. Radiat Res 147:541–550
Xia Y, Choi HK, Lee K (2012) Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem 49:24–40
Yamamoto N, Renfrew AK, Kim BJ et al (2012) Dual targeting of hypoxic and acidic tumor environments with a cobalt(III) chaperone complex. J Med Chem 55:11013–11021
Yin J, Glaser R, Gates KS (2012) On the reaction mechanism of tirapazamine reduction chemistry: unimolecular N-OH homolysis, stepwise dehydration, or triazene ring-opening. Chem Res Toxicol 25:634–645
Zeman EM, Brown JM, Lemmon MJ et al (1986) SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int J Radiat Oncol Biol Phys 12:1239–1242
Zeman EM, Hirst VK, Lemmon MJ et al (1988) Enhancement of radiation-induced tumor cell killing by the hypoxic cell toxin SR 4233. Radiother Oncol 12:209–218
Zeman EM, Baker MA, Lemmon MJ et al (1989) Structure-activity relationships for benzotriazine di-N-oxides. Int J Radiat Oncol Biol Phys 16:977–981
Zhao Y, Thomas HD, Batey MA et al (2006) Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res 66:5354–5362
Zhong H, Chiles K, Feldser D et al (2000) Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 60:1541–1545
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this paper
Cite this paper
Hay, M.P., Hicks, K.O., Wang, J. (2014). Hypoxia-Directed Drug Strategies to Target the Tumor Microenvironment. In: Koumenis, C., Hammond, E., Giaccia, A. (eds) Tumor Microenvironment and Cellular Stress. Advances in Experimental Medicine and Biology, vol 772. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5915-6_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5915-6_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5914-9
Online ISBN: 978-1-4614-5915-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)