
Abstract
Metabolic interactions between adipose tissue and the heart may play an active role in progression of heart failure (HF). The aim of the study was to examine changes in myocardial and adipose tissue metabolism and gene expression in a rat HF model induced by chronic volume overload. HF was induced by volume overload from aorto-caval fistula (ACF) in 3-month-old male Wistar rats and animals were studied in the phase of decompensated HF (22nd week). HF rats showed marked eccentric cardiac hypertrophy, pulmonary congestion, increased LV end-diastolic pressure, and intraabdominal fat depletion. HF rats had preserved glucose tolerance, but increased circulating free fatty acids (FFA) and attenuated insulin response during oral glucose challenge. Isolated organ studies showed preserved responsiveness of adipose tissue lipolysis and lipogenesis to epinephrine and insulin in ACF. The heart of HF animals had markedly reduced triglyceride content (almost to half of controls), attenuated anti-oxidative reserve (GSH/GSSG), upregulated HF markers (ANP, periostin, thrombospondin-4), specific signaling pathways (Wnt, TGF-β), and downregulated enzymes of mitochondrial fatty acid oxidation, citric acid cycle, and respiratory chain. Adipose tissue transcription profiling showed upregulated receptor for gastric inhibitory polypeptide. In conclusion, ACF-induced HF model displays several deregulations of systemic metabolism. Despite elevation of systemic FFAs, myocardial triglycerides are low and insulin levels are attenuated, arguing against a role of lipotoxicity or insulin resistance in this model. Attenuated postprandial insulin response and relative lack of its antilipolytic effects may facilitate intraabdominal fat depletion observed in ACF-HF animals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure is often associated with derangements of systemic and myocardial fat metabolism that may have a role in the disease progression [1–5]. Fatty acids (FA) represent the predominant energetic substrate for the heart, covering 50–70% of myocardial ATP need [2]. The majority of FA supplied to the heart originates from adipose tissue. FA are released from adipocytes by lipolysis and transported in the bloodstream as free FA (FFA). After the entry into a cardiomyocyte, FA is either imported into mitochondria and oxidized or re-esterified into triglycerides (TG) and stored [6].
In chronic HF, excessive fat mobilization and weight loss is associated with poor prognosis [7]. Characteristics of adipose tissue in HF and the mechanisms of adverse effects of fat mobilization on cardiac function remain poorly understood. Circulating FFA are often elevated [8, 9] due to increased levels of prolipolytic hormones (catecholamines, TNF-α, and angiotensin-II) [10] that dominate over of antilipolytic factors, primarily insulin. Elevated circulating FFA can also contribute to insulin resistance [11] that is independently associated with adverse prognosis in HF [12].
Myocardial metabolism in HF is characterized by downregulation of enzymes of β-oxidation of FA and of other mitochondrial enzymes [3, 13, 14]. It has been suggested that imbalance between myocardial FFA delivery and utilization may lead to myocardial FA overload, TG over-accumulation, altered gene transcription due to lipid-responsive regulatory elements [3], and to deposition of toxic lipid intermediates [15] that can promote cardiac dysfunction. Evidence for cardiac “lipotoxicity” was found in obese rats [16], in transgenic animals [17] and in diabetic or obese end-stage HF patients [15]. The extent of myocardial lipid accumulation in HF due to hemodynamic overload, without concomitant diabetes or obesity, has never been addressed even in animal models.
Despite volume overload is clinically common cause or contributing factor of HF, it is far less studied than pressure overload or chronic myocardial infarction. Importantly, mechanisms and consequences of cardiac adaptation may differ according to type of overload [18]. Chronic volume overload due to aorto-caval fistula (ACF) in rats is an established model of chronic HF [19–22]. The creation of ACF leads rapidly to development of compensatory cardiac hypertrophy to maintain elevated, but largely ineffective cardiac output. After long asymptomatic stage, HF signs gradually develop and overt HF is present in 80% of animals at 21st week post-fistula [22]. The ACF-HF model recapitulates many features of human advanced HF, including gradual onset, elevated cardiac filling pressures [21, 23–25], diminished “effective” cardiac output with splanchnic hypoperfusion [21], neurohumoral activation [23], and altered calcium handling with diminished cardiac energetic efficiency [26]. Moravec et al. [14, 27] had documented that myocardial long-chain FA oxidation is impaired in ACF rats [2], but substrate metabolism at the whole body level has not yet been further studied in this model. The aim of the study was to examine changes in myocardial and adipose tissue metabolism and gene expression in a rat HF model induced by chronic volume overload.
Materials and methods (for details see online Suppl. 1)
Chronic heart failure model
Volume overload HF was induced in male Wistar rats (300-350 g) by creating ACF using 1.2 mm needle from laparotomy in general anesthesia, as described previously [19, 22, 28]. The animals were kept on a 12/12-h light/dark cycle, fed normal salt/protein diet (0.45% NaCl, 19–21% protein, SEMED, and CR) and were weighted weekly until the end of the experiment in the 22nd week. The investigation conformed to the NIH Guide for the care and use of laboratory animals (NIH Publication No. 85-23, 1996), Animal protection laws of the Czech Republic (311/1997) and was approved by the ethics committee of IKEM.
Echocardiography and hemodynamics
Echocardiography was performed in general anesthesia with 10 MHz probe (Vivid System 5, GE, USA) 10th week post-ACF. End-systolic and end-diastolic LV volumes were derived by cubic equation [20] and stroke volume as their difference. Relative wall thickness was defined as sum of end-diastolic anterior wall + posterior wall LV thickness, divided by end-diastolic LV diameter. Hemodynamics was measured at the study end (22th week) with 2F micro-manometer catheter (Millar Instruments) inserted into the aorta and LV via carotid artery in general anesthesia. The presence of ACF was verified from laparotomy and the animals were exsanguinated. The coronary tree of the excised heart was rapidly flushed with cardioplegic solution. The organs were weighted and normalized to body weight or tibial length.
Myocardial morphology and fat staining
Basal portions of the left ventricle were fixed in 4% paraformaldehyde, embedded into TissuTek, cut and stained with Sudan Black for lipids. Staining with anti-α actinin antibody together with Alexa488-conjugated wheat germ agglutinin (WGA) was performed to evaluate myocyte size and proportion of fibrosis. Images were acquired from Olympus BX51 microscope and Leica SPE confocal system and quantitatively analyzed using ImageJ (NIH) software.
Gene expression and GSEA pathway analysis in the myocardial and adipose tissue
Samples of LV tissue and epididymal fat (n ≥ 6 in each group) were immediately harvested into RNA preserving solution (RNA-Later, Ambiogen, USA). Total RNA was isolated (RNeasy-MicroKit, Qiagen, USA), checked for integrity, amplified, and hybridized on Illumina RatRef-12v1 Expression BeadChip (Illumina, USA). The raw data were analyzed and processed using beadarray package of the Bioconductor, as described before [29]. Analysis of differential expression was performed with the Limma package [30] and annotated against RatRef_12_V1_0_R3_11222119_A.bgx maniphest (Illumina, USA). Cut-off level for differential regulation was set to fold change >2 or <0.5, with Storey q < 0.01. The data are MIAME-compliant and are deposited in the ArrayExpress database (accession #: E-MTAB-190). Gene set enrichment analysis (GSEA) was performed on the samples from LV tissue on gene pathways defined by the KEGG database (release 57.0) [31]. Only the pathways with false discovery rate < 0.05 are reported.
Quantitative real-time PCR
Reverse transcription was performed by QuantiTect® Reverse Transcription Kit (QIAGEN Inc., USA). The qRT-PCR was performed on LightCycler 2.0 System using LightCycler® 480 DNA SYBR Green I Master kit (Roche Diagnostics, Germany) and results were analyzed by LightCycler software. Crossing point values were further determined using the R environment (R Development Core Team 2007). Detailed description of the analysis and the list of amplicons/primers of target and housekeeping genes are provided in Supplementary file 1 and 3, respectively.
Biochemical analyses
Oral glucose tolerance tests (OGTT, 300 mg glucose/100 g BW after overnight fasting) were performed at the 21st week by sampling tail vein at 0, 30, 60, and 120 min. Serum glucose was measured by enzymatic assay (Pliva-Lachema, CR), FFA with colorimetry (Roche Diagnostics, Germany), and insulin with rat-specific ELISA (Mercodia, Sweden). Tissue TG were measured after N2 pulverization, and chloroform/methanol extraction with enzymatic assay (Pliva-Lachema, CR) was also used for serum TG. The activity of SOD was analyzed by the reaction of nitrotethrazolium blue reduction and nitrophormasane formation [32]. The reduced (GSH) and oxidized form of glutathione (GSSG) was determined by high performance liquid chromatography with fluorescent detection (Chromsystems, Germany). The level of thiobarbituric acid-reactive substances (TBARS) was determined by the reaction with thiobarbituric acid [33].
Metabolic assessment of isolated adipose tissue
Basal and epinephrine-stimulated epididymal adipose tissue lipolysis was examined during a 2-h in vitro fat tissue incubation in Krebs–Ringer buffer with or without epinephrine (0.25 mg ml−1) as described previously [34]. Lipolysis was quantified as FFA release into the medium. Basal and insulin-stimulated lipogenesis was quantified by incorporation of 14C-U-glucose into neutral, chlorophorm-extracted lipids during 2 h incubation in Krebs–Ringer bicarbonate buffer with or without insulin (250 μU ml−1) as described previously [34].
Statistics
Animals that died during the experiment (18% total mortality including ACF operation) were excluded from the analyses. Sample size for echocardiography was 12–14/group, for other methods 5–8/group. Data are expressed as means ± SD, in graphs as means ± SE. Student’s t test statistics was used for comparisons, and P value < 0.05 was considered significant. Gene expression differences were compared with false discovery rate (FDR) and described with Storey’s q value.
Results
Echocardiography, hemodynamics, and organ weights
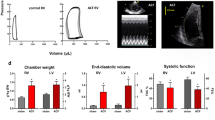
ACF animals had similar growth rate and final body size as controls (Fig. 1). Echocardiography (Table 1) confirmed cardiac enlargement, reduced relative wall thickness (0.33 ± 0.05 vs. 0.45 ± 0.05, P < 0.001), mildly reduced fractional shortening, and markedly increased stroke volume and cardiac output (+196%) in ACF. By the end of experiment (22th week), 65% of ACF animals showed clinical HF signs (lethargy, jagged fur, and labored breathing). Hemodynamics (Table 1) showed similar systemic arterial pressure, LV dP/dt max and maximal LV pressures, but markedly elevated LV end-diastolic pressure (+263%), longer systolic duration (+21%), or LV relaxation time constant τ (+33%), and slower relaxation rate dP/dt min (+24%, P = 0.07) in ACF. ACF animals had a marked increase in heart (+148%), lung (+72%), and liver weights (+17%) (Table 2). Despite similar body weights, the ACF group had surprisingly lower epididymal fat pad weight (−29%) (Fig. 6). Subcutaneous (inguinal) fat pad and kidney weights were similar in both groups.

Upper panel Body weight during the course of experiment, and the weight of the whole heart normalized to body weight. Lower panel Concentration of triglycerides (TG) in heart, or liver tissue and in serum, correlation between myocardial TG content and epididymal fat. Gray controls, black aorto-caval fistula (ACF)
Myocardial morphology and biochemistry
LV mid-wall histology (Fig. 2) showed biventricular myocyte hypertrophy due to myocyte elongation [5], with no increase in morphologic measure of fibrosis (WGA-positive area: ACF: 25 ± 0.8%, sham: 30 ± 4%, P = 0.39). Sudan Black staining for lipids, performed to examine possible lipid overload, showed no visible fat vacuoles and no difference in stain intensity (ACF: 205 ± 22, sham 208 ± 23 intensity units, P = 0.13). Myocardial TG concentration was by 53% lower in ACF (2.2 ± 1.2 vs. 4.3 ± 2.2 μmol g−1, P = 0.006, Fig. 1), but the liver TG content was similar (ACF: 4.2 ± 0.8 vs. 4.2 ± 0.8 μmol g−1, P = 0.9). Myocardial TG correlated (r = 0.64, P = 0.002) with epididymal fat pad weight (Fig. 1). Parameters of redox state and oxidative stress were also analyzed in the myocardium. ACF group had a similar myocardial level of reduced glutathione (GSH: 4.4 ± 1.4 vs. 4.3 ± 0.3 μmol g−1, P = 0.8), a trend toward more of oxidized glutathione (GSSG: 0.39 ± 0.11 vs. 0.32 ± 0.03 μmol g−1, P = 0.12), and significantly lower redox reserve (GSH/GSSG ratio, Fig. 3) than controls. Superoxide dismutase (SOD) tended to be lower in ACF hearts (P = 0.07), and there was no difference in myocardial TBARS concentration (P = 0.44) as a marker of oxidative stress.

Myocardial morphology. a Transversal section of the heart illustrates marked biventricular enlargement in the ACF animal (left) compared to control (right). b Confocal microscopic image of mid-section of the left ventricle. Red myocytes stained with α-actinin Ab, Green tissue fibrosis stained with Alexa488-conjugated wheat germ agglutinin, Blue Hoechst nuclear counterstaining. Note wider and longer myocytes in ACF, but similar extent of fibrosis. c Sudan Black staining for fat. Note similar density of staining and absence of lipid inclusions in both groups. Figure in color is available in online version

Markers of myocardial oxidative stress. Left The ratio of reduced to oxidized glutathione (GSH/GSSG), middle superoxide dismutase activity (SOD), right thiobarbituric acid-reactive substances (TBARS). Gray controls, black aorto-caval fistula (ACF)
Myocardial gene expression
Exploratory principal component analysis (PCA) and hierarchical clustering (HC) revealed pronounced differences in gene expression between groups. Although the variation among ACF animals was large, the samples formed a well-defined and separated cluster. Of 23401 transcripts analyzed, 249 were differentially regulated (fold change >2 or <0.5, q < 0.01), 186 upregulated and 63 downregulated in ACF (heat maps in Supplementary file 2). GSEA analysis using KEGG database [31] was used to identify systematic changes of transcription in pathways (Table 4). The list of the most differentially expressed genes (q < 0.01, fold change > 3.8 or <0.38) in those pathways are presented in the Table 3. Fatty acid metabolism, oxidative phosphorylation, glycolysis, citric acid cycle, branched amino-acid degradation, and PPAR signaling pathway were the most downregulated pathways. In contrast, pathways of complement cascade, extracellular matrix–receptor interaction, cell adhesion, phagosome/lysosome, and antigen presentation and cell adhesion molecules were the most upregulated. Quantitative RT-PCR confirmed upregulation of heart failure markers (natriuretic peptide precursor A), EC-matrix components (periostin, thrombospondin-4) and components of regulatory WNT1 pathway (WISP2), along with downregulation of enzymes of glycolysis (enolase 3) and FA oxidation (hydroxyacyl-CoA dehydrogenase) (Fig. 4).

Validation of gene expression changes in cardiomyocytes (a) and adipocytes (b) using quantitative real-time polymerase chains reaction (qRT-PCR). Results are normalized to housekeeping genes (see methods in supplementary file 1) and expressed as fold change in ACF versus Sham animals. *P < 0.01
Adipose tissue gene expression
Gene expression analysis was also performed in epididymal adipose tissue to identify changes induced by the presence of HF. PCA and HC revealed well-separated clusters of control and ACF rats. As ACF animals showed larger variation, only eight genes were differentially regulated at fold-change level > 2 or <0.5, q < 0.01 (all downregulated in ACF). The differences between groups were less pronounced than in myocardial tissue, so a GSEA was not performed. The list of the most differentially expressed genes (q < 0.01, fold change >1.5 or <0.5) is presented in Table 5. Full lists of differentially expressed genes and pathways (q or FDR < 0.05) are provided in online (Suppl. 2 and 3).
Oral glucose tolerance test
To assess the dynamics of metabolic substrates in the blood and its coordination by insulin, oral glucose tolerance test was performed. ACF and controls had similar baseline serum glucose (4.5 ± 0.6 vs. 4.6 ± 0.7 mmol l−1, P = 0.5) and insulin (26 ± 20 vs. 37 ± 30 pmol ml−1, P = 0.25), but increased serum FFA (1.57 ± 0.3 vs. 1.15 ± 0.2 mmol l−1, P = 0.001, Fig. 5). Glycaemia at 30′, 60′, and 120′ (P = 0.12; 0.11; and 0.40) and its AUC area (819 ± 101 vs. 761 ± 63 mmol s l−1, P = 0.2) were similar between groups. Interestingly, serum insulin response during OGTT was markedly attenuated in HF animals—insulin at 60′and 120′ were significantly lower in ACF (43 ± 27 vs. 105 ± 91, P = 0.007; 58 ± 44 vs. 185 ± 151 pmol ml−1, P = 0.001). FFA at 60′ OGTT remained elevated in ACF by 63% (P = 0.0007).

Oral glucose tolerance test. Upper panel serum glucose and FFA. Lower panel insulin and area under the curve (AUC) of glucose–time plot. 0 min = administration of glucose. Note attenuated increase of insulin after glucose challenge in ACF group
Adipose tissue metabolism
To understand the mechanism for ACF-associated intraabdominal fat depletion and to assess responsiveness of fat tissue to humoral stimuli, epididymal adipose tissue was examined in vitro. Basal and epinephrine-stimulated of lipolysis was intact in ACF animals. Similarly, the lipogenesis was normal both at baseline and after insulin stimulation (Fig. 6), indicating preserved responsiveness of adipocytes.

Epididymal adipose tissue weight and metabolism. Epididymal fat weight, lipogenesis, and lipolysis in basal and epinephrine-stimulated state. Note intraabdominal–epididymal fat tissue depletion and preserved responsiveness of adipocytes to stimuli
Discussion
Presented study revealed several specific, previously not described metabolic abnormalities in rats with HF due to ACF. The heart of HF animals had diminished redox reserve (GSH/GSSG ratio), upregulated HF markers, and downregulated mitochondrial metabolic pathways, including β-oxidation of FA. Despite circulating FFA were elevated, myocardial TG content was reduced in ACF group compared to sham. Body fat distribution was also affected by the presence of HF-ACF animals demonstrated selective depletion of intraabdominal adipose tissue, probably due to enhanced fat mobilization. The latter finding might be linked to abnormally attenuated insulin response to glucose challenge in ACF rats. The results argue against the role of myocardial lipid overload in this particular HF model and indicate that hypoinsulinaemia can contribute to intraabdominal fat tissue depletion observed in HF. The results of the study help to understand mechanisms leading to cardiac cachexia and suggest possible targets for an intervention.
Cardiac remodeling due to ACF
Creation of aorto-caval fistula led to volume overload with an increase of cardiac output by ~200% and to marked eccentric biventricular hypertrophy. Increased LV end-diastolic filling pressures and lung weights confirmed decompensated HF by the end of the study (22nd week post-ACF). Invasive hemodynamics showed relatively preserved LV contractility (dP/dt max), although to fully exclude latent contractile dysfunction we would have to use truly load-independent contractile measures [35]. Diastolic function was already impaired, probably due to combined effect of eccentric chamber remodeling, increased myocardial mass, and upregulation of ECM proteins that may increase passive ventricular stiffness (lysyl oxidase, fibronectin, collagen VIII, and thrombospondin-4). The findings are in line with previous reports from ACF-HF model indicating that intense neurohumoral activation precedes the gross decline of myocardial performance [36]. Myocardial gene expression analysis did confirm robust changes typical for HF phenotype, with more than 1% of targeted transcripts being differentially expressed. The highest overexpression in ACF hearts (Table 3) was found for natriuretic peptide precursors, thrombospondin-4 [37], Wnt-pathway [18], TGF-β pathway components, periostin [38], and thyrotropin-releasing hormone [39] that were already identified as markers of stress-induced myocardial remodeling. Gene set enrichment analysis (GSEA) that detects concordant changes in transcription in functional pathways [31], indicated widespread upregulation of genes of extracellular matrix–receptor interaction, cell adhesion molecules (Table 4). This probably reflects increased matrix turnover due to stretch-activated transcription in ACF hearts, although the histology did not show excessive ventricular fibrosis, confirming previous reports [5, 18, 23, 24, 28]. The upregulation of complement cascade components, phagosome/lysosome, and cytokine–cytokine interaction pathways may reflect inflammatory cell infiltration, yet active synthesis of complement components in stressed cardiomyocytes is also possible [40]. The most significant change detected by GSEA was downregulation of pathways for FA oxidation, citric acid cycle, respiratory electron chain, and branched amino-acid catabolism (Table 4). Similar broad downregulation of mRNAs of metabolic enzymes had been described also in other models of advanced HF [3] and it results from attenuated activity of PPARα/RXRα/PGC-1α regulatory complex [2]. Despite the functional impact of diminished transcription of these important genes is attenuated by redundancy in enzymatic activities [41], ACF hearts do show altered substrate use with selective decrease in oxidation of long-chain FA [14, 27, 42] and the critical limit apparently is intramitochondrial import of fatty acyls [14].
Decreased intramyocardial TG content in ACF hearts
In ACF group, myocardial TG content was reduced almost to a half compared to control despite circulating FFA levels were elevated. This important finding suggest that myocardial TG content does not merely reflect prevailing serum FFA concentration as suggested [3, 43], but it is also affected by the presence of heart failure or perhaps by the cause of heart hypertrophy. Similarly to the study, 30% reduction in myocardial TG pool was described by Lewandowski’s group in pressure-overloaded rat hearts [44]. The reduction of myocardial TG content was explained by an increase of “anaplerotic” influx of carbons into the citric acid cycle via malic enzyme [45] that competes with other NADPH-consuming reactions, like TG synthesis or glutathione reduction. This may occur in ACF as well, since we noticed a parallel reduction of GSH/GSSG ratio and TG content in ACF hearts. Low TG content might also be explained by limited intracellular FFA import, because several transport genes were downregulated in ACF hearts (CD36/FAT, FABP3, and FATP3, fold change: 0.7–0.8, q = 0.02–0.004). On the other side, physiologic, exercise-induced cardiac hypertrophy is associated with an increase of myocardial TG content [46]. Myocardial TG content in rodents might have implications for cardiac reserve [4], as 10–30% of ATP production in heart comes from oxidation of intramyocardial TG [47]. Conversely, excessive myocardial TG accumulation is linked to “lipotoxicity” [15, 48]. Recent evidence suggests that not TG itself, but rather diacylglycerols, ceramides, or acylcarnitines are responsible for these toxic effects [49]. The observation of diminished myocardial TG content in pathological heart hypertrophy speaks against causal involvement of lipotoxicity in non-diabetic, non-obese HF.
Attenuated response of insulin to oral glucose challenge in ACF animals
Insulin has a key role in coordination of substrate metabolism [50], but whether ACF-induced HF has an impact on insulin levels has never been addressed. Against the expectations, HF animals showed normal basal serum insulin, but attenuated increase of insulin after oral glucose loading. The reasons for attenuated postprandial dynamics of insulin in ACF are not clear. Diminished glucose absorption is ruled out, because post-load glycaemia was even higher in ACF. A steal effect of ACF is also improbable because the shunt is distal to the pancreas. Plausible explanation could be in insufficient insulin release due to HF-induced splanchnic vasoconstriction [21, 51] and/or due to protracted exposure of pancreatic β-cells to increased FFA that impairs insulin secretion [52]. Despite of postprandial hypoinsulinaemia, the whole body glucose tolerance (glucose-time AUC area) was maintained in ACF rats by an unknown mechanism. Extrapancreatic effects of incretins or increased insulin sensitivity of peripheral tissues could be involved and need further investigation. The findings suggest that drugs aiming to improve cardiac metabolism by improving insulin sensitivity may not be effective in ACF-induced volume overload HF. Because secretion of insulin after a meal suppresses adipose tissue lipolysis [53], relative postprandial hypoinsulinaemia in ACF may contribute to increased FFA release from adipose tissue and to body fat loss.
Depletion of intraabdominal adipose tissue in ACF
Animals with ACF had significantly reduced epididymal fat weight, although total body weight was similar, probably due to more pronounced fluid retention. Epididymal fat is highly vascularized and dynamic fat pool and represents 30–50% of confined intraabdominal depots in rats. Depletion of intraabdominal (retroperitoneal) fat was also reported in rat model of cardiac volume overload from aortic regurgitation [54]. In the study, adipose tissue of ACF animals showed several differentially expressed genes (Table 5), but the changes were less pronounced than in cardiac tissue. Interestingly, the most upregulated gene in HF adipose tissue was a receptor for gastric inhibitory polypeptide (GIP), a gut-produced hormone (incretin) with insulin-releasing and extrapancreatic glucoregulatory actions that also regulates fat deposition in adipocytes [55]. Recent genome-wide association study found that genetic variation GIP receptor significantly influences insulin and glucose responses to an oral glucose challenge in humans [56]. An involvement of GIP receptor in HF-induced changes of adipose tissue metabolism has never been reported and it requires further investigation. In vitro studies of adipose tissue metabolism showed preserved lipolysis and lipogenesis in ACF hearts, with normal responsiveness to epinephrine or insulin stimulation. Increased catecholamines or angiotensin-II [10] can explain increased FFA release in ACF. Previously, it has been shown in rats that chronic angiotensin-II infusion leads to adipose tissue lipolysis and to weight loss and these effects are attenuated by angiotensin or β-adrenergic receptor blockade [10]. Postprandial hypoinsulinemia and attenuated antilipolytic effects can also contribute to excessive intraabdominal fat mobilization in HF due to ACF. Although HF in general is associated with elevated insulin levels and impaired whole body insulin sensitivity [9, 12], attenuated insulin secretion has been described in patients with decompensated advanced HF [51]. The study raises the question whether correction of hypoinsulinemia might favorably modulate fat depletion, or whether long-term inhibition of lipolysis could be beneficial in HF. Lipolysis in adipose tissue can be inhibited by GPR109A receptor agonists (nicotinic acid and acipimox) [57]. Acute administration of acipimox acutely reduced cardiac work and oxygen efficiency [58], but long-term effects on cardiac function of in advanced HF have not yet been tested. Lipolysis can also be inhibited by angiotensin receptor antagonists or β-adrenergic receptor blockers [10] and their antilypolytic effects might contribute to favorable effects of these drugs on weight loss and prognosis of patients with HF.
The study has several limitations. First, we performed metabolic characterization of ACF state only in male rats. It has been shown previously that sex and estrogen status are important determinants of cardiac adaptation to volume overload, with less eccentric hypertrophy in females than in males [59, 60], but no study examined yet sex-related differences in myocardial metabolism in ACF. Second, cardiac and metabolic remodeling of the heart is time-dependent, dynamic process, but we studied just one time-point—advanced HF stage. Further studies in different stages of HF development are warranted. Third, statistical methods that we used for gene expression analysis may not discern modest, but still potentially relevant changes in transcriptional regulation bellow the pre-specified cut-off of gene expression. Fourth, to fully quantify the impact of ACF on intrinsic LV contractility, we would have to subject animals to simultaneous invasive pressure–volume analysis of the heart and its lack is a limitation of the study.
In conclusion, animals with HF due to ACF displayed several specific alterations of FA metabolism and fat distribution. Circulating FFA were higher but cardiac TG content was reduced in HF group, arguing against the role of lipid overload. The observation of intraabdominal fat depletion and attenuated postprandial insulin dynamics may help to understand mechanisms of HF-induced fat loss and cachexia.
References
Opie LH, Knuuti J (2009) The adrenergic-fatty acid load in heart failure. J Am Coll Cardiol 54:1637–1646
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90:207–258
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
van Bilsen M, van Nieuwenhoven FA, van der Vusse GJ (2009) Metabolic remodelling of the failing heart: beneficial or detrimental? Cardiovasc Res 81:420–428
Benes J Jr, Melenovsky V, Skaroupkova P, Pospisilova J, Petrak J, Cervenka L, Sedmera D (2011) Myocardial morphological characteristics and proarrhythmic substrate in the rat model of heart failure due to chronic volume overload. Anat Rec (Hoboken) 294:102–111
Sorokina N, O’Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, Ballal K et al (2007) Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation 115:2033–2041
Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, Harrington D et al (1997) Wasting as independent risk factor for mortality in chronic heart failure. Lancet 349:1050–1053
Lommi J, Kupari M, Yki-Jarvinen H (1998) Free fatty acid kinetics and oxidation in congestive heart failure. Am J Cardiol 81:45–50
Paolisso G, De Riu S, Marrazzo G, Verza M, Varricchio M, D’Onofrio F (1991) Insulin resistance and hyperinsulinemia in patients with chronic congestive heart failure. Metabolism 40:972–977
Cabassi A, Coghi P, Govoni P, Barouhiel E, Speroni E, Cavazzini S, Cantoni AM et al (2005) Sympathetic modulation by carvedilol and losartan reduces angiotensin II-mediated lipolysis in subcutaneous and visceral fat. J Clin Endocrinol Metab 90:2888–2897
Shulman GI (2000) Cellular mechanisms of insulin resistance. J Clin Invest 106:171–176
Doehner W, Rauchhaus M, Ponikowski P, Godsland IF, von Haehling S, Okonko DO et al (2005) Impaired insulin sensitivity as an independent risk factor for mortality in patients with stable chronic heart failure. J Am Coll Cardiol 46:1019–1026
Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H (2001) Metabolic gene expression in fetal and failing human heart. Circulation 104:2923–2931
Christian B, Alaoui-Talibi Z, Moravec M, Moravec J (1998) Palmitate oxidation by the mitochondria from volume-overloaded rat hearts. Mol Cell Biochem 180:117–128
Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H (2004) Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18:1692–1700
Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA 97:1784–1789
Listenberger LL, Schaffer JE (2002) Mechanisms of lipoapoptosis: implications for human heart disease. Trends Cardiovasc Med 12:134–138
Toischer K, Rokita AG, Unsold B, Zhu W, Kararigas G, Sossalla S, Reuter SP, Becker A et al (2010) Differential cardiac remodeling in preload versus afterload. Circulation 122:993–1003
Garcia R, Diebold S (1990) Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc Res 24:430–432
Cantor EJ, Babick AP, Vasanji Z, Dhalla NS, Netticadan T (2005) A comparative serial echocardiographic analysis of cardiac structure and function in rats subjected to pressure or volume overload. J Mol Cell Cardiol 38:777–786
Flaim SF, Minteer WJ, Nellis SH, Clark DP (1979) Chronic arteriovenous shunt: evaluation of a model for heart failure in rat. Am J Physiol 236:H698–H704
Brower GL, Janicki JS (2001) Contribution of ventricular remodeling to pathogenesis of heart failure in rats. Am J Physiol Heart Circ Physiol 280:H674–H683
Ruzicka M, Yuan B, Leenen FH (1994) Effects of enalapril versus losartan on regression of volume overload-induced cardiac hypertrophy in rats. Circulation 90:484–491
Ryan TD, Rothstein EC, Aban I, Tallaj JA, Husain A, Lucchesi PA, Dell’Italia LJ (2007) Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol 49:811–821
Brower GL, Henegar JR, Janicki JS (1996) Temporal evaluation of left ventricular remodeling and function in rats with chronic volume overload. Am J Physiol 271:H2071–H2078
Takewa Y, Chemaly ER, Takaki M, Liang LF, Jin H, Karakikes I, Morel C, Taenaka Y, Tatsumi E, Hajjar RJ (2009) Mechanical work and energetic analysis of eccentric cardiac remodeling in a volume overload heart failure in rats. Am J Physiol Heart Circ Physiol 296:H1117–H1124
Alaoui-Talibi Z, Landormy S, Loireau A, Moravec J (1992) Fatty acid oxidation and mechanical performance of volume-overloaded rat hearts. Am J Physiol 262:H1068–H1074
Ruzicka M, Yuan B, Harmsen E, Leenen FH (1993) The renin-angiotensin system and volume overload-induced cardiac hypertrophy in rats. Effects of angiotensin converting enzyme inhibitor versus angiotensin II receptor blocker. Circulation 87:921–930
Strnad H, Lacina L, Kolar M, Cada Z, Vlcek C, Dvorankova B, Betka J, Plzak J, Chovanec M et al (2010) Head and neck squamous cancer stromal fibroblasts produce growth factors influencing phenotype of normal human keratinocytes. Histochem Cell Biol 133:201–211
Smyth GK (2005) Limma: linear models for microarray data. In: Gentleman V, Careyand S, Dudoid S, Irizarry R, Huber W (eds) Bioinformatics and computational biology solutions using R and Bioconductor. Springer, New York, pp 397–420
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Concetti A, Massei P, Rotilio G, Brunori M, Rachmilewitz EA (1976) Superoxide dismutase in red blood cells: method of assay and enzyme content in normal subjects and in patients with beta-thalassemia (major and intermedia). J Lab Clin Med 87:1057–1064
Yokode M, Kita T, Kikawa Y, Ogorochi T, Narumiya S, Kawai C (1988) Stimulated arachidonate metabolism during foam cell transformation of mouse peritoneal macrophages with oxidized low density lipoprotein. J Clin Invest 81:720–729
Pravenec M, Kazdova L, Maxova M, Zidek V, Mlejnek P, Simakova M, Kurtz TW (2008) Long-term pioglitazone treatment enhances lipolysis in rat adipose tissue. Int J Obes (Lond) 32:1848–1853
Carabello BA (2002) Evolution of the study of left ventricular function: everything old is new again. Circulation 105:2701–2703
Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L et al (2005) Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation 112:3280–3288
Schellings MW, van Almen GC, Sage EH, Heymans S (2009) Thrombospondins in the heart: potential functions in cardiac remodeling. J Cell Commun Signal 3(3–4):201–213
Stansfield WE, Andersen NM, Tang RH, Selzman CH (2009) Periostin is a novel factor in cardiac remodeling after experimental and clinical unloading of the failing heart. Ann Thorac Surg 88:1916–1921
Jin H, Fedorowicz G, Yang R, Ogasawara A, Peale F, Pham T, Paoni NF (2004) Thyrotropin-releasing hormone is induced in the left ventricle of rats with heart failure and can provide inotropic support to the failing heart. Circulation 109:2240–2245
Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, Singh M, Yang J et al (2009) Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest 119:986–996
Morgan EE, Chandler MP, Young ME, McElfresh TA, Kung TA, Rennison JH, Tserng KY, Hoit BD, Stanley WC (2006) Dissociation between gene and protein expression of metabolic enzymes in a rodent model of heart failure. Eur J Heart Fail 8:687–693
Benes J, Kazdova L, Drahota Z, Houstek J, Medrikova D, Kopecky J, Kovarova N et al (2011) The effect of metformin therapy on cardiac function and survival in volume-overload model of heart failure in rats. Clin Sci (Lond) 121:29–41
Hammer S, van der Meer RW, Lamb HJ, Schar M, de RA, Smit JW, Romijn JA (2008) Progressive caloric restriction induces dose-dependent changes in myocardial triglyceride content and diastolic function in healthy men. J Clin Endocrinol Metab 93:497–503
O’Donnell JM, Fields AD, Sorokina N, Lewandowski ED (2008) The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J Mol Cell Cardiol 44:315–322
Pound KM, Sorokina N, Ballal K, Berkich DA, Fasano M, La Noue KF, Taegtmeyer H, O’Donnell JM, Lewandowski ED (2009) Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res 104:805–812
Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ (2009) DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem 284:36312–36323
Saddik M, Lopaschuk GD (1991) Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J Biol Chem 266:8162–8170
Wende AR, Abel ED (2010) Lipotoxicity in the heart. Biochim Biophys Acta 1801:311–319
Son NH, Yu S, Tuinei J, Arai K, Hamai H, Homma S, Shulman GI, Abel ED, Goldberg IJ (2010) PPARgamma-induced cardiolipotoxicity in mice is ameliorated by PPARα deficiency despite increases in fatty acid oxidation. J Clin Invest 120:3443–3454
Cahova M, Vavrinkova H, Kazdova L (2007) Glucose–fatty acid interaction in skeletal muscle and adipose tissue in insulin resistance. Physiol Res 56:1–15
Sharma B, Majid PA, Pakrashi BC, Dykes JR, Taylor SH (1970) Insulin secretion in heart failure. Br Med J 2:396–398
Grill V, Bjorklund A (2000) Dysfunctional insulin secretion in type 2 diabetes: role of metabolic abnormalities. Cell Mol Life Sci 57:429–440
Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, De Fronzo RA (1989) Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest 84:205–213
Bouchard-Thomassin AA, Lachance D, Drolet MC, Couet J, Arsenault M (2011) A high-fructose diet worsens eccentric left ventricular hypertrophy in experimental volume overload. Am J Physiol Heart Circ Physiol 300:H125–H134
Irwin N, Flatt PR (2009) Therapeutic potential for GIP receptor agonists and antagonists. Best Pract Res Clin Endocrinol Metab 23:499–512
Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, Lyssenko V et al (2010) Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 42:142–148
Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S (2003) PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 9:352–355
Tuunanen H, Engblom E, Naum A, Nagren K, Hesse B, Airaksinen KE, Nuutila P, Iozzo P, Ukkonen H, Opie LH, Knuuti J (2006) Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 114:2130–2137
Brower GL, Gardner JD, Janicki JS (2003) Gender mediated cardiac protection from adverse ventricular remodeling is abolished by ovariectomy. Mol Cell Biochem 251:89–95
Gardner JD, Murray DB, Voloshenyuk TG, Brower GL, Bradley JM, Janicki JS (2010) Estrogen attenuates chronic volume overload induced structural and functional remodeling in male rat hearts. Am J Physiol Heart Circ Physiol 298:H497–H504
Acknowledgments
This study was supported by Ministry of Heath [MZO-00023001 to V.M. and L.C., IGA MZCR NS-10300-3 to J.P., NS10497-3/2009 to V.M.]; Ministry of Education [MSMT-1MO510 to V.M., J.B. and L.C., VZ 0021620806 to D.S., 0021620806 to J.P., 1M6837805002 to H.S. and M.K.]; the Grant agency [305/09/1390 to V.M.]; and the Academy of sciences [AV0Z50520514 to H.S. and M.K, AV0Z50110509 to D.S.] of the Czech Republic.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Melenovsky, V., Benes, J., Skaroupkova, P. et al. Metabolic characterization of volume overload heart failure due to aorto-caval fistula in rats. Mol Cell Biochem 354, 83–96 (2011). https://doi.org/10.1007/s11010-011-0808-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-011-0808-3