
Abstract
Tumour hypoxia has been pursued as a cancer drug target for over 30 years, most notably using bioreductive (hypoxia-activated) prodrugs that target antineoplastic agents to low-oxygen tumour compartments. Despite compelling evidence linking hypoxia with treatment resistance and adverse prognosis, a number of such prodrugs have recently failed to demonstrate efficacy in pivotal clinical trials; an outcome that demands reflection on the discovery and development of these compounds. In this review, we discuss a clear disconnect between the pathobiology of tumour hypoxia, the pharmacology of hypoxia-activated prodrugs and the manner in which they have been taken into clinical development. Hypoxia-activated prodrugs have been evaluated in the manner of broad-spectrum cytotoxic agents, yet a growing body of evidence suggests that their activity is likely to be dependent on the coincidence of tumour hypoxia, expression of specific prodrug-activating reductases and intrinsic sensitivity of malignant clones to the cytotoxic effector. Hypoxia itself is highly variable between and within individual tumours and is not treatment-limiting in all cancer subtypes. Defining predictive biomarkers for hypoxia-activated prodrugs and overcoming the technical challenges of assaying them in clinical settings will be essential to deploying these agents in the era of personalised cancer medicine.
Similar content being viewed by others


Main
Initial evidence for the existence of hypoxia within tumours was contemporaneous with the discovery of the structure of DNA and preceded the discovery of cellular oncogenes by two decades. In the subsequent years, the prevalence of hypoxia and its association with and contribution to poor prognosis (Brizel et al, 1996; Hockel et al, 1996) and treatment failure (Nordsmark et al, 2005) in multiple indications provided a compelling rationale for clinical-stage drug development programmes targeting or exploiting this feature of the tumour microenvironment. The majority of these studies have explored prodrugs of antineoplastic agents that are activated in cells by oxygen-inhibited enzymatic reduction (hypoxia-activated prodrugs; HAPs) (Wilson and Hay, 2011; Phillips, 2016). The clinical development of six of these compounds (tirapazamine, apaziquone, banoxantrone, porfiromycin, PR-104 and RH1) has been discontinued, whereas two remain active (evofosfamide/TH-302 and tarloxotinib bromide/TH-4000). However, it was recently reported that evofosfamide did not achieve primary overall survival endpoints in combination with chemotherapy in two large phase 3 studies in advanced unresectable or metastatic pancreatic adenocarcinoma (MAESTRO; NCT01746979) and in soft tissue sarcoma (TH-CR-406/SARC021; NCT01440088), whereas a phase 2 trial in NSCLC (NCT02093962) was discontinued for futility.
The conspicuous failure of two major registrational programs—those of tirapazamine and evofosfamide—calls for a considered reflection on the rationale for drugging hypoxia, molecule-specific liabilities that may be corrected in subsequent drug discovery efforts, and the manner in which these agents are taken into development. Despite its antiquity, HAP development has yet to fully leverage the advancements in molecular profiling that have enabled targeted treatment of defined tumour subtypes in other areas of medical oncology (Chapman et al, 2011; Mateo et al, 2015). Indeed, the task of translating these compounds into the clinic has been confounded by the complex biology and heterogeneity of the target and failure to clearly define and access—using diagnostic assays fit for commercial drug development—clinical indications in which hypoxia is present or treatment-limiting. The latter is particularly imperative in the context of the HAPs of DNA-reactive cytotoxins, the toxicity profiles of which have frequently required dose reductions in combination with standard of care (Rischin et al, 2001; McKeage et al, 2012). Of equal importance is the need to develop diagnostics for predicting the sensitivity of individual tumours to HAPs; an effort that may be informed by an increasing understanding of the molecular pharmacology of these agents (Figure 1). In this sense, the challenges for HAPs are reflective of wider trends in the development of precision medicine. In this review, we outline recent developments in the study of tumour hypoxia and the mechanism of action of HAPs and how these may inform the clinical deployment of these prodrugs in the context of personalised cancer medicine.

Molecular pharmacology of HAPs. Aromatic nitro (e.g. evofosfamide, tarloxotinib, PR-104, nitroCBI) and N-oxide (e.g. tirapazamine, SN30000) HAPs are activated in tumours by a process that is initiated by enzymatic one-electron reduction to yield a prodrug radical anion. In well-oxygenated tissue, the unpaired electron is rapidly scavenged from this reducing radical by molecular oxygen, resulting in a futile redox cycle. In the absence of oxygen, the radical anion either fragments or is reduced further to produce a cytotoxic effector species that engages a pharmacological target (e.g. by kinase inhibition or damaging DNA through alkylation, oxidation or poisoning of topoisomerase II). Collectively, this understanding of the molecular pharmacology of HAPs implies that presence of tumour hypoxia, expression of one-electron reductases in addition to cell-intrinsic modifiers of sensitivity of the cytotoxic effector may influence the efficacy of HAPs in individual malignancies.
Hypoxia and Outcome: Finding the Point of Maximal Impact
The initiating premise for HAP development was the identification of hypoxia in many types of tumours and its association with treatment resistance and poor prognosis. The strongest such evidence comes from clinical trials incorporating oxygen needle electrode-based measurements in accessible tumours of the head and neck (Nordsmark et al, 2005), cervix (Fyles et al, 1998, 2002), and prostate (Milosevic et al, 2012). Similar corroborating evidence in these and many other tumour types has been obtained using indirect assessments of hypoxia. These include exogenous nitroimidazole-based hypoxia probes used in either PET imaging (e.g. [18F]-FAZA, [18F]-MISO, [18F]-HX4) or immunohistochemistry assays (EF5, pimonidazole). Indirect assessments of hypoxia have also been conducted using mRNA or protein-based expression incorporating single or multiple genes (signatures) induced by hypoxia. Based on all such assessments, the levels of hypoxia in virtually every cancer type examined have demonstrated a remarkable diversity across patients. This variation is important, in part because it provides a tool to differentiate patients with distinct clinical prognoses. For example, high levels of hypoxia are associated with local-regional failure of patients with head and neck squamous cell carcinoma (HNSCC) in a meta-analysis of 397 patients treated with definitive radiotherapy (Nordsmark et al, 2005). Similarly, high levels of hypoxia predict for worse overall survival in cervix (Fyles et al, 1998) and prostate cancer (Milosevic et al, 2012) treated with radiotherapy. Levels of hypoxia vary from near zero to almost 100%; however, no clinical factors (e.g. size, stage) or genomic features have been identified that are predictive of these values.
Despite these clinical observations, none of the large HAP clinical registration trials have incorporated biomarkers to identify patients expected to benefit from such therapies. Thus, one explanation for the lack of success of these trials has been the inclusion of patients that have little chance to benefit simply because their tumours have low levels of hypoxia. There are some retrospective studies that support this conclusion. Rischin et al (2006) used [18F]-MISO PET to assess hypoxia in a sub-study of 45 patients within a large randomised phase 2 study comparing tirapazamine plus chemoradiation to chemoradiation alone for HNSCC. They demonstrated a significant benefit of tirapazamine in patients containing hypoxic tumours, with only one of 19 patients experiencing a local-regional failure, compared to 8 of 13 in the control arm. Data from Toustrup et al (2012b) have similarly demonstrated that biomarkers of hypoxia can identify subsets of hypoxic HNSCC that benefit from treatments targeting hypoxic cells. A retrospective study using a 15-gene hypoxia signature (Toustrup et al, 2011) to classify patients into ‘more hypoxic’ and ‘less hypoxic’ subsets, demonstrated that the benefit of nimorazole (a hypoxic radiosensitiser) in the Danish Head And Neck Cancer 5 trial (DAHANCA 5) was observed only within the hypoxic group (Toustrup et al, 2012b). Five-year local-regional control rates improved from 18 to 49% with nimorazole in the hypoxic group of 114 patients, whereas there was no benefit in the less hypoxic group of 209 patients.
Compared with head and neck cancer, it is more difficult to estimate the proportion of patients with hypoxic tumours expected to benefit from evofosfamide in the recent phase 3 trials in pancreatic adenocarcinoma and soft tissue carcinoma. Intraoperative oxygen needle electrode measurements were made in seven pancreas carcinoma patients and showed high levels of hypoxia (Koong et al, 2000). However, a more recent study in 10 patients using pimonidazole showed that like other cancers, hypoxic levels show a ride range of values from 0–26% (Dhani et al, 2015). This is particularly relevant, given that pimonidazole activation and binding occurs through a similar oxygen-dependent enzymatic reaction as evofosfamide (described below). Although completion of larger ongoing trials in pancreatic cancer using pimonidazole and/or [18F]-FAZA PET will be needed to fully understand the variation in hypoxia, it is highly likely that many patients in the MAESTRO trial will have had levels of hypoxia insufficient to benefit from evofosfamide.
In addition to the need for hypoxia, there are other important considerations relevant to the potential efficacy of HAPs in different disease contexts. It has become clear that hypoxia, even when present, is not equally important in all patients. This is particularly relevant in curative settings where HAPs are expected to be beneficial only when hypoxic cells are actually limiting therapy outcome. As described above, this has been shown definitively in HNSCC treated by radiotherapy. However, even in this case, the importance of hypoxia is not equivalent in all patients. HNSCC consist of multiple disease subtypes, including those caused by infection with human papillomavirus (HPV). HPV infection is associated with significantly improved overall survival and the aetiology of this disease, including the underlying causal genetic mutations, is distinct from HPV-negative disease (Ang et al, 2010; TCGA Network, 2015). Interestingly, the range and levels of hypoxia, as assessed by the 15-gene signature described above, appear similar in HPV-positive and HPV-negative HNSCC (Toustrup et al, 2012a, 2012b). However, a retrospective analysis of the DAHANCA 5 study showed that nimorazole benefitted patients with hypoxic HPV-negative tumours but had no benefit in HPV-positive HNSCC (Lassen et al, 2010). Trinkaus et al (2014) similarly demonstrated that the addition of tirapazamine to chemoradiation for HNSCC was beneficially only in the [18F]-MISO-avid, HPV-negative subset. These data demonstrate that hypoxic cells, although present, likely do not represent the treatment-limiting population in the HPV-positive subset of this disease.
Another distinction of the importance of hypoxia in disease subtypes was recently revealed in prostate cancer. An earlier study of 247 patients used oxygen needle electrodes to assess hypoxia and found that high levels are associated with rapid biochemical failure in patients treated with curative intent by radiotherapy (Milosevic et al, 2012). A follow-up study in this same patient cohort identified two distinct genetic subtypes and although hypoxia was present in both at similar levels, it was associated with poor prognosis only in the subtype with high genomic instability (Lalonde et al, 2014). This distinction was validated using a hypoxic gene signature in an independent cohort of patients treated by surgery alone. Again, one must conclude that like HNSCC, hypoxic cells in prostate cancer may be treatment-limiting in some patients and not in others.
This prostate study illustrates a second important context that should be considered when evaluating HAPs. In HNSCC, the association of hypoxia with local-regional failure is consistent with the direct resistance of hypoxic cells to radiation. However, in prostate and cervix cancer the influence of hypoxia on prognosis is associated with regional or distal metastasis (Milosevic et al, 2012). Hypoxia has been shown to drive properties associated with aggressive disease, including increased angiogenesis, genetic instability, metastasis, and stemness (Wouters and Koritzinsky, 2008). In these cases, there is a need to distinguish the role of hypoxia in contributing to disease progression from its role in limiting treatment response. Future personalised approaches to cancer therapy using HAPs will thus need to include not only assessment of hypoxia, but also an additional evaluation of the genetic subtypes and an understanding of the role of hypoxia in patient prognosis.
Finally, it is important to distinguish the use of HAPs in curative vs non-curative treatment settings. As discussed above, the benefit from HAPs in definitive regimens is expected to be limited to patients where hypoxic cells are in fact responsible for treatment failure. In non-curative settings, HAPs may prove to be beneficial in tumours even when this is not the case. Evofosfamide was developed as a chemotherapeutic agent that could exploit the presence of hypoxic cells in advanced cancers. The success of this approach is dependent on sufficient levels of hypoxia coupled with a bystander effect due to diffusion of the active effector to other cells in the tumour as discussed below. In this case the hypoxic cells may or may not be contributing to disease progression or prognosis. To date, evofosfamide has only been tested in this context and the potential benefit of this agent in curative combination settings remains unknown.
Identification and Profiling of Prodrug-Activating Reductases
The requirement for enzymatic reduction of HAPs to facilitate their therapeutic effect (Figure 1) implies that the expression of activating reductases may be an important source of variability between individual tumours in sensitivity to HAPs. For this reason, significant effort has focused on identifying enzymes that are competent for HAP activation and on characterising their expression in cancer. Most HAPs (including nitroaromatics, quinones and benzotriazine di-oxides) are activated via a mechanism that begins with one-electron reduction by flavin-dependent oxidoreductases to generate an oxygen-sensitive radical (Figure 1). Ninety annotated human flavoproteins catalyse two-electron transfer from NAD(P)H to flavin nucleotide cofactors, which can effect one-electron reduction of endogenous and xenobiotic substrates (Lienhart et al, 2013). HAPs with one-electron reduction potentials above ∼–400 mV can compete with the flavin cofactors for electron acceptance and are activated in this manner (Wardman, 2001).
Identifying the specific flavoenzymes that are responsible for activation of HAPs has proven difficult in light of functional redundancy in the flavoproteome, in addition to the low-substrate specificities of these proteins as xenobiotic metabolising enzymes. Studies utilising recombinant proteins or forced expression in isogenic cell lines have identified a number of flavoproteins to be competent for HAP activation in vitro. These include POR, MTRR, NDOR1, NOS isoforms, FOXRED2 and CYB5R3 (Patterson et al, 1997; Papadopoulou et al, 2003; Guise et al, 2007; Chandor et al, 2008; Guise et al, 2012; Meng et al, 2012; Wang et al, 2014; Hunter et al, 2014b). However, these gain-of-function approaches do not necessarily implicate specific enzymes as major mediators of HAP activation at endogenous levels of expression. Emerging technology for efficient and cost-effective generation of definitive gene knockout models and loss-of-function genetic screens (Shalem et al, 2014) will facilitate a more precise description of this aspect of HAP pharmacology. Indeed, we recently utilised reductase-focused and genome-scale RNAi libraries to identify POR (P450 oxidoreductase) as the single most important modifier of sensitivity to the benzotriazine di-oxide HAP SN30000 in hypoxic cell culture (Figure 2A) (Hunter et al, 2015). Knockdown or knockout of POR significantly (but incompletely) abrogated SN30000 activity in vitro, and expression of POR correlated with activation of SN30000 and tirapazamine in a panel of diverse carcinoma cells. Importantly, we found POR to be highly expressed in a subset of HNSCC (Figure 2B) with much lower intratumour heterogeneity than the hypoxia marker carbonic anhydrase 9, suggesting that measuring POR expression by diagnostic biopsy may be a viable biomarker strategy. We also identified strong POR expression in some HNSCC found to harbour regions of hypoxia (Figure 2C); the coincidence of these two features implies a tumour phenotype likely to benefit from HAPs. A critical question is the extent to which reductase expression will influence tumour response to HAPs when weighed against other putative biomarkers such as hypoxia and modifiers of effector sensitivity. This question has been difficult to address, in part because the specific binding of 2-nitroimidazole bioreductive probes such as EF5, pimonidazole, FAZA, misonidazole or HX4 is also dependent on both hypoxia and one-electron reductase activity (Wang et al, 2012). Although confounding in the context of discriminating individual sensitivity determinants, the demonstration that EF5 and benzotriazine di-oxide HAPs are activated by the same reductases (Wang et al, 2012, 2014) suggests potential for 2-nitroimidazole PET imaging radiopharmaceuticals as dual reporters of both hypoxia and one-electron reductase activity, at least for this class of HAPs.

HAP activation in tumours. (A) Identification of POR as a major determinant of SN30000 sensitivity by high-throughput shRNA screening of HT-29 and PANC-1 cells. The metric plotted is the Z-transformed log2 of hairpin enrichment factor (treated/control) following exposure to SN30000. (B) Immunohistochemical staining for POR in a tissue section from an HPV-negative hypopharyngeal squamous carcinoma. Intense, relatively homogenous POR staining is observed in the malignant epithelium with minimal reactivity in the tumour-associated stroma. (C) Pre-treatment [18F]-misonidazole PET imaging for hypoxia in the same patient showing an avid mass in the posterior pharynx. Reproduced, with permission, from (Hunter et al, 2015).
In contrast to the abovementioned agents, aliphatic (tertiary amine) N-oxides such as banoxantrone are activated via two-electron reduction that is catalysed by CYP isozymes. The basis for oxygen sensitivity in this mechanism is presumed to be competitive oxidation by O2 of reducing centres in the CYP catalytic cycle. Some quinone and nitroaromatic HAPs are also substrates for obligate two-electron reductases that are not inhibited by oxygen. For example, the significant single-agent activity of PR-104 in H460 xenografts (Patterson et al, 2007) was subsequently found to be caused by strong expression of the two-electron reductase AKR1C3 in this model (Guise et al, 2010). Although such effects can be considered an off-target liability, they may be exploitable in malignancies that strongly express the implicated two-electron reductase. The latter consideration has led to interest in evaluating PR-104 for the treatment of acute leukaemias (Houghton et al, 2011; Konopleva et al, 2015), some of which highly express AKR1C3 (Birtwistle et al, 2009; Jamieson et al, 2014). Indeed, Moradi Manesh et al (2015) recently identified strong AKR1C3 expression in primary paediatric T-lineage ALL xenografts, in which the AKR1C3-dependent activity of PR-104 was superior to the clinical combination of vincristine, dexamethasone and l-aspariginase. Although these findings point to an opportunity for PR-104, observations of hypoxia-related activity of HAPs in leukaemia models (Benito et al, 2015), presumably mediated by one-electron reduction, suggest a need for a more comprehensive set of predictive biomarkers in this setting.
Determinants of Intrinsic Sensitivity to Prodrug Metabolites
The above determinants address the potential for HAPs to release antineoplastic agents (effectors) selectively within the tumour microenvironment. The next requirement is to identify biomarkers that predict sensitivity of the tumour cells to the effector, an issue that has received relatively little attention in the HAP context. For the DNA-reactive cytotoxics generated by most HAPs, DNA damage response (DDR) pathways will obviously be important although with differences between pharmacophores. Several studies have evaluated effects of knockout or knockdown of DNA repair genes on HAP sensitivity in vitro. Tirapazamine was shown to have a two- to five-fold higher potency against hypoxic human and Chinese hamster ovary (CHO) cells in which homologous recombination repair (HRR) genes (BRCA1, BRCA2, Rad51d, Ercc1, Ercc4/Xpf, Xrcc2, Xrcc3) were mutated or suppressed by RNAi (Evans et al, 2008). CHO cell lines with mutations in HRR genes (Ercc1, Ercc4/Xpf, Rad51d) also showed marked hypersensitivity to PR-104A and its active metabolite PR-104H (Gu et al, 2009), as also reported for evofosfamide, which showed increased potency against lines with defects in Xrcc3, Ercc4/Xpf, BRCA1, Brca2 and FANCA (Meng et al, 2012).
Direct comparison of HRR dependence of different HAP classes showed cytotoxicity of prodrugs of DNA crosslinkers (PR-104A, evofosfamide) to be more dramatically potentiated by HRR defects than for benzotriazine di-oxides (tirapazamine, SN30000 and SN29751) (Hunter et al, 2012). This same pattern has been observed in human triple-negative breast and colon carcinoma lines with suppressed HRR, including demonstration of notable activity of PR-104 and evofosfamide as monotherapy or in combination with radiotherapy against BRCA2-null DLD-1 xenografts (Hunter et al, 2014a). These findings point to the potential for use of HAPs such as evofosfamide against HRR-defective tumours, potentially including BRCA-related breast and ovarian carcinoma. A rational biomarker strategy may be use 2-nitroimidazole PET imaging to evaluate hypoxia plus one-electron reductase activity in conjunction with the assessment of HRR status by mutational profiling as validated clinically for PARP-1/2 inhibitors (Mateo et al, 2015). Although attractive in principle, further work is needed to validate DDR biomarkers specific to DNA crosslinking metabolites. It is disappointing, in that respect, that there has been little systematic effort to develop predictive biomarkers for crosslinking agents, other than mutation or expression of a single DNA repair gene (ERCC1) (Gao and Liu, 2015), despite the ongoing clinical importance of this class. Even for the most widely used crosslinker, cisplatin, biomarkers that are predictive of sensitivity or resistance are still not in routine clinical use (Bowden, 2014)—a problem that is symptomatic of a broader neglect of determinants of sensitivity to cytotoxic chemotherapy.
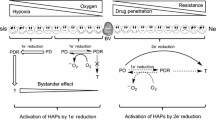
A central issue in developing biomarkers of intrinsic sensitivity is that of spatial heterogeneity of hypoxia within tumours, coupled with the allied question of whether the critical target population comprises only the hypoxic cells themselves. In relation to DDR biomarkers, it is noteworthy that persistent hypoxia transcriptionally or translationally downregulates most DNA repair pathways, including HRR (Scanlon and Glazer, 2015), which is thus expected to confer additional sensitivity to HAPs just as it does to conventional crosslinking agents (Chan et al, 2008) and PARP-1/2 inhibitors (Chan et al, 2010). However, although HAPs are designed for activation within hypoxic microenvironments, diffusion of effectors out of hypoxic zones has the potential to extend activity to cells that have not experienced hypoxic stress. This ‘bystander effect’, illustrated in Figure 3, has been suggested to contribute to the single-agent activity of PR-104 (Foehrenbacher et al, 2013) and evofosfamide (Sun et al, 2012) in tumour xenografts. Thus for HAPs with relatively stable effectors, intrinsic sensitivity biomarkers in the oxic compartment may be important determinants of response. Clear evidence for bystander effects of HAPs is provided by the killing of activation-deficient ‘target’ cells by activation-competent ‘activator’ cells in anoxic 3D multicellular layer (MCL) co-cultures; bystander killing is seen with dinitrobenazamide mustards related to PR-104 but not with tirapazamine (Wilson et al, 2007). In addition, the active metabolites PR-104H and PR-104M diffuse out of anoxic 3D multicellular layers exposed to PR-104A at rates high enough to account for much of the monotherapy activity of PR-104 in xenograft models (Foehrenbacher et al, 2013).

Hypothetical transverse section of tumour vascular chords showing steady state hypoxia gradients resulting from spatial heterogeneity of perfusion. Therapeutically significant zones are indicated in the oxygen concentration gradient, demarking cells that are classified as euoxic (O2>25 μ M), moderately hypoxic (O2<25 μ M, typically sufficient to induce a HIF-1 response), radiobiologically hypoxic (O2<5 μ M) and essentially anoxic (O2<0.5 μ M, enabling efficient activation of nitroaromatic HAPs) beyond which necrosis ensues. Numbers indicate intravascular O2 concentrations (μ M), which can vary temporally as well as spatially. Perivascular hypoxia is shown associated with two poorly perfused vessels in the centre of the section. Chemoresistant and radioresistant cells at moderate hypoxia (‘HIF activation’+‘radiobiological hypoxia’) are accessible to bystander effects resulting from diffusion of effectors from nitroaromatic HAPs (arrows), but only if contiguous with severely hypoxic (anoxic) regions. Hypoxic regions that are beyond the bystander range of nitroaromatic prodrugs (asterisks) may be more effectively treated by HAPs such as benzotriazine di-oxides that are less sensitively inhibited by oxygen.
This bystander effect is likely also to be important for killing of moderately hypoxic chemoresistant and radioresistant cells in tumours (Figure 3) given that nitroaromatic HAPs are activated only under severe hypoxia in cell culture (Koch, 1993; Hicks et al, 2007; Meng et al, 2012). However, exploiting hypoxia in this way depends on the chemo- and radioresistant hypoxic populations being contiguous with regions that are essentially anoxic. Given the macroregional variations in perfusion and hypoxia that are typical within tumours, many regions of moderate hypoxia (asterisks in Figure 3) will not meet this criterion. In such cases, direct targeting of moderately hypoxic cells with benzotriazine di-oxide HAPs (tirapazamine, SN30000) that are activated at ∼10-fold higher pO2 than nitroaromatic HAPs (Hicks et al, 2007, 2010) may be a preferable strategy. These considerations point to a need to understand the spatial distribution of hypoxia and other key biomarkers in order to rationally select an optimal HAP for an individual tumour; this will likely call on emerging capabilities for functional imaging of intratumour heterogeneity (O'Connor et al, 2015a) and its integration with molecular profiling and histopathology.
Conclusions
An emerging paradigm in cancer medicine is to personalise therapy using defined biomarkers, with small molecule and antibody-based therapeutics targeting known mutant oncogenes in individual tumours being the best examples. Identification and validation of predictive biomarkers for HAPs is a considerably more challenging prospect: determinants of sensitivity are multifactorial, and in most cases the primary target is a transient hypoxic subpopulation that limits treatment outcomes with current standards of care—although additional utility may be achievable if bystander effects enable targeting of relatively well-oxygenated cells in tumours. Our current ability to identify specific clinical settings in which hypoxia can be leveraged for therapeutic advantage is still surprisingly weak, and the molecular profile of the key treatment-limiting subpopulations is incompletely understood. Despite these limitations, there have been important advances in relation to each aspect of the challenge, including development of high-spatial resolution hypoxia imaging in tumours by oxygen-enhanced MRI (O'Connor et al, 2015b) and the advent of efficient forward genetic screening for discovery of drug sensitivity genes using whole-genome CRISPR/Cas9 knockout libraries (Shalem et al, 2014). The latter can be expected to provide more complete, pharmacophore-specific, predictive biomarker sets to complement detection of tumour hypoxia.
One of the additional challenges in the clinical development of HAPs is that validation of predictive biomarkers depends on identifying responsive tumours, yet monotherapy activity based on conventional response endpoints is an unrealistic expectation in most cases. Thus, although not discussed in this short review, pharmacodynamic biomarkers that demonstrate activity in the critical target subpopulation (e.g. loss of hypoxic cells as assessed by functional imaging) are also needed to justify advancement of HAPs to combination settings. From that perspective, it is likely that the long-anticipated goal of overcoming (and exploiting) tumour hypoxia with prodrugs will require further development of both response biomarkers and predictive biomarkers, drawing on the increasingly powerful tools of molecular oncology. It is encouraging that first steps towards that goal are being taken in two multi-centre phase 2 trials of the hypoxia-activated pan-ERBB inhibitor tarloxotinib (NCT02454842 and NCT02449681) in which pre-treatment and post-treatment EGFR status and baseline HX4 PET imaging for hypoxia is being evaluated at select sites.
References
Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, Kim H, Axelrod R, Silverman CC, Redmond KP, Gillison ML (2010) Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med 363: 24–35.
Benito J, Ramirez MS, Millward NZ, Velez J, Harutyunyan KG, Lu H, Shi YX, Matre P, Jacamo R, Ma H, Konoplev S, McQueen T, Volgin A, Protopopova M, Mu H, Lee J, Bhattacharya PK, Marszalek JR, Davis RE, Bankson JA, Cortes JE, Hart CP, Andreeff M, Konopleva M (2015) Hypoxia-activated prodrug TH-302 targets hypoxic bone marrow niches in pre-clinical leukemia models. Clin Cancer Res doi:10.1158/1078-0432.CCR-14-3378.
Birtwistle J, Hayden RE, Khanim FL, Green RM, Pearce C, Davies NJ, Wake N, Schrewe H, Ride JP, Chipman JK, Bunce CM (2009) The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: Implications for leukemogenesis. Mutat Res 662: 67–74.
Bowden NA (2014) Nucleotide excision repair: why is it not used to predict response to platinum-based chemotherapy? Cancer Lett 346: 163–171.
Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, Dewhirst MW (1996) Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res 56: 941–943.
Chan N, Koritzinsky M, Zhao H, Bindra R, Glazer PM, Powell S, Belmaaza A, Wouters B, Bristow RG (2008) Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res 68: 605–614.
Chan N, Pires IM, Bencokova Z, Coackley C, Luoto KR, Bhogal N, Lakshman M, Gottipati P, Oliver FJ, Helleday T, Hammond EM, Bristow RG (2010) Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res 70: 8045–8054.
Chandor A, Dijols S, Ramassamy B, Frapart Y, Mansuy D, Stuehr D, Helsby N, Boucher JL (2008) Metabolic activation of the antitumor drug 5-(Aziridin-1-yl)-2,4-dinitrobenzamide (CB1954) by NO synthases. Chem Res Toxicol 21: 836–843.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507–2516.
Dhani NC, Serra S, Pintilie M, Schwock J, Xu J, Gallinger S, Hill RP, Hedley DW (2015) Analysis of the intra- and intertumoral heterogeneity of hypoxia in pancreatic cancer patients receiving the nitroimidazole tracer pimonidazole. Br J Cancer 113: 864–871.
Evans JW, Chernikova SB, Kachnic LA, Banath JP, Sordet O, Delahoussaye YM, Treszezamsky A, Chon BH, Feng Z, Gu Y, Wilson WR, Pommier Y, Olive PL, Powell SN, Brown JM (2008) Homologous recombination is the principal pathway for the repair of DNA damage induced by tirapazamine in mammalian cells. Cancer Res 68: 257–265.
Foehrenbacher A, Patel K, Abbattista MR, Guise CP, Secomb TW, Wilson WR, Hicks KO (2013) The role of bystander effects in the antitumor activity of the hypoxia-activated prodrug PR-104. Front Oncol 3: article 263.
Fyles A, Milosevic M, Hedley D, Pintilie M, Levin W, Manchul L, Hill RP (2002) Tumor hypoxia has independent predictor impact only in patients with node-negative cervix cancer. J Clin Oncol 20: 680–687.
Fyles AW, Milosevic M, Wong R, Kavanagh MC, Pintilie M, Sun A, Chapman W, Levin W, Manchul L, Keane TJ, Hill RP (1998) Oxygenation predicts radiation response and survival in patients with cervix cancer. Radiother Oncol 48: 149–156.
Gao Y, Liu D (2015) The roles of excision repair cross-complementation group1 in objective response after cisplatin-based concurrent chemoradiotherapy and survival in head and neck cancers: a systematic review and meta-analysis. Oral Oncol 51: 570–577.
Gu Y, Patterson AV, Atwell GJ, Chernikova SB, Brown JM, Thompson LH, Wilson WR (2009) Roles of DNA repair and reductase activity in the cytotoxicity of the hypoxia-activated dinitrobenzamide mustard PR-104A. Mol Cancer Ther 8: 1714–1723.
Guise CP, Abbattista MR, Singleton RS, Holford SD, Connolly J, Dachs GU, Fox SB, Pollock R, Harvey J, Guilford P, Donate F, Wilson WR, Patterson AV (2010) The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res 70: 1573–1584.
Guise CP, Abbattista MR, Tipparaju SR, Lambie NK, Su J, Li D, Wilson WR, Dachs GU, Patterson AV (2012) Diflavin oxidoreductases activate the bioreductive prodrug PR-104A under hypoxia. Mol Pharmacol 81: 31–40.
Guise CP, Wang AT, Theil A, Bridewell DJ, Wilson WR, Patterson AV (2007) Identification of human reductases that activate the dinitrobenzamide mustard prodrug PR-104A: a role for NADPH:cytochrome P450 oxidoreductase under hypoxia. Biochem Pharmacol 74: 810–820.
Hicks KO, Myint H, Patterson AV, Pruijn FB, Siim BG, Patel K, Wilson WR (2007) Oxygen dependence and extravascular transport of hypoxia-activated prodrugs: comparison of the dinitrobenzamide mustard PR-104A and tirapazamine. Int J Radiat Oncol Biol Phys 69: 560–571.
Hicks KO, Siim BG, Jaiswal JK, Pruijn FB, Fraser AM, Patel R, Hogg A, Liyanage HD, Dorie MJ, Brown JM, Denny WA, Hay MP, Wilson WR (2010) Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clin Cancer Res 16: 4946–4957.
Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P (1996) Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res 56: 4509–4515.
Houghton PJ, Lock R, Carol H, Morton CL, Phelps D, Gorlick R, Kolb EA, Keir ST, Reynolds CP, Kang MH, Maris JM, Wozniak AW, Gu Y, Wilson WR, Smith MA (2011) Initial testing of the hypoxia-activated prodrug PR-104 by the pediatric preclinical testing program. Pediatr Blood and Cancer 57: 443–453.
Hunter FW, Hsu HL, Su J, Pullen SM, Wilson WR, Wang J (2014a) Dual targeting of hypoxia and homologous recombination repair dysfunction in triple-negative breast cancer. Mol Cancer Ther 13: 2501–2514.
Hunter FW, Jaiswal JK, Hurley DG, Liyanage HD, McManaway SP, Gu Y, Richter S, Wang J, Tercel M, Print CG, Wilson WR, Pruijn FB (2014b) The flavoprotein FOXRED2 reductively activates nitro-chloromethylbenzindolines and other hypoxia-targeting prodrugs. Biochem Pharmacol 89: 224–235.
Hunter FW, Wang J, Patel R, Hsu HL, Hickey AJ, Hay MP, Wilson WR (2012) Homologous recombination repair-dependent cytotoxicity of the benzotriazine di-N-oxide CEN-209: comparison with other hypoxia-activated prodrugs. Biochem Pharmacol 83: 574–585.
Hunter FW, Young RJ, Shalev Z, Vellanki RN, Wang J, Gu Y, Joshi N, Sreebhavan S, Weinreb I, Goldstein DP, Moffat J, Ketela T, Brown KR, Koritzinsky M, Solomon B, Rischin D, Wilson WR, Wouters BG (2015) Identification of P450 oxidoreductase as a major determinant of sensitivity to hypoxia-activated prodrugs. Cancer Res 75: 4211–4223.
Jamieson SM, Gu Y, Manesh DM, El-Hoss J, Jing D, Mackenzie KL, Guise CP, Foehrenbacher A, Pullen SM, Benito J, Smaill JB, Patterson AV, Mulaw MA, Konopleva M, Bohlander SK, Lock RB, Wilson WR (2014) A novel fluorometric assay for aldo-keto reductase 1C3 predicts metabolic activation of the nitrogen mustard prodrug PR-104A in human leukaemia cells. Biochem Pharmacol 88: 36–45.
Koch CJ (1993) Unusual oxygen concentration dependence of toxicity of SR-4233, a hypoxic cell toxin. Cancer Res 53: 3992–3997.
Konopleva M, Thall PF, Yi CA, Borthakur G, Coveler A, Bueso-Ramos C, Benito J, Konoplev S, Gu Y, Ravandi F, Jabbour E, Faderl S, Thomas D, Cortes J, Kadia T, Kornblau S, Daver N, Pemmaraju N, Nguyen HQ, Feliu J, Lu H, Wei C, Wilson WR, Melink TJ, Gutheil JC, Andreeff M, Estey EH, Kantarjian H (2015) Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica 100: 927–934.
Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, Bastidas AJ, Vierra M (2000) Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys 48: 919–922.
Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N, Dunning MJ, Halim S, Lamb AD, Moon NC, Zafarana G, Warren AY, Meng X, Thoms J, Grzadkowski MR, Berlin A, Have CL, Ramnarine VR, Yao CQ, Malloff CA, Lam LL, Xie H, Harding NJ, Mak DY, Chu KC, Chong LC, Sendorek DH, P'ng C, Collins CC, Squire JA, Jurisica I, Cooper C, Eeles R, Pintilie M, Dal Pra A, Davicioni E, Lam WL, Milosevic M, Neal DE, van der Kwast T, Boutros PC, Bristow RG (2014) Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol 15: 1521–1532.
Lassen P, Eriksen JG, Hamilton-Dutoit S, Tramm T, Alsner J, Overgaard J (2010) HPV-associated p16-expression and response to hypoxic modification of radiotherapy in head and neck cancer. Radiother Oncol 94: 30–35.
Lienhart WD, Gudipati V, Macheroux P (2013) The human flavoproteome. Arch Biochem Biophys 535: 150–162.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, Seed G, Jain S, Ralph C, Protheroe A, Hussain S, Jones R, Elliott T, McGovern U, Bianchini D, Goodall J, Zafeiriou Z, Williamson CT, Ferraldeschi R, Riisnaes R, Ebbs B, Fowler G, Roda D, Yuan W, Wu YM, Cao X, Brough R, Pemberton H, A'Hern R, Swain A, Kunju LP, Eeles R, Attard G, Lord CJ, Ashworth A, Rubin MA, Knudsen KE, Feng FY, Chinnaiyan AM, Hall E, de Bono JS (2015) DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373: 1697–1708.
McKeage MJ, Jameson MB, Ramanathan RK, Rajendran J, Gu Y, Wilson WR, Melink TJ, Tchekmedyian NS (2012) PR-104 a bioreductive pre-prodrug combined with gemcitabine or docetaxel in a phase Ib study of patients with advanced solid tumours. BMC Cancer 12: 496.
Meng F, Evans JW, Bhupathi D, Banica M, Lan L, Lorente G, Duan JX, Cai X, Mowday AM, Guise CP, Maroz A, Anderson RF, Patterson AV, Stachelek GC, Glazer PM, Matteucci MD, Hart CP (2012) Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol Cancer Ther 11: 740–751.
Milosevic M, Warde P, Menard C, Chung P, Toi A, Ishkanian A, McLean M, Pintilie M, Sykes J, Gospodarowicz M, Catton C, Hill RP, Bristow R (2012) Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin Cancer Res 18: 2108–2114.
Moradi Manesh D, El-Hoss J, Evans K, Richmond J, Toscan CE, Bracken LS, Hedrick A, Sutton R, Marshall GM, Wilson WR, Kurmasheva RT, Billups C, Houghton PJ, Smith MA, Carol H, Lock RB (2015) AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood 126: 1193–1202.
Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, Becker A, Adam M, Molls M, Dunst J, Terris DJ, Overgaard J (2005) Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol 77: 18–24.
O'Connor JP, Rose CJ, Waterton JC, Carano RA, Parker GJ, Jackson A (2015a) Imaging intratumor heterogeneity: role in therapy response, resistance, and clinical outcome. Clin Cancer Res 21: 249–257.
O'Connor JP, Boult JK, Jamin Y, Babur M, Finegan KG, Williams KJ, Little RA, Jackson A, Parker GJ, Reynolds AR, Waterton JC, Robinson SP (2015b) Oxygen-enhanced MRI accurately identifies, quantifies, and maps tumor hypoxia in preclinical cancer models. Cancer Res 76 (4): 787–795.
Papadopoulou MV, Ji M, Rao MK, Bloomer WD (2003) Reductive metabolism of the nitroimidazole-based hypoxia-selective cytotoxin NLCQ-1 (NSC 709257). Oncol Res 14: 21–29.
Patterson AV, Ferry DM, Edmunds SJ, Gu Y, Singleton RS, Patel K, Pullen SM, Hicks KO, Syddall SP, Atwell GJ, Yang S, Denny WA, Wilson WR (2007) Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res 13: 3922–3932.
Patterson AV, Saunders MP, Chinje EC, Talbot DC, Harris AL, Strafford IJ (1997) Overexpression of human NADPH:cytochrome c (P450) reductase confers enhanced sensitivity to both tirapazamine (SR 4233) and RSU 1069. Br J Cancer 76: 1338–1347.
Phillips RM (2016) Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother Pharmacol 77 (3): 441–457.
Rischin D, Hicks RJ, Fisher R, Binns D, Corry J, Porceddu S, Peters LJ (2006) Prognostic significance of [18F]-misonidazole positron emission tomography-detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group Study 98.02. J Clin Oncol 24: 2098–2104.
Rischin D, Peters L, Hicks R, Hughes P, Fisher R, Hart R, Sexton M, D'Costa I, von Roemeling R (2001) Phase I trial of concurrent tirapazamine, cisplatin, and radiotherapy in patients with advanced head and neck cancer. J Clin Oncol 19: 535–542.
Scanlon SE, Glazer PM (2015) Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair (Amst) 32: 180–189.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343: 84–87.
Sun JD, Liu Q, Wang J, Ahluwalia D, Ferraro D, Wang Y, Duan JX, Ammons WS, Curd JG, Matteucci MD, Hart CP (2012) Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res 18: 758–770.
TCGA Network (2015) Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517: 576–582.
Toustrup K, Sorensen BS, Alsner J, Overgaard J (2012a) Hypoxia gene expression signatures as prognostic and predictive markers in head and neck radiotherapy. Semin Radiat Oncol 22: 119–127.
Toustrup K, Sorensen BS, Lassen P, Wiuf C, Alsner J, Overgaard J (2012b) Gene expression classifier predicts for hypoxic modification of radiotherapy with nimorazole in squamous cell carcinomas of the head and neck. Radiother Oncol 102: 122–129.
Toustrup K, Sorensen BS, Nordsmark M, Busk M, Wiuf C, Alsner J, Overgaard J (2011) Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res 71: 5923–5931.
Trinkaus ME, Hicks RJ, Young RJ, Peters LJ, Solomon B, Bressel M, Corry J, Fisher R, Binns D, McArthur GA, Rischin D (2014) Correlation of p16 status, hypoxic imaging using [18F]-misonidazole positron emission tomography and outcome in patients with loco-regionally advanced head and neck cancer. J Med Imaging Radiat Oncol 58: 89–97.
Wang J, Foehrenbacher A, Su J, Patel R, Hay MP, Hicks KO, Wilson WR (2012) The 2-nitroimidazole EF5 is a biomarker for oxidoreductases that activate the bioreductive prodrug CEN-209 under hypoxia. Clin Cancer Res 18: 1684–1695.
Wang J, Guise CP, Dachs GU, Phung Y, Hsu HL, Lambie NK, Patterson AV, Wilson WR (2014) Identification of one-electron reductases that activate both the hypoxia prodrug SN30000 and diagnostic probe EF5. Biochem Pharmacol 91: 436–446.
Wardman P (2001) Electron transfer and oxidative stress as key factors in the design of drugs selectively active in hypoxia. Curr Med Chem 8: 739–761.
Wilson WR, Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11: 393–410.
Wilson WR, Hicks KO, Pullen SM, Ferry DM, Helsby NA, Patterson AV (2007) Bystander effects of bioreductive drugs: Potential for exploiting pathological tumor hypoxia with dinitrobenzamide mustards. Radiat Res 167: 625–636.
Wouters BG, Koritzinsky M (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8: 851–864.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hunter, F., Wouters, B. & Wilson, W. Hypoxia-activated prodrugs: paths forward in the era of personalised medicine. Br J Cancer 114, 1071–1077 (2016). https://doi.org/10.1038/bjc.2016.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.79
- Springer Nature Limited
Keywords
This article is cited by
-
Photochemically-driven highly efficient intracellular delivery and light/hypoxia programmable triggered cancer photo-chemotherapy
Journal of Nanobiotechnology (2023)
-
Cell proliferation, drug distribution and therapeutic effects in relation to the vascular system of solid tumours
British Journal of Cancer (2023)
-
Dual hypoxia-responsive supramolecular complex for cancer target therapy
Nature Communications (2023)
-
Exploiting Therapeutic Vulnerabilities in Triple-Negative Breast Cancer: Successes, Challenges, and Opportunities
Current Breast Cancer Reports (2023)
-
Therapeutic targeting of the hypoxic tumour microenvironment
Nature Reviews Clinical Oncology (2021)