
Abstract
In this chapter we review antenatal diagnosis and obstetric management of fetal conditions that are relevant to paediatric surgeons. We restrict our discussion to fetal conditions that have been shown to, or have the potential to, benefit from in utero surgical therapy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introducing the Fetal Patient
The ascendancy of antenatal imaging and genetic biotechnology has transformed fetal medicine in the modern era. In current clinical practice, women are offered a raft of early pregnancy screening options for aneuploidy, placental disorders and increasingly earlier diagnosis of fetal abnormalities. The introduction of first trimester ultrasound for nuchal translucency measurement as part of Down syndrome screening also potentiates early diagnosis of several major structural malformations [1,2,3,4,5,6]. For pregnancies categorized as higher risk based on this early screening or on past or familial history, invasive genetic diagnostic testing can be pursued and further highly specialized diagnostic fetal ultrasound and echocardiography can be offered from 16 weeks gestation [7,8,9]. Furthermore, fetal magnetic resonance offers additional imaging capabilities to further define anatomical defects detected on ultrasound screening. Most recently, the stage is now set for non-invasive prenatal diagnosis, whereby free fetal DNA is extracted from maternal plasma during pregnancy, to radically change our approach to antenatal diagnosis as high resolution genome wide evaluation becomes available [10]. Multidisciplinary counselling, therefore, must assume even greater clinical importance if we are to effectively assist pregnant women to navigate through the increasingly complex information and myriad of choices that come with improved antenatal diagnosis.
In this regard, for some very selected abnormalities, fetal surgical intervention may prove beneficial. It is indeed possible that early antenatal diagnosis will allow timely in utero surgical correction of an anatomic defect or reverse a pathophysiologic process, which may then permanently alter the trajectory of growth and development in a positive way. For this to be successful, however, strict criteria have been proposed as a means of justifying the implicit risks to the mother and fetus by surgically invading the amniotic cavity during pregnancy (Table 4.1, [11]). Of paramount importance in this selection is accurate antenatal diagnosis with a clearly defined antenatal and postnatal course. In recent years, we have made some headway in defining these parameters for certain pathologies. Furthermore, detailed sonographic diagnostic criteria, often complemented by fetal MRI, have developed novel predictive indices to better delineate the likely in utero progression of disease, health at birth and even in the neonatal period. This has allowed patient selection for fetal intervention to be refined [12]. In addition, the literature has transcended from heterogeneous, small observational studies of fetal surgical interventions to successful multicentre randomised trials for instance on twin-twin transfusion syndrome [13], myelomeningocoele [14], congenital diaphragmatic hernia [15, 16] and lower urinary tract obstruction [17]. Notwithstanding the maternal burden, we are now in a better position to provide counselling to parents to determine whether fetal surgery is likely to rescue abnormal fetal development or not. There remains more to be done. It should be remembered the inspiration and translation of fetal surgery has all come from the careful antenatal examination and meticulous description of the fetal patient.
Herein lies the focus of this chapter, where we review antenatal diagnosis and obstetric management of fetal conditions that are relevant to paediatric surgeons. We restrict our discussion to fetal conditions that have been shown to, or have the potential to, benefit from in utero surgical therapy.
2 Lower Urinary Tract Obstruction
2.1 Definition and Epidemiology
Lower urinary tract obstruction (LUTO) refers to a heterogeneous, pathological group of disorders that directly affect the urethra. The presence of posterior urethral valves (PUV) is most common, whereby a Mullerian or cloacal embryological membranous remnant is responsible for the obstruction. Obstruction can also be caused by urethral atresia, urethral stenosis, ectopic insertion of a ureter, perivesical tumours and prune belly syndrome [18]. The incidence of LUTO is reported at 2.2 cases in 10,000 births, with PUV occurring in 1 in 7031 births [19]. Most cases are in males (PUV are exclusively male), whereby in females urethral atresia is more common and consideration should be given to rarer cloacal plate pathologies such as megacystis-microcolon-hypoperistalsis-syndrome (MMHS).
2.2 Genetics
Most cases are sporadic; in some, cases have been associated with polymorphisms of genes expressed during development of the urinary tract [20]. Recurrence risk is low. MHHS syndrome is an exception, which has an autosomal recessive inheritance pattern. Chromosomal abnormalities, in particular trisomies 13, 18 and 21 have been reported in 12% of cases [21]. Genetic testing is therefore mandatory.
2.3 Pathophysiology and Natural History
LUTO results in bladder distension with compensatory smooth muscle hypertrophy of the bladder wall. With increasing pressure and distension, the bladder wall eventually loses its natural elasticity and poor tone and function develops. Increasing backpressure from vesico-ureteric reflux drives bilateral hydronephrosis above, with enlarging pyelectasis and calyectasis then responsible for progressive compression of the vulnerable developing renal parenchyma. This pressure-induced mechanism of injury may act in concert with premature activation of the renin-angiotensin system causing vasoconstriction and further hypoxic injury [22]. Fibrosis to the renal medulla and renal cortex results in renal dysplasia and eventually renal insufficiency. Some have suggested dysplasia may even result from other abnormal embryological communication unrelated to the obstruction [23]. Regardless, urine production fails and oligohydramnios ensues. This results in the development of pulmonary hypoplasia [24]. Oligohydramnios inhibits lung expansion up to 20%, primarily a result of altered fetal posture and narrowing of the fetal thorax, with reduced fetal respiration [24]. Respiration is considered an important factor in stimulating lung growth. Decreased airway branching may occur and alveoli numbers are reduced and may be structurally immature with reduced collagen and elastin [25]. Depending on gestational age and severity, respiratory embarrassment at birth is the norm, yet the condition can also be lethal.
Prognosis is dependent on the degree of renal insufficiency and the presence and degree of pulmonary hypoplasia. Mortality for antenatally diagnosed cases is 45%, but rises to 95% for those with persistent oligohydramnios [26]. For survivors at birth, one third will develop renal failure necessitating dialysis and is the leading cause of childhood renal transplantation [27]. Voiding problems happen as well.
2.4 Antenatal Diagnosis
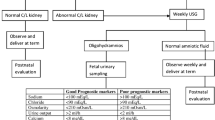
The sensitivity of ultrasound for detecting LUTO is 95% with 80% specificity [27]. The diagnosis is usually made at the 18–20 week ultrasound, although in some may be evident at the 11–13 week ultrasound and confirmed, if persistent, at 16 weeks [28]. The classical sonographic feature is the “key hole” sign (Fig. 4.1), whereby the dilated proximal urethra above the obstruction extends into the dilated and thick walled bladder (greater than 2 mm is considered pathologic) [22]. The dilated bladder, or megacystis, may occupy much of the abdomen, and even spontaneously rupture and produce urinary ascites. Ureterectasis, caliectasis and hydronephrosis are all evident sonographically (Fig. 4.2). Bilateral renal dysplasia appears as increased echogenicity of the renal parenchyma and subcortical cysts are indicative of particularly poor prognosis [26]. Similarly, the presence of oligohydramnios signifies major obstruction, and if present prior to 24 weeks is associated with a higher prevalence of renal dysplasia and pulmonary hypoplasia [21]. Oligohydramnios with an absence of caliectasis may suggest such severely damaged dysplastic kidneys that they are no longer capable of producing demonstrable urine [22]. Where possible, the bladder should be observed during voiding for the presence of vesicoureteric reflux. Fetal sex should also be determined. Of note, Potter’s facies and club feet can be seen in chronic cases. A thorough search for features of trisomy 13, 18 and 21 must also be undertaken and a detailed examination for the presence of other abnormalities.

Lower urinary tract obstruction : key hole sign. The fetal abdomen is distended by an anechogenic cystic structure, extending into the proximal urethra. Image: courtesy and copyright UZ Leuven

Lower urinary tract obstruction later in gestation with bilateral ureterectasis and hydronephrosis and caliectasis, clearly displayed by 3Dinverted rendering. This modality extracts the fluid filled higher urinary tract, dilated into its intrarenal portions. Image: courtesy and copyright UZ Leuven
The limitation of ultrasound is in accurately defining the specific aetiology of the LUTO as well as to predict renal function. Amnioinfusion may be required to explore structural integrity. Fetal MRI may offer additional information, particularly when oligohydramnios prevents adequate resolution. Fetal MRI has been shown to modify the diagnosis of LUTO in 5 of 16 cases in the third trimester [29] and to better define subcortical cysts, potentially aiding prenatal counselling.
2.5 Antenatal Prediction of Prognosis
Much effort has been expended to define antenatal determinants of fetal and postnatal renal (and therefore respiratory) function, with a view to identify candidates for in utero intervention. Unfortunately, in the present day, the waters remain somewhat muddied. Momentum developed in the late 1980s following experimental animal studies that demonstrated rescuing renal function by decompressing the obstructed bladder in utero [30, 31]. The rationale for this approach is that renal development is complete at birth, rendering postnatal surgical strategies largely ineffective. This raised the possibility that relieving the obstruction in utero, particularly during the peak of nephrogenesis (20–30 weeks gestation), may be beneficial [22]. Several observational series have since highlighted the importance of appropriate patient selection, whereby the risks of surgical invasion (most commonly preterm prelabour rupture of membranes, premature birth and chorioamnionitis) are applied only to a fetus that has potentially salvageable renal function. To predict this, there is now a large body of work examining the performance of urinary and serum electrolytes to quantify fetal renal function (Table 4.2) [18]. It has become apparent that serial measurements of urinary electrolytes (via vesicocentesis) are more representative of fetal renal function, as urinary stasis may confound the initial measurement [32]. However, in a 2007 systematic review, Morris and colleagues [33] argued that no particular fetal urinary analyte could accurately predict poor postnatal renal function. Profiling fetal serum obtained by cordocentesis for beta 2 microglobulin may improve this assessment. It is not possible to measure directly fetal urea and creatinine due to their small molecular size and placental filtration between fetal and maternal circulations, however beta 2 microglobulin has a larger molecular size and appears to reflect fetal glomerular filtration rate more reliably [34, 35]. Furthermore, repeated measurements are possible even after the placement of a shunt, which is not possible for urinary electrolytes [36]. Some groups have even suggested renal biopsy [37]. Our group uses a combined algorithm of sonographic features (kidney echogenicity and presence of cortical cysts, amniotic fluid volume), urinary electrolytes and serum beta 2 microglobulin. We simultaneously perform amniocentesis for karyotype, vesicocentesis for urinary electrolytes and cordocentesis for beta 2 microglobulin to help select potential candidates for a vesicoamniotic shunt (Fig. 4.3) and inform our prenatal counselling. In some cases of severe oligohydramnios, an amnioinfusion is required to complete this diagnostic work up. Candidates for prenatal intervention are generally considered if they are male fetuses with “good” prognostic features and worsening oligohydramnios. Regrettably, there are probably some fetuses with a favourable renal function profile but with normal amniotic fluid that, although currently not considered for shunting, do deteriorate later, sometimes even in infancy. Predicting this group remains problematic. Furthermore, shunting in utero intervention in female fetuses is usually not successful owing to the complexity of LUTO in this group [18]. Overall, several observational studies in males with LUTO have now suggested shunting seems to prevent neonatal death from lethal pulmonary hypoplasia, but the effect on improving renal function in survivors is less clear [21, 32, 38]. The most recent systematic review was in 2010 by Morris and colleagues [39], who examined 20 studies including 369 fetuses with LUTO. At that time there were no randomised trials and several studies were of poor methodological quality in accounting for bias and in reporting the techniques used. In their meta-analysis of 12 studies, they found that antenatal bladder drainage improved perinatal survival compared with no treatment (OR 3.86, 95% CI 2.00–7.45) and appeared most beneficial in a subgroup of fetuses with a poor predicted prognosis (OR 12.85, 95% CI 1.25–153.03). However, when examining survival with normal renal function in seven studies, a key component in prenatal counselling with parents, the analysis favoured no intervention. This may suggest that the reduced number of survivors with normal renal function following antenatal intervention compared to those without fetal treatment, may represent a group of fetuses with severe renal disease who would have died in utero from pulmonary hypoplasia if they had not received intervention. This may also suggest, as discussed above, that additional mechanisms are responsible for renal parenchymal damage other than an entirely pressure-mediated process from obstruction. When the authors examined a subgroup of fetuses with LUTO and good predicted renal function from urinalysis, there was a trend to favour intervention but this did not reach statistical significance. No fetuses with poor predicted prognosis survived. Caution must be applied in interpreting this data however, particularly subgroup analysis, given the observational nature of this research. With regard to longer term follow up, Biard and colleagues [40] recently reported long-term follow up of 20 male fetuses with LUTO and oligohydramnios who were shunted antenatally. Renal function was acceptable in 45% of cases, with 22% having renal impairment and 33% with end-stage renal disease necessitating transplantation. Of note is that the self-perceived qualtiy of life of survivors fell in the normal range. Hence, despite several decades of research, a robust prospective randomised trial was still in the making.

Schematic drawing of vesico-amniotic shunt in place. Insert: double pigtail shunt connects the bladder to the amniotic cavity (reproduced with permission from the UZ Leuven, Belgium)
The Birmingham group in 2007 designed the PLUTO trial [17], whereby fetuses with LUTO were randomized to either vesicoamniotic shunt placement or observation when the treating clinician was uncertain of the need for a shunt. Key to this study design was the demonstration of clinical equipoise, which was attempted by surveying paediatric nephrologists, paediatric urologists and maternal fetal medicine subspecialists [41]. After demonstrating clinical equipoise, the PLUTO trial study design did not stipulate an algorithm to formally evaluate fetal renal function prior to randomisation and instead relied on the clinician’s uncertainty as to whether the fetus would benefit from a vesicoamniotic shunt placement or not. Disappointingly, the trial was stopped early in 2012 due to poor recruitment and many questions remain unresolved [42]. They concluded that survival seemed to be higher after shunting, but the size and direction of the effect remain uncertain, such that benefit could not be conclusively proven. Also, the chance of newborn babies surviving with normal renal function seems very low irrespective of whether or not vesicoamniotic shunting is done. A further advance may yet come from fetal cystoscopy (Fig. 4.4). Cystoscopy potentially offers an attractive combination of improved diagnosis, and thereby improved patient selection, and potentially even definitive treatment. Fetoscopic antegrade catheterization and hydro- or laser ablation of urethral valves has now been described, but technical challenges of instrument size and manoeuvrability limits percutaneous access to the bladder neck. Nevertheless, Ruano and colleagues have recently reported the largest prospective series of 23 fetuses with LUTO managed with the option of fetal cystoscopy [43]. The procedure, which can be done as early as 16 weeks of gestation, allows a more robust diagnosis, and in case of urethral valves, definitive treatment by laser fulguration may be attempted. A recent systematic review of four fetal cystoscopy studies [44] demonstrated that the initial diagnosis of posterior urethral valves changed in 32% (6/19) typically towards urethral atresia. In terms of postnatal survival, cystoscopic ablation of the valves was superior to expectant management yet comparable to shunting. The latter procedure is neither without any risk for collateral damage to pelvic floor structures.

Schematic drawing of fetal cystoscopy. Insert: Through a 7 Fr cannula a semi-flexible endoscope has been inserted into the bladder to visualize the bladder outlet (reproduced with permission from the UZ Leuven, Belgium)
2.6 Obstetric Management
Patients should be referred to a fetal medicine referral centre for diagnosis and multidisciplinary counselling with maternal fetal medicine specialists, paediatric urologists, paediatric nephrologists and neonatologists all considered essential. In most cases, delivery at a tertiary centre is appropriate. A detailed sonographic assessment should be undertaken as described above, and given the presence of chromosomal abnormalities in 12% of cases [21], fetal karyotype should be offered. As described earlier, for mild cases where fetuses have normal amniotic fluid and no evidence of renal dysplasia on ultrasound, they should be observed closely for evidence of reducing amniotic fluid or signs of renal dysplasia. If renal function remains stable, there should be no respiratory problems at birth, and thus timing and mode of birth should be guided by usual obstetric considerations. Post-natal surgical evaluation and correction is then undertaken. Should there be sonographic signs of failing renal function, then management is guided by gestational age. If the pregnancy is more than 32 weeks, elective delivery (covered by glucocorticoids for fetal lung maturity when appropriate) is advised to pursue postnatal repair [22]. When less than 32 weeks gestation, we would offer investigational fetal MRI to aid in the examination for signs of renal dysplasia and recommend formal evaluation of fetal renal function by both urinary electrolytes and serum beta 2 microglobulin. In the good prognosis group, we will offer parents the option of vesicoamniotic shunting and/or fetal cystoscopy after through counselling of the limitations discussed earlier and close ongoing surveillance. In the poor prognosis group, or in those who present early with severe chronic obstruction, oligo- or anhydramnios and renal dysplasia evident on ultrasound, we would counsel the parents regarding termination of pregnancy or expectant care. Expectant care should be clearly defined by obstetric and neonatal physicians, including advising no intervention for fetal heart rate abnormalities in labour, providing intermittent auscultation of the fetal heart rate at maternal request only, no indication for cardiotocograph monitoring in labour and no active resuscitation at birth. Parents should be well counselled regarding comfort care of the neonate after birth, post mortem and diagnostic investigations and support services for the family should be initiated. The place of active management is as described above.
3 Sacrococcygeal Teratoma
3.1 Definition and Epidemiology
Sacrococcygeal teratomas (SCT) are tumours derived from the totipotent Hensen node of the primitive streak, thereby retaining a full differentiation repertoire down ectodermal, endodermal or mesodermal cell pathways [45]. It is the most common tumour in the fetus with an incidence of one in 40,000 births with more females than males affected (4:1) [46], although malignant change is more common in males. The American Academy of Pediatrics Surgical Section classification system is shown in Table 4.3, [47] which describes SCTs according to their relationship with the sacral region. Note that this classification is a surgical description and does not reflect prognosis.
3.2 Genetics
Considered sporadic. Rare autosomal dominant inheritance in some families has been reported [26]. Rarely associated with aneuploidy.
3.3 Pathophysiology and Natural History
Given SCT’s are of totipotent origin, cells can differentiate into embryonic (mature and immature teratomas) or extraembryonic (choriocarcinoma and yolk sac teratoma) tumours. Most commonly, gastrointestinal, respiratory and nervous tissue material is present [26]. SCT’s show a paraxial or midline distribution usually from the brain to the sacral region, thereby reflecting aberrant migration of the primordial germ cells. Other less common sites are the anterior mediastinum, pineal area, retroperitoneal area, neck, stomach, and vagina [18]. SCT’s are the most common presentation at birth. They represent an extragonadal form that arise in the presacral area, which, as demonstrated by the classification presented in Table 4.3, can extend into the pelvic and abdominal cavity or develop more exteriorly. They are heralded for imposing vascularity and the propensity for rapid growth in utero, which can risk fetal survival through the development of high output cardiac failure and/or fetal anaemia, non-immune hydrops, destructive processes to adjacent viscera or obstetric complications. The condition can also cause maternal problems (Ballentyne Syndrome). Most SCT’s are benign, however can recur following surgical resection and do have malignant potential in around 10% of neonates born with SCT, particularly in more immature and in type IV tumours [22].
3.4 Antenatal Diagnosis
SCT is usually diagnosed at the 18–20 week ultrasound, although occasionally reported at the 12-week ultrasound [6]. Alternatively, patients may present later with the clinical sequelae of polyhydramnios, such as large for dates, uterine irritability, preterm labour or preterm prelabour rupture of membranes. Sonography reveals a mixed solid and cystic mass, often with calcification, arising from the distal spine and sacral region (Fig. 4.5). A SCT is considered destructive to the spine, whereas as a myelomeningocoele will widen the spine and is likely to have additional cerebral signs [26]. A thorough description of the external and internal pelvic components of the tumour is necessary. The latter may benefit from fetal MRI to more accurately delineate the degree of pelvic extension and distortion of local anatomy, where acoustic shadowing from pelvic bones on 2D ultrasound may be bothersome [48]. Bladder elevation, ureteral dilatation and hydronephrosis with the potential for renal dysplasia from compression forces may occur. Similarly, gastrointestinal dilatation may result from obstruction. Rectovaginal fistula and imperforate anus may also develop from direct tumour spread. Danzer and colleagues (2006) [48] evaluated their experience with additional fetal MRI compared with sonography alone. In their series of 24 patients, MRI was superior to sonography for assessing colon displacement (n = 11), urinary tract dilatation (n = 9), hip dislocation (n = 4), intraspinal extension (n = 2), and vaginal dilation (n = 1). In fetuses with sacrococcygeal teratoma types II and III, MRI was again superior to sonography in demonstrating the cephalic extent of the tumor. In two referred cases, MRI led to a changed diagnosis (healthy, n = 1; myelomeningocele, n = 1). They concluded additional anatomic resolution from MRI resulted in more accurate antenatal counselling and preoperative planning for surgical resection.

Sacrococcygeal teratoma . Sagittal section on the midline with the lumbosacral vertebrae visible. A tumor orginates at its distal end, and with Doppler ultrasound the feeding vessels from the presacral area. The echogenicity of the tumor varies, with cysts as well as within the solid parts. Image: courtesy of and copyright UZ Leuven
SCT’s are renowned for their impressive vascularity, often jeopardising fetal cardiac integrity. This can be simply visualised with 2D colour Doppler or more recently has been evaluated with 3D power Doppler, allowing calculation of the vascular volume of the tumour relative to the heart [49]. Cardiac failure and non-immune hydrops may develop, usually from high output cardiac failure following rapid growth of the tumour with extensive arteriovenous shunting. Furthermore, if the tumour outgrows its blood supply and becomes necrotic it can rupture, resulting in haemorrhage and fetal anaemia [22]. Fetal anaemia can be observed by an increase in the middle cerebral artery Doppler peak systolic velocity [50]. High output failure can be diagnosed sonographically and with fetal echocardiography, demonstrating increased inferior vena cava diameter (reflecting increased venous return), dilated ventricles, increased cardiac output and descending aortic flow velocity, and absent or reversed end diastolic flow of the umbilical artery from vascular “steal” from the umbilical artery to the placenta [22, 26, 51]. Olutoye and colleagues (2004) [52] suggested reversal of end diastolic flow may indicate “backwash” of flow into the low resistance vascular bed of the tumor. Polyhydramnios, placentomegaly, hepatomegaly, increased skin thickness and fluid within fetal compartments may then be recognised consistent with hydrops. Fetal hydrops is considered a pre-terminal sign. Fetal growth restriction may also be apparent. A thorough examination for additional abnormalities should be undertaken, although most are usually the direct result of tumour extension.
3.5 Antenatal Prediction of Prognosis
Overall, fetal SCT confers a reasonable prognosis, however a particular subset of fetuses is at great risk. The mortality rate for those who present for the first time as neonates is around 5%, considerably better when compared to those who present antenally who have 50% mortality [53]. Similarly, those who present earlier in gestation have a higher mortality rate of up to 90% less than 30 weeks, compared to 15% in later pregnancy [26, 54]. Accordingly, reliable antenatal prediction of disease progression and survival is imperative to counsel parents. A rapidly growing tumour places enormous burden on the fetal heart and the presence of hydrops should confer grave concerns for the survival of the fetus. Flake and colleagues (1986) [53] described the presence of hydrops and polyhydramnios being associated with a mortality of seven out of seven fetuses and in a series by Bond and colleagues (1990) [54], they reported ten out of ten hydropic fetuses and nine out of nine fetuses with placentomegaly died. There have been some occasional cases of survival with early delivery and post-natal resection reported since then [55, 56] (Robertson 1995, Nakayama 1991), however this appears uncommon. Some fetal deaths may also occur in fetuses without demonstrable hydrops [57], although abnormalities of absent or reversed end diastolic flow of umbilical artery Doppler may herald impending fetal loss [52].
It would seem reasonable that tumour size would be predictive of the risk of cardiac failure and thus, mortality. Wilson and colleagues (2009) [58] reported tumour growth rates approaching 150 cm3 per week as a predictor of increased perinatal mortality. However, Westerburg and colleagues (2000) [59] did not confirm that tumor size to be predictive of demise, but instead found the most vascular tumours, regardless of size, had the worst prognosis. In 2006, Benachi and colleagues [60] devised a combined classification system based on tumour diameter, vascularity and rate of growth determined by ultrasound (Table 4.4 ). This was from a series of 44 fetuses with SCT mostly diagnosed on second trimester ultrasound. The fetal and neonatal loss rates were similar (12% and 13% respectively) and half were born preterm. Group one and three did very well, delivering at term with all fetuses surviving. Group two had the worse prognosis, delivering at 31 weeks on average with a 52% perinatal mortality rate; about half dying in utero and half dying in the neonatal period. Surprisingly in this series, most who died were not hydropic. It is worth mentioning that in survivors, morbidities included intraventricular hemorrhage, pulmonary hypertension (and other steal syndromes), acute renal failure and three infants had a rectal perforation or sepsis requiring colostomy. For smaller tumours, a good prognosis is generally expected, presumably due to less circulatory demand. Makin and colleagues [61] reported in 2006 on 29 cases of SCT diagnosed antenatally. The long-term outcome for fetuses not requiring intervention (n = 17) was excellent, with constipation in one child the only reported outcome at 39 months. Of note in this series, six of the seven cases of hydrops resulted in fetal or neonatal demise.
Most recently, Rodriquez and colleagues (2011) [62] have proposed tumour volume to fetal weight ratio (TFR) as an early prognostic classification for fetal SCT. In a retrospective series of ten cases, a TFR greater than 0.12 measured at less than 24 weeks gestation either by ultrasound or fetal MR, was predictive of poor outcomes with a sensitivity of 100% and a sensitivity of 83%. A TFR less than 0.12 predicted an uncomplicated course with 100% survival. All hydropic fetuses and all fetuses that died had a TFR greater than 0.12, with just 20% of fetuses having an uncomplicated outcome above this threshold.
3.6 Obstetric Management
All patients with suspected SCT should be referred to a tertiary fetal medicine centre for evaluation and multidisciplinary counselling. Maternal fetal medicine specialists, neonatologists and paediatric surgeons are essential members of this team. A detailed sonogram, echocardiogram and fetal MRI are recommended to confirm the diagnosis and exclude additional abnormalities. If SCT is confirmed, a detailed evaluation of the extent of internal pelvic extension, additional visceral involvement and fetal haemodynamic state is required. Classification according to Benachi [60] and the TFR discussed above [62] could be considered. Karyotype is generally not required, unless additional abnormalities are present. Termination of pregnancy should be discussed with parents, particular for a large or rapidly growing SCT, the presence of dysplastic renal involvement and if cardiovascular compromise is evident on ultrasound. The presence of hydrops should be conveyed as a particularly ominous prognostic feature. If termination of pregnancy delivery is planned, it is important that the delivery team consider possible labour dystocia. An intact fetus is preferable for thorough post mortem pathological examination.
For ongoing pregnancies, close surveillance is mandatory. Serial sonography is recommended every 1–2 weeks to identify progression of disease. It must be remembered that SCT’s can grow very rapidly and the fetal cardiovascular system may come quickly into jeopardy. Sonographic and echocardiographic signs of hydrops should be pursued, including measurements of a high output circulation such as the inferior vena caval diameter (>1 cm), descending aorta flow velocity (>120 cm/s) and the umbilical artery Doppler waveform [22]. The middle cerebral artery Doppler peak systolic velocity should be examined for signs of anaemia [50]. The renal and gastrointestinal tract should be examined for signs of obstruction. Amniotic fluid volume should be measured for polyhydramnios, and if present, a cervical length measurement to predict preterm labour is advised. Obstetric complications may evolve with advancing pregnancy. In a series by Hedrick and colleagues in 2004 [57], in 26 women with fetal SCT who did not undertake termination of pregnancy, 81% experienced obstetric complications. These included polyhydramnios in seven women, oligohydramnios if four women, preterm labour in 13 women, pre-eclampsia in four women and gestational diabetes, HELLP syndrome and hyperemesis each occurring in one woman. Maternal mirror syndrome has also been reported in cases of SCT with hydrops.
Antenatal fetal intervention for SCT may be an option for selected cases. Dr. Flake and colleagues discuss fetal surgical resection for SCT in detail in Chap. 10. For our purposes in this Chapter, we will briefly discuss additional interventions in the antenatal period, and how this is informed by antenatal diagnosis. In general terms, antenatal intervention is reserved for fetuses at high risk that are still remote from term. Drainage of polyhydramnios is a simple and safe procedure that may help resolve uterine irritability and maternal discomfort. Similarly, if fetal anaemia is detected by increased peak systolic velocity of the middle cerebral artery Doppler signal, intrauterine blood transfusion is a feasible option [63]. In cases of renal tract obstruction from pelvic extension of the SCT, a vesicoamniotic shunt may protect the renal parenchyma from injury in much the same way as fetuses with LUTO [60, 61, 64]. Clinical experience, however, is scarce to formally evaluate these strategies. Open resection with cardiac monitoring, thermocoagulation and radiofrequency ablation of the tumour is discussed elsewhere in this book.
Timing of birth is usually dictated by either fetal or maternal complications. The presence of fetal haemodynamic compromise mandates immediate delivery. For small tumours in healthy fetuses, vaginal delivery may be possible. Delivery in a tertiary centre is essential. Cyst aspiration and decompression does not seem beneficial to aid vaginal delivery [26]. There have, however, been reports of fetal death after tumour rupture, avulsion and asphyxia following complications of vaginal birth [22]. Close surveillance in labour with continuous cardiotocography monitoring and the presence of senior obstetric and neonatal staff are necessary. For tumours more than 4.5 cm, or in cases of fetal haemodynamic compromise, caesarean section is advised to prevent labour dystocia and reduce the chance of tumour rupture and fatal haemorrhage [26]. This approach requires meticulous planning with obstetric, anaesthetic, neonatal, haematologic and paediatric surgical teams. The caesarean should take place in an elective setting with fetal lung maturity assisted by glucocorticoids when less than 37 weeks gestation [65]. Depending on tumour size, consideration should be given to maternal general anaesthesia to aid uterine relaxation for careful manipulation and delivery of the fetus. The uterine incision should be generous to facilitate an atraumatic delivery of the fetus, particularly in cases with a large mass. A classical (vertical) uterine incision may be advisable to increase the surgical field of exposure, especially in more preterm gestations where a lower uterine segment may not be sufficiently formed and where delivery is anticipated to be difficult. Intraoperative cyst aspiration of 2 L in one case has also been reported to aid delivery [66]. Uterine bleeding from extension of the incision during delivery of the fetus or from atony must be anticipated in advance and oxytocics be used readily, if not prophylactically. Neonatal intensivists and haematologists should be prepared for resuscitation of the neonate and extreme care must be taken when handling the tumour, especially if a “stalk” is present with a risk of torsion [26]. Even superficial bleeding can result in life threatening haemorrhage [26]. Tumour rupture and neonatal death have been reported even from handling during caesarean delivery [67]. In the absence of haemorrhage at birth, surgical excision can generally be delayed to allow stabilisation of neonatal circulation and further imaging to be undertaken in the neonatal intensive care to inform definitive surgical plans. When there is hemodynamic instability, immediate neonatal surgery, even restricted to debulking, may be required to arrest the steal effect. In that case an adjacent operation suite must be available.
4 Fetal Congenital Thoracic Malformations
4.1 Definition and Epidemiology
Fetal congenital thoracic malformations (CTMs) constitute a heterogeneous group of pathologies involving the airways and/or lung parenchyma. Fur our purposes in this chapter, we will focus our attention on congenital cystic adenomatoid malformation (CCAM), and the related pathology, bronchopulmonary sequestration (BPS). CCAM’s are defined as benign, multicystic, dysplastic lung tumours with an overgrowth and proliferation of terminal bronchioles that receive their blood supply from pulmonary vessels. BPS on the other hand, refers to a distinct mass of non-functioning lung tissue that is not in communication with the normal lung architecture and receives its blood supply from an anomalous systemic vessel. Both lesions may also coexist together referred to as a hybrid CCAM-BPS [22, 68].
The European Surveillance of Congenital Anomalies (EUROCAT) population-based registry [69], in 2008 reported on 222 fetuses with CTMs, with an incidence of 4.44/10,000 (i.e. including live births, fetal deaths and terminations of pregnancy). Of these 222 cases, 52 had CCAM alone with an incidence of 1.04/10,000. With regard to live births, the incidence was 3.52 and 0.94 per 10,000 live births in 2008 for all CTMs and CCAMs respectively in EUROCAT countries. The reported annual incidence of BPS ranges between 0.15 and 6.45% of all CTMs [68].
4.2 Genetics
Sporadic inheritance. When isolated, not associated with aneuploidy.
4.3 Pathophysiology and Natural History
CCAM’s (Fig. 4.6) may be the result of failed maturational processes during the psuedoglandular stage of lung development [70], representing focal dysplastic regions [71] or related to airway obstruction [72]. Importantly, to help differentiate them from BPS, they derive their vasculature directly from pulmonary vessels. CCAM’s are almost always unilobular, with bilateral tumours occurring in less than 2% of cases. About one in four are associated with additional fetal abnormalities, such as pectus excavatum, renal agenesis, congenital diaphragmatic hernia, bowel atresia and non-immune hydrops [26]. The natural history and progression is rather variable but overall prognosis is good. Small lesions are largely asymptomatic, while larger lesions can result in mediastinal shift, compression of systemic venous return, resulting in hydrops and risking fetal death. Pulmonary hypoplasia may also be evident, but difficult to predict. The haemodynamic state may in turn be worsened by a leakage of proteins into the amniotic fluid and thereby reducing systemic oncotic pressure [73]. Polyhydramnios may occur due to mechanical compression of the fetal oesophagus inhibiting swallowing, and occurs in 70% of cases [74]. The risk of preterm prelabour rupture of membranes and/or preterm birth naturally follows. However, in some fetuses, the CCAM may reduce in size and resolution of the lesion may occur. This has been observed in between 5 and 10% of cases [74]. CCAM’s tend to plateau in their rate of growth around 26 weeks gestation, and furthermore, resolution via involution may eventually occur if the tumour outgrows its vascular supply.

Congenital Cystic Adenomatoid Malformation . Left: microcystic type, with uniform more echogenic lung tissue; right: macrocystic type, next to remnant lung tissue measured with the calipers. Images courtesy and copyright UZ Leuven
BPS is generally thought to represent ectopic pulmonary buds, which can be either intralobular or extralobular (Fig. 4.7). This difference is explained embryologically. Intralobular sequestration develops early, before the development of the pleura, and the ectopic buds become incorporated within the adjacent lung, almost exclusively in the lower lobe (98%). Extralobular sequestration develops later and is less common (25%), whereby the pulmonary bud is separated from the adjacent lung and has its own pleural covering. This is more likely to be associated with additional abnormalities than intralobular sequestration (50% versus 10%), particularly congenital heart disease, and is most common on the left side of the thorax (90%) [26]. In both cases, the ectopic bud is thought to develop caudal to the lung and migrates in a caudal direction along with the oesophagus [22]. Up to 10% of extralobular sequestrations may lie inferior to the diaphragm [26]. Around 75% of antenatally diagnosed BPS may resolve, most likely from outgrowing their blood supply or infarction mediated by vascular torsion of their pedicle. However, in some fetuses, there exists a considerably poorer outlook. Torsion of the extralobular pedicle may then obstruct venous and lymphatic drainage and result in ipsilateral hydrothorax, which may then place the mediastinum under tension. Without intervention, hydrops and fetal death are likely. If there is an abdominal extralobular BPS, hydrops is less likely, however polyhydramnios may occur from impaired swallowing due to gastro-oesophageal compression. Similarly, for intralobular BPS, hydrops may be the result of high output circulation from a “left to left” ateriovenous shunting between the anomalous arterial supply and pulmonary veins [22].

Bronchopulmonary sequestration: hyperechogenic pathologic lugn tissue, feeding vessel being demonstrated by Doppler ultrasound. Images courtesy and copyright UZ Leuven
4.4 Antenatal Diagnosis
In the antenatal period, there has been a shift in recent years away from pathological diagnosis to descriptive appearances based on imaging. The original histological classification system for CCAM was by Stocker and colleagues in 1977 [70], which they more recently updated in 2002 [75] (Fig. 4.6). In their most recent classification, five types of lesions are suggested that attempt to present a spectrum of abnormalities across consecutive airway types (Table 4.5 ). In other words, lesions are described as moving down the bronchial tree from bronchial, bronchiolar, alveolar and to peripheral. However, this system has not been embraced universally. Applying a histopathological classification system to a condition that is increasingly recognized and diagnosed antenatally, may not be useful in predicting prognosis and guiding clinical management decisions. Furthermore, it appears likely that types 0, 3 and 4 represent different pathogenetic processes. More recently, the Children’s Hospital of Philadelphia published a simplified classification of microcystic (solid sonographic appearance) and macrocystic (single or multiple cysts >5 mm) types. It is likely this can be more useful clinically [76]. Sonographic examples of this classification of CCAM are shown in Fig. 4.6. It is important to visualise the vascular supply by colour Doppler to help differentiate with BPS, whereby an anomalous systemic vessel commonly arising from the descending aorta will be seen feeding a solid echogenic mass (Fig. 4.7). In both cases of CTM, sonographic features of hydrops, mediastinal shift and the presence of additional fetal abnormalities should be sought. Polyhydramnios is commonly encountered, in which case a cervical length measurement may be informative. Fetal MRI may have additional value is cases where the diagnosis is uncertain, in particular with CCAM versus congenital diaphragmatic hernia, or when they coexist [77].
4.5 Antenatal Prediction of Prognosis
A significant proportion of lung lesions may regress; nevertheless cautious frequent observation is required to identify a large or enlarging CCAM that imposes pulmonary compression and risks the development of hydrops. The presence of fetal hydrops alerts a poor prognosis and its detection is essential to guide counselling and management. This is demonstrated by a large series reported by Adzick and colleagues [74] in 1998 of 134 fetuses diagnosed antenatally with CCAM. In 25 fetuses with a large CCAM causing hydrops, there was a 100% mortality rate. This is in stark contrast to non-hydropic fetuses, who experienced 100% postnatal survival. In 2002 Crombleholme and colleagues [78] developed a prognostic measure for the development of hydrops. They defined the ratio of the mass area to head circumference as the CCAM volume ratio (CVR). This is sonographically measured (in milliliters) by using the formula for an ellipse: CCAM volume/Head Circumference = (length × height × width × 0.52)/HC (Fig. 4.6). It is gestational age independent. They found when the CVR is higher than 1.6, there was an 80% risk for fetal hydrops. Sonographic follow-up frequency based on the CVR has been also proposed, with weekly follow-up for CVR less than 1.2, twice a week for CVR 1.2–1.6, or even more for CVR greater than 1.6, but this protocol remains to be validated in larger studies.
4.6 Obstetric Management
All cases of a suspected CTM should be referred to a tertiary fetal medicine referral centre for diagnostic work up, counselling and management. The multidisciplinary team should comprise maternal fetal medicine specialists, paediatric surgeons and neonatologists. A detailed ultrasound and echocardiographic examination should be performed to define the CTM, its vascular supply, the presence of mediastinal shift or signs of hydrops. The potential for coexisting abnormalities should be excluded, in particular cardiac abnormalities such as truncus arteriosus and Tetralogy of Fallot [22]. Amniocentesis for karyotype is not necessary when isolated. We suggest performing a CVR for cases of CCAM and followed serially as described above. Fetal MRI may be required when the diagnosis is uncertain, in hybrid CCAM-BPS cases or in the presence of CDH. To our knowledge there are no series demonstrating predictive value of lung size in terms of pulmonary hypoplasia, which overall is uncommon. Termination of pregnancy is an option when there are additional severe abnormalities or in the presence of hydrops.
Alternatively, for hydropic fetuses with macrocystic CCAM (unilocular or multilocular), fetal intervention should be discussed with parents as an option to try to reverse hydrops, potentially protect residual fetal lung parenchyma. Expectant management offers a nearly hopeless prognosis and risks maternal complications. The rationale for intervention is to decompress a dominant cyst or resect or involute a larger solid mass to resolve mediastinal compression, restore fetal haemodynamic equilibrium and thus improve cardiac function. Minimally invasive intrauterine puncture or shunting of macrocystic masses (Fig. 4.8) is the treatment of choice when possible, now that its efficacy has been demonstrated.

Schematic drawing of thoracic shunt being deployed in the chest. First the pig tail loops in the effusion are deployed, and thereafter the tail is deployed in the amniotic cavity. Drawing Myrthe Boymans; copyright UZ Leuven
Despite an evidence gap of randomised trials, the systematic review by Knox and colleagues [79] showed in utero therapy was associated with significantly improved survival in hydropic fetuses (OR 19.28, 95% CI 3.67–101.27), particularly at preterm gestations. With regard to thoracoamniotic shunting in particular, Wilson and colleagues (2006) [80] described 23 cases from CHOP at a mean gestational age of 21–22 weeks. The mean CVR in this group was 2.4, which reduced considerably to 0.7 after shunting. The survival was 74%, with one fetal and five neonatal deaths. In a review by Witlox and colleagues [81] in 2011, summarizing 68 shunted cases including 44 hydropic and 24 non-hydropic fetuses, whereby a large cyst was causing major mediastinal shift. For hydropic fetuses, 89% (39/44) were live born, nine infants died in the neonatal period, giving an overall perinatal survival of 68% (30/44). For non-hydropic fetuses (n = 24), all were live-born, three died in the neonatal period due to pulmonary hypoplasia, giving an overall perinatal survival of 87.5%. Thoracic deformation has been reported but seems rare [82]. Shunting is typically performed until 32 weeks gestation, although more recently has been reported for hydropic fetuses up until 37 weeks [81]. The risk of intervention at later gestations, mostly preterm prelabour rupture of membranes, is less troublesome when compared to the considerable neonatal risks of delivering a preterm hydropic infant. The potential to mitigate hydrops in utero and thereby gain further lung maturation before birth would seem likely to translate into improved respiratory function during transition to air breathing and even during postnatal surgery. This approach requires validation.
Not all hydropic fetuses will have a dominant or large cyst(s) amenable to drainage, in particular fetuses with microcystic CCAM. However, most interestingly, several authors have now observed resolution of hydrops following glucocorticoid therapy [83, 84]. This has occurred at standard dosages given for fetal lung maturation. Curran and colleagues (2010) [85] recently updated Tsao’s initial series from 2003 and reported 13 fetuses with microcystic CCAM and hydrops or a CVR>1.6 receiving glucocorticoids. Hydrops resolved in 78% of cases and survival was 85%. It is hypothesised that glucocorticoids may accelerate maturation or involution of the tumour. The same group has now embarked on a randomized controlled trial (clinical-trials.gov NCT00670956) to investigate this more specifically, however in the meantime, glucocorticoids seem a reasonable first-line therapy or medical adjunct for these hydropic fetuses. Their role in non-hydropic fetuses has not been examined. Other fetuses may have solid lesions not suitable for drainage. These are best treated by open fetal surgery and lobectomy, as reviewed by Flake et al. in this book. In the largest fetal surgery series available today, survival rate was 50% [76].
Hydropic fetuses with BPS theoretically may benefit from minimally invasive techniques as well, although this is less defined. In particular occlusion to the anomalous systemic feeding vessel using thermocoagulation by laser or electrosurgery, or sclerosing agents have been reported in a very small number of high-risk fetuses with good outcomes [81, 86,87,88,89].
In non-hydropic fetuses, mode of delivery is determined by usual obstetric factors, with spontaneous vaginal delivery at term in a tertiary centre generally favoured.
5 Fetal Hydrothorax/Pleural Effusion
5.1 Definition and Epidemiology
Pleural effusion can be defined as primary or secondary. Primary pleural effusion is more correctly termed hydrothorax antenatally and is due to lymphatic or “chylous” leak with resultant fluid accumulation in the thorax. This may be unilateral or bilateral. After birth, chylothorax becomes the more common terminology. In the more common secondary pleural effusion, the effusion harbingers underlying pathology often part of widespread fluid accumulation from immune or non-immune hydrops. The overall incidence is around 1 in 15,000 pregnancies [26].
5.2 Genetics
Chromosomal abnormalities can be present. In the most recent series, Ruano and colleagues (2011) [90] reported 23 of 56 (41%) fetuses with pleural effusion had underlying chromosomal abnormalities. Turner’s syndrome (45 XO) was the most common in 15 of 56 fetuses (65%) followed by Down syndrome in five fetuses (22%). However, Yinon and colleagues (2010) reported a much smaller association with anuploidy, with four from 88 fetuses being abnormal [91]. A number of genetic syndromes have also been reported, such as Caffey’s cortical hyperostosis (autosomal dominant) and Opitz-Frias hypertelorism hypospadius syndrome (autosomal recessive) [26]. Males appear twice as likely to be affected than females.
5.3 Pathophysiology and Natural History
Small effusions may remain stable or even regress. Aubard and colleagues [92] observed spontaneous resolution in 22% of 204 cases of primary fetal hydrothorax (Fig. 4.9). However, large (or enlarging) effusions have the potential to cause mediastinal shift, compromised venous return and lung compression, risking pulmonary hypoplasia, hydrops fetalis and intrauterine death (Fig. 4.9). Bigras and colleagues [93] demonstrated by echocardiography a cardiac tamponade effect from rising intrathoracic pressure, whereby ventricular dimensions decreased and the inferior vena cava dimensions consequently increased, reflecting impaired ability of the heart to accommodate venous drainage. Oesophageal compression and impaired fetal swallowing will result in polyhydramnios, which in turn increases the likelihood of preterm prelabour rupture of membranes and preterm birth. When bilateral, pulmonary lymphangiectasia should be expected and the prognosis is poor even despite fetal treatment, because abnormal lymphatics preclude normal gas exchange in the lung [18]. Furthermore, lung development appears most sensitive to compression effects during the cannalicular phase of lung development; this corresponds to an increased risk of pulmonary hypoplasia between 16 and 24 weeks gestation [94].

Bilateral hydrothorax. Image: courtesy UZ Leuven
Ruano (2011) [90] attempted to define the natural history of fetal pleural effusions without antenatal intervention. In their observational study on 56 fetuses, they divided their cohort into three groups: Group 1 included 14 (25%) fetuses with isolated pleural effusion without other structural abnormalities; Group 2 included 19 fetuses (34%) with a pleural effusion and additional anomalies but normal karyotype; Group 3 included 23 fetuses (41%) with a pleural effusion and an abnormal karyotype. The overall perinatal mortality was 42/56 (75.0%), with fetal death observed in 38 (68%) and neonatal death in 6 (10.7%) cases. Group I demonstrated a significantly higher rate of neonatal survival (64%), less fetal death (29%) and less neonatal death (7.1%) when compared to Group II (0, 78.9 and 21.1%, respectively) and Group III (13.0, 82.6 and 4.3%, respectively).
5.4 Antenatal Diagnosis
Pleural effusions may be evident on ultrasound from as early as the first trimester, however these are more commonly associated with aneuploidy. Most cases are diagnosed in the third trimester. Effusions are seen as anechoic regions surrounding and usually compressing the lung, which can be unilateral or bilateral. A classic “bat wing” appearance may become evident. The diaphragm may appear flattened or everted. The mediastinum can come under tension with shifting to the contralateral hemithorax. In this case, ventricular diameters decrease, inferior vena cava diameters widen and pulmonary artery Doppler peak velocities increase [93]. An effusion ratio has been reported, which is a ratio of the cross-sectional area of the effusion to that of the thorax [94]. Furthermore, the severity of left ventricular compression correlates with the effusion ratio [93].
Once an effusion is identified, differentiating between primary and secondary causes is important. Primary pleural effusion is a diagnosis of exclusion. Additional structural abnormalities are found in over a third of cases [90] and a detailed fetal echocardiography is mandatory. A large effusion with mediastinal shift may make visualisation of cardiac anatomy difficult, however this may improve after drainage. Concomitant pulmonary pathologies such as CDH, CCAM and BPS are secondary causes that should be especially sought. Signs of hydrops should be looked for in other fetal compartments and the amniotic fluid index measured for polyhydramnios. Sonographic features of congenital infection, aneuploidy and anaemia should be examined. In practice however, a fetal thoracocentesis (pleural aspiration) is required to confirm the diagnosis. This can be performed in a single pass following amniocentesis for karyotype. A pleural fluid that is predominantly lymphocytic (>80%) is considered pathognomic for chylothorax or primary pleural effusion [22, 94].
5.5 Antenatal Prediction of Prognosis
The presence of hydrops has consistently shown to predict poor prognosis [90, 92, 94]. In the presence of fetal hydrops, the estimated survival rate falls steeply from 80% to 30%. Fetuses with a secondary pleural effusion fare considerably worse than those with a primary hydrothorax. In Ruano and colleagues’ recent series [90], mortality for hydropic fetuses in Group 1 was 50% versus 100% mortality for hydropic fetuses in Group 2. Other predictors have been suggested, but do not appear consistently across studies: bilateral effusions [90], effusion ratio [95] and gestation at diagnosis [96]. Furthermore, prognosis after thoracoamniotic shunting is often related to the resolution of hydrops [97].
Most recently, fetal total lung volumes as assessed by three-dimensional ultrasonography were used to predict perinatal outcomes in cases of primary pleural effusion. In a cohort of 19 fetuses, the observed to expected total lung volumes were significantly correlated with perinatal outcomes such as respiratory morbidity and perinatal death, and to bilateral effusions and hydrops [98]. The place for this measurement in clinical management will need to be examined in larger studies.
5.6 Obstetric Management
A referral to a dedicated fetal medicine tertiary centre is required. The diagnostic work up is similar to that of hydrops. A detailed sonogram and echocardiogram for structural abnormalities and signs of aneuploidy and infection is performed. The middle cerebral artery peak systolic velocity is examined for anaemia. Maternal blood is analysed for serological evidence of infection (toxoplasmosis, rubella, cytomegalovirus, herpes, parvovirus B19, varicella, syphilis), red cell group and antibodies for immune hydrops, haemoglobin electrophoresis, and Kleihauer–Betke test of fetomaternal haemorrhage. Parents should be counselled for amniocentesis for genetic testing and congenital infectious screen (culture and PCR) [94]. Thoracocentesis should be considered at the same time, especially in cases of presumed primary hydrothorax. If a large effusion is evident, then delivery should occur at a tertiary centre. Multidisciplinary counseling with maternal fetal medicine specialists, neonatologists, paediatric surgeons and geneticists are appropriate.
Small effusions nevertheless require close follow up every 1–2 weeks to watch for volume expansion, mediastinal shift, hydrops, polyhydramnios and cervical shortening. A small stable effusion can be managed conservatively with good outcome (survival between 75–100%) [92, 99]. The same cannot be said, however, for fetuses that become hydropic. Survival rates are universally poor without intervention. Hydrops therefore represents the principal indication for fetal intervention. The systematic review of Knox [79] mentioned earlier concluded that in the presence of hydrops therapy seems warranted. Survival rates improve to 45–66% [79, 94, 97]. One may start with thoracocentesis however the effusion may be expected to reaccumulate over several days. Thoracoamniotic shunting can be performed using a double-pigtail catheter (Fig. 4.10). Alternatively, serial thoracenteses can be performed [100]. There is no evidence of a difference in outcome between the two approaches [101]. The complication rate of shunting is about 15% for iatrogenic rupture of membranes [102] and 5–10% for direct fetal loss. Shunt dislodgment has been reported, however posterior insertion may prevent the fetus from dislodging it. Preterm birth is common, with a mean gestational age at birth of 34–35 weeks. At birth the shunt is usually left, however needs obviously to be clamped.

ultrasound images after thoraco-amniotic shunt placement. (a) On the left the free part in the amniotic cavity is seen, and (b) on the right the intra-thoracic part. Image: courtesy UZ Leuven
Larger effusions risk pulmonary hypoplasia and respiratory distress at birth and ventilatory support may be needed. There is some discussion in the literature about thoracocentesis immediately prior to delivery to improve the transition to air breathing and facilitate resuscitation measures at birth. Others advocate thoracocentesis after birth, as they believe fluid reaccumulation can be rapid and render the fetus hypovolaemic and compromise resuscitation efforts at birth. Thoracocentesis can instead be performed once circulatory access has been established and fluid therapy initiated [22, 103].
6 Congenital Diaphragmatic Hernia
6.1 Definition and Epidemiology
The prevalence of congenital diaphragmatic hernia (CDH) ranges between 1 and 4 per 10,000 births, qualifying it as a rare disease. The exact etiology of the condition remains unknown. The vast majority of cases are left sided (LCDH), 13% are right sided (RCDH), and bilateral lesions, complete agenesis and other rarities comprise less than 2%. CDH can occur in association with other anomalies (in which case the mortality is over 85%) (Table 4.6), or as an isolated condition [104, 105].
6.2 Genetics
CDH most likely is caused by disruption of common developmental pathways, by several genes spread across the genome [106]. We recently reviewed the genetic factors underlying CDH, including evidence from genetic & teratogenic animal models of CDH and differential expression analysis [107]. Though all these fetuses with isolated CDH are routinely karyotyped, identification of (novel) submicroscopic imbalances and novel genes and/or therapeutic targets, requires the use of high resolution diagnostic methods. Several large centers do so, both from a clinical perspective as well as for research. We custom designed a high resolution array for comparative genomic hybdrization (CGH) [108]. The next step will be to use exome sequencing techniques in selected familial cases. Given the high probability of a mutation(s) segregating with the CDH phenotype, this approach will likely reveal the underlying gene(s) involved in the pathogenesis of CDH for each family studied. Advanced genetic testing in large populations with (apparently) isolated CDH, will lead to a better understanding of the genetics of this condition [109].
6.3 Pathophysiology and Natural History
Isolated CDH refers to the surgically correctable defect in the diaphragm, but the key problem is its consequence for lung development. Already from the first trimester the abdominal content herniates into the thorax, interfering with lung development. This causes hypoplasia of both lungs, i.e. fewer airway branches and abnormal pulmonary vessels, as well as a lesser lung compliance. At birth this causes ventilatory insufficiency and pulmonary hypertension, which can be lethal before the defect can be surgically repaired. The condition remains lethal in up to 30%, despite prenatal referral to a high volume center offering standardized neonatal care [110,111,112]. Survivors may have several morbidities, such as bronchopulmonary dysplasia and persistent pulmonary hypertension, gastro-esophageal reflux and other feeding problems, less frequently they have thoracic deformations after successful repair or other issues. Eventually most lead a life very close to normal provided when managed in a multidisciplinary follow up program [113, 114].
6.4 Antenatal Diagnosis
As ultrasound screening programmes have become widely implemented, one would expect the diagnosis is made before birth. In reality around two out of three cases are picked up before birth [115, 116]. The most striking signs become obvious on a cross section of the thorax, with compression and in left sided cases obvious displacement of the heart, by abdominal organs. In left CDH the stomach is often more posterior, viscera as well as spleen may be herniated (Fig. 4.11). An important feature is the presence of liver into the thorax. Right CDH is more difficult to recognize, because only the liver may be herniated, and echogenicity of the liver may be confused with that of the lung. Assessment of liver position is done by Doppler interrogation of the umbilical vein and liver vessels, as well as the position of the gall bladder (Fig. 4.11). Indirect signs of CDH are polyhydramnios and a smaller abdominal circumference. Differential diagnosis includes essentially other pulmonary pathology with cystic features, such as cystic adenomatoid malformation, bronchogenic cysts, rarely enteric or neuroenteric cysts, mediastianal teratoma and thymic cysts, or bronchopulmonary sequestration or bronchial atresia.

Fetus with left CDH before and after fetal therapy. (a) lung measurement at the four chamber view with the so-called “longest axis method” as well as “tracing method”. These dimensions will be used to calculate the Lung to Head Ratio. (b) measurement of the lung 1 day after balloon insertion, with changed echogenicity and dimensions. (c) Herniation of the liver. (d) visualization of the major vessels help in its identification. Copyright UZ Leuven; (From Deprest et al., Treatment of Congenital Diaphragmatic Hernia. In: Fetal MRI. Prayer D (Ed). Springer Verlag, Heidelberg 2011, 528 pp.)
Prenatal diagnosis of CDH should prompt referral to a tertiary centre experienced in assessing this anomaly and managing CDH in the perinatal period. A comprehensive diagnostic and prognostic work up comprises advanced imaging, genetic testing and multidisciplinary counselling, so that parents can take a well informed decision. In view of the options, an individualized prediction of outcome is crucial.
6.5 Antenatal Prediction of Prognosis
Most frequent quoted predictors of outcome are the presence of associated anomalies, and for isolated cases, lung size, position of the liver and the stomach, and to a lesser extent assessment of lung vasculature. Herein we will focus on the best validated predictors; however research on improved prediction is actively ongoing.
The Lung-to-Head Ratio (LHR) consists of measurement of the lung contralateral to the defect, at the level of the four chamber view. It is divided by the head circumference as measured in the standard biparietal view [117] (Fig. 4.11). The most accurate method for measuring is by tracing the lung contours [118, 119]. The LHR is a function of gestational age, because the lung grows four times more than the head over the entire gestation. To overcome this problem, we proposed to express the LHR of the index case as a function of what is expected in a gestational aged control (observed [O]/expected [E] LHR). The expected value can be calculated using formulas specific for the side of the lesion [120], which are also available on line (www.totaltrial.eu). The prognostic value of the O/E LHR was validated in 354 fetuses with unilateral isolated CDH, that were evaluated between 18 and 38 weeks gestation; later meta-analyses have confirmed this [121]. The O/E LHR is predicting mortality as well as early neonatal morbidity (Fig. 4.12) [122, 123].

Survival rates of fetuses with isolated left-sided congenital diaphragmatic hernia, depending on measurement of the observed/expected lung:head ratio (O/E LHR) and position of the liver position as in the antenatal congenital diaphragmatic hernia registry. From: Jan A. Deprest et al., Antenatal prediction of lung volume and in-utero treatment by fetal endoscopic tracheal occlusion in severe isolated congenital diaphragmatic hernia, Seminars in Fetal & Neonatal Medicine (2009), doi:https://doi.org/10.1016/j.siny.2008.08.010 (Deprest, Flemmer et al. 2009); with permission of authors and publisher
Lung size can also be estimated by volumetric techniques, either using 3D ultrasound or fetal Magnetic Resonance Imaging (MRI). Because the ipsilateral lung is not always visible on 3D US, we do prefer MRI [124] (Fig. 4.13). Again measured fetal lung volume must be expressed as a function of what one expects for a gestational age- or weight-matched control [125, 126]. A recent meta-analysis has shown its predictive value [127]. Though we feel fetal MRI will become the method of choice, there is currently no proof for superiority over ultrasound yet [128].

T2 weighted images of fetus with left sided CDH at 26 weeks without liver herniation. Top left, sagittal section with tracing of the body contours. Top right: coronal view of the fetus demonstrating the level at which the two axial images are made (bottom). Lung tracing (dotted line) on the two axial views. Scale: white bar in right lower corner is 1 cm (From Deprest et al., Treatment of Congenital Diaphragmatic Hernia. In: Fetal MRI. Prayer D (Ed). Springer Verlag, Heidelberg 2011, 528 pp.)
The presence of liver herniation is also predictive, but whether it can be used as a sole feature remains a matter of debate [127, 129]. Liver herniation is easily visualized by ultrasound, using the umbilical vein as a landmark. We and others have proposed to use MRI to quantify the degree of liver herniation more accurately, but we refer to the literature for further details [130] (Fig. 4.14). Position of the stomach is in Japan accepted as a predictor of outcome, based on its position as up or down [131, 132] or more specifically on its location in the thoracic cavity [133]. Several classification systems have been devised for standardized determinations of the stomach position such as by Kitano et al. [133], and more recently by Cordier et al., yet the latter on ultrasound [134]. Another less studied factor is pulmonary vascular assessment, for which again we refer to the literature (reviewed in [130]). Eventually more accurate prediction can be expected by combining techniques [135, 136], especially when they assess different aspects of the disease, such as parenchymal lung measurement and evaluation of the pulmonary vascularization.

T2 weighted images of fetus with left sided CDH at 26 weeks with liver herniation, as best shown on coronal view (left image). Sagittal view (middle) demonstrating the reference line (full line) at the level of the xyphoid process (white arrow) and above (dashed line), which correspond to the axial images on the right. Liver tracing (dotted line) and contours of the thoracic cavity (full and dashed lines) shown on both axial views. Scale: white bar in right lower corner is 1 cm (From Deprest et al., Treatment of Congenital Diaphragmatic Hernia. In: Fetal MRI. Prayer D (Ed). Springer Verlag, Heidelberg 2011, 528 pp.)
6.6 Antenatal Management
CDH has been the sentinel condition proposed for fetal surgery. We refer to a recent review for the history of fetal surgery for this condition [137, 138]. Initially in utero anatomical two step repair was proposed. Since this includes reduction of the liver which causes umbilical vein kinking, the majority of cases are not amenable for fetal surgery. Now prenatal intervention consists of percutaneous Fetoscopic Endoluminal Tracheal Occlusion (FETO) (Fig. 4.15). Tracheal occlusion prevents egress of lung fluid, increasing airway pressure, causing proliferation, increased alveolar airspace and maturation of pulmonary vasculature (reviewed in [139]). The current timing of insertion and removal of the balloon is based on observations in sheep experiments, with a recent trend for later insertion to reduce the impact of preterm delivery, mainly due to ruptured membranes. Sustained TO, though inducing lung growth, reduces the number of type II pneumocytes hence surfactant expression. This can be improved by in utero release (“plug-unplug sequence”) [140]. Perinatal steroid administration has also been shown to beneficial [141]. Ideal balanced lung growth and maturation is however obtained experimentally by 47 resp. 1 h cycles of occlusion and release, but this is yet clinically impossible [142].

Schematic drawing of percutaneous fetoscopic endoluminal tracheal occlusion. Drawing Myrthe Boymans; copyright UZ Leuven.
Several occlusion methods have been described, but an endovascular balloon is what currently is used [143]. This technique is amenable for a percutaneous approach under local anesthesia [144]. Also purpose designed instruments were developed with support of the European Commission (reviewed in [145]). Over time, invasiveness was reduced by moving away from general over loco-regional to local anesthesia, with fetal analgesia and immobilization [146]. The FETO task force proposes for severe cases insertion of the balloon at around 28 weeks, and reversal of occlusion at 34 weeks. In utero reversal is achieved either by fetoscopy (50%) or ultrasound guided puncture (19%). Prenatal (>24 h) removal of the balloon has been shown to increase survival and decrease morbidity [147]. Removal at birth can be done on placental circulation or less ideally after birth. Peri- or postnatal retrieval should not be underestimated and problems with this have been the cause of neonatal death [146, 148, 149].
We have reported outcomes of 210 interventions, in fetuses with liver herniation and an O/E LHR <27–28%. Compared to historical controls from the antenatal CDH registry, FETO increased survival in severe cases with left-sided CDH from 24.1 to 49.1%, and in right-sided from 0 to 35.3% (p < 0.001) [122]. The strongest predictors of survival were observed/expected LHR prior to the procedure (OR, 1.490; P = 0.019) and gestational age at delivery (OR 1.024; P = 0.007). Early delivery typically is the consequence of Preterm Premature Rupture Of the Membranes (PPROM), occurring within 3 weeks occurred in 16.7% cases. Mean gestation at delivery was 35.3 weeks; only one in three delivers prior to 34 weeks. Interestingly, survival for those delivering at 32–33 weeks is equal to that at 34 weeks or later (60%; left sided cases only) (Fig. 4.16). Short term morbidity in survivors is better than expected: it is close to that of cases with moderate pulmonary hypoplasia, that were managed expectantly during pregnancy [147].

Graphical display of number of patients delivering (light blue bars) and number of fetuses surviving (dark blue bars) as a function of gestational age. Modified from Deprest J, Nicolaides K, Done E, et al.: Technical aspects of fetal endoscopic tracheal occlusion for congenital diaphragmatic hernia. J Pediatr Surg 46:22–32, 2011 (Deprest, Nicolaides et al. 2011), with permission from the authors and the publisher
The early clinical experience has shown few demonstrable clinical side effects of the balloon on the developing trachea, except in very early occlusions and complications arising at the time of removal [48]. However, the neonates and infants do have obvious tracheomegaly, that does not seem to have a clinical impact, except for a barking cough on effort [48, 49]. Over time, the widening seems to become less important [49]. Most newborns require surgical patching of the diaphragm, indicating the rather large size of the defect in this selected group. The use of patch has previously been shown to be a predictor of outcome. High patch rates will increase the number of patch related complications, so that we felt research on engineering a diaphragmatic patch is now really required [150].
6.7 Trials on FETO Versus Expectant Management
Our initial experience has meanwhile been confirmed in other hands [151,152,153]. There is even one Brazilian randomized trial that showed an increased survival following FETO, though the survival rate in expectantly managed cases was very low (5%), which is not representative for most institutions [154]. Also right and left sided cases were pooled, which does not seem appropriate. In Europe there are now two trials ongoing (www.totaltrial.eu) (Table 4.7). One is in severe hypoplasia, with insertion of the balloon at 27–30 weeks and its removal at 34 weeks (NCT01240057). This protocol is based on our existing experience. A few items are worth mentioning. (1) We have slightly moved the time point of insertion from 26–28 to 27–30 weeks. This will lessen the risk for delivery prior to 32 weeks, the latter having a negative impact on survival (Fig. 4.16). The reason why we do not insert the balloon later, is that we have evidence for a less vigorous lung response with insertion beyond 30 weeks [155]. (2) We chose for in utero balloon removal (3) at 34 weeks. Prenatal removal was associated with a higher survival [145, 147]. Moreover survival does not increase with gestation at delivery, once beyond 34 weeks. Timely and elective balloon removal avoids unexpected emergency balloon retrieval. However, in the two Brazilian series there was no apparent difference in survival with balloon removal at the time or prior to birth. The trial in foetuses with moderate hypoplasia has started earlier (NCT00763737)(Table 4.7). In this group occlusion is done at 30–32 weeks. For both trials, our neonatal colleagues from all over Europe designed a standardised consensus postnatal management protocol [156] (Table 4.8). In right sided cases, the cut off is at O/E LHR <45%.
7 Fetal Cardiac Interventions
Antenatal interventions for congenital heart disease remain experimental, despite a well-defined rationale, with only a few expert centres worldwide developing expertise. The Children’s Hospital of Boston has led this research. In this Chapter, we will review critical valvular aortic stenosis with evolving hypoplastic left heart syndrome (HLHS) in the fetus, which is considered the sentinel pathology for potential intervention. For readers interested in the experimental intervention for pulmonary atresia and hypoplastic right ventricle, we refer to the Boston group’s two recent publications as well as one from Linz [157,158,159,160].
7.1 Definition and Epidemiology
Critical aortic stenosis is a congenital obstruction to the left ventricular outflow tract. It occurs most commonly as a result of fusion or one or both aortic commissures, thereby reducing leaflet mobility [26]. The incidence is 3.5 in 10,000 births, and represents 5% of childhood cardiac disease. HLHS is a lethal congenital heart abnormality, whereby the left ventricle is unable to support the systemic circulation. It usually occurs due to obstruction to left ventricular outflow, with aortic atresia the most common (Fig. 4.17). The degree of the ventricular hypoplasia is proportional to the degree of outlet obstruction, unless an escape mechanism exists such as a ventricular septal defect [161].

Hypoplastic left heart syndrome
7.2 Genetics
Aortic stenosis is associated with Turner, Noonan and William’s syndrome. Aneuploidy is present in 5% of cases of congenital heart disease [22]. It is more common in males, 4:1. Recurrence is 2% with one affected sibling and 6% with two affected siblings. HLHS occurs in 10,000 births, with males twice as likely as females to be affected [22]. It is most commonly sporadic.
7.3 Pathophysiology and Natural History
Most fetuses with aortic valve stenosis will not sustain fetal compromise during pregnancy. However, a subset with critical stenosis (ductus dependent) may result in decreased right to left shunting between atria and thereby retard normal left ventricular development and result in HLHS. A pressure effect from the obstructed outflow tract first enlarges the left ventricle, coronary perfusion falls and cardiac function is impaired. Chronically raised ventricular pressures cause endocardial fibroelastosis and left sided structures such as the aortic root, mitral valve and left ventricle then fail to develop with advancing gestation [22, 162, 163]. Indeed, a preferential return of oxygenated blood towards the right ventricle and lower body rather than towards the left ventricle and the brain may also lead to suboptimal brain oxygenation in utero, which may be in part responsible for poorer neurodevelopmental outcomes [164, 165]. Significant restriction of the foramen ovale is present in 22% of patients with HLHS [166] and in 6% the atrial septum is completely closed. This drives increased pulmonary vein pressure and left atrial hypertension, as well as to pulmonary venous arterialization (abnormal vascular musculature) and hydrops. After birth, pulmonary venous return to the left atrium is increased, and obstruction to pulmonary venous drainage results in a further rise in pulmonary pressure, with severe hypoxemia, pulmonary oedema and haemorrhage. Emergency atrial septostomy is required.
About 5% of HLHS develop in the second trimester and are progressive [167]. Intrauterine growth restriction and hydrops may similarly develop. In Sharland and coauthors’ series of 30 fetuses with left ventricular dysfunction, five fetuses with aortic stenosis developed into HLHS. There appears to be an overlap of diseases of primary left ventricular endocardial fibroelastosis, critical stenosis of the aortic valve and the HLHS [163].
Despite improvements in neonatal surgical and intensive care, the outcome of fetuses with hypoplastic left heart syndrome (HLHS) remains poor. Postnatal surgery, which for many results in a far from perfect single ventricle Fontan-type circulation [168], has a significant mortality rate, leading to a total survival of less than 65% [167]. Without it survival is not possible. The surgical choice is Norwood’s three staged procedure or neonatal transplantation. For some fetuses with critical aortic stenosis, biventricular repair remains a possibility, with the therapeutic options including open and balloon valvulotomy [169].
7.4 Antenatal Diagnosis
Critical aortic valvular stenosis may be diagnosed antenatally, however sonographic features for milder forms may be unreliable [22]. An early clue may include dilatation of the ascending aorta [26]. With critical obstruction, the left ventricle becomes hypokinetic and may appear dilated and hypoplastic on a standard 2D four chamber view. With advancing gestation however, ventricular size will become relatively smaller. Endocardial fibroelastosis may become evident. In cases of aortic and mitral atresia, the ventricle has a “slit” like appearance or may even appear absent, whereas aortic stenosis with mitral stenosis there is a small and very round or “tense” left ventricle, with the apex of the heart formed by the right ventricle [170]. Colour Doppler will reveal poor or absent filling of the diminutive ventricle. The aortic valve leaflets may be thickened with reduced motion and reveal turbulent and increased antegrade velocity on Doppler imaging. An absence of antegrade flow across the aortic valve confirms atresia. With severe stenosis, there may be hypoplasia of the mitral valve and aortic arch, or mitral stenosis and coarctation [26]. Mitral regurgitation can be determined by colour Doppler. The right ventricle and tricuspid valve should be inspected for regurgitation. The patency of the foramen ovale should be demonstrated by colour Doppler. Flow may become reversed across the atria, from left to right, with consequences for the pulmonary vasculature [26]. Doppler examination of the pulmonary veins then complements this assessment. In fetuses with a restrictive foramen ovale, the venous a wave is increased and peaked, with reversal of flow in the pulmonary veins during atrial systole [170]. Moving the transducer to the sagittal view will demonstrate retrograde flow in the ascending aorta and transverse aortic arch. Here, a coarctation may be visualised. At the three-vessel view, colour Doppler will show flow in the opposite direction of the adjacent pulmonary artery and arterial duct, confirming retrograde flow in the transverse aorta from the arterial duct. The Doppler profile in the arterial duct will similarly be altered. It is important to note here that the brain receives oxygenated blood via retrograde filling of the head and neck vessels from the arterial duct. Accordingly, the middle cerebral artery Doppler pulsatility index should be examined for central redistribution, or a fall in resistance, in an attempt to maintain fetal cerebral perfusion and guard against cerebral hypoxia. Fetal growth and amniotic fluid index may indicate IUGR. Sonographic evidence of additional abnormalities, aneuploidy and hydrops should all be meticulously sought.
7.5 Antenatal Prediction of Prognosis
For fetuses with critical valvular aortic stenosis, accurate prediction of evolving HLHS is imperative for parental counselling regarding continuation of the pregnancy, antenatal care, timing and patient selection for potential fetal intervention, to plan resuscitation and postnatal surgery. Mäkikallio and co-authors [171] from the Children’s Hospital in Philadelphia, recently published anatomic and physiological variables in the mid gestation fetus with aortic stenosis and normal left ventricle size that are predictive of progression to HLHS. In their study on 43 fetuses diagnosed with aortic stenosis and normal left ventricular length at less than 30 weeks gestation, there were 23 live-born patients of which 17 developed HLHS and six having a postnatal biventricular circulation. All fetuses that progressed to HLHS had retrograde flow in the transverse aortic arch, 88% had left-to-right flow across the foramen ovale, 91% had monophasic mitral inflow, and 94% had significant LV dysfunction. With regard to all six fetuses who successfully achieved a biventricular circulation postnatally, they had antegrade flow in the transverse aortic arch, biphasic mitral inflow and normal LV function. These echocardiographic indices may help refine patient selection and assist prenatal counselling for progression of aortic stenosis to HLHS.
As with other fetal pathologies, the presence of hydrops suggests a bleak outlook. Similarly, the presence of tricuspid regurgitation conveys a poor prognosis for both survival and postnatal surgical success [170]. An added layer of complexity comes with a restrictive foramen ovale. Fetuses with a restrictive or intact foramen have a worse prognosis and may not respond to medical therapy in the postnatal period. This is a result of abnormal pulmonary vasculature that develops from high atrial pressures. Hypoxaemia can develop quickly at birth and cardiovascular collapse is likely without immediate balloon atrial septostomy or surgery. Antenatal prediction of emergency atrial septostomy (or fetal intervention described below) is therefore essential to plan resuscitation and cardiac intervention at birth. The Cincinnati group have developed a pulmonary venous Doppler forward/reverse velocity time integral (FR VTI) ratio, that when less than three, optimizes specificity for predicting emergency atrial septostomy [158]. Others have performed a maternal hyperoxygenation challenge to study the response of the fetal pulmonary vasculature. Less than a10% decrease in the branch pulmonary artery pulsatility index after 10 min of 60% inspired oxygen administered to the mother is a non-reactive test and is predictive of emergency intervention at birth [172].
Furthermore, as introduced earlier, half of the long-term survivors have poor neurodevelopmental outcome [173], which may in part have an antenatal origin. Reduced middle cerebral artery pulsatility index may be a marker of such cerebral hypoxia, however this has not been specifically correlated with neurodevelopmental follow up in infants with HLHS.
7.6 Obstetric Management
Referral of suspected cases to an expert tertiary centre is required. Multidisciplinary counselling with maternal fetal medicine specialists, neonatologists, paediatric cardiologists and cardiothoracic surgeons are necessary. The diagnosis should be confirmed by fetal echocardiography as discussed above, including predictive indices. Additional abnormalities should be excluded and an amniocentesis performed to evaluate chromosomes. After counselling, termination of pregnancy should be offered to parents due to the poor prognosis and significant surgical morbidity that is necessary to sustain life. Expectant management may also be offered, whereby the pregnancy is conservatively cared for without intervention to spontaneous labour. At birth, comfort care to the neonate is offered. A clear plan from neonatology colleagues and parental support is clearly necessary. This may become more of an option as the pregnancy continues and repeat echocardiograms from 28 weeks gestation reveal progressive HLHS.
In some cases of evolving HLHS due to outlet valve obstruction, timely fetal balloon valvuloplasties offer the attractive rationale of ventricular recovery in utero and further growth (Fig. 4.18). Antenatal intervention theoretically reduces intraventricular pressure, improves coronary perfusion, reduces pulmonary pressures, promotes ventricular growth, and avoids development of myocardial fibroelastosis, thus all enabling improved cardiac function and potential biventricular postnatal repair [18].

Schematic drawing of in utero percutaneous aortic valve dilatation. Drawing: Myrthe Boymans. Reproduced, with permission from the UZ Leuven
Standardised needle-based procedures to access the fetal heart have been developed from groups in Boston (Massachusetts, US) [174] and Linz (Austria) [159]. A cardiocentesis is performed using a Seldinger technique to advance the working sheath into the target area under ultrasound guidance. Most cases can safely be performed under maternal local or locoregional anesthesia with fetal analgesia and immobilization. Maternal laparotomy rates are now therefore uncommon, falling from 27 to 10% in the Boston experience [160]. For the procedure, an 18 or 19 gauge needle is inserted into the fetal left ventricle at the level of the apex and in alignment with the left ventricular outflow tract (Fig. 4.18). A guide wire and a catheter with a coronary dilatation balloon are advanced through the aortic valve, which is dilated to 120% of the valve annulus. Fetal valves differ to children’s valves, in that they are less complex structures, difficult to dilate and likely to re-stenose. Fetal resuscitation with inotropic medication, other vasoactive agents, or blood must be anticipated early to correct fetal acidosis or anaemia. Fetal complications include bradycardia requiring fetal resuscitation (17–38%), hemopericardium (13%), ventricular thrombosis (15–20%) and fetal death (8–13%) [159]. Maternal morbidity is rare, except when uterine exposure is needed for the procedure or emergency delivery. Rupture of membranes occurs in 2–7% of cases [175, 176].
In their largest series published in 2009 [174], the Boston group examined prenatal aortic valvuloplasty in 70 fetuses with aortic stenosis and evolving HLHS. The intervention was technically successful in 52 (74%) of treated fetuses, of whom 17 (33%) achieved biventricular circulation postnatally. However, prenatal aortic valvuloplasty altered the growth and function of some left heart structures (aortic valve, ascending aorta, mitral valve), but surprisingly did not change the growth velocity of the left ventricle. This may alter the approach to fetuses with evolving HLHS. Nevertheless, in fetuses with a larger left ventricle at enrolment, aortic valvuloplasty did increase the likelihood of a biventricular circulation. The authors developed a multivariate threshold scoring system of valvular and ventricular size and pressure to predict fetuses able to survive postnatally with a biventricular circulation. This may improve patient selection for fetal intervention. Of note, in utero atrial septoplasty for a highly restrictive foramen ovale has also been described, however the numbers remain small at this stage [177].
For otherwise continuing pregnancies, the fetal heart should be examined at 28 weeks gestation and then fortnightly thereafter for progression and the development of hydrops. The pregnancy should otherwise be allowed to continue to term. Delivery should occur in an experienced tertiary centre with immediate neonatal, cardiology and cardiothoracic surgery teams present. There appears to be no significant advantage of caesarean delivery when compared to vaginal birth. The Children’s Hospital of Philadelphia compared 79 cases of HLHS delivered either vaginally or by caesarean section [178]. They found those delivered by elective caesarean had lower pH and higher partial pressure of CO2 on arterial cord blood gas analysis, however no other differences in 1- and 5-min Apgar scores, markers of end organ function, echocardiographic parameters, length of hospitalization, and survival to discharge. It is even possible that vaginal delivery may prove advantageous, given that the fetus has a relatively fixed stroke volume and relies on heart rate to drive cardiac output. The physiological demands from vaginal delivery may protect the fetus by augmenting cardiac output from the single functioning ventricle [178]. Some centres plan induction of labour close to term to ensure a neonatal intensive care bed and appropriate staff are available and prepared to care for the neonate.
Others however, have advocated planned caesarean section for cases with restricted foramen ovale. It is argued a scheduled delivery facilitates immediate transfer of the neonate to an already well-prepared team for emergency atrial septoplasty [158, 179]. An ex-utero intrapartum treatment (EXIT) procedure has also been reported to initiate extracorporeal membrane oxygenation before placental separation. Once stabilized, the neonate then underwent radiofrequency perforation of the atrial septum with a good outcome [180].
Prostaglandin E1 therapy, often in conjunction with inotropes and correction of acidosis, is required for severe stenosis and HLHS [26]. If the stenosis is less severe and left ventricular function appears adequate and the condition is not duct dependent, the ductus arteriosus is allowed to close under close supervision.
8 Amniotic Band Syndrome
8.1 Definition and Epidemiology
Amniotic band syndrome (ABS) is a group of disruptive abnormalities and limb amputations; the likely consequence of early rupture of the amnion [26]. The reported incidence is 1 in 1300 live births.
8.2 Genetics
Considered sporadic. Associations with Ehlers-Danlos syndrome and osteogenesis imperfecta have been reported [22, 26]. There is no known risk of recurrence in future pregnancies.
8.3 Pathophysiology and Natural History
At first glance of our simple definition of early amnion rupture, one might expect the pathophysiology to be rather simple. However, there is a wide range of clinical manifestations and several theories have been developed to try and understand the observed complexity. Indeed there is a spectrum of many anomalies involving limbs, craniofacial structures, trunk, constrictive bands, and visceral and body wall abnormalities [22]. It is fair to say the exact cause remains unclear.
Of the proposed theories, Streeter in 1930 [181] suggested an intrinsic defect in the embryonic germinal disk and amniotic cavity, which resulted in later amniotic bands. This explains central anomalies of viscera, limb body wall complex and craniofacial structures, but is less satisfactory for extremities [182]. Torpin in 1965 [183] described an extrinsic theory, whereby early rupture of the amnion partially dislodges the fetus to the extra-amniotic space, with secondary amniochorionic mesodermal bands developing. A vascular disruption theory has also been proposed [184]. Most recently Moerman and colleagues [185] in Leuven, synthesised this work by proposing three distinct lesions: constrictive amniotic bands, amniotic adhesions and limb-body wall complex. This was based on their pathological study of 18 cases of ABS. In this series, four cases had constrictive bands presumed to be from amnion rupture that resulted in limb entanglement in shrivelled amniotic strands. This type may also be associated with cord entanglement and fetal death. In other cases, adhesive bands were observed to be the result of a broad fusion between disrupted fetal parts (mostly cephalic) and an intact amniotic membrane, while the limb-body wall complex is likely the result of vascular disruption. The authors suggest that the observed craniofacial defects (encephalocoeles and/or facial clefts) in these fetuses were not caused by constrictive amniotic bands, but were the result of a vascular disruption with or without cephalo-amniotic adhesion. Accordingly, the theories of Streeter and Torpin are not mutually exclusive and in fact may overlap. Timing of amnion rupture may also help to explain the variation in clinical manifestations. Huang and colleagues [186], suggested more severe visceral and skull abnormalities are likely to occur if rupture occurs before 45 days gestation.
Chorioamniotic separation, although considered normal until 16 weeks gestation, may occur either spontaneously, or following extrachorionic haemorrhage or invasive procedures such as amniocentesis, amnioreduction or fetal surgery [22, 187]. This may also increase the likelihood of ABS [188].
The natural history is difficult to predict. The degree of developmental disruption will determine the clinical outcome. Not unexpected with a definition of ruptured membranes is preterm birth, with an average gestational age of 32 weeks in cases of ABS [22]. Low birth weight is also more common [189].
8.4 Antenatal Diagnosis
Sonographic features may be either isolated or appear in combination, reflecting the numerous forms of ABS. The bands themselves may be evident sonographically (Fig. 4.19), or more commonly, their effect on the fetal tissue can be seen. For example, bands may be inferred by absent or amputated digits or portions of limbs and constrictive bands from swollen distal limbs (Fig. 4.20). Similarly Doppler ultrasound may reveal compromised flow. Clubfeet or hands may occur. Craniofacial abnormalities may include cleft lip and palate, microphthalmia, encepholocoele and anencephaly. Encephalocoele can be attributed to ABS when they occur off the midline and anencephaly when some portion of the calvarium is present [22]. Gastroschisis, omphalocoele (less commonly) and limb body wall complex can all be associated with ABS [26]. Severe spinal defects, particularly when associated with abdominal wall defects, are consistent with ABS. Umbilical cord Doppler flow is essential to exclude cord involvement.

Amniotic band syndrome , evidenced by ultrasound. Image: courtesy UZ Leuven


Limb in Amniotic band syndrome. (a) Right forearm at 23 weeks: significant edema at the distal part and visualization of two notches created by the amniotic band (arrows). (b) Ultrasound image of the right arm and hand 1 month after fetoscopic release of the band. There is a decrease of the edema, with still two notches visible due to the amniotic band (arrows). c. At birth, there is still amniotic band tissue present around the arm, infiltrating into the subcutis. Images: courtesy UZ Leuven
The sonographic presence of amniotic bands is not necessary to complete a diagnosis of ABS however, provided characteristic features are present. Fetal MRI has also been shown to be complementary to routine ultrasound [190].
8.5 Antenatal Prediction of Prognosis
Prediction of prognosis is not well established, however is likely to be proportionate to the complexity of the ABS. Tadmor and colleagues (1997) [191] serially observed lower limb amputation from 21 weeks gestation, with evolving sonographic signs of a constriction ring, oedema, patent then absent limb arterial Doppler, bending, breaking and finally resorption of both tibia and fibula. However, other reports have shown resolution [192]. Serial Doppler ultrasound forms an important part of clinical assessment, particularly with regard to cord involvement. Reversed end diastolic flow of the umbilical artery Doppler may become evident with worsening constriction [193]. Hüsler and colleagues (2009) [194] proposed a prenatal classification to reflect stages of ABS involving extremities, which is based on Weinzweig’s postnatal classification [195] (Table 4.9).
8.6 Obstetric Management
Referral to a dedicated fetal medicine centre is appropriate. A detailed sonographic survey and fetal echocardiogram is essential. Amniocentesis for karyotype is recommended for cases of diagnostic uncertainty. Obstetric management is dependent on whether the ABS is isolated or more complex. For isolated cases, expectant management is the norm. Weekly Doppler surveillance in comparison to the contralateral side for constrictive progression seems appropriate. When progression is identified, fetoscopic intervention may salvage the developing limb. However, given the natural history is not well established, patient selection is not established. Compromised but present flow is probably the appropriate time for intervention. Richter and colleagues (2012) [182] recently reviewed all published cases of intrauterine release of amniotic bands to rescue fetal limbs. The outcomes were generally favourable, although it might not always be possible to visualize and/or dissect and cut the bands, without risk for collateral damage. The bands may be covered within a constrictive ring in an oedematous area. After the procedure partial to full disappearance of the signs, flow and/or functional recovery may be obtained, but postnatal surgery remains often necessary. In their series of ten cases, preterm prelabour rupture of the membranes occurred in 78% prior to 34 weeks, with 67% of cases delivering preterm. Ruptured membrane rates were higher following fetoscopic band release when compared with fetoscopy for other indications, which may be related to the inherent pathology of ABS. In 90% of cases (8/9), the band could be cut to some extent, but only in three cases (33.3%) there was complete release possible. Functional salvage of the limb occurred in 6/9 cases (67%). In the other three, secondary postnatal amputation was required in one and in two cases there was persistent reduced mobility.
For more complex ABS with multiple abnormalities, multidisciplinary counselling with geneticists, neonatologists and paediatric surgeons are essential. Options of expectant management and termination of pregnancy should be discussed with parents.
9 In Conclusion: From Tinkering to Translation
For the field of fetal intervention and surgery to continue to translate into improved and meaningful outcomes for our fetal and neonatal patients, training and research collaborations between maternal-fetal medicine specialists, neonatologists and paediatric surgeons must become fluid across multiple centres. Effective collaboration is necessary to ensure cohorts of fetal patients with rare diseases are identified and streamlined into formalised trials in specialised centres to evaluate our novel interventions. This chapter is but one example of the necessary dialogue needed across different medical specialties if we are to help improve the lives of our unborn fetal patients.
References
Gilmore, L., et al., Arginine functionalization of hydrogels for heparin binding-a supramolecular approach to developing a pro-angiogenic biomaterial. Biotechnol Bioeng, 2012.
Altman D, et al. Anterior colporrhaphy versus transvaginal mesh for pelvic-organ prolapse. N Engl J Med. 2011;364(19):1826–36.
Falcon O, et al. Screening for trisomy 21 by fetal tricuspid regurgitation, nuchal translucency and maternal serum free beta-hCG and PAPP-A at 11 + 0 to 13 + 6 weeks. Ultrasound Obstet Gynecol. 2006;27(2):151–5.
Stanford E, Moen M, Cassidenti A. Traditional native tissue vs mesh-augmented pelvic organ prolapse repairs: providing an accurate interpretation of current literature. Reply. Int Urogynecol J. 2012;23(9):1319–20.
Dane B, et al. Ultrasound screening for fetal major abnormalities at 11–14 weeks. Acta Obstet Gynecol Scand. 2007;86(6):666–70.
Shepherd JP, et al. Uniaxial biomechanical properties of seven different vaginally implanted meshes for pelvic organ prolapse. Int Urogynecol J. 2012;23(5):613–20.
Hodges RJ, Wallace EM. Testing for Down syndrome in the older woman: a risky business? Aust N Z J Obstet Gynaecol. 2005;45(6):486–8.
Morris JK, Waters JJ, de Souza E, The population impact of screening for Down syndrome. audit of 19 326 invasive diagnostic tests in England and Wales in. Prenat Diagn. 2008;32(6):596–601.
Lose G, Gras S. While we wait for a new regulatory framework for surgical mesh. Int Urogynecol J. 2012;23(8):969–70.
Hillman, S.C., et al. Microarray comparative genomic hybridization in prenatal diagnosis: a review. Ultrasound Obstet Gynecol. 2012;40(4):385–91.
Harrison MR, Adzick NS. The fetus as a patient. Surgical considerations. Ann Surg. 1991;213(4):279–91. discussion 277–8
Deprest JA, et al. Fetal surgery is a clinical reality. Semin Fetal Neonatal Med. 2010;15(1):58–67.
Senat MV, et al. Endoscopic laser surgery versus serial amnioreduction for severe twin-to-twin transfusion syndrome. N Engl J Med. 2004;351(2):136–44.
Adzick NS, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364(11):993–1004.
Harrison MR, et al. A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia. N Engl J Med. 2003;349(20):1916–24.
Rodrigues HC, Deprest J, v d Berg PP. When referring physicians and researchers disagree on equipoise: the TOTAL trial experience. Prenat Diagn. 2011;31(6):589–94.
Kilby M, et al. PLUTO trial protocol: percutaneous shunting for lower urinary tract obstruction randomised controlled trial. BJOG. 2007;114(7):904–5. e1–4
Creasy RKR, Iams R, Lockwood JD, Moore CJ, Creasy TR. Resnik’s maternal-fetal medicine. Principles and practice, 6th edn. Invasive fetal therapy. Philadelphia: Saunders Elsevier; 2009.
Anumba DO, et al. Diagnosis and outcome of fetal lower urinary tract obstruction in the northern region of England. Prenat Diagn. 2005;25(1):7–13.
Feist, A., et al., Increased incidence of cutaneous squamous cell carcinoma in lung transplant recipients taking long-term voriconazole. J Heart Lung Transplant, 2012.
Escamilla J, Lane MA, Maitin V. Cell-free supernatants from probiotic Lactobacillus casei and Lactobacillus rhamnosus GG decrease colon cancer cell invasion in vitro. Nutr Cancer. 2012;64(6):871–8.
Wondergem B, et al. Expression of the PTTG1 oncogene is associated with aggressive clear cell renal cell carcinoma. Cancer Res. 2012;72(17):4361–71.
Lor KW, et al. Plerixafor as first- and second-line strategies for autologous stem cell mobilization in patients with non-Hodgkin’s lymphoma or multiple myeloma. Pharmacotherapy. 2012;32(7):596–603.
Weigert O, et al. Molecular ontogeny of donor-derived follicular lymphomas occurring after hematopoietic cell transplantation. Cancer Discov. 2012;2(1):47–55.
Muller, P.A., et al., Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene, 2012.
Ng YZ, et al. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res. 2012;72(14):3522–34.
Weinger, J.G., et al., MHC Mismatch Results in Neural Progenitor Cell Rejection Following Spinal Cord Transplantation in a Model of Viral-Induced Demyelination. Stem Cells, 2012.
Thobakgale CF, et al. Frequent and strong antibody-mediated natural killer cell activation in response to HIV-1 Env in individuals with chronic HIV-1 infection. J Virol. 2012;86(12):6986–93.
Tang QQ, Lane MD. Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem. 2012;81:715–36.
Styles L, et al. Refining the value of secretory phospholipase A2 as a predictor of acute chest syndrome in sickle cell disease: results of a feasibility study (PROACTIVE). Br J Haematol. 2012;157(5):627–36.
Roberts NA, et al. Rank signaling links the development of invariant gammadelta T cell progenitors and Aire(+) medullary epithelium. Immunity. 2012;36(3):427–37.
Johnson MP, et al. Sequential urinalysis improves evaluation of fetal renal function in obstructive uropathy. Am J Obstet Gynecol. 1995;173(1):59–65.
Morris RK, et al. Systematic review of accuracy of fetal urine analysis to predict poor postnatal renal function in cases of congenital urinary tract obstruction. Prenat Diagn. 2007;27(10):900–11.
Lane LV, et al. Pathology in practice. Nonepitheliotropic B-cell lymphoma of the nasal cavity with associated suppurative rhinitis and epidermal ulceration and lymphoma of the right kidney. J Am Vet Med Assoc. 2012;240(6):677–9.
Soghoian, D.Z., et al., HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci Transl Med. 2012;4(123):123ra25.
Menkhorst EM, et al. Decidual-secreted factors alter invasive trophoblast membrane and secreted proteins implying a role for decidual cell regulation of placentation. PLoS One. 2012;7(2):e31418.
Campbell JM, et al. Insulin Increases epiblast cell number of in vitro cultured mouse embryos via the PI3K/GSK3/p53 pathway. Stem Cells Dev. 2012;21(13):2430–41.
Levy Y, et al. Effect of intermittent interleukin-2 therapy on CD4+ T-cell counts following antiretroviral cessation in patients with HIV. AIDS. 2012;26(6):711–20.
Morris RK, et al. Systematic review of the effectiveness of antenatal intervention for the treatment of congenital lower urinary tract obstruction. BJOG. 2010;117(4):382–90.
Lane JT. Does the fat cell have something to say to the platelet about keeping thrombosis in check in diabetes? Transl Res. 2012;159(1):12–4.
Lane AA, et al. Risk factors for development of pneumonitis after high-dose chemotherapy with cyclophosphamide, BCNU and etoposide followed by autologous stem cell transplant. Leuk Lymphoma. 2012;53(6):1130–6.
Morris RK, et al. Percutaneous vesicoamniotic shunting versus conservative management for fetal lower urinary tract obstruction (PLUTO): a randomised trial. Lancet. 2013;382(9903):1496–506.
Ruano R, et al. Early fetal cystoscopy for first-trimester severe megacystis. Ultrasound Obstet Gynecol. 2011;37(6):696–701.
Morris RK, Ruano R, Kilby MD. Effectiveness of fetal cystoscopy as a diagnostic and therapeutic intervention for lower urinary tract obstruction: a systematic review. Ultrasound Obstet Gynecol. 2011;37(6):629–37.
Partridge, E.A. and A.W. Flake, Maternal-fetal surgery for structural malformations. Best Pract Res Clin Obstet Gynaecol. 2012;26(5):669–82.
Schropp KP, et al. Sacrococcygeal teratoma: the experience of four decades. J Pediatr Surg. 1992;27(8):1075–8; discussion 1078–9
Altman RP, Randolph JG, Lilly JR. Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section Survey-1973. J Pediatr Surg. 1974;9(3):389–98.
Nygaard I, et al. Summary of research recommendations from the Inaugural American Urogynecologic Society Research Summit. Female Pelvic Med Reconstr Surg. 2011;17(1):4–7.
Feola A, et al. Impact of pregnancy and vaginal delivery on the passive and active mechanics of the rat vagina. Ann Biomed Eng. 2011;39(1):549–58.
Yamaguchi Y, et al. Spontaneous rupture of sacrococcygeal teratoma associated with acute fetal anemia. Ultrasound Obstet Gynecol. 2006;28(5):720–2.
Olutoye OO, et al. Abnormal umbilical cord Dopplers may predict impending demise in fetuses with sacrococcygeal teratoma. A report of 2 cases. Fetal Diagn Ther. 2003;18(6):428–31.
Higgins, E.W., et al., Effect of estrogen replacement on the histologic response to polypropylene mesh implanted in the rabbit vagina model. Am J Obstet Gynecol. 2009;201(5):505 e1–9.
Flake AW, et al. Fetal sacrococcygeal teratoma. J Pediatr Surg. 1986;21(7):563–6.
Bond SJ, et al. Death due to high-output cardiac failure in fetal sacrococcygeal teratoma. J Pediatr Surg. 1990;25(12):1287–91.
Pierce, L.M., et al. Biomechanical properties of synthetic and biologic graft materials following long-term implantation in the rabbit abdomen and vagina. Am J Obstet Gynecol. 2009;200(5):549 e1–8.
Letouzey, V., et al., Is degradable antibiotic coating for synthetic meshes provide protection against experimental animal infection after fascia repair?. J Biomed Mater Res B Appl Biomater, 2011.
Hedrick HL, et al. Sacrococcygeal teratoma: prenatal assessment, fetal intervention, and outcome. J Pediatr Surg. 2004;39(3):430–8. discussion 430–8
Wilson RD, et al. Sacrococcygeal teratomas: prenatal surveillance, growth and pregnancy outcome. Fetal Diagn Ther. 2009;25(1):15–20.
Westerburg B, et al. Sonographic prognostic factors in fetuses with sacrococcygeal teratoma. J Pediatr Surg. 2000;35(2):322–5. discussion 325–6
Benachi A, et al. Prenatally diagnosed sacrococcygeal teratoma: a prognostic classification. J Pediatr Surg. 2006;41(9):1517–21.
Makin EC, et al. Outcome of antenatally diagnosed sacrococcygeal teratomas: single-center experience (1993–2004). J Pediatr Surg. 2006;41(2):388–93.
Rodriguez MA, et al. Tumor volume to fetal weight ratio as an early prognostic classification for fetal sacrococcygeal teratoma. J Pediatr Surg. 2011;46(6):1182–5.
Mari G. Middle cerebral artery peak systolic velocity: is it the standard of care for the diagnosis of fetal anemia? J Ultrasound Med. 2005;24(5):697–702.
Jouannic JM, et al. Successful intrauterine shunting of a sacrococcygeal teratoma (SCT) causing fetal bladder obstruction. Prenat Diagn. 2001;21(10):824–6.
Stutchfield P, Whitaker R, Russell I. Antenatal betamethasone and incidence of neonatal respiratory distress after elective caesarean section: pragmatic randomised trial. BMJ. 2005;331(7518):662.
Subramanian D, et al. Rate, type, and cost of pelvic organ prolapse surgery in Germany, France, and England. Eur J Obstet Gynecol Reprod Biol. 2009;144(2):177–81.
Hoehn T, et al. Fatal rupture of a sacrococcygeal teratoma during delivery. J Perinatol. 1999;19(8 Pt 1):596–8.
Kotecha S, et al. Antenatal and postnatal management of congenital cystic adenomatoid malformation. Paediatr Respir Rev. 2012;13(3):162–71.
Garne E, et al. EUROCAT website data on prenatal detection rates of congenital anomalies. J Med Screen. 2010;17(2):97–8.
Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977;8(2):155–71.
Chen CC, Ridgeway B, Paraiso MF. Biologic grafts and synthetic meshes in pelvic reconstructive surgery. Clin Obstet Gynecol. 2007;50(2):383–411.
Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12(1):17–37.
Hernanz-Schulman M, et al. Pulmonary sequestration: diagnosis with color Doppler sonography and a new theory of associated hydrothorax. Radiology. 1991;180(3):817–21.
Adzick NS, et al. Fetal lung lesions: management and outcome. Am J Obstet Gynecol. 1998;179(4):884–9.
Stocker J. Congenital pulmonary airway malformation: a new name and an expanded classification of congenital cystic adenomatoid malformation of the lung. Histopathology. 2002;41:424–31.
Adzick NS. Management of fetal lung lesions. Clin Perinatol. 2003;30(3):481–92.
Hubbard AM, et al. Congenital chest lesions: diagnosis and characterization with prenatal MR imaging. Radiology. 1999;212(1):43–8.
Crombleholme TM, et al. Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung. J Pediatr Surg. 2002;37(3):331–8.
Knox EM, et al. In-utero pulmonary drainage in the management of primary hydrothorax and congenital cystic lung lesion: a systematic review. Ultrasound Obstet Gynecol. 2006;28(5):726–34.
Wilson RD, et al. Cystic adenomatoid malformation of the lung: review of genetics, prenatal diagnosis, and in utero treatment. Am J Med Genet A. 2006;140(2):151–5.
Witlox RS, Lopriore E, Oepkes D. Prenatal interventions for fetal lung lesions. Prenat Diagn. 2011;31(7):628–36.
Merchant AM, et al. Postnatal chest wall deformities after fetal thoracoamniotic shunting for congenital cystic adenomatoid malformation. Fetal Diagn Ther. 2007;22(6):435–9.
Tsao K, et al. Resolution of hydrops fetalis in congenital cystic adenomatoid malformation after prenatal steroid therapy. J Pediatr Surg. 2003;38(3):508–10.
Peranteau WH, et al. Effect of maternal betamethasone administration on prenatal congenital cystic adenomatoid malformation growth and fetal survival. Fetal Diagn Ther. 2007;22(5):365–71.
Curran PF, et al. Prenatal steroids for microcystic congenital cystic adenomatoid malformations. J Pediatr Surg. 2010;45(1):145–50.
Bermudez C, et al. Percutaneous ultrasound-guided sclerotherapy for complicated fetal intralobar bronchopulmonary sequestration. Ultrasound Obstet Gynecol. 2007;29(5):586–9.
Oepkes D, et al. Successful ultrasound-guided laser treatment of fetal hydrops caused by pulmonary sequestration. Ultrasound Obstet Gynecol. 2007;29(4):457–9.
Ruano, R., et al. Percutaneous laser ablation under ultrasound guidance for fetal hyperechogenic microcystic lung lesions with hydrops: a single center cohort and a literature review. Prenat Diagn. 2012;32(12):1127–32.
Baud D, et al. Minimally invasive fetal therapy for hydropic lung masses: three different approaches and review of the literature. Ultrasound Obstet Gynecol. 2013;42(4):440–8.
Ruano R, et al. Prenatal diagnosis and natural history of fetuses presenting with pleural effusion. Prenat Diagn. 2011;31(5):496–9.
Su KC, et al. Abdominovaginal sacral colpoperineopexy: patient perceptions, anatomical outcomes, and graft erosions. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(5):503–11.
Gainey HL. Postpartum observation of pelvic tissue damage: further studies. Am J Obstet Gynecol. 1955;70(4):800–7.
Bigras JL, et al. Echocardiographic evaluation of fetal hydrothorax: the effusion ratio as a diagnostic tool. Ultrasound Obstet Gynecol. 2003;21(1):37–40.
Chang J, et al. Port insertion and removal techniques to minimize premature rupture of the membranes in endoscopic fetal surgery. J Pediatr Surg. 2006;41(5):905–9.
Handa VL, et al. Pelvic floor disorders 5–10 years after vaginal or cesarean childbirth. Obstet Gynecol. 2011;118(4):777–84.
Longaker MT, et al. Primary fetal hydrothorax: natural history and management. J Pediatr Surg. 1989;24(6):573–6.
Yinon, Y., et al., Perinatal outcome following fetal chest shunt insertion for pleural effusion. Ultrasound Obstet Gynecol. 2010;36(1):58–64.
Ruano, R., et al., Three-dimensional ultrasonographic assessment of fetal total lung volume as a prognostic factor in primary pleural effusion. J Ultrasound Med. 2012;31(11):1731–9.
Rustico MA, et al. Fetal pleural effusion. Prenat Diagn. 2007;27(9):793–9.
Tanemura M, et al. A case of successful fetal therapy for congenital chylothorax by intrapleural injection of OK-432. Ultrasound Obstet Gynecol. 2001;18(4):371–5.
Deurloo KL, et al. Isolated fetal hydrothorax with hydrops: a systematic review of prenatal treatment options. Prenat Diagn. 2007;27(10):893–9.
Picone O, et al. Thoracoamniotic shunting for fetal pleural effusions with hydrops. Am J Obstet Gynecol. 2004;191(6):2047–50.
Wilson RH, et al. Prenatal pleural effusion associated with congenital pulmonary lymphangiectasia. Prenat Diagn. 1985;5(1):73–6.
Graham G, Devine PC. Antenatal diagnosis of congenital diaphragmatic hernia. Semin Perinatol. 2005;29(2):69–76.
Dott MM, Wong LY, Rasmussen SA. Population-based study of congenital diaphragmatic hernia: risk factors and survival in Metropolitan Atlanta, 1968–1999. Birth Defects Res A Clin Mol Teratol. 2003;67(4):261–7.
Holder AM, et al. Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet. 2007;80(5):825–45.
Brady, P.D., et al., Recent Developments in the Genetic Factors Underlying Congenital Diaphragmatic Hernia. Fetal Diagn Ther, 2010.
Srisupundit K, et al. Targeted array comparative genomic hybridisation (array CGH) identifies genomic imbalances associated with isolated congenital diaphragmatic hernia (CDH). Prenat Diagn. 2010;30(12–13):1198–206.
Brady PD, et al. Identification of dosage-sensitive genes in fetuses referred with severe isolated congenital diaphragmatic hernia. Prenat Diagn. 2013;33(13):1283–92.
Grushka JR, et al. Effect of hospital case volume on outcome in congenital diaphragmatic hernia: the experience of the Canadian Pediatric Surgery Network. J Pediatr Surg. 2009;44(5):873–6.
van den Hout L, et al. Actual outcome in infants with congenital diaphragmatic hernia: the role of a standardized postnatal treatment protocol. Fetal Diagn Ther. 2011;29(1):55–63.
Hayakawa M, et al. Effect of hospital volume on the mortality of congenital diaphragmatic hernia in Japan. Pediatr Int. 2013;55(2):190–6.
Delacourt, C., et al., Long term respiratory outcomes of congenital diaphragmatic hernia, esophageal atresia, and cardiovascular anomalies. Seminars in fetal & neonatal medicine, 2012.
van den Hout L, et al. Can we improve outcome of congenital diaphragmatic hernia? Pediatr Surg Int. 2009;25(9):733–43.
Gallot D, et al. Antenatal detection and impact on outcome of congenital diaphragmatic hernia: a 12-year experience in Auvergne, France. Eur J Obstet Gynecol Reprod Biol. 2006;125(2):202–5.
Gallot D, et al. Prenatal detection and outcome of congenital diaphragmatic hernia: a French registry-based study. Ultrasound Obstet Gynecol. 2007;29(3):276–83.
Metkus AP, et al. Sonographic predictors of survival in fetal diaphragmatic hernia. J Pediatr Surg. 1996;31(1):148–51. discussion 151–2
Peralta CF, et al. Assessment of lung area in normal fetuses at 12–32 weeks. Ultrasound Obstet Gynecol. 2005;26(7):718–24.
Jani J, et al. Assessment of lung area in fetuses with congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2007;30(1):72–6.
Dekoninck P, et al. Results of fetal endoscopic tracheal occlusion for congenital diaphragmatic hernia and the set up of the randomized controlled TOTAL trial. Early Hum Dev. 2011;87(9):619–24.
Knox E, et al. Prenatal detection of pulmonary hypoplasia in fetuses with congenital diaphragmatic hernia: a systematic review and meta-analysis of diagnostic studies. J Matern Fetal Neonatal Med. 2010;23(7):579–88.
Jani J, et al. Observed to expected lung area to head circumference ratio in the prediction of survival in fetuses with isolated diaphragmatic hernia. Ultrasound Obstet Gynecol. 2007;30(1):67–71.
Jani JC, et al. Prenatal prediction of neonatal morbidity in survivors with congenital diaphragmatic hernia: a multicenter study. Ultrasound Obstet Gynecol. 2009;33(1):64–9.
Jani JC, et al. Lung volumes in fetuses with congenital diaphragmatic hernia: comparison of 3D US and MR imaging assessments. Radiology. 2007;244(2):575–82.
Cannie M, et al. Prenatal prediction of survival in isolated diaphragmatic hernia using observed to expected total fetal lung volume determined by magnetic resonance imaging based on either gestational age or fetal body volume. Ultrasound Obstet Gynecol. 2008;32(5):633–9.
Cannie M, et al. Fetal body volume: use at MR imaging to quantify relative lung volume in fetuses suspected of having pulmonary hypoplasia. Radiology. 2006;241(3):847–53.
Mayer, S., et al. The correlation between lung volume and liver herniation measurements by fetal MRI in isolated congenital diaphragmatic hernia: a systematic review and meta-analysis of observational studies. Prenat Diagn, 2011.
Jani J, et al. Value of prenatal magnetic resonance imaging in the prediction of postnatal outcome in fetuses with diaphragmatic hernia. Ultrasound Obstet Gynecol. 2008;32(6):793–9.
Cannie M, et al. Quantification of intrathoracic liver herniation by magnetic resonance imaging and prediction of postnatal survival in fetuses with congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2008;32(5):627–32.
Claus F, et al. Prenatal anatomical imaging in fetuses with congenital diaphragmatic hernia. Fetal Diagn Ther. 2011;29(1):88–100.
Goodfellow T, et al. Congenital diaphragmatic hernia: the prognostic significance of the site of the stomach. Br J Radiol. 1987;60(718):993–5.
Hatch E Jr, Kendall J, Blumhagen J. Stomach position as an in utero predictor of neonatal outcome in left-sided diaphragmatic hernia. J Pediatr Surg. 1992;27(6):778–9.
Kitano Y, et al. Re-evaluation of stomach position as a simple prognostic factor in fetal left congenital diaphragmatic hernia: a multicenter survey in Japan. Ultrasound Obstet Gynecol. 2011;37(3):277–82.
Cordier, A.G., et al. Stomach position versus liver-to-thoracic volume ratio in left-sided congenital diaphragmatic hernia. J Matern Fetal Neonatal Med, 2014.
Done, E., et al. Maternal hyperoxygenation test in fetuses undergoing FETO for severe isolated congenital diaphragmatic hernia. Ultrasound Obstet Gynecol, 2010.
Cruz-Martinez R, et al. Contribution of intrapulmonary artery Doppler to improve prediction of survival in fetuses with congenital diaphragmatic hernia treated with fetal endoscopic tracheal occlusion. Ultrasound Obstet Gynecol. 2010;35(5):572–7.
Deprest JA, et al. The making of fetal surgery. Prenat Diagn. 2010;30(7):653–67.
Deprest JA, Nicolaides K, Gratacos E. Fetal surgery for congenital diaphragmatic hernia is back from never gone. Fetal Diagn Ther. 2011;29(1):6–17.
Khan PA, Cloutier M, Piedboeuf B. Tracheal occlusion: a review of obstructing fetal lungs to make them grow and mature. Am J Med Genet C Semin Med Genet. 2007;145C(2):125–38.
Flageole H, et al. The plug-unplug sequence: an important step to achieve type II pneumocyte maturation in the fetal lamb model. J Pediatr Surg. 1998;33(2):299–303.
Davey, M., et al. Pulmonary arteriole muscularization in lambs with diaphragmatic hernia after combined tracheal occlusion/glucocorticoid therapy. Am J Obstet Gynecol. 2007;197(4):381e1–7.
Nelson SM, et al. Rescue of the hypoplastic lung by prenatal cyclical strain. Am J Respir Crit Care Med. 2005;171(12):1395–402.
Bealer JF, et al. The ‘PLUG’ odyssey: adventures in experimental fetal tracheal occlusion. J Pediatr Surg. 1995;30(2):361–4. discussion 364–5
Deprest J, Gratacos E, Nicolaides KH. Fetoscopic tracheal occlusion (FETO) for severe congenital diaphragmatic hernia: evolution of a technique and preliminary results. Ultrasound Obstet Gynecol. 2004;24(2):121–6.
Deprest J, et al. Technical aspects of fetal endoscopic tracheal occlusion for congenital diaphragmatic hernia. J Pediatr Surg. 2011;46(1):22–32.
Jani JC, et al. Severe diaphragmatic hernia treated by fetal endoscopic tracheal occlusion. Ultrasound Obstet Gynecol. 2009;34(3):304–10.
Done, E., et al. Predictors of neonatal morbidity in fetuses with severe isolated congenital diaphragmatic hernia undergoing fetoscopic tracheal occlusion. Ultrasound Obstet Gynecol, 2013.
Wegrzyn P, et al. Premature labor after fetal endoscopic tracheal occlusion for congenital diaphragmatic hernia: post-procedure management problems. Ultrasound Obstet Gynecol. 2010;36(1):124–5.
McHugh K, et al. Tracheomegaly: a complication of fetal endoscopic tracheal occlusion in the treatment of congenital diaphragmatic hernia. Pediatr Radiol. 2010;40(5):674–80.
Fauza DO, et al. Fetal tissue engineering: diaphragmatic replacement. J Pediatr Surg. 2001;36(1):146–51.
Ruano R, et al. Comparison between fetal endoscopic tracheal occlusion using a 1.0-mm fetoscope and prenatal expectant management in severe congenital diaphragmatic hernia. Fetal Diagn Ther. 2011;29(1):64–70.
Peralta CF, et al. Fetoscopic endotracheal occlusion for severe isolated diaphragmatic hernia: initial experience from a single clinic in Brazil. Fetal Diagn Ther. 2011;29(1):71–7.
Kohl T, et al. Encouraging early clinical experience with deliberately delayed temporary fetoscopic tracheal occlusion for the prenatal treatment of life-threatening right and left congenital diaphragmatic hernias. Fetal Diagn Ther. 2006;21(3):314–8.
Ruano R, et al. A randomized controlled trial of fetal endoscopic tracheal occlusion versus postnatal management of severe isolated congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2012;39(1):20–7.
Cannie MM, et al. Evidence and patterns in lung response after fetal tracheal occlusion: clinical controlled study. Radiology. 2009;252(2):526–33.
Reiss I, et al. Standardized postnatal management of infants with congenital diaphragmatic hernia in Europe: the CDH EURO Consortium consensus. Neonatology. 2010;98(4):354–64.
Tworetzky W, et al. In utero valvuloplasty for pulmonary atresia with hypoplastic right ventricle: techniques and outcomes. Pediatrics. 2009;124(3):e510–8.
Divanovic, A., et al. Prediction and perinatal management of severely restrictive atrial septum in fetuses with critical left heart obstruction: clinical experience using pulmonary venous Doppler analysis. J Thorac Cardiovasc Surg. 2011;141(4):988–94.
Arzt W, et al. Intrauterine aortic valvuloplasty in fetuses with critical aortic stenosis: experience and results of 24 procedures. Ultrasound Obstet Gynecol. 2011;37(6):689–95.
Oepkes D, et al. 2010 Report from the ISPD Special Interest Group fetal therapy: fetal cardiac interventions. Prenat Diagn. 2011;31(3):249–51.
Glazener CM, et al. New postnatal urinary incontinence: obstetric and other risk factors in primiparae. BJOG. 2006;113(2):208–17.
Sharland G, et al. Hypoplastic left-heart syndrome. Lancet. 2001;357(9257):722.
Sharland GK, et al. Left ventricular dysfunction in the fetus: relation to aortic valve anomalies and endocardial fibroelastosis. Br Heart J. 1991;66(6):419–24.
Kaltman JR, et al. Impact of congenital heart disease on cerebrovascular blood flow dynamics in the fetus. Ultrasound Obstet Gynecol. 2005;25(1):32–6.
Limperopoulos C, et al. Brain volume and metabolism in fetuses with congenital heart disease: evaluation with quantitative magnetic resonance imaging and spectroscopy. Circulation. 2010;121(1):26–33.
Rychik J, et al. The hypoplastic left heart syndrome with intact atrial septum: atrial morphology, pulmonary vascular histopathology and outcome. J Am Coll Cardiol. 1999;34(2):554–60.
Rychik J, et al. Perinatal and early surgical outcome for the fetus with hypoplastic left heart syndrome: a 5-year single institutional experience. Ultrasound Obstet Gynecol. 2010;36(4):465–70.
Gewillig M. The Fontan circulation. Heart. 2005;91(6):839–46.
Gaynor JW, Elliott MJ. Congenital left ventricular outflow tract obstruction. J Heart Valve Dis. 1993;2(1):80–93.
Nguyen, T., et al. Echocardiography of hypoplastic left heart syndrome. Cardiol Young. 2011;21(Suppl 2):28–37.
Makikallio, K., et al. Fetal aortic valve stenosis and the evolution of hypoplastic left heart syndrome: patient selection for fetal intervention. Circulation. 2006;113(11):1401–5.
Viktrup L, Lose G. The risk of stress incontinence 5 years after first delivery. Am J Obstet Gynecol. 2001;185(1):82–7.
Tabbutt S, et al. Neurodevelopmental outcomes after staged palliation for hypoplastic left heart syndrome. Pediatrics. 2008;121(3):476–83.
McElhinney DB, et al. Predictors of technical success and postnatal biventricular outcome after in utero aortic valvuloplasty for aortic stenosis with evolving hypoplastic left heart syndrome. Circulation. 2009;120(15):1482–90.
Gardiner HM, Kumar S. Fetal cardiac interventions. Clin Obstet Gynecol. 2005;48(4):956–63.
Mizrahi-Arnaud A, et al. Pathophysiology, management, and outcomes of fetal hemodynamic instability during prenatal cardiac intervention. Pediatr Res. 2007;62(3):325–30.
Marshall AC, et al. Results of in utero atrial septoplasty in fetuses with hypoplastic left heart syndrome. Prenat Diagn. 2008;28(11):1023–8.
Tegerstedt G, et al. Obstetric risk factors for symptomatic prolapse: a population-based approach. Am J Obstet Gynecol. 2006;194(1):75–81.
Hartmann K, et al. Outcomes of routine episiotomy: a systematic review. JAMA. 2005;293(17):2141–8.
Olivieri, L., et al. Hypoplastic left heart syndrome with intact atrial septum sequelae of left atrial hypertension in utero. J Am Coll Cardiol. 2011;57(20):e369.
Sandvik H, et al. Validation of a severity index in female urinary incontinence and its implementation in an epidemiological survey. J Epidemiol Community Health. 1993;47(6):497–9.
Richter, J., et al. Fetoscopic release of an amniotic band with risk of amputation: case report and review of the literature. Fetal diagnosis and therapy, 2012.
Torpin R. Amniochorionic Mesoblastic Fibrous Strings and Amnionic Bands: Associated Constricting Fetal Malformations or Fetal Death. Am J Obstet Gynecol. 1965;91:65–75.
Van Allen MI. Fetal vascular disruptions: mechanisms and some resulting birth defects. Pediatr Ann. 1981;10(6):219–33.
Moerman P, et al. Constrictive amniotic bands, amniotic adhesions, and limb-body wall complex: discrete disruption sequences with pathogenetic overlap. Am J Med Genet. 1992;42(4):470–9.
Thom DH, et al. Parturition events and risk of urinary incontinence in later life. NeurourolUrodyn. 2011;30(8):1456–61.
Rodrigues, A., et al. Limb constriction secondary to pseudoamniotic band syndrome after selective fetoscopic laser surgery: report of a case with a favorable outcome. Fetal Diagn Ther. 2012;32(4):288–91.
Fritel X, et al. Pelvic floor disorders 4 years after first delivery: a comparative study of restrictive versus systematic episiotomy. BJOG. 2008;115(2):247–52.
Wehbeh H, et al. The relationship between the ultrasonographic diagnosis of innocent amniotic band development and pregnancy outcomes. Obstet Gynecol. 1993;81(4):565–8.
Urwitz-Lane R, Ozel B. Sexual function in women with urodynamic stress incontinence, detrusor overactivity, and mixed urinary incontinence. Am J Obstet Gynecol. 2006;195(6):1758–61.
Tadmor O, et al. Analysis of umbilical artery flow parameters during fetal variable decelerations using computerized Doppler waveforms. Fetal Diagn Ther. 1999;14(1):2–10.
Pedersen TK, Thomsen SG. Spontaneous resolution of amniotic bands. Ultrasound Obstet Gynecol. 2001;18(6):673–4.
Kanayama MD, Gaffey TA, Ogburn PL Jr. Constriction of the umbilical cord by an amniotic band, with fetal compromise illustrated by reverse diastolic flow in the umbilical artery. A case report. J Reprod Med. 1995;40(1):71–3.
Husler MR, et al. When is fetoscopic release of amniotic bands indicated? Review of outcome of cases treated in utero and selection criteria for fetal surgery. Prenat Diagn. 2009;29(5):457–63.
Weinzweig N. Constriction band-induced vascular compromise of the foot: classification and management of the “intermediate” stage of constriction-ring syndrome. Plast Reconstr Surg. 1995;96(4):972–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer-Verlag London Ltd., part of Springer Nature
About this chapter

Cite this chapter
Hodges, R., De Catte, L., Devlieger, R., Lewi, L., Van Mieghem, T., Deprest, J. (2018). Antenatal Diagnosis: Current Status for Paediatric Surgeons. In: Losty, P., Flake, A., Rintala, R., Hutson, J., lwai, N. (eds) Rickham's Neonatal Surgery. Springer, London. https://doi.org/10.1007/978-1-4471-4721-3_4
Download citation
DOI: https://doi.org/10.1007/978-1-4471-4721-3_4
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-4720-6
Online ISBN: 978-1-4471-4721-3
eBook Packages: MedicineMedicine (R0)