
Abstract
Cardiac adaptation to increased volume load is encountered in physiologic scenarios such as growth, pregnancy and aerobic exercise training and in pathological conditions like atrio-ventricular valve insufficiency or systemic arterio-venous shunt. Experimental models of pure volume overload due to chronic mitral regurgitation in dogs or aorto-caval fistula in rodents show that this type of remodelling differs in many ways from pressure overload. The early response is characterised by increased diastolic wall stress, activation of specific signalling pathways (Akt, Wnt), cardiomyocyte elongation, mast cell infiltration and degradation of extracellular matrix which allows increasing the chamber volume. Volume overload leads to eccentric cardiac hypertrophy with increased ventricular diameter but reduced relative wall thickness, enhanced diastolic function and relatively preserved systolic function. Myocardial collagen content is normal, in contrast to fibrosis due to pressure overload. Although initially well tolerated, pathological volume overload is an inevitably progressive condition. As a result of neurohumoral activation, cardiomyocyte dysfunction, abnormal Ca2+ handling and cell loss due to apoptosis, overt end-stage heart failure develops after long latent period. Understanding the pathophysiology of volume-overload cardiac remodelling is a mandatory step for development and proper timing of novel surgical and pharmaceutical therapies which may prevent or postpone the onset of heart failure.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Heart failure
- Volume overload
- Mitral regurgitation
- Aorto-caval fistula
- Diastolic function
- Systolic function
- Cardiac remodelling
- Cardiac hypertrophy
- Experimental models
- Hemodynamics
1 Introduction
The heart has just a few options of how to handle an increased volume load and how to provide a larger-than-normal cardiac output. First, it can use Frank-Starling autoregulation, by which an increase of end-diastolic sarcomere length instantly boosts contractile performance. This mechanism operates on a beat-to-beat basis and adjusts ventricular contractility to subtle variations in loading but has limited ability to contain larger fluctuations. Second, an increased preload with some delay activates the neurohumoral autonomic response that augments cardiac output by increasing heart rate, relaxation and contractility of the ventricle, mediated by sympathetic stimulation. Third, longer exposure to an increased load initiates changes in chamber ventricular geometry and increases the mass of the heart, both of which represent the main long-standing compensation [1]. Chamber remodelling and hypertrophy are initially adaptive allowing the organism to survive in an altered hemodynamic state, but over the long-term, these have adverse effects on survival. In contrast to cardiac remodelling due to myocardial infarction or pressure overload, cardiac adaptation to increased volume load is also encountered more frequently in physiologic scenarios such as growth, pregnancy and aerobic exercise training.
At the tissue level, cardiac hypertrophy is defined as an increase in ventricular or heart mass produced by the increased cell size of differentiated cardiomyocytes. Cardiac cells represent only a third of the total cardiac cell number but they occupy more than 70 % of cardiac volume. Therefore, changes in cardiomyocyte cell volume translate into changes of heart weight. In mammals, heart growth before birth is characterised by cardiomyocyte proliferation (hyperplasia) which ceases postnatally. Then the number of cardiomyocytes in the heart remain almost constant for the rest of life (2–4 × 109) and all further postnatal heart growth occurs through cardiomyocyte size expansion (hypertrophy) [2]. The remarkable increase in cardiomyocyte cell volume during postnatal ontogenesis (by up to 40×) is often associated with multiplication of genetic material within the nucleus (polyploidy). Adult human cardiomyocytes are mostly (75 %) tetraploid, but the ploidy may further increase with pathological heart growth [3]. Adult cardiomyocytes are terminally differentiated cells and posses only negligible (or no) mitotic capacity. Recent studies of carbon isotope composition of nuclear DNA of cardiomyocytes provided evidence for slow intrinsic postnatal cardiomyocyte renewal (replacement 0.45–1 % of cardiomyocytes/year) [4]. The presence of cardiac progenitor cells or the possibility of dividing more differentiated cardiomyocytes still remains a matter of intensive debate [3]. At the level of organism, the definition of hypertrophy is more complicated, because heart weight responds to changes of body weight, body composition and the degree of physical activity. Scaling to lean body mass is probably the most accurate way how to assess appropriateness of heart weight.
Cardiac hypertrophy can be functionally categorised as physiologic or pathologic. The cardinal features of both types are summarised in Table 9.1. Physiological hypertrophy responds to metabolic factors or peptide factors governing normal growth (Insulin-like growth factor-1, IGF-1) [5] and is driven by signalling pathways (protein kinase Akt) that are distinctive from those activated in pathological heart growth. Physiological hypertrophy is characterised by preserved chamber geometry, by absence of maladaptive features (fibrosis, foetal gene re-expression, apoptosis, metabolic remodelling), and by its full reversibility [6]. Pathological hypertrophy is a consequence of a persistent abnormal elevation of the hemodynamic load, combined with pro-hypertrophic effects of elevated circulating neurohormones (norepinephrine, angiotensin-II). In large prospective studies in a human population, pathological cardiac hypertrophy has been associated with increased risk of heart failure and arrhythmic death [7].
2 Physiological Volume Load-Induced Hypertrophy
2.1 Aerobic Exercise Training
An ample case of physiological hypertrophy is aerobic exercise training. While the maximal heart rate is intrinsically dependent on age and cannot be increased by training [8], increased stroke volume by training is the only mechanism for boosting maximal cardiac output and exercise performance. The mode of exercise and ensuing response of systemic circulation govern the pattern of cardiac adaptation. Strength training (isometric exercise) increases systemic vascular resistance, so the left ventricle (LV) must perform against it as a pressure pump, a situation which favours concentric LV remodelling. In contrast, endurance training (isometric exercise) increases cardiac output, while systemic vascular resistance is either normal of reduced [8]. In this situation, the heart operates as a flow pump and the whole organ adopts to handle increased venous return, developing hypertrophy with either preserved or mildly eccentric geometry. Highly trained endurance athletes (rowers, long-distance runners, cyclists, swimmers) have increased LV mass in the range of 10–20 % [9, 10], but sometimes up to 37 % [11]. The increase in LV mass is proportional to increases in LV volume and is associated with preservation or even enhancement of indexes of systolic and diastolic function (systolic and early diastolic longitudinal velocities or strains) [12]. Long-term studies in Olympic athletes performing uninterrupted high-intensity training over prolonged periods of time (~8 years) did not show any deterioration of LV function over time [13]. Other cardiac chambers adapt to exercise induced volume load as well-endurance training leads to a parallel increase of right ventricular volume [14] and left atrial size [15]. In contrast to the left ventricle, left atrial diameter regresses less with detraining—atrial dilatation may explain the higher incidence of atrial fibrillation in older endurance athletes. Exercise-induced changes of LV are also affected by various cofounders such as ethnicity, gender and genetic background. As a result, some trained subjects may end up in a shadow zone where physiological adaptation overlaps with pathologic hypertrophy [16]. In a cross-sectional study of 947 elite athletes performing various types of aerobic activity, 38 % exceeded the normal LV chamber value for the population (<54 mm), but only 4 % had clearly abnormal end-diastolic dimension (>60 mm), while abnormal septum thickness (>13 mm) was even more rare (1.3 %) [17]. In subjects with abnormal hypertrophy, prolonged detraining led to a 28 % reduction of LV mass and to normalisation of LV thickness in all. However, the resolution of LV diameter was incomplete and substantial chamber dilatation persisted in more than 20 % of detrained athletes, particularly in those who gained weight or engaged in recreational sport activities [18].The prognostic impact of residual heart hypertrophy or chamber dilatation after cessation of intensive training remains unknown.
2.2 Pregnancy
Pregnancy leads to expansion of blood volume by 40–45 % and to an increase of cardiac output by 30–50 % that starts in the 6th week, reaches a plateau by the 2nd trimester and then stays until delivery [19]. This temporary increase in volume loading leads to mild dilatation of the left ventricle, a decrease in wall thickness/diameter ratio and to a 10–20 % increase in LV mass [20], that is fully reversible within 8th weeks post-delivery [21]. Molecular mechanisms responsible for heart hypertrophy during pregnancy remain understudied. In addition to mechanical stress, the rise of estrogens towards the end of pregnancy seems to contribute to pregnancy-related heart hypertrophy by increased activity of stretch-responsive c-Src kinase activity [22]. The heart response to pregnancy-induced overload is also modulated by the neuroregulin/ErbB system which has cardioprotective and cardioregenerative properties. Inhibition of this system with kinase inhibitor lapatinib in rodents led to pregnancy-associated maternal LV dysfunction and death [23].
2.3 Postprandial Hypertrophy
An extreme and intriguing form of physiological adaptation to increased volume and metabolic demands is the postprandial cardiac hypertrophy in large carnivorous snakes. The Burmese python’s heart increases its mass by 40 % within 48 h after consumption of a large meal, driven by a large increase in cardiac output and a 7-fold increase in oxygen consumption related to digestion of the prey [24]. Postprandial heart enlargement is not due to cardiomyocyte division but purely due to hypertrophy which is fully reversible in the postprandial period. As recently demonstrated, heart growth occurs due to activation of physiological signal transduction pathways (Akt and AMPK kinases) and is paralleled by high upregulation of fatty acid transport and oxidation enzymes (CD36/fatty acid translocase, medium-chain acyl-coenzyme A dehydrogenase). This massive growth response is triggered not only by mechanotransduction and metabolic hormones, but also by a specific combination of circulating fatty acids (nutrient signalling) that directly promote cardiomyocyte hypertrophy even in a mammalian heart [25].
3 Pathological Volume-Overload Heart Hypertrophy
Pathological volume overload-induced cardiac hypertrophy is often a result of structural defect that creates an inefficiency of circulation, such as mitral regurgitation or a shunt between the systemic vein and artery [1]. Similar cardiac remodelling can also develop in high cardiac output states due to low systemic arterial resistance. The causes of non-physiological cardiac overload are summarised in Table 9.2.
3.1 Valve Regurgitation
Frequent causes of mitral regurgitation are age-related or myxomatous degeneration, infective endocarditis, rheumatic disease or altered valve geometry by other cardiac heart pathology (“functional mitral regurgitation”). In mitral regurgitation, a part of the stroke volume escapes back into the atrium and then re-enters the ventricle in the following cardiac cycles. Because regurgitation occurs into the low pressure chamber, the contracting ventricle is exposed to pure volume overload which triggers an increase in LV diameter, mass and compliance. Mitral regurgitation is initially well tolerated, but if severe enough (regurgitant fraction >50 %) it leads to heart failure or premature death [26, 27]. Isolated volume overload can occur also in the right ventricle (due to atrial septal defect or tricuspid regurgitation) which tolerates it better than pressure overload [1]. Detailed mechanisms of early and late remodelling of the heart due to mitral regurgitation will be discussed below. Aortic insufficiency is noted only briefly here, because it represents a state of mixed volume and pressure overload—the left ventricle is exposed to higher systolic and diastolic pressures than in pure volume overload due to mitral regurgitation or systemic arterio-venous shunt [1].
3.2 Systemic Arterio-Venous Shunt
Another structural defect that leads to volume overload of both ventricles is an abnormal connection (shunt, fistula) between the large systemic artery and vein. Small arterio-venous fistula is often created surgically in patients with renal failure to allow easy vascular access for haemodialysis. Sometimes the feeding artery undergoes eccentric remodelling so that fistula flow considerably increases which may cause considerable volume overload for the heart. A shunt flow of at least 20–50 % of cardiac output is needed to develop heart failure [28]. Arterio-venous fistula can be traumatic due to a penetrating injury of proximal limb [29] or as a consequence catheterisation in the groin. Intraabdominal arterio-venous fistula can also develop after ligation of vascular stub during nephrectomy [30] or adnexectomy. Rarely, significant shunting develops as a consequence of large vascular malformations, either as isolated haemangioma or multiple arterio-venous malformations often affecting the liver (hereditary telangiectasia, Rendu-Weber-Osler disease) [31].
3.3 High Cardiac Output States
Volume overload can also occur as a consequence of a high-output circulatory state (cardiac index >3.9 l min m−2 in humans) [32], driven primarily by excessive systemic vasodilatation. Cardiac output redistribution and relative hypotension due to low systemic vascular resistance trigger neurohumoral activation, fluid retention and development of clinical heart failure [32, 33]. In sepsis, low systemic vascular resistance is as a consequence of endotoxin-induced, NO-mediated systemic vasodilatation. In some patients with pulmonary disease or hypoventilation, low systemic vascular resistance is a consequence of vasodilatory properties of retained carbon dioxide [34]. Low vascular resistance can also be observed in some severely obese subjects [35] who may have simultaneously an alveolar hypoventilation resulting in increased cardiac output and congestion [36]. In anaemia, increased cardiac output compensates the diminished oxygen-carrying capacity of the blood. In severe chronic anaemia, diminished inactivation of endothelium-derived NO by erythrocytes may additionally contribute to excessive vasodilatation [37]. Other states of high cardiac output state include thyrotoxicosis, severe thiamine deficiency and advanced Paget’s bone disease (Table 9.2).
4 Animal Models of Volume Overload
4.1 Models of Mitral Regurgitation
Carabello’s group developed a model of chronic volume overload due to mitral regurgitation in dogs, created by partial destruction of mitral valve chordae using forceps under fluoroscopic control [38]. After 3 months of severe mitral regurgitation (regurgitant fraction >50 %), end-diastolic volume increased by 46 %, left ventricular mass increased by 36 %, mean end-diastolic LV pressure increased to 25 mmHg and cardiac output was reduced [38, 39]. This model was used in seminal studies that studied the relations between cardiac remodelling, wall stress and indices of ventricular function in chronic mitral regurgitation [40]. A similar model was later adopted also in pigs [41]. Due to high costs and ethical concerns of research use of dogs, a rodent model of chronic mitral regurgitation in rats has been recently developed. Mitral regurgitation is created by a puncture of mitral valve leaflets with a needle that is advanced from the cardiac apex and exposed from a small left thoracotomy. The amount of regurgitation created can be visually checked during the procedure with a human intracardiac echocardiographic (ICE) probe inserted into rat’s oesophagus [42–44]. The procedure has acceptable mortality rate of 17–27 %. Twelve weeks after the procedure, LV end-diastolic dimension increased by 26 %, LV end-diastolic volume doubled, LV mass index increased by 35 % while LV end-diastolic pressure also increased in a part of the animals. The myocardium of rats with mitral regurgitation demonstrated a myosin heavy chain isotype switch (from α to β) and downregulation of sarcoplasmic reticulum Ca2+ ATP-ase 2 (SERCA2), confirming the presence of features of maladaptive remodelling. One-year survival of rats with mitral regurgitation was only 38 % [42].
4.2 Systemic Arterio-Venous Shunt Models
Systemic shunt due to artificial aorto-caval fistula (ACF) in rodents is now the most widely used volume overload model. Previously, a surgical anastomosis approach was used in rats [45] and pigs [46, 47]. More recently, Garcia and Diebold developed a simplified needle-puncture technique in which infrarenal aorta and adjacent vena cava inferior is punctured with a 18G phlebotomy needle (1.2 mm outer diameter) from midline laparotomy [48]. The puncture site is sealed with cyanoacrylate glue during brief abdominal aortic clamping and the abdominal cavity is then closed with a suture. The procedure is rapid (<20 min) and has low procedural mortality (<10 %). Shortly after ACF creation, pronounced cardiac hypertrophy with chamber dilatation develops: the heart weight/body weight increases by 52 % by the 2nd week and by 95 % by the 16th week, compared to sham-operated animals [49]. Volume overload is initially well tolerated, but after 12–15 weeks, congestion and heart failure signs such as tachypnoea, lethargy, piloerection and ascites, gradually develop [50]. The slow progress of symptoms mimics well the insidious onset of heart failure in humans [51]. After 21 weeks, 80 % animals are either in heart failure or dead [52]; 1 year mortality approaches to 72 % [50].
The course of morbidity after ACF induction depends on the shunt size, gender, genetic background [53] and strain differences [54]. The large fistula from a large needle leads quickly to heart failure, while small fistula from thinner needle induces only milder, less symptomatic hypertrophy [55, 56]. Females seem to tolerate volume overload better than males and develop less pronounced heart hypertrophy. This protective gender effect is removed by ovariectomy [57] and is restored back by oestrogen administration. The beneficial effects of oestrogens can be explained by their inhibitory effects on mast cell-mediated extracellular matrix degradation [58]. Oestrogens also increase the active form of Bcl-2 protein that confers the resistance to apoptosis [59].
In a small fraction of animals after ACF procedure (<10 %), the fistula closes itself over time, with resulting cardiac mass that falls within normal limits (<mean + 2*SD of control group); these animals should probably be post hoc excluded from experiments. The needle technique for ACF creation was later adopted also for mice [60, 61], which has higher surgical mortality and ACF failure rates.
4.3 Models of Aortic Insufficiency
Magid at all developed a rabbit model of aortic insufficiency created by retrograde transcarotid puncture of aortic leaflets under echocardiographic guidance [62, 63]. Arsenaut et al. more recently adopted a similar technique for rats with a favourable procedure mortality <20 % [64]. The procedure leads to more isolated hypertrophy of the LV (LV weight/body weight increase by 60 % 26 week) [65], but it does not result in overt of heart failure. The model has been successfully used in worthy studies documenting beneficial effects of beta-blockade [66], moderate exercise [67] or angiotensin-converting enzyme (ACE) inhibition [68]. However, these findings can be condition-specific and cannot be generalised to other volume overload states.
5 Differences Between Volume and Pressure Overload
The principal differences between volume overload and pressure overload-induced cardiac adaptation and remodelling are summarised in Table 9.3.
5.1 Wall stress and Chamber Geometry
According to Laplace’s law, the mechanical wall stress in the LV wall can be approximated as σ = (LV pressure × LV radius)/(2 × LV wall thickness). In a landmark study, Grossmann invasively measured LV wall stress in 30 patients with various forms of overload due to valve lesions [69]. Patients with pressure overload, most often due to aortic stenosis, had higher systolic LV pressure, more concentric LV hypertrophy, but normal systolic and diastolic wall stress. In contrast, patients with volume overload, in this study most often due to aortic regurgitation, had an eccentric hypertrophy, normal systolic wall stress, normal LV thickness/diameter ratio, but increased diastolic wall stress. The authors proposed that increased systolic tension leads to cardiomyocyte thickening and feedback return of systolic wall stress to normal. On the other hand, increased diastolic tension leads to cardiomyocyte elongation that helps to maintain stroke volume, but it does not fully normalise diastolic wall stress.
By direct comparisons of invasive data from dogs with pressure or volume overload with matched levels of the LV stroke work, Carabello et al. showed that for the same work demand, LV hypertrophic response to volume is smaller than to pressure [40]. This implicates that in mitral regurgitation, each unit of myocardium performs more work and the dysfunction occurs at smaller degree of LV hypertrophy than in pressure overload. Patients with mitral regurgitation have reduced LV mass/volume ratio, supporting the experimental observations of limited hypertrophic response to volume overloading [70].
Based on the Grossmann study [69], concentric hypertrophy due to pressure overload was long viewed as an necessary adaptation, allowing to normalise elevated systolic wall stress [69] and to prevent early failure of the ventricle. This notion has been more recently challenged by experiments where hypertrophy in pressure-overloaded hearts was abolished by gene manipulations [71] or by antihypertrophic drugs [72]. In these models, heart failure did not develop despite persistent LV load elevation, suggesting that hypertrophy itself, independently of wall stress, is the factor responsible for progression towards heart failure. This idea has some support in clinical data from patients with aortic stenosis, where the degree of hypertrophic response for a given hemodynamic overload predicts the presence of systolic dysfunction [73]. Whether similar concept applies also to volume overload remains to be studied.
5.2 Mechanosensing
The driving force for cardiac hypertrophy are mechanical forces that act on cardiomyocytes and activate mechanosensing signalling pathways [69, 74]. Mechanical forces are sensed both inside and at surface of the cardiac cell. At the sarcolemma, stress is sensed by stretch-sensitive transient receptor potential channels (TRP channels) and by membrane-spanning integrins that interconnect intracellular load-bearing cytoskeleton to the extracellular matrix and interact with sub-sarcolemmal focal adhesion kinases (FAK). Intracellularly, mechanical forces are sensed by protein complexes at Z-disc (muscle-LIM protein and others) and by titin, a gigantic protein that spans from Z-disc to M-band as a single molecule (~1 μm distance) and forms a myofibrillar backbone important for regulation of diastolic stretch [75].
Mechanical forces differ between pressure and volume overload, being responsible for two distinctive patterns of remodelling response. Not only the magnitude, but also the timing, of wall stress within cardiac cycle is important. In tissue culture experiments, Yamamoto showed that cardiomyocytes exposed to strain during systolic phase had a more prominent activation of mitogen-activated protein kinase (MAPK), increased B-type natriuretic peptide (BNP) expression and protein synthesis rate than cardiomyocytes exposed to strain during diastolic phase [76]. Imposing a deformation during systole (when cell stiffness is greater) could deliver more strain on cardiomyocyte and increase the stretch-responsive signalling. Alternatively, the coincidence of mechanical strain with electrical activation or intracellular calcium level of cell may be important in determining the difference between pressure overload and volume overload.
5.3 Signalling Pathway Activation
The molecular biology of these two different remodelling patterns has been examined in detail by Toisher et al. who studied mice with severe pressure overload (transaortic constriction) or volume overload (ACF) [60]. Mean LV wall stress/cycle early after surgery was equally elevated in both groups of animals (+69 and +67 %). Increased systolic or diastolic wall stress (measured immediately after surgery) led in both cases to similar cardiac hypertrophy (+22 % and +29 % by 1st week), but otherwise largely different molecular myocardial phenotypes. Pressure overload led to concentric hypertrophy with increased diffuse myocardial fibrosis, inflammatory cell infiltration (CD45+) and cardiomyocyte apoptosis measured by activated caspase-3 and terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL). In contrast, volume overload led to eccentric hypertrophy without increased fibrosis, without inflammation and with less cardiomyocyte apoptosis. In the long-term, pressure-overload hypertrophy resulted in more severe LV dysfunction and higher mortality compared with shunt. The analysis demonstrated striking differences in signalling pathways that were activated by different overload conditions. Pressure overloaded hearts showed increased activation of extracellular-signal-regulated kinase (ERK) and even more persistent activation of calcium/calmodulin-dependent protein kinase II (CaMKII). On the other hand, volume overload showed early activation of Wnt signalling pathway and persistent activation of Akt-kinase (protein kinase B). Increased CaMKII activation in pressure overload resulted in altered Ca2+ cycling, increased L-type calcium current, calcium transients, fractional sarcoplasmic reticulum calcium release and calcium spark frequency. None of these changes happened in the shunt model. This important experimental study indicates that differences in the loading pattern result in distinct load-dependent regulation of gene expression and therefore may require different overload-specific pharmacological interventions.
5.4 Protein Turnover
Both types of overload have also a diverse impact on protein synthetic rate. Myocardial contractile proteins have rapid turnover (5–10 days) so stable heart mass is preserved by maintaining a constant equilibrium between synthesis and degradation rate [1]. By measuring 3H-labelled leucine incorporation into a heavy chain of myosin, Carabello et al. showed that acute pressure overload in dogs lead to a 30 % increase in synthesis of this sarcomeric protein and returns to normal when LV mass is increased. Interestingly, in volume overload, myosin synthesis is not increased either in the acute [77] or chronic phase [78]. Therefore, myocardial growth after induction of mitral regurgitation is accounted for by a decrease in the fractional rate of protein degradation [78]. Similar findings were also reported in rabbit model of aortic regurgitation. Total cardiac protein synthesis contributed only to the early phase of hypertrophy (3 days), whereas progressive chronic eccentric hypertrophy (at 1 month) was due to suppression of protein degradation [63]. Reduction of protein degradation rate implicates a longer life-span of myocardial contractile proteins and also longer exposure to oxidative damage that may contribute to functional impairment [1, 79].
5.5 Coronary Circulation
Both overload states may differ in the impact on coronary circulation. In pressure overload, coronary perfusion is typically diminished due to decreased coronary flow reserve [80], capillary rarefaction [81], decreased capillary density [82] and insufficient angiogenesis [83]. Substantially less information is available on volume overload states. In a dog model of severe chronic mitral regurgitation, coronary blood flow and flow reserve is preserved [84] and abnormalities in coronary blood flow do not explain the resting contractile dysfunction. However, a progressive reduction of subendocardial capillary density was also found in ACF model of volume overload [82, 85].
6 Function of Volume-Overloaded Ventricle
Assessment of ventricular performance in volume overloaded states is complicated by changed chamber geometry, mass and loading conditions. Most studies that used accurate load-independent measures of LV function were performed with the canine mitral regurgitation model [38–40, 78, 86]. More recently, similar data became available from the ACF model in rodents [60, 87].
6.1 Afterload
Immediately after creation of experimental mitral regurgitation, afterload (expressed as wall stress) is reduced due to partial emptying of left ventricle into the low pressure left atrium. Within hours, afterload returns to normal due to adaptive changes in chamber geometry [88]. In chronic compensated mitral regurgitation, mean systolic wall stress is similar to controls. With progression of the disease and with development of LV dysfunction, systolic wall stress actually increases, which may have further deleterious effects on chamber performance [70]. Volume overload due to advanced stage ACF is also characterised by increased systolic wall stress [89].
6.2 Diastolic Function
In compensated chronic volume overload, diastolic wall stress is increased [1, 40, 89], but the diastolic properties of the left ventricle are enhanced due to increased LV distensibility [90]. Corin et al. performed a detailed study of diastolic indexes in patients with mitral regurgitation using pressure-volume analysis [91]. In patients with compensated mitral regurgitation, the adaptations to chronic volume overload led to supernormal diastolic function due to the right-shift of end-diastolic pressure-volume relationship and enhanced diastolic compliance, enabling the left ventricle to operate at lower levels of filling pressures. However, in patients with mitral regurgitation and LV dysfunction, both myocardial and chamber stiffness were again increased, indicating that the late increase of LV stiffness may contribute to the clinical manifestations of congestive heart failure [91].
6.3 Acute Changes of Systolic Function
Berko et al. examined LV function in acute severe mitral regurgitations in dogs. After creation of valve insufficiency, LV stroke work and ejection fraction increased, but the slope of end-systolic pressure-volume relation (end-systolic elastance, Ees) was reduced or unchanged if Ees was normalised to end-diastolic volume [88]. The post-procedural increase in LV ejection fraction was not due to increased contractility or decreased afterload (that quickly returned to normal), but rather a consequence of increased preload. The observed disparity between LV ejection fraction and Ees supports the concept that LV function is overestimated by ejection indexes in the presence of mitral regurgitation and that ejection indexes are poor predictors of myocardial function in hearts with mitral regurgitation. Numerous clinical studies demonstrated a drop of ejection fraction in patients undergoing mitral valve replacement for severe mitral regurgitation, suggesting that after the surgery, irreversible myocardial dysfunction may become unmasked when the regurgitation is gone [88].
6.4 Chronic Changes of Systolic Function
Prolonged volume overload is associated with the insidious onset of intrinsic myocardial dysfunction. In a dog model of mitral regurgitation lasting for 3 months, Urabe et al. examined myocardial tissue composition, global LV function and contractility of isolated cardiomyocytes [92]. Reduced LV function (Ees) correlated with impaired sarcomere shortening velocity of isolated cardiomyocytes from the same individual, indicating that the contractile defect is present both at the ventricular and cellular level. Quantitative morphology showed no difference in LV interstitial volume fraction, but decreased myofibrillar volume fraction. Therefore, LV ventricular function occurs due to a combination of cardiomyocyte cell loss, intrinsic cardiomyocyte contractile dysfunction and perhaps inadequate compensatory hypertrophy.
The cause of gradual cardiomyocyte loss in a volume overloaded heart can be an increased rate of apoptosis, not matched by adequate cell replacement [93]. In patients with advanced mitral regurgitation, cardiomyocytes show an activation of pro-apoptotic signalling (Bax protein, protein p53, caspase 3, 8 and 9), although no evidence of DNA fragmentation [94]. Activation of apoptosis (increased Bax protein, caspase 3/9, TNF-α) has also been demonstrated in the advanced stage of volume overload due to ACF in rats [59, 60]. When directly compared, volume overload induces apoptosis less than a pressure overload of similar degree [60].
Intrinsic abnormalities of cardiomyocyte function due to impaired Ca2+ homeostasis and excitation/contraction coupling contributes to systolic dysfunction due to volume overload. Myocardial strips from patients undergoing mitral valve replacement demonstrated severe depression of tension generation and blunted force-frequency curve with a negative slope in exercise range of heart rate [95] and had downregulated sarcoplasmic Ca2+ -ATPase (SERCA) and Na+/Ca2+ exchanger (NCX) genes [96]. Similarly, in shunt-induced volume overload, rats with advanced ACF (10–28 weeks post-procedure) have reduced systolic function, reduced Ca2+ release from sarcoplasmic reticulum and decreased protein levels of ryanodine receptor 2 (RyR2) [97, 98] or SERCA [99]. Activated caspases [100] or calpain proteases [101] can negatively impact cardiomyocyte contractility due to partial proteolysis of contractile proteins. Increased oxidative stress from xanthine oxidase-derived ROS can also contribute to myofibrillar damage [102].
7 Time Course of Cardiac Adaptation and Remodelling in Response to Volume Overload
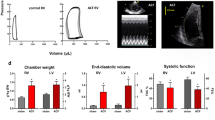
The temporal pattern of volume overload-induced cardiac remodelling and the transition of compensated cardiac hypertrophy into heart failure is best described in the rat ACF model [51, 52, 103, 104]. Severe mitral valve insufficiency induces similar cardiac changes [42, 105, 106], but heart failure syndrome is less pronounced than in shunt-induced overload. The progressive development from the early, adaptive phase to decompensated end-stage heart failure will be described in three phases for both models below (Fig. 9.1).

7.1 Early, Adaptive Phase of Volume Overload (0–2 Weeks of ACF Model)
Immediately after ACF creation, cardiac output is increased by 30–100 %, leading to enhanced venous return and an increase in cardiac filling pressures. A marked increase of diastolic wall stress activates stretch-sensitive responses which turn on pro-hypertrophic signalling pathways (Akt kinase and Wnt signalling pathway), increases ROS generation and leads to inflammatory cell infiltration [60]. The confirmed source of ROS is mitochondrial xanthine oxidase, which activity considerably increases within 24 h after ACF induction [107]. Gene expression analysis demonstrated increased transcription of genes regulating extracellular matrix, cell cycle, inflammation and apoptosis [103] which is followed by eccentric hypertrophy and cardiomyocyte elongation.
7.1.1 Extracellular Matrix Loss
The most rapid and profound changes in the early phase occurs in the extracellular matrix [104]. Partial degradation and remodelling of the extracellular matrix is responsible for enhanced diastolic ventricular compliance and biventricular enlargement, allowing to accommodate increased volume load and to maintain effective cardiac output. Ryan et al. [89] showed that immediately after ACF creation, eccentric LV remodelling is mediated by loss of interstitial collagen (by −50 % at 2nd day) and by loosening of extracellular matrix scaffold by activated matrix metalloproteinases. After several days and after the enlargement of the ventricle, the collagen volume fraction returns to normal. Bradykinin inhibitors prevented extracellular matrix loss, but ACE inhibitors potentiate it via increased bradykinin production. The lack of effect of ACE inhibitors on early volume overload-induced remodelling [89, 108] is in striking contrast to pressure overload-related or infarct-related remodelling.
7.1.2 Inflammatory and Mast Cells Infiltration
An infiltration of the myocardium with inflammatory cells, particularly mast cells, seems to play a central role in triggering the early response to volume overload. Brower et al. documented a close association between myocardial mast cell density and matrix metalloproteinase (MMP) activity after creation of ACF [109]. Mast cell accumulation after ACF induction occurs due to in situ maturation and differentiation of resident mast cells [110] and due to mast cell recruitment driven by tumour necrosis factor α (TNF-α) produced by cardiomyocytes in response to stretch [111]. Inhibition of mast cell degranulation with cromolin or nedocromyl prevented the increase in mast cell number, MMP activation and volume-induced structural changes of the heart [109, 112]. Mast cells-deficient rats displayed reduced MMP-2 activity, higher collagen content and less LV dilatation after ACF [113]. Mast cells store and release many biologically active mediators, including TNF-α, and proteolytic enzymes (tryptase, chymase) capable of activating MMPs. TNF-α neutralisation with infliximab (TNF-α antibody) attenuated myocardial inflammatory response, mast cell recruitment and prevented MMP activation and collagen depletion although it did not completely prevented cardiac remodelling [111].
This sequence of events in the early phase is not unique to the rat ACF model. Early mast cell infiltration, increased chymase and MMP-2 activity and ensuing extracellular collagen depletion has also been described in a dog model of mitral regurgitation [114]. Gene expression profiling showed marked upregulation of MMP-1, MMP-9 and extensive downregulation of pro-fibrotic growth factors (TGF-β, CTGF) and non-collagen components of extracellular matrix such as decorin, fibulin and fibrillin [105]. Patients undergoing valve replacement for severe chronic mitral regurgitation have increased myocardial and plasma levels of TNF-α and its receptors. The study also found a relationship between myocardial TNF-α expression and LV remodelling [115]. In summary, early response to volume overload is quite specific and distinctive than other causes of abnormal loading. However, it remains a question whether this phase represents a good target for therapy—lack of adaptive increase in LV end-diastolic volume after acute volume overload might be associated with poorer short-term survival [109].
7.2 Intermediate Stage of Compensated Ventricular Hypertrophy (3–10 Week in ACF Model)
After several weeks, early phase of remodelling resolves, leaving the heart in the stage of compensated eccentric hypertrophy with still relatively preserved systolic function and enhanced diastolic function. Inflammatory response in the myocardium is less intense, MMP-2 activity is lower and the extracellular matrix has become more stabilised. At this stage, animals show no clinical signs of heart failure [50], but neurohumoral activation gradually develops. By the 9th week, ACF rats have a significant increase in plasma renin concentration and activity, 50 % increase in norepinephrine, 4-times higher vasopressin concentration [116], increased aldosterone and natriuretic peptides [117]. Persistent elevation of circulating norepinephrine contributes to catecholamine-driven β-adrenergic receptor pathway desensitisation [46].
The sequence of events responsible for neurohumoral activation and transition from compensated hypertrophy into overt heart failure is summarised in Fig. 9.2. The reason for neurohumoral activation is the “underfilling” of the arterial part of vascular tree due to low systemic vascular resistance which triggers volume baroreceptor activation [33]. In addition, ACF rats have an abnormal distribution of cardiac output. Using radioactive microspheres, Flaim et al. showed that ACF rats have increased cardiac output due to high shunt flow (37 % of total output), but simultaneously have splanchnic and renal hypoperfusion [118]. Diminished renal perfusion and baroreceptor activation triggers neurohumoral response, causing sodium and water retention. Renal resistance to endogenous natriuretic peptides contributes to fluid retention—kidneys of rats with ACF have attenuated natriuretic response to acute volume loading [119] and to exogenous atrial natriuretic peptide administration [120]. The ACF model is therefore suitable for studies of cardio-renal interactions in chronic heart failure [117].

7.3 Late Stage of Decompensated Heart Failure (>15 Week of ACF Model)
Over time, the indexed heart mass (heart/body weight ratio) increases more than twice in comparison to sham-operated animals [121], the ventricle become markedly dilated but relatively thin, with decreased LV diameter/wall thickness ratio [52]. Chamber dilatation in the advanced stage is a consequence of the reappearance of local inflammatory response [103]. Total collagen content in myocardium is not increased, but MMP-2 activity is again increased, similarly as in early phase [103]. Ventricular filling pressures, lung and liver wet weights are increased due to congestion in pulmonary and systemic circulation [52]. The increase of LV wall stress and simultaneous intense neurohumoral activation promotes the vicious cycle of adverse myocardial remodelling. As a result of apoptosis-mediated cardiomyocyte cell loss [122] and of intrinsic cardiomyocyte dysfunction, LV contractile dysfunction now becomes obvious [49, 103].
In ACF model, the right ventricle became dilated and dysfunctional as well, because the right heart has to pump similar surplus of volume. Moreover, rats with advanced ACF develop moderate pulmonary hypertension (RV systolic pressure ~40–50 mmHg) [123] due to a combination of post-capillary venous hypertension and increased pulmonary arterial resistance. From that reason, right ventricular myocardium is exposed not only to excessive volume but also to pressure, leading to more right ventricular collagen accumulation [108] and higher angiotensin-II and endothelin levels than in the left ventricular myocardium [47]. Ventricular dilatation leads to functional mitral and tricuspid regurgitations that further exacerbate volume overload. Molecular features show typical heart failure fingerprint—a switch of myosin heavy chain expression from α- to β-isoform [49] and decreased calcium handling proteins (SERCA, RyR2) [99, 124]. Quantitative histology showed myocardial perivascular fibrosis, leukocyte infiltration [103], apoptosis (by TUNEL assay) and decreased subendocardial capillary density [82]. Gene expression analysis from the late stage of ACF shows the reappearance inflammatory response genes and an increased expression of genes controlling non-collagen components of the extracellular matrix [103]. One of the most upregulated proteins, both at the mRNA and protein level, was matricellular protein periostin which modulates interactions between the cardiomyocyte and extracellular matrix [103, 125]. Unbiased proteomic analysis demonstrated increased expression ROS generating enzyme monoamine oxidase A (MAO-A) and attenuated expression of two NADPH-generating enzymes (nicotinamide nucleotide transhydrogenase-NNT and isocitrate dehydrogenase-IDH2) that may contribute to an observed reduction of redox reserve, reflected by the ratio of reduced and oxidised glutathione (GSH/GSSG ratio) [124].
7.3.1 Metabolic Remodelling
Characteristic features of advanced stage heart failure are metabolic and bioenergetic abnormalities of the myocardium [103, 125]. Progressive downregulation of enzymes controlling myocardial fatty acid transport and β-oxidation have been observed in most experimental models and in patients with moderate-to-advanced heart failure [126]. Attenuated myocardial palmitate oxidation has recently been demonstrated in a volume overload ACF model [125, 127, 128]. Changes in myocardial substrate utilisation may represent a return to foetal metabolic programme when glucose is the predominant energetic source [129], or can be a consequence of secondary humoral influences due to elevated neurohormones and cytokines (angiotensin II, TNF-α) [130]. Whether downregulation of fatty acid oxidation in the failing heart has adaptive, maladaptive or neutral consequences for the course of heart failure is still intensively debated [131]. Interestingly, the increase in myocardial fatty acid utilisation induced by chronic administration of metformin in rats with ACF-induced heart failure was not associated with any change of cardiac function or prognosis [121]. A time-series examination of the gene transcription profile in the ACF model found that intense upregulation of NFκB-driven inflammatory genes in advanced ACF stage (15th week) coincided with downregulation of transcription factor PPAR-α which controls fatty acid metabolism [103]. Despite gene expression analysis which suggested downregulation of several components of respiratory chain and of ATP biogenesis, functional studies of cardiac mitochondria from compensated, intermediary stage (12 weeks) [132] or from decompensated heart failure stage (22nd week) [127] showed a preserved resting mitochondrial respiratory function, but increased vulnerability to anoxia-reoxygenation injury [132].
The advanced stage of ACF also served to explore changes in systemic metabolism induced by the presence of heart failure. Rats with ACF and established heart failure (22ndweek) had elevated circulating levels of free fatty acids and a depletion of intraabdominal fat as consequence of increased fatty acid mobilisation. However, glucose tolerance and insulin sensitivity was preserved and postprandial insulin levels were actually attenuated in ACF animals, perhaps due to splanchnic and pancreatic hypoperfusion. Despite elevated free fatty acid levels in serum, ACF animals had markedly reduced myocardial triglyceride content (by 50 % compared to sham), indicating that myocardial triglyceride levels are not dependent on fatty acid delivery and that myocardial lipid overload does not occur in this heart failure model [125].
7.3.2 Electric Remodelling
Volume-overload induced heart hypertrophy is associated with an increased risk of arrhythmias. In long-term survival studies in the ACF model, a third of rats with ACF died suddenly [50, 52], presumably due to arrhythmic death. Given the known lack of fibrosis, myocardial substrate for increased electrical instability is unknown. Recent immunohistochemic analysis of junctions in rats with ACF demonstrated normal localisation and distribution of myocardial connexins, but a 60 % reduction of phosphorylated connexin-43 isoform which may be responsible for slower conduction velocity and an increased risk of re-entrant arrhythmias [121]. The connexin-43 phosphorylation state was normal in earlier ACF stage (11th week), suggesting that heart failure, but not cardiac hypertrophy itself, contributed to connexin hypophosphorylation.
7.3.3 Advanced stage of Mitral Regurgitation
Only a few studies have examined the molecular characteristics of late stages of mitral regurgitation. Pu et al. showed that chronic mitral regurgitation in rats led to a myosin heavy chain shift, SERCA2 downregulation and impaired LV contractility. One year survival in the rats with mitral regurgitation was only 38 %. From echocardiographic variables, increased end-diastolic LV diameter at 6th week, but not LV mass or fractional shortening, was the first parameter that significantly predicted mortality [42]. Kim et al. recently reported a more detailed description of the course of heart changes of the same model at 4-month duration. Immediately after creation of mitral regurgitation, an increase of LV end-diastolic dimension and ejection fraction was observed, followed by LV thinning, progressive LV dilatation and finally by a modest decrease in ejection fraction by the 12th week. By the end of the study, end-systolic elastance was reduced and lung weights were increased, suggesting the onset of heart failure. Structural analysis of the myocardium showed perivascular fibrosis, increased apoptosis and upregulation of inflammatory genes (interleukin-6 and interleukin-16). Despite the fact that the authors did not provide any evidence for phosphodiesterase-5 upregulation in volume overloaded heart, chronic sildenafil therapy in this experiment reduced LV dilatation and LV mass, improved LV contractility, increased exercise tolerance, attenuated apoptosis (by 50 %) and inflammatory response [44]. This study suggests that enhancement of cyclic guanosine monophosphate (cGMP)-dependent signalling deactivates pro-hypertrophic pathways similarly as in pressure overload [72].
8 Possible Therapeutic Targets
Because volume overload is primarily a defect of hemodynamics, the most effective therapy is surgical correction of increased hemodynamic stress. In an experimental model of severe chronic mitral regurgitation in dogs, valve replacement restored previously depressed load-independent indexes of contractile function [133]. Similarly, Hutchinson et al. recently reported a reversal of LV ventricular structure and function at 4 weeks of volume overload due to aorto-caval fistula using pressure-volume analysis. After closure of the shunt with aortic stentgraft, LV dimensions returned early to normal within 4 weeks, but LV systolic and diastolic function needed 11 weeks to normalise [87]. In clinical scenarios, a radical repair of hemodynamic defect is sometimes not possible and attempts to alleviate unfavourable course of the disease with drugs are then mandated. Potential pharmaceutical targets for therapy are briefly summarised in Table 9.4. It should be noted that the beneficial effects of drugs may be specific for a particular disease phase. For example, ACE inhibitors have no direct effect on the early phase of volume remodelling, but they attenuate renin-angiotensin system activation and decrease mortality in the late stage of ACF [134].
9 Conclusions
Cardiac adaptation to a pathological increase in volume load differs in many ways from pressure overload or post-myocardial remodelling. The early response to volume overload is characterised by increased diastolic wall stress, activation of specific signalling pathways, cardiomyocyte elongation, inflammatory infiltration and degradation of extracellular matrix which allows for the increase of the chamber size. Volume overload leads to eccentric cardiac hypertrophy with increased ventricular volume, reduced relative wall thickness, enhanced diastolic function and relatively preserved systolic function. Although initially well tolerated, pathological volume overload is inevitably a progressive condition. As a result of neurohumoral activation, cardiomyocyte dysfunction and cell loss due to apoptosis, overt end-stage heart failure develops in the end. Understanding the pathophysiology of volume-overload cardiac remodelling is a mandatory step for the new development and proper timing of novel surgical and pharmaceutical therapies which may prevent or postpone the onset of heart failure.
Abbreviations
- LV:
-
Left ventricle
- ACF:
-
Aorto-caval fistula
- Ees:
-
End-systolic elastance
- ROS:
-
Reactive oxygen species
- ACE:
-
Angiotensin-converting enzyme
- MMP:
-
Matrix metalloproteinase
- TNF:
-
Tumour necrosis factor
References
Carabello BA (2012) Volume overload. Heart Fail Clin 8:33–42
Adler CP, Costabel U (1975) Cell number in human heart in atrophy, hypertrophy, and under the influence of cytostatics. Recent Adv Stud Cardiac Struct Metab 6:343–355
Laflamme MA, Murry CE (2011) Heart regeneration. Nature 473:326–335
Bergmann O, Bhardwaj RD, Bernard S et al (2009) Evidence for cardiomyocyte renewal in humans. Science 324:98–102
Dorn GW 2nd (2007) The fuzzy logic of physiological cardiac hypertrophy. Hypertension 49:962–970
Kehat I, Molkentin JD (2010) Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 122:2727–2735
Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP (1990) Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322:1561–1566
Baggish AL, Wood MJ (2011) Athlete’s heart and cardiovascular care of the athlete: scientific and clinical update. Circulation 123:2723–2735
duManoir GR, Haykowsky MJ, Syrotuik DG, Taylor DA, Bell GJ (2007) The effect of high- intensity rowing and combined strength and endurance training on left ventricular systolic function and morphology. Int J Sports Med 28:488–494
Baggish AL, Wang F, Weiner RB et al (2008) Training-specific changes in cardiac structure and function: a prospective and longitudinal assessment of competitive athletes. J Appl Physiol 104:1121–1128
Milliken MC, Stray-Gundersen J, Peshock RM, Katz J, Mitchell JH (1988) Left ventricular mass as determined by magnetic resonance imaging in male endurance athletes. Am J Cardiol 62:301–305
Baggish AL, Yared K, Wang F et al (2008) The impact of endurance exercise training on left ventricular systolic mechanics. Am J Physiol Heart Circ Physiol 295:H1109–H1116
Pelliccia A, Kinoshita N, Pisicchio C, et al. (2010) Long-term clinical consequences of intense, uninterrupted endurance training in olympic athletes. J Am Coll Cardiol 55:1619–1625
Scharhag J, Schneider G, Urhausen A et al (2002) Athlete’s heart: right and left ventricular mass and function in male endurance athletes and untrained individuals determined by magnetic resonance imaging. J Am Coll Cardiol 40:1856–1863
Pelliccia A, Maron BJ, Di Paolo FM et al (2005) Prevalence and clinical significance of left atrial remodeling in competitive athletes. J Am Coll Cardiol 46:690–696
Maron BJ, Pelliccia A, Spirito P (1995) Cardiac disease in young trained athletes. Insights into methods for distinguishing athlete’s heart from structural heart disease, with particular emphasis on hypertrophic cardiomyopathy. Circulation 91:1596–15601
Pelliccia A, Maron BJ, Spataro A, Proschan MA, Spirito P (1991) The upper limit of physiologic cardiac hypertrophy in highly trained elite athletes. N Engl J Med 324:295–301
Pelliccia A, Maron BJ, De Luca R et al (2002) Remodeling of left ventricular hypertrophy in elite athletes after long-term deconditioning. Circulation 105:944–949
Libby P (2008) Braunwald’s heart disease: a textbook of cardiovascular medicine. Saunders/Elsevier, Philadelphia
Katz R, Karliner JS, Resnik R (1978) Effects of a natural volume overload state (pregnancy) on left ventricular performance in normal human subjects. Circulation 58:434–441
Schannwell CM, Zimmermann T, Schneppenheim M et al (2002) Left ventricular hypertrophy and diastolic dysfunction in healthy pregnant women. Cardiology 97:73–78
Eghbali M, Deva R, Alioua A et al (2005) Molecular and functional signature of heart hypertrophy during pregnancy. Cir Res 96:1208–1216
Lemmens K, Doggen K, De Keulenaer GW (2011) Activation of the neuregulin/ErbB system during physiological ventricular remodeling in pregnancy. Am J Physiol Heart Circ Physiol 300:H931–H942
Andersen JB, Rourke BC, Caiozzo VJ, Bennett AF, Hicks JW (2005) Physiology: postprandial cardiac hypertrophy in pythons. Nature 434:37–38
Riquelme CA, Magida JA, Harrison BC et al (2005) Fatty acids identified in the Burmese python promote beneficial cardiac growth. Science 334:528–531
Ling LH, Enriquez-Sarano M, Seward JB et al (1966) Clinical outcome of mitral regurgitation due to flail leaflet. N Engl J Med 335:1417–1423
Ahmed MI, McGiffin DC, O’Rourke RA, Dell’Italia LJ (2009) Mitral regurgitation. Curr Probl Cardiol 34:93–136
Anderson CB, Codd JR, Graff RA et al (1976) Cardiac failure and upper extremity arteriovenous dialysis fistulas. Case reports and a review of the literature. Arch Intern Med 136:292–297
Epstein FH, Post RS, McDowell M (1953) The effects of an arteriovenous fistula on renal hemodynamics and electrolyte excretion. J Clin Invest 32:233–241
Glaser R, Kramer RJ, Hamby RI, Aintablian A, Zeldis SM (1978) Renal arteriovenous fistula masquerading as severe valvar heart disease. Brit Heart J 40:972–975
Peery WH (1987) Clinical spectrum of hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease). Am J Med 82:989–997
Anand IS, Florea VG (2001) High output cardiac failure. Curr Treat Options Cardiovasc Med 3:151–159
Schrier RW (1988) Pathogenesis of sodium and water retention in high-output and low-output cardiac failure, nephrotic syndrome, cirrhosis, and pregnancy (1). N Engl J Med 319:1065–7210
Anand IS, Chandrashekhar Y, Ferrari R et al (1992) Pathogenesis of congestive state in chronic obstructive pulmonary disease. Studies of body water and sodium, renal function, hemodynamics, and plasma hormones during edema and after recovery. Circulation 86:12–21
Messerli FH, Sundgaard-Riise K, Reisin E et al (1983) Disparate cardiovascular effects of obesity and arterial hypertension. Am J Med 74:808–812
Kaltman AJ, Goldring RM (1976) Role of circulatory congestion in the cardiorespiratory failure of obesity. Am J Med 60:645–653
Anand IS, Chandrashekhar Y, Wander GS, Chawla LS (1995) Endothelium-derived relaxing factor is important in mediating the high output state in chronic severe anemia. J Am CollCardiol 25:1402–1407
Kleaveland JP, Kussmaul WG, Vinciguerra T, Diters R, Carabello BA (1988) Volume overload hypertrophy in a closed-chest model of mitral regurgitation. Am J Physiol 254:H1034–H1041
Carabello BA, Nakano K, Corin W, Biederman R, Spann JF Jr (1989) Left ventricular function in experimental volume overload hypertrophy. T Am J Physiol 256:H974–H981
Carabello BA, Zile MR, Tanaka R (1992) Cooper Gt. Left ventricular hypertrophy due to volume overload versus pressure overload. Am J Physiol 263:H1137–H1144
Neilan TG, Ton-Nu TT, Kawase Y, et al. (2008) Progressive nature of chronic mitral regurgitation and the role of tissue Doppler-derived indexes. American journal of physiology. Heart Circ Physiol 294:H2106–H2111
Pu M, Gao Z, Zhang X, Liao D et al (2009) Impact of mitral regurgitation on left ventricular anatomic and molecular remodeling and systolic function: implication for outcome. Am J Physiol Heart Circ Physiol 296:H1727–H1732
Pu M, Gao Z, Li J, Sinoway L, Davidson WR Jr (2005) Development of a new animal model of chronic mitral regurgitation in rats under transesophageal echocardiographic guidance. J Am Soc Echocardiogr y 18:468–474
Kim KH, Kim YJ, Ohn JH et al (2012) Long-Term effects of sildenafil in a rat model of chronic mitral regurgitation: benefits of ventricular remodeling and exercise capacity. Circulation 125:1390–1401
Liu Z, Hilbelink DR, Crockett WB, Gerdes AM (1991) Regional changes in hemodynamics and cardiac myocyte size in rats with aortocaval fistulas. 1. Developing and established hypertrophy. Circ Res 69:52–58
Hammond HK, Roth DA, Insel PA et al (1992) Myocardial beta-adrenergic receptor expression and signal transduction after chronic volume-overload hypertrophy and circulatory congestion. Circulation 85:269–280
Modesti PA, Vanni S, Bertolozzi I et al (2004) Different growth factor activation in the right and left ventricles in experimental volume overload. Hypertension 43:101–108
Garcia R, Diebold S (1990) Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc Res 24:430–432
Wang X, Ren B, Liu S et al (2003) Characterization of cardiac hypertrophy and heart failure due to volume overload in the rat. J Appl Physiol 94:752–763
Melenovsky V, Skaroupkova P, Benes J et al (2011) The course of heart failure development and mortality in rats with volume overload due to Aorto-Caval Fistula. Kidney Blood Press Res 35:167–173
Brower GL, Henegar JR, Janicki JS (1996) Temporal evaluation of left ventricular remodeling and function in rats with chronic volume overload. Am J Physio 271:H2071–H2078
Brower GL, Janicki JS (2001) Contribution of ventricular remodeling to pathogenesis of heart. Am J Physiol Heart Circ Physiol 280:H674–H683
Souzeau E, Llamas B, Belanger S, Picard S, Deschepper CF (2006) A genetic locus accentuates the effect of volume overload on adverse left ventricular remodeling in male and female rats. Hypertension 47:128–133
Oliver-Dussault C, Ascah A, Marcil M et al (2010) Early predictors of cardiac decompensation in experimental volume overload. Mol Cell Biochem 338:271–282
Langenickel T, Pagel I, Hohnel K, Dietz R, Willenbrock R (2000) Differential regulation of cardiac ANP and BNP mRNA in different stages of experimental heart failure. Am J Physiol Heart Circ Physiol 278:H1500–H1506
Huang M, LeBlanc MH, Hester RL (1994) Evaluation of the needle technique for producing an arteriovenous fistula. J Appl Physio 77:2907–2911
Brower GL, Gardner JD, Janicki JS (2003) Gender mediated cardiac protection from adverse ventricular remodeling is abolished by ovariectomy. Mol Cell Biochem 251:89–95
Chancey AL, Gardner JD, Murray DB, Brower GL, Janicki JS (2005) Modulation of cardiac mast cell-mediated extracellular matrix degradation by estrogen. Am J Physiol Heart Circ Physiol 289:H316–H321
Dent MR, Tappia PS, Dhalla NS (2010) Gender differences in apoptotic signaling in heart failure due to volume overload. Apoptosis 15:499–510
Toischer K, Rokita AG, Unsold B et al (2010) Differential cardiac remodeling in preload versus afterload. Circulation 122:993–1003
Scheuermann-Freestone M, Freestone NS, Langenickel TH (2001) A new model of congestive heart failure in the mouse due to chronic volume overload. Eur J Heart Fail 3:535–543
Magid NM, Young MS, Wallerson DC et al (1988) Hypertrophic and functional response to experimental chronic aortic regurgitation. J Mol Cell Cardiol 20:239–246
Magid NM, Borer JS, Young MS, Wallerson DC, DeMonteiro C (1993) Suppression of protein degradation in progressive cardiac hypertrophy of chronic aortic regurgitation. Circulation 87:1249–1257
Arsenault M, Plante E, Drolet MC, Couet J (2002) Experimental aortic regurgitation in rats under echocardiographic guidance. J Heart Valve Dis 11:128–134
Champetier S, Bojmehrani A, Beaudoin J et al (2009) Gene profiling of left ventricle eccentric hypertrophy in aortic regurgitation in rats: rationale for targeting the beta-adrenergic and renin-angiotensin systems. Am J Physiol Heart Circ Physiol 296:H669–H677
Zendaoui A, Lachance D, Roussel E, Couet J, Arsenault M (2011) Usefulness of carvedilol in the treatment of chronic aortic valve regurgitation. Circ Heart Fail 4:207–213
Lachance D, Plante E, Bouchard-Thomassin AA et al (2009) Moderate exercise training improves survival and ventricular remodeling in an animal model of left ventricular volume overload. Circ Heart Fail 2:437–445
Plante E, Gaudreau M, Lachance D et al (2004) Angiotensin-converting enzyme inhibitor captopril prevents volume overload cardiomyopathy in experimental chronic aortic valve regurgitation. Can J Physio Pharmacol 82:191–199
Grossman W, Jones D, McLaurin LP (1975) Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest 56:56–64
Corin WJ, Monrad ES, Murakami T et al (1987) The relationship of afterload to ejection performance in chronic mitral regurgitation. Circulation 76:59–67
Esposito G, Rapacciuolo A, Naga Prasad SV et al (2002) Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation 105:85–92
Takimoto E, Champion HC, Li M et al (2005) Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 11:214–222
Kupari M, Turto H, Lommi J (2005) Left ventricular hypertrophy in aortic valve stenosis: preventive or promotive of systolic dysfunction and heart failure? Eur Heart J 26:1790–1796
Meerson FZ (1961) On the mechanism of compensatory hyperfunction and insufficiency of the heart. Cor et vasa 3:161–177
Buyandelger B, Ng KE, Miocic S et al (2011) Genetics of mechanosensation in the heart. J Cardiovasc Transl Res 4:238–244
Yamamoto K, Dang QN, Maeda Y et al (2001) Regulation of cardiomyocyte mechanotransduction by the cardiac cycle. Circulation 103:1459–1464
Imamura T, McDermott PJ, Kent RL et al. (1994) Acute changes in myosin heavy chain synthesis rate in pressure versus volume overload. Circ Res 75:418–425
Matsuo T, Carabello BA, Nagatomo Y et al (1998) Mechanisms of cardiac hypertrophy in canine volume overload. Am J Physiol 275:H65–H74
Prochniewicz E, Lowe DA, Spakowicz DJ et al (2008) Functional, structural, and chemical changes in myosin associated with hydrogen peroxide treatment of skeletal muscle fibers. Am J Physiol Cell physio 294:C613–C626
Marcus ML, Doty DB, Hiratzka LF, Wright CB, Eastham CL (1982) Decreased coronary reserve: a mechanism for angina pectoris in patients with aortic stenosis and normal coronary arteries. New Eng J Med 307:1362–1366
Perrino C, Naga Prasad SV, Mao L et al (2006) Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest 116:1547–1560
Michel JB, Salzmann JL, Ossondo Nlom M et al (1986) Morphometric analysis of collagen network and plasma perfused capillary bed in the myocardium of rats during evolution of cardiac hypertrophy. Basic Res i Cardiol 81:142–154
Sano M, Schneider MD (2002) Still stressed out but doing fine: normalization of wall stress is superfluous to maintaining cardiac function in chronic pressure overload. Circulation 105:8–10
Carabello BA, Nakano K, Ishihara K et al (1991) Coronary blood flow in dogs with contractile dysfunction due to experimental volume overload. Circulation 83:1063–1075
Rakusan K, Moravec J, Hatt PY (1980) Regional capillary supply in the normal and hypertrophied rat heart. Microvasc Res 20:319–326
Tsutsui H, Spinale FG, Nagatsu M et al (1994) Effects of chronic beta-adrenergic blockade on the left ventricular and cardiocyte abnormalities of chronic canine mitral regurgitation. J Clin Invest 93:2639–2648
Hutchinson KR, Guggilam A, Cismowski MJ, Galantowicz ML, West TA, Stewart JA Jr, Zhang X, Lord KC, Lucchesi PA (2011) Temporal pattern of left ventricular structural and functional remodeling following reversal of volume overload heart failure. J Appl Physiol 111:1778–1788
Berko B, Gaasch WH, Tanigawa N, Smith D, Craige E (1987) Disparity between ejection and end- systolic indexes of left ventricular contractility in mitral regurgitation. Circulation 75:1310–1319
Ryan TD, Rothstein EC, Aban I et al (2007) Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede ardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol 49:811–821
Zile MR, Tomita M, Nakano K et al (1991) Effects of left ventricular volume overload produced by mitral regurgitation on diastolic function. Am J Physiol 261:H1471–H1480
Corin WJ, Murakami T, Monrad ES, Hess OM, Krayenbuehl HP (1991) Left ventricular passive diastolic properties in chronic mitral regurgitation. Circulation 83:797–807
Urabe Y, Mann DL, Kent RL et al (1992) Cellular and ventricular contractile dysfunction in experimental canine mitral regurgitation. Circ Res 70:131–147
Wencker D, Chandra M, Nguyen K et al (2003) A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 111:1497–1504
Moorjani N, Westaby S, Narula J et al (2009) Effects of left ventricular volume overload on mitochondrial and death-receptor-mediated apoptotic pathways in the transition to heart failure. J Am Coll Cardiol 103:1261–1268
Mulieri LA, Leavitt BJ, Martin BJ, Haeberle JR, Alpert NR (1993) Myocardial force-frequency defect in mitral regurgitation heart failure is reversed by forskolin. Circulation 88:2700–2704
Leszek P, Szperl M, Klisiewicz AJ et al (2007) Alteration of myocardial sarcoplasmic reticulum Ca2+ -ATPase and Na+ -Ca2+ exchanger expression in human left ventricular volume overload. Eur J Heart Fail 9:579–586
Juric D, Yao X, Thandapilly S, Louis X et al (2010) Defects in ryanodine receptor function are associated with systolic dysfunction in rats subjected to volume overload. Experi Physiol 95:869–879
Ding YF, Brower GL, Zhong Q et al (2008) Defective intracellular Ca2 + homeostasis contributes to myocyte dysfunction during ventricular remodelling induced by chronic volume overload in rats. Clin Exp Pharmacol Physiol 35:827–835
Takewa Y, Chemaly ER, Takaki M et al (2009) Mechanical work and energetic analysis of eccentric cardiac remodeling in a volume overload heart failure in rats. Am J Physiol Heart Circ Physiol 296:H1117–H1124
Communal C, Sumandea M, de Tombe P et al (2002) Functional consequences of caspase activation in cardiac myocytes. Proc Natl Acad Sci USA 99:6252–6256
Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM Jr et al (2001) Preload induces troponin I degradation independently of myocardial ischemia. Circulation 103:2035–2037
Ahmed MI, Gladden JD, Litovsky SH et al (2010) Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction >60 %. J Am Coll Cardiol 55:671–679
Chen YW, Pat B, Gladden JD et al (2011) Dynamic molecular and histopathological changes in the extracellular matrix and inflammation in the transition to heart failure in isolated volume overload. Am J Physiol Heart Circ Physiol 300:H2251–H2260
Hutchinson KR, Stewart JA Jr, Lucchesi PA (2010) Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol 48:564–569
Zheng J, Chen Y, Pat B et al (2009) Microarray identifies extensive downregulation of noncollagen extracellular matrix and profibrotic growth factor genes in chronic isolated mitral regurgitation in the dog. Circulation 119:2086–2095
Kim KH, Kim YJ, Lee SP et al (2011) Survival, exercise capacity, and left ventricular remodeling in a rat model of chronic mitral regurgitation: serial echocardiography and pressure-volume analysis. Korean Circ J 41:603–611
Gladden JD, Zelickson BR, Wei CC et al (2011) Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free Radic Biol Med 51:1975–1984
Ruzicka M, Keeley FW, Leenen FH (1994) The renin-angiotensin system and volume overload- induced changes in cardiac collagen and elastin. Circulation 90:1989–1996
Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS (2002) Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol 283:H518–H525
Levick SP, Melendez GC, Plante E et al (2011) Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovasc Res 89:12–19
Chen Y, Pat B, Zheng J et al (2010) Tumor necrosis factor-alpha produced in cardiomyocytes mediates a predominant myocardial inflammatory response to stretch in early volume overload. J Mol Cell Cardiol 49:70–78
Brower GL, Janicki JS (2005) Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J Card Fail 11:548–556
Levick SP, Gardner JD, Holland M et al (2008) Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J Mol Cell Cardiol 45:56–61
Stewart JA Jr, Wei CC, Brower GL et al (2003) Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J Mol Cell Cardiol 35:311–319
Oral H, Sivasubramanian N, Dyke DB et al (2003) Myocardial proinflammatory cytokine expression and left ventricular remodeling in patients with chronic mitral regurgitation. Circulation 107:831–837
Riegger GA, Liebau G, Bauer E, Kochsiek K (1985) Vasopressin and renin in high output heart ailure of rats: hemodynamic effects of elevated plasma hormone levels. J Cardiovasc Pharmacol 7:1–5
Abassi Z, Goltsman I, Karram T, Winaver J, Hoffman A (2011) Aortocaval fistula in rat: a unique model of volume-overload congestive heart failure and cardiac hypertrophy. J Biomed Biotechnol 2011:729497
Flaim SF, Minteer WJ (1980) Ventricular volume overload alters cardiac output distribution in rats during exercise. J Appl Physiol 49:482–490
Willenbrock R, Pagel I, Scheuermann M et al (1999) Renal function in high-output heart failure in rats: role of endogenous natriuretic peptides. J Am Soc Nephrol 10:572–580
Wilkins MR, Settle SL, Stockmann PT, Needleman P (1999) Maximizing the natriuretic effect of endogenous atriopeptin in a rat model of heart failure. Proc Natl Acad Sci USA 87:6465–6469
Benes J Jr, Melenovsky V, Skaroupkova P, Pospisilova J, Petrak J, Cervenka L, Sedmera D (2011) Myocardial morphological characteristics and proarrhythmic substrate in the rat model of heart failure due to chronic volume overload. Anat Rec (Hoboken) 294:102–111
Dent MR, Das S, Dhalla NS (2007) Alterations in both death and survival signals for apoptosis in heart failure due to volume overload. J Mol Cell Cardiol 43:726–732
Liu Z, Hilbelink DR, Gerdes AM (1991) Regional changes in hemodynamics and cardiac myocyte size i n rats with aortocaval fistulas. 2. Long-term effects. Circ Res 69:59–65
Petrak J, Pospisilova J, Sedinova M et al (2011) Proteomic and transcriptomic analysis of heart f ailure due to volume overload in a rat aorto-caval fistula model provides support for new potential therapeutic targets—monoamine oxidase A and transglutaminase 2. Proteome Sci 9:69
Melenovsky V, Benes J, Skaroupkova P et al (2011) Metabolic characterization of volume overload heart failure due to aorto-caval fistula in rats. Mol Cell Biochem 354:83–96
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
Benes J, Kazdova L, Drahota Z, et al. (2011) Effect of metformin therapy on cardiac function and survival in a volume-overload model of heart failure in rats. Clin Sci (London) 121:29–41
Christian B, El Alaoui-Talibi Z, Moravec M, Moravec J (1998) Palmitate oxidation by the mitochondria from volume-overloaded rat hearts. Mol Cell Biochem 180:117–128
Razeghi P, Young ME, Alcorn JL et al (2001) Metabolic gene expression in fetal and failing human heart. Circulation 104:2923–2931
Pellieux C, Montessuit C, Papageorgiou I, Lerch R (2009) Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumour necrosis factor- alpha. Cardiovas Res 82:341–350
van Bilsen M, van Nieuwenhoven FA, van der Vusse GJ (2009) Metabolic remodelling of the failing heart: beneficial or detrimental? Cardiovasc Res 81:420–428
Marcil M, Ascah A, Matas J et al (2006) Compensated volume overload increases the vulnerability of heart mitochondria without affecting their functions in the absence of stress. J Mol Cell Cardiol 41:998–1009
Nakano K, Swindle MM, Spinale F, Ishihara K, Kanazawa S, Smith A, Biederman RW, Clamp L, Hamada Y, Zile MR et al (1991) Depressed contractile function due to canine mitral regurgitation improves after correction of the volume overload. J Clin Invest 87:2077–2086
Oka T, Nishimura H, Ueyama M, Kubota J, Kawamura K (1993) Lisinopril reduces cardiac hypertrophy and mortality in rats with aortocaval fistula. Eur J Pharmacol 234:55–60
Spinale FG, Ishihra K, Zile M et al (1993) Structural basis for changes in left ventricular function and geometry because of chronic mitral regurgitation and after correction of volume overload. J thoracic Cardiovasc Surg 106:1147–1157
Gerdes AM, Clark LC, Capasso JM (1995) Regression of cardiac hypertrophy after closing an aortocaval fistula in rats. Am J Physiol 268:H2345–H2351
Abassi ZA, Brodsky S, Karram T et al (2001) Temporal changes in natriuretic and antinatriuretic systems after closure of a large arteriovenous fistula. Cardiovasc Res 51:567–576
Ruzicka M, Yuan B, Leenen FH (1994) Effects of enalapril versus losartan on regression of volume overload-induced cardiac hypertrophy in rats. Circulation 90:484–491
Perry GJ, Wei CC, Hankes GH et al (2002) Angiotensin II receptor blockade does not improve left ventricular function and remodeling in subacute mitral regurgitation in the dog. J Am Coll Cardiol 39:1374–1379
Karram T, Abbasi A, Keidar S et al (2005) Effects of spironolactone and eprosartan on cardiac remodeling and angiotensin-converting enzyme isoforms in rats with experimental heart failure. Am J Physiol Heart Circ Physiol 289:H1351–H1358
Francis B, Winaver J, Karram T, Hoffman A, Abassi Z (2004) Renal and systemic effects of chronic blockade of ET(A) or ET(B) receptors in normal rats and animals with experimental heart failure. J Cardiovasc Pharmacol 44(Suppl 1):S54–S58
Wei CC, Lucchesi PA, Tallaj J et al (2003) Cardiac interstitial bradykinin and mast cells modulate pattern of LV remodeling in volume overload in rats. Am J Physiol Heart Circ Physiol 285:H784–H792
Ulasova E, Gladden JD, Chen Y et al (2011) Loss of interstitial collagen causes structural and functional alterations of cardiomyocyte subsarcolemmal mitochondria in acute volume overload. J Mol Cell Cardiol 50:147–156
Sabri A, Rafiq K, Seqqat R, Kolpakov MA, Dillon R, Dell’italia LJ (2008) Sympathetic activation causes focal adhesion signaling alteration in early compensated volume overload attributable to isolated mitral regurgitation in the dog. Circ Res 102:1127–1136
Shyu KG, Lu MJ, Chang H et al (2005) Carvedilol modulates the expression of hypoxia- inducible factor-1 alpha and vascular endothelial growth factor in a rat model of volume- overload heart failure. J Card Fail 11:152–159
Pat B, Killingsworth C, Chen Y et al (2010) Mast cell stabilization decreases cardiomyocyte and LV function in dogs with isolated mitral regurgitation. J Card Fail 16:769–776
Pat B, Chen Y, Killingsworth C et al (2010) Chymase inhibition prevents fibronectin and myofibrillar loss and improves cardiomyocyte function and LV torsion angle in dogs with isolated mitral regurgitation. Circulation 122:1488–1495
Melendez GC, Li J, Law BA et al (2011) Substance P induces adverse myocardial remodelling via a mechanism involving cardiac mast cells. Cardiovasc Res 92:420–429
Jobe LJ, Melendez GC, Levick SP, Du Y, Brower GL, Janicki JS (2009) TNF-alpha inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am J Physiol Heart Circ Physiol 297:H1462–H1468
Abassi ZA, Barac YD, Kostin S et al (2011) TVP1022 attenuates cardiac remodeling and kidney dysfunction in experimental volume overload-induced congestive heart failure. Circ Heart Fail 4:463–473
Kawase Y, Ly HQ, Prunier F et al (2008) Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol 51:1112–1119
Wegner M, Hirth-Dietrich C, Stasch JP (1996) Role of neutral endopeptidase 24.11 in AV fistular rat model of heart failure. Cardiovas Res 31:891–898
Abassi ZA, Yahia A, Zeid S et al (2005) Cardiac and renal effects of omapatrilat, a vasopeptidase inhibitor, in rats with experimental congestive heart failure. Am J Physiol Heart Circ Physiol 288:H722–H728
Abassi Z, Burnett JC Jr, Grushka E et al (1991) Atrial natriuretic peptide and renal cGMP in rats with experimental heart failure. Am J Physiol 261:R858–R864
Bishara B, Shiekh H, Karram T et al (2008) Effects of novel vasopressin receptor antagonists on renal function and cardiac hypertrophy in rats with experimental congestive heart failure. J Pharmacol Exp Ther 326:414–422
Gardner JD, Murray DB, Voloshenyuk TG et al (2010) Estrogen attenuates chronic volume overload induced structural and functional remodeling in male rat hearts. Am J Physiol Heart Circ Physiol 298:H497–H504
Goltsman I, Wang X, Lavallie ER et al. (2011) Effects of chronic rosiglitazone treatment on renal. Circ Heart Fail 4:345–354
Nemoto S, Razeghi P, Ishiyama M et al (2005) PPAR-gamma agonist rosiglitazone ameliorates ventricular dysfunction in experimental chronic mitral regurgitation. Am J Physiol Heart Circ Physiol 288:H77–H82
El Alaoui-Talibi Z, Guendouz A, Moravec M, Moravec J (1997) Control of oxidative metabolism in volume-overloaded rat hearts: effect of propionyl-L-carnitine. Am J Physiol 272:H1615–H1624
Acknowledgments
Special thanks to Thomas T. O´Hearn, II for proofreading and editing the text. This work was supported by the project (Ministry of Health, Czech Republic) for development of research organization 00023001 (IKEM, Prague, Czech Republic)—Institutional support, by grant of Ministry of Education (MSMT-1MO510 and KONTAKT LH12052), EU Operational Program Prague—Competitiveness (project “CEVKOON” CZ.2.16/3.1.00/22126) and the Grant Agency of the Academy of Sciences of the Czech Republic (305/09/1390).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Melenovsky, V. (2013). Cardiac Adaptation to Volume Overload. In: Ostadal, B., Dhalla, N. (eds) Cardiac Adaptations. Advances in Biochemistry in Health and Disease, vol 4. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5203-4_9
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5203-4_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5202-7
Online ISBN: 978-1-4614-5203-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)