
Abstract
Distillation is the process in which the separation of components in a mixture is achieved due to differences in volatility, that is differences in vapour pressure, of the components in the mixture to be separated. The simplest kind of batch distillation process results in only a limited increase in the concentration of the more volatile component (MVC) in the distillate; a greater degree of separation can be obtained by using a fractionating column. In the food industry, distillation is used in the purification of raw alcohol and the production of beverage alcohol from wheat; the separation of flavour components, for example from the condensed vapour resulting from evaporation of fruit juices; as an important step in the production of bioethanol from sugar beet; and in the production of food grade white oils which are used as binders or coating materials. Other applications of distillation are the recovery, concentration and fractionation of aromas; the recovery of solvents from miscella; and the concentration and recovery of solvents used in extraction process such as the recovery of isopropanol used in the extraction of pectin from fruit peel.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
15.1 Introduction to Distillation
Distillation is the process in which the separation of components in a mixture is achieved due to differences in volatility, that is differences in vapour pressure, of the components in the mixture to be separated. The simplest kind of batch distillation process results in only a limited increase in the concentration of the more volatile component (MVC) in the distillate; a greater degree of separation can be obtained by using a fractionating column. In the food industry, distillation is used in the purification of raw alcohol and the production of beverage alcohol from wheat; the separation of flavour components, for example from the condensed vapour resulting from evaporation of fruit juices; as an important step in the production of bioethanol from sugar beet; and in the production of food grade white oils which are used as binders or coating materials. Other applications of distillation are the recovery, concentration and fractionation of aromas; the recovery of solvents from miscella; and the concentration and recovery of solvents used in extraction process such as the recovery of isopropanol used in the extraction of pectin from fruit peel.
Steam distillation is used where the temperatures encountered in conventional distillation would result in thermal damage to the food material; an example is the production of hop extracts used in brewing. An alternative method of reducing the temperature is to operate the column under vacuum as in the preservation of aromas during gin distillation. In some instances, vacuum and steam distillation can be combined.
15.2 Batch Distillation
The principles of vapour–liquid equilibrium were set out in Section 3.3. Example 3.9 showed the difference between the concentration of an ideal solution and the concentration of the vapour above the solution at equilibrium. It is this difference which is the basis for distillation as a separation process for mixtures of volatile liquids.
Figure 15.1 illustrates a batch (or differential) distillation system. The vapour formed on boiling the liquid in the still is removed and condensed. The vapour is in equilibrium with the liquid from which it rises and is richer in the MVC. The remaining liquid therefore becomes progressively weaker in the MVC and therefore the total quantity of vapour which is generated (and which is subsequently condensed) is not in equilibrium with the residual liquid.

Batch (or differential) distillation
Now, if at any given time the number of moles of liquid in the still is F, with a composition x (mole fraction of the MVC), and dF is the quantity of vapour with a composition y, then a component material balance for the MVC gives
and
which on integration gives
where W and x W represent the residual conditions. Thus
Equation (15.4) is the Rayleigh equation which must be solved graphically by plotting \(\frac{1}{{y - x}}\) against x and measuring the area under the curve in order to obtain \(\frac{F}{W}\).
15.2.1 Example 15.1
A batch containing a binary mixture of 55 mol% ethanol in water is distilled until the residue contains 10 mol% ethanol. Determine the mean distillate composition. The equilibrium data for the ethanol–water system (mole fraction of ethanol) are as follows:
From the equilibrium data the following table can be constructed:
x | \(\dfrac{1}{{y - x}}\) |
|---|---|
0.05 | 3.45 |
0.1 | 3.13 |
0.2 | 3.03 |
0.3 | 3.57 |
0.4 | 4.55 |
0.5 | 6.25 |
0.6 | 10.0 |
If now \(\frac{1}{{y - x}}\) is plotted against x, the area under the curve between \({x_{\textrm{W}}} = 0.10\), the residue composition, and \({x_{\textrm{F}}} = 0.55\), the feed composition, is 1.94. Thus \(\ln \left( {\frac{F}{W}} \right) = 1.94\) and therefore \(\frac{F}{W} = 6.96\). Putting \(F = 100\) kmol gives \(W = 14.3\) kmol. A material balance for the system can be written as
where x D is the mean distillate composition. Thus
from which \({x_{\textrm{D}}} = 0.625\). In other words the distillate contains 62.5 mol% ethanol.
The method outlined above can be simplified in two ways.
15.2.2 Linear Equilibrium Relationship
The equilibrium relationship is assumed to be represented by a straight line, \(y = ax + b\), over the range of x of interest. Substituting this into Eq. (15.4) and integrating gives
and
and therefore
Alternatively, Eq. (15.7) can be put into the form
15.2.2.1 Example 15.2
An equimolar mixture of acetone and water is distilled until 50% of the feed is removed from the still, the vapour being withdrawn and condensed continuously. If the vapour–liquid equilibrium relationship can be approximated by \(y = 0.1\;x + 0.8\), where y and x are the mole fractions of acetone in vapour and liquid, respectively, determine the composition remaining in the still and the average composition of the distillate.
From the given data, \({x_{\textrm{F}}} = 0.50\) and the corresponding equilibrium concentration is \({y_{\textrm{F}}} = 0.85\). For 100 kmol of feed, 50 kmol remains in the still and therefore Eq. (15.8) gives
Thus \({x_{\textrm{W}}} = 0.163\). From an overall material balance
where x D is the mean distillate composition. Hence \({x_{\textrm{D}}} = 0.837\) and the average distillate composition is 83.7 mol% acetone.
15.2.3 Constant Relative Volatility
The volatility of a substance is defined as the partial pressure divided by the mole fraction in the liquid phase. The relative volatility α in a binary system of A and B (where A is the MVC) is therefore, by definition,
Substituting from Dalton’s law [Eq. (3.20)] for the partial pressures of both A and B gives
Recognising that \({y_{\textrm{B}}} = 1 - {y_{\textrm{A}}}\) and \({x_{\textrm{B}}} = 1 - {x_{\textrm{A}}}\), for a binary mixture, Eq. (15.10) becomes
and, on re-arrangement, in the general case,
For the case of constant relative volatility, Eq. (15.12) can be substituted into Eq. (15.4) thus avoiding the need for graphical integration. The details of the consequent derivation are too lengthy to be included here but the result is Eq. (15.13) which relates the molar quantities of feed and residue to their respective compositions:
In the foregoing description of a batch distillation column the distillate composition changes with time and therefore different product batches can be collected over the period of operation. Therefore if the column is operated with reflux, the reflux ratio is kept constant. On the other hand, if it is desired to keep the product composition constant it is necessary to continuously change the reflux ratio and there will be a consequent decrease in the product rate to balance the increasing concentration of the less volatile component (LVC) in the still.
15.3 Ideal Stages and Equilibrium
The design of a continuous distillation column is based upon the calculation of the number of ideal, or equilibrium, stages required to bring about a given separation, in other words the number of stages required to generate product streams of a particular concentration. The number of actual stages, for example the number of plates in a distillation column, is related to the number of ideal stages by an efficiency factor and hence the size of the column can be specified. The concept of an ideal stage, which then determines the size of actual process equipment, applies to all mass transfer operations, including leaching which is covered in Section 15.6. However, it is perhaps most easily explained in relation to distillation.
Figure 15.2 is a plot (for an ideal solution) of the equilibrium values of the mole fraction in liquid and vapour phases against temperature, forming the bubble point and dew point curves, respectively. Consider a liquid of composition x n brought to its boiling point (point A in Fig. 15.2). The vapour in equilibrium with this liquid has a concentration y n . If this vapour is now condensed the concentration of the resulting liquid will be \({x_{n + 1}}\) and thus it is richer in the more volatile component. The step ABC is an ideal stage or equilibrium stage. Successive stages of partial vaporisation followed by condensation allow a distillate to be produced which is progressively richer in the MVC. This process is known as rectification and it allows a much greater increase in concentration of the MVC than is possible in the batch distillation process described in Section 15.2.

Concept of ideal stage: partial vaporisation and condensation
A fractionating column (Fig. 15.3) consists of a series of plates or trays which are designed to bring liquid and vapour into intimate contact allowing the two phases to approach as close as possible to equilibrium. Plates may be as simple as a series of perforated horizontal discs, occupying the column cross section and evenly spaced throughout its height, across which liquid flows and through which vapour bubbles pass upwards. As vapour passes up through the column it becomes progressively richer in the MVC and as liquid flows down the column, from plate to plate, it becomes progressively less rich in the MVC. There are many complex designs of plate some of which include a weir to maintain a given level of liquid on the plate and a downcomer to allow liquid to pass to the plate below. Instead of plates, distillation columns may be fitted with a continuous high surface area matrix, known as ‘packing’, which both provides intimate contact between liquid and vapour and allows free passage for each phase through the column.

Continuous fractionating column
The mixture to be separated is fed onto an intermediate plate, usually close to the middle of the column, the exact position depending upon the concentration and temperature of the feed stream. At the bottom of the column is a reboiler which consists of a conventional tubular heat exchanger usually heated by process steam. The liquid in the reboiler is at its bubble point and liquid product, known as residue, is withdrawn from here at a concentration x W.
The section of the column above the feed plate is known as the rectifying section and that below the feed plate as the stripping section. Vapour leaves the reboiler and partially condenses into the liquid on the next plate above. The latent heat which is liberated when condensation takes place then generates an equivalent quantity of vapour richer in the MVC. Mass transfer takes place on each plate because the vapour passing into liquid on the tray above is not in equilibrium with it. However, as mass transfer proceeds, vapour and liquid approach equilibrium although in practice this is not attained and the enrichment on each plate is less than that of an ideal stage. The degree to which equilibrium is attained is a function of the efficiency of vapour-liquid mixing on the plate and therefore of its mechanical design.
The less volatile component condenses from vapour into the liquid and therefore increases the concentration of the MVC in the vapour. The MVC is vaporised from the liquid on a plate thereby decreasing the MVC in the liquid (or stripping out the MVC from the liquid, hence stripping section). The number of moles of vapour that condense into liquid and the number of moles of liquid generating vapour, within respective sections of the column, is approximately equal. This condition is known as equimolar counter diffusion.
The vapour and liquid streams throughout the column are at their respective bubble and dew points and therefore the temperature in the column decreases from reboiler to condenser.
The condenser is a heat exchanger which condenses the overhead vapour to give a liquid at its bubble point. Some of this liquid is removed as product, the distillate at a concentration x D, but a proportion is returned to the column as reflux in order to increase the concentration of MVC in the overhead vapour. This increase in the degree of separation at the top of the column is at the cost of an increased energy supply to the reboiler because the overall vapour load in the column is increased.
The ratio of refluxed liquid, which is returned to the column, to the distillate, which is withdrawn as product, is called the reflux ratio. At its minimum value (which is readily determined) an infinite number of stages is needed to change the feed composition to that of the required distillate concentration. Any increase in reflux ratio above the minimum value gives a workable column but with a large number of stages. Increasing the ratio further reduces the number of stages and therefore reduces the capital cost of the distillation column. However, at high reflux ratios the capital cost increases again because a larger diameter column is needed to handle the increased vapour flow. The optimum reflux ratio is a balance between the capital costs of the column and the operating costs, which are a function of the steam and cooling water usage, and which increase with reflux ratio.
15.4 Continuous Fractionation: The McCabe–Thiele Method
The simplest design method for a continuous fractionating distillation column is a graphical stage-to-stage calculation known as the McCabe–Thiele method. This relies upon comparing the actual change in composition throughout the column, in terms of operating lines which are derived from material balances, with the equilibrium concentrations on a vapour–liquid equilibrium diagram such as the example in Fig. 3.8.
15.4.1 Material and Energy Balances
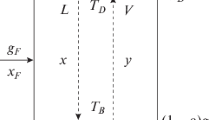
Figure 15.4 represents a continuous distillation column with two representative plates in each half of the column. F, D and W are the molar flow rates of the feed, distillate and residue streams, respectively, and x F, x D and x W are the corresponding mole fractions of MVC in the liquid phase. The overall material balance over the column is then
and the component balance for the more volatile component is

Material balances across a continuous distillation column
The enthalpy balance over a single plate can be written as
where L and V are the molar flow rates of liquid and vapour, respectively, and h L and h V are the corresponding liquid and vapour enthalpies. Equation (15.16) can be simplified by adopting a number of assumptions: (i) the column is well insulated and the heat losses may be omitted; (ii) that the system is ideal and therefore the heat of mixing may be assumed to be zero; (iii) that for mixtures, the molar latent heat of vaporisation is constant and therefore
and finally (iv) that temperature changes between stages are small and therefore
Thus, Eq. (15.16) reduces to
This is the assumption of ‘constant molal overflow’, that is, that the vapour and liquid flow rates are constant in a given half of the column.
15.4.2 Derivation of Operating Lines
McCabe–Thiele is a graphical method based upon drawing operating lines for the top and bottom sections of the column and then drawing in the ideal stages between the operating lines and the equilibrium curve. The operating lines are based on material balances in the respective sections of the column.
(a) Rectifying section: The overall material balance, drawing the envelope between plates n and \(n + 1\), and assuming constant molal overflow, is
and the component balance for the MVC is
On re-arrangement this gives the equation of the operating line
which relates the composition of the vapour rising to a plate to the composition of liquid on that plate. It is more convenient to write Eq. (15.22) in terms of the reflux ratio R which is defined as
and therefore the equation of the operating line becomes
The most convenient way of drawing the line is by establishing boundary conditions. First, for the distillate \({x_{n + 1}} = {x_{\textrm{D}}}\) and therefore
which simplifies to
Second, putting \({x_{n + 1}} = 0\) gives
and therefore the operating line for the rectifying section (the top operating line) passes through the co-ordinates (x D, x D) and \(\left( {0,\frac{{{x_{\textrm{D}}}}}{{R + 1}}} \right)\).
(b) Stripping section: For the stripping section, below the feed plate, the assumption of constant molal overflow gives \({L_m} = {L_{m + 1}}\) and therefore, the overall material balance is
The component balance for the MVC yields
The relevant boundary condition is \({x_{m + 1}} = {x_{\textrm{W}}}\), for the residue, and therefore Eq. (15.29) becomes
and thus
The operating line is now drawn through the co-ordinate \(({x_{{\textrm{W}},}}\;{x_{\textrm{W}}})\) with a gradient equal to \(\frac{{{L_m}}}{{{V_m}}}\).
(c) Intersection of operating lines: In practice the operating line for the rectifying section is drawn in first, followed by that for the stripping line from \(({x_{{\textrm{W}},}}\;{x_{\textrm{W}}})\) to the point of intersection which has the general co-ordinates \(({x_{q,}}\;{x_q})\). The location of this point depends upon the state of the feed. Using the general co-ordinates, the rectifying and stripping operating lines become, respectively,
and
Subtracting Eq. (15.19) from (15.20) gives
and, from a material balance across the feed plate (Fig. 15.4), noting that \({L_{m + 1}} = {L_m}\),
Let the enthalpy per kmol of feed be hʹ, the enthalpy per kmol of feed at its boiling point be h f and the molar latent heat of vaporisation be \({h_{{\textrm{fg}}}}\). Thus the enthalpy change to bring the feed up to the boiling point from its initial condition is \(F({h_{\textrm{f}}} - h')\). The molar mass of vapour which must be condensed to provide this heat is then \(\frac{{F({h_{\textrm{f}}} - h')}}{{{h_{{\textrm{fg}}}}}}\) and this in turn is equal to the net upward flow of vapour. Thus
Substituting this into Eq. (15.35) results in
Defining the quantity q as the heat to vaporise 1 kmol of actual feed divided by the molar latent heat of the feed,
It follows from Eq. (15.37) that
Thus substituting into Eq. (15.34) gives
which can be re-arranged to give the equation of the q-line, on which the two operating lines intersect,
This line passes through the co-ordinates \(({x_{{\textrm{F}},}}\,{x_{\textrm{F}}})\) and has a gradient equal to \(\left( {\frac{q}{{q - 1}}} \right)\). Thus for a feed stream at its bubble point \(q = 1\) and the gradient of the q-line line is ∞. If the feed is a saturated vapour then \(q = 0\) and the gradient of the q-line line is 0. For other cases the value of q must be calculated from Eq. (15.38).
The McCabe–Thiele procedure is best explained by referring to Example 15.3.
15.4.2.1 Example 15.3
A binary mixture of volatile liquids containing 55% A and 45% B is to be separated by continuous fractionation into a distillate with a composition of 96% A and a residue containing no more than 5% A. All compositions are on a molar basis. The vapour–liquid equilibrium data for this system (in terms of mole fraction of A) are as follows:
How many ideal stages are required if the feed liquid is at its bubble point as it enters the distillation column and the column is operated at a reflux ratio of 1.4?
The steps in the graphical solution are as follows:
-
(i)
Plot the vapour–liquid equilibrium data. It is also convenient to draw in the line \(y = x\).
-
(ii)
Draw in the q-line. The feed is at its bubble point and therefore, from Eq. (15.38), \(q = 1\). The gradient is therefore equal to ∞ and the line is drawn from the co-ordinates (0.55, 0.55) vertically upwards. In Fig. 15.5, for clarity, the q-line has been extended beyond the equilibrium curve but this is not a necessary part of the solution.
-
(iii)
The distillate composition is \({x_{\textrm{D}}} = 0.96\) and therefore \(\frac{{{x_{\textrm{D}}}}}{{R + 1}} = \frac{{0.96}}{{2.4}} = 0.40\). Hence the operating line for the rectifying section has the co-ordinates (0, 0.40) and (0.96, 0.96). Again, it is not necessary to extend this below the intersection with the q-line and it is drawn so in Fig. 15.5 solely for clarity.
-
(iv)
The residue composition is \({x_{\textrm{W}}} = 0.05\) and thus the operating line for the stripping section passes through the co-ordinate (0.05, 0.05) and intersects with the top operating line on the q-line (Fig. 15.5).
-
(v)
Beginning at the top of the column, \({x_{\textrm{D}}} = 0.96\), draw in the ideal stages (Fig. 15.6). A total of six ideal stages are required to give a residue composition of \({x_{\textrm{W}}} = 0.05\).

Construction of operating lines in Example 15.3

Construction of ideal stages in Example 15.3
The number of actual plates in the column is calculated from the number of ideal stages and a figure for the overall plate efficiency. Overall plate efficiency is defined as the number of ideal (or theoretical) plates divided by the number of actual plates expressed as a percentage. However, it is assumed that the reboiler acts as an ideal stage and this must be subtracted from the number of stages determined by the graphical construction.
15.4.2.2 Example 15.4
Determine the number of actual plates required in the distillation column in Example 15.3 if the stage efficiency is 75%.
The number of ideal plates is 6–1 = 5 and therefore the number of actual plates is \(\frac{5}{{0.75}} = 6.7\). This must be rounded up to the nearest integer and therefore seven actual plates are required in the column plus the reboiler.
15.4.3 Minimum Reflux Ratio
Changing the reflux ratio R changes the gradient of the operating line for the rectifying section and therefore changes the number of stages required for a given separation. At the condition known as total reflux no distillate is removed from the column, the reflux ratio is at its maximum and the operating line coincides with \(y = x\). Reducing R reduces the gradient of the operating line and increases the number of stages. At minimum reflux conditions, when \(R = {R_{\min }}\), an infinite number of stages is required to increase the feed concentration x F to that of the distillate x D (Fig. 15.7). Now, from Eq. (15.24) the gradient at minimum reflux is \(\frac{{{R_{\min }}}}{{{R_{\min }} + 1}}\) and from inspection of Fig. 15.7
where y F is the value of y at the point where the q-line intersects the equilibrium line. The minimum reflux ratio is thus determined graphically and columns are often operated at some multiple of \({R_{\min }}\).

Determination of minimum reflux ratio
15.4.3.1 Example 15.5
Determine the minimum reflux ratio for the column in Example 15.3.
Referring to Fig. 15.7 and Eq. (15.42),
from which \({R_{\min }} = 0.32\).
15.5 Steam Distillation
Steam distillation is a technique used in cases where the substance which is to be distilled has a high boiling point, where its solubility in water is very low, and where conventional direct distillation cannot be used because the high temperatures necessary may result in decomposition of the constituents of the substance. Steam distillation allows the separation of a high boiling point substance from small quantities of non-volatiles impurities at safe temperatures and therefore it finds use in the food industry, for example in the deodorisation of edible fats and oils.
Steam is passed directly into the liquid in the still. Where both liquid water (A) and a second, insoluble, liquid phase (B), for example the impurity, are present then if the pressure is fixed (at, say, atmospheric pressure) the temperature will also be fixed. Each liquid phase will exert its pure component vapour pressure and the mixture will boil when the sum of the vapour pressures equals atmospheric pressure, that is when
For as long as two liquid phases are present the mixture will boil at a constant temperature, well below the boiling point of B, and will produce a vapour of constant composition. The boiling point of the mixture is that temperature which satisfies Eq. (15.43) and the composition of the vapour which is generated is given by
The vapour containing A and B is then condensed and because the two liquid phases are immiscible they can be separated relatively easily. When all the water has been evaporated the temperature will rise to the boiling point of B. However, the ability to distil at a temperature low enough to avoid thermal damage to the original mixture comes at the cost of the high heat input required to vaporise large quantities of water.
The material balance over the bottom of the column is now represented by Fig. 15.8 where S is the flow rate of steam into the column. The overall material balance is now

Material balance for steam distillation
If constant molal overflow is assumed, that is \({V_m} = S\) and \({L_m} = {L_{m + 1}} = W\), then the operating line for the stripping section is given by Eq. (15.29). The operating line has a gradient equal to \(\frac{{{L_m}}}{{{V_m}}}\) but it is more convenient to draw the line between the intersection with
-
(i)
\(y = x\)
Putting \(y = {x_{m + 1}}\) Eq. (15.29) becomes
$${x_{m + 1\;}} = \frac{{{L_m}}}{{{V_m}}}{x_{m + 1}} - \frac{W}{{{V_m}}}{x_{\textrm{W}}}$$((15.46))and substituting from Eq. (15.45), and eliminating L and V, yields
$${x_{m + 1\;}} = \frac{{{x_{\textrm{W}}}W}}{{W - S}}$$((15.47)) -
(ii)
\(y = 0\)
Putting \(y = 0\) gives \(x = {x_W}\). Therefore the operating line is drawn as in Fig. 15.9 and the first stage is constructed starting at \(y = 0\), \(x = {x_W}\).

Operating line for steam distillation
Thus the use of live steam injected directly into the column eliminates the need for a reboiler but increases the number of plates required in the lower part of the column.
15.6 Leaching
15.6.1 Introduction
Extraction processes encompass both liquid–liquid extraction and solid–liquid extraction; the latter is often referred to as leaching. In liquid–liquid extraction a solute, initially dissolved in one liquid, is preferentially dissolved in a second solvent when the two liquids are brought into intimate contact. This usually results in two immiscible liquid phases which are then separated and the desirable solute is recovered from the preferential solvent, for example by distillation. A further kind of extraction process uses a supercritical fluid as the solvent; this is covered in Section 15.7.
Leaching is defined as the extraction of a soluble constituent from solid material using a selective liquid solvent. The desirable product may be either a solid which is free of the soluble component or, more usually, a solution of the solute in the solvent. Leaching is a mass transfer operation in which the driving force is the difference in concentration of the solute between the feed material and the solvent which is initially solute-free.
The following list gives an idea of the range of applications of leaching in the food industry:
-
(i)
vegetable oil (e.g. olive oil, peanut oil, sunflower oil, cottonseed oil) from seeds and nuts using organic solvents such as hexane, alcohols or acetone;
-
(ii)
sugar from sugar beet using water;
-
(iii)
caffeine from coffee beans using water;
-
(iv)
caffeine from tea using methylene chloride or water;
-
(v)
fish oil from waste fish using organic solvents.
15.6.2 Process Description
Three stages may be identified in a leaching process:
-
(i)
the dissolution of the solute in a selective solvent, thus separating the solute from the solid matrix and which involves a change of phase;
-
(ii)
diffusion of the solute, in solution, through the pores of the solid to the external surface of the particle; and
-
(iii)
transfer of the solute into the bulk liquid solvent.
In the case of oil extraction from seeds a mechanical pressing stage may be employed before the leaching stage. This is for two reasons. First, pressed oil usually has different characteristics to extracted oil and will sell at a premium price (olive oil is an example) and, second, expression followed by leaching increases the overall efficiency of the oil recovery process.
There are four principal factors which influence the rate of leaching.
-
(a)
Particle size: Ideally the initial solid material should be in a particulate form with a narrow particle size distribution which will result in an equal residence time and an equal extraction time for all particles. The fine particles in a wide particle size distribution can lodge inside the larger pores of the larger particles and thus reduce the extraction rate. If the solute is concentrated in pockets within the raw material then crushing may be necessary as a preliminary step.
Mean particle size also affects the rate of leaching. Smaller particles have a greater specific surface and will give higher extraction rates. Similarly, a smaller particle diameter reduces the time required for diffusion through the internal pores of the particle. However, the circulation of solvent within the extractor, and the separation of inert solids from the fluid, may be more difficult with smaller particles.
Vegetable material may need to be cut or shredded before the extraction stage and leaves, for example tea leaves, may first need to be dried and ground. Cellular materials have the disadvantage of low extraction rates due to the resistance at the cell walls and therefore a grinding or flaking step is often necessary to break down cell walls. In the extraction of vegetable oil from seeds, the seeds are often steamed and flaked prior to leaching. The flaked seeds should result in a particle bed that is well drained and allows good contact between solids and solvent.
-
(b)
Solvent: Clearly the solvent must be selective and remove only those components which it is desired to remove. If the solvent has a low viscosity this will aid flow in the extraction equipment. However, viscosity will increase as the concentration of solute increases and consequently the extraction rate decreases with concentration because of the lower driving force for mass transfer.
-
(c)
Temperature: The extraction rate will increase with temperature because both the solubility of the solute in the selective solvent and the diffusivity increase with temperature. An increase in temperature will also reduce the viscosity of both the solvent and the concentrated product solution and will therefore help to improve fluid circulation. However, there may well be limits to the temperature which can be used for food materials and, in fact, many processes operate at ambient temperature.
-
(d)
Agitation: Agitation of the contents of the extractor will increase the rate of mass transfer from the particle surface into the bulk solution. It also prevents the sedimentation of fine solids and makes greater use of the available particle surface area.
15.6.3 Types of Equipment
-
(a)
Fixed bed: The simplest form of contact between the solids to be leached and the solvent is the single stage batch operation in which coarse, free-draining, solids are placed in a tank and solvent is added and left for a pre-determined time or until extraction is judged to be complete. The liquid is then drained off and the solids removed. This operation may be made semi-batch by recirculating the solvent through the bed to increase the percentage recovery of solute.
Increased recovery can be obtained by using an extraction battery in which a number of leaching tanks are arranged in series allowing for counter-current flow of solution. In this kind of operation fresh solvent is added to the solids from which most solute has been extracted and the fresh raw material is in contact with the most concentrated solution which is about to be discharged. The leaching tanks are designed to allow liquid to drain from the bottom and for solids to be discharged when extraction is complete. In what is known as the Shanks process (Fig. 15.10) tanks 1, 2, 3 and 4 are full of solids (tank 1 having being filled first and tank 4 most recently). Tank 5 is empty, the leached solids having just been discharged. Therefore tank 4 contains the most concentrated solution and fresh solvent is added to tank 1. The solution from tank 4 is drained and removed as product and that in 3 drained and added to 4, 2 transferred to 3, and 1 to 2. Fresh solid is now added to tank 5 and the leached solids are discharged from tank 1. The process continues with liquid again being transferred from tank to tank in a forward direction.
-
(b)
Dispersed solid: Particulate solids which form impermeable beds can be agitated within a tank of solvent, either as a batch or with recirculation of the solvent. Such equipment can again be operated either batchwise or as a semi-batch process as with the fixed bed described above. For fine solids the use of a draught tube baffle (DTB), which imposes a more regular circulation pattern, may be appropriate. As an alternative to a mechanical impeller, compressed air may be fed to the base of a tank fitted with a DTB and air bubbles then lift the solids up through the central region of the vessel before they descend in the annular space.
-
(c)
Moving bed: In these devices the solids are moved through the solvent, for example by means of a bucket elevator. A number of designs of continuous extraction units are in industrial use and two are worthy of particular mention. The Rotacell is a horizontal cylindrical tank with up to 18 perforated cells which rotate on top of a stationary compartmentalised tank of the same size. This allows a continuous feed of solids to fill each cell in turn as it passes the feed point. The leached solids are discharged after one revolution of the tank. Solution is sprayed onto the top of bed of solids, drains through the cell and is collected in the compartment below before being pumped to a spray positioned over the adjacent cell. Fresh solvent is fed only to the last compartment before the discharge point.
The Bollman extractor is used for seeds and vegetable material which do not disintegrate during leaching. It consists of a series of perforated baskets, perhaps 20–30, which rotate on a vertically aligned loop within a solvent-tight enclosure. Solids are fed to the top basket on the downward part of the cycle and are sprayed with ‘half miscella’, that is, an oil–solvent solution of intermediate concentration. The solids and liquid are therefore in co-current flow. At the bottom of the loop the solids have been fully extracted and the liquor, known as ‘full miscella’, is removed as product. The baskets now move upwards and fresh solvent is sprayed onto the solids, at the top of the loop, which are about to be discharged. Thus, for this part of the cycle, solids and liquid are in counter-current flow and the liquor removed at the bottom of the loop is the ‘half miscella’ which is then pumped to the top of the plant and sprayed onto the fresh solids entering the process.

Leaching: Shanks system
15.6.4 Counter-Current Leaching: Representation of Three-Component Systems
Referring to Fig. 15.11, the net flow of material to the right of the system is given by
where W is the total mass flow rate of the underflow stream,w is the total mass flow rate of the overflow stream and H, known as a ‘difference point’, represents that stream which when added to w n will give W n .

Material balance across continuous counter-current leaching system
An overall balance, across all stages, gives
and the corresponding component balance is
where x is the mass fraction of a given component in the underflow stream and y represents the mass fraction in the overflow stream. The net flow to the right for any given component is \({x_n}\;{W_n} - {y_n}\;{w_n}\) and therefore the composition of the net flow to the right (with respect to that component) is given by
If the net flow of a component is to the right then x d will be positive. If the net flow is to the left then x d will be negative. For example, the net flow of solvent is to the left and therefore \({x_{\textrm{d}}}_{\textrm{S}}\) is negative.
The three-component system of a leaching process is most easily represented by a right-angled triangle which can be plotted on conventional rectilinear graph paper. This is illustrated in Fig. 15.12 in which the vertices of the triangle represent the three pure components, respectively: solute (A), inert solids (B) and solvent (S). Thus the sides of the triangle represent two-component mixtures and points inside the triangle represent three-component mixtures. Lines parallel to the hypotenuse represent lines of constant mass fraction of inert solids x B.

Representation of a three-component leaching system
The lever rule applies to these representations of three-component systems and can be summarised as follows:
-
(i)
where two streams are mixed together the point representing the composition of the mixture lies on a straight line joining the components. Thus in Fig. 15.13 γ represents a mixture of α and β and its position can be found from
$$\frac{{{\textrm{mass of }}\alpha }}{{{\textrm{mass of }}\beta }} = \frac{{{\textrm{length }}\gamma \beta }}{{{\textrm{length }}\alpha \beta }}$$((15.52)) -
(ii)
if one stream is subtracted from another then the resultant lies on an extension of the line joining the two points; and
-
(iii)
if too much is subtracted from the stream then the resultant lies outside the triangle and the composition is imaginary.

Lever rule
The line PQT in Fig. 15.12 represents the composition of all the underflow streams: P the composition when the solution in the underflow is pure solvent, T when it is a mixture of solute and inert solids, and Q when it is a mixture of inserts and a solution of composition R. If now K is defined as the mass of solution in the underflow per unit mass of inert solids in the underflow, then
By definition
where x A, x B and x S are the mass fractions in the underflow of solute, inert solids and solvent, respectively. Thus combining Eqs. (15.53) and (15.54) gives the equation of PQT, the locus of underflows,
Equation (15.55), the equation of a straight line parallel to the hypotenuse, is valid when the mass of solution per unit mass of inert solids is constant. However, if the quantity of solution in the underflow stream is a function of the solution concentration then the locus of underflows is a curve represented by the dotted line in Fig. 15.12.
15.6.5 Procedure to Calculate the Number of Ideal Stages
Referring to Fig. 15.14, the general procedure to calculate the number of ideal stages n and the overflow product composition y 1 can be set out. Let us assume that the following quantities are given:
-
(i)
the composition of the solid to be extracted, x 1
-
(ii)
the composition of the washed solid, \({x_{n + 1}}\)
-
(iii)
the composition of the feed solvent, \({y_{n + 1}}\), and
-
(iv)
the relationship between solution and inert solids in the underflow, which allows the line PQT to be drawn.
-
(1)
The difference point H must lie on
-
(a)
a straight line drawn through x 1 and y 1 because H represents the difference between the flow to the right and the flow to the left. Further, x 1 is located on the side of the triangle where the solvent composition is zero (i.e. the co-ordinates are \({x_{{{\textrm{S}}_1}}} = 0\) and \({x_{{{\textrm{A}}_1}}}\)) and y 1 is calculated from an overall material balance and must lie on the hypotenuse, and on
-
(b)
a straight line drawn between \({x_{n + 1}}\) and \({y_{n + 1}}\). \({x_{n + 1}}\) is the composition of the washed solids and must lie on the locus of underflows (i.e. on the line PQT in the case of constant underflow) and \({y_{n + 1}}\) is the solvent composition and must lie at the apex of the triangle which represents 100% solvent (assuming that pure solvent is used, which is normally the case).
-
(a)
-
(2)
Locate the point H at the intersection of the two lines in (1).
-
(3)
The underflow stream of composition x 2 contains both solids and solution. The solution has the same concentration as y 1, with which it is in equilibrium. Therefore x 2 lies on both the straight line from the origin to y 1 and the line PQT because it is an underflow stream.
-
(4)
Locate x 2 at the intersection of PQT and the line joining 0 to y 1.
-
(5)
The composition y 2 of the overflow from stage 2 must lie on both the straight line joining H and x 2 and the hypotenuse.
-
(6)
Locate y 2.
-
(7)
Repeat this procedure until x A is less than the desired value of \({x_{n + 1}}\) and then count the number of ideal stages required.

Construction of ideal stages for a leaching system
15.6.5.1 Example 15.6
A solvent-free solid containing 40% solute by mass is to be leached using 2 kg of pure solvent per 3 kg of fresh feed. Experimental measurements indicate that each kg of inert solids in the underflows retains an equal mass of solution. Determine the number of ideal stages required to produce an extract solution containing 65% solute by mass.
A material balance is required in order to determine the various unknown compositions. It is often more convenient to solve the material balance by constructing a table of compositions and mass low rates for the four input and output streams than to use the conventional algebraic approach. A basis for the calculation is required; in this case a fresh feed rate of 3 kg s–1 is appropriate. Thus
Overflow product \(({w_1})\) | ◂ | Overflow feed \(({w_{n + 1}})\) | ||||
|---|---|---|---|---|---|---|
y (b) | Mass (kg s–1) | y (a) | Mass (kg s–1) | |||
A | 0.65 | 0.91(g) | A | – | – | |
B | – | – | B | – | – | |
S | 0.35 | 0.49(j) | S | 1.00 | 2.00(c) | |
Total | 1.00 | 1.40(f) | Total | 1.00 | 2.00(c) | |
Underflow feed \(({W_1})\) | ▸ | Underflow product \(({W_{n + 1}})\) | ||||
x (b) | Mass (kg s–1) | x | Mass (kg s–1) | |||
A | 0.40 | 1.20(j) | A | 0.081(h) | 0.29(h) | |
B | 0.60 | 1.80(c) | B | 0.500(d) | 1.80(e) | |
S | – | – | S | 0.419(j) | 1.51(j) | |
Total | 1.00 | 3.00(c) | Total | 1.0 | 3.60(e) | |
From the given data \(K = 1\) and thus from Eq. (15.55) \({x_{\textrm{S}}} = 0.5 - {x_{\textrm{A}}}\). Now (i) when \({x_{\textrm{A}}} = 0\), \({x_{\textrm{S}}} = 0.5\) and (ii) when \({x_{\textrm{S}}} = 0\), \({x_{\textrm{A}}} = 0.5\). Therefore the locus of underflows can be drawn in and from the subsequent construction four ideal stages are required to reduce the underflow product composition to \({x_{{{\textrm{A}}_{n + 1}}}} = 0.081\).
15.6.5.2 Example 15.7
A counter-current process is to be specified to extract an edible oil from vegetable seeds. The seeds contain 20% oil by mass and 90% of the oil is to be recovered in a solution of 50% concentration (by mass). The mass of solution removed in the underflow per kg of inert solid material K is a function of the mass fraction of solute in the overflow stream y A and is given by \(K = 0.7 + 0.5{y_{\textrm{A}}} + 3y_{\textrm{A}}^2\). The seeds are extracted with fresh solvent. Prepare a material balance for this process and determine the number of actual stages required if the stage efficiency is 75%.
Overflow product \(({w_1})\) | ◂ | Overflow feed \(({w_{n + 1}})\) | ||||
|---|---|---|---|---|---|---|
y (b) | Mass (kg s–1) | y (a) | Mass (kg s–1) | |||
A | 0.50 | 18(d) | A | – | – | |
B | – | – | B | – | – | |
S | 0.50 | 18(d) | S | 1.00 | ||
Total | 1.00 | 36(d) | Total | 1.00 | ||
Underflow feed \(({W_1})\) | ▸ | Underflow product \(({W_{n + 1}})\) | ||||
x (b) | Mass (kg s–1) | x | Mass (kg s–1) | |||
A | 0.20 | 20(c) | A | 2(e) | ||
B | 0.80 | 80(c) | B | 80(e) | ||
S | – | – | S | |||
Total | 1.00 | 100(c) | Total | 1.0 | ||
The locus of underflows is obtained as follows: From the definition of K the mass fraction of solute in the underflow x A is
and the mass fraction of solvent in the underflow x S is
Thus, selecting values of y A and using \(K = 0.7 + 0.5{y_{\textrm{A}}} + 3y_{\textrm{A}}^2\), the following table can be constructed.
y A | K | x A | x S |
|---|---|---|---|
0 | 0.70 | 0 | 0.4418 |
0.1 | 0.78 | 0.0438 | 0.3944 |
0.2 | 0.92 | 0.0958 | 0.3833 |
0.3 | 1.12 | 0.1585 | 0.3700 |
0.4 | 1.38 | 0.2319 | 0.3479 |
0.5 | 1.70 | 0.3148 | 0.3148 |
0.6 | 2.08 | 0.4052 | 0.2701 |
The mass ratio of oil to inerts in the underflow is \({y_{\textrm{A}}}K\) which for this problem is equal to \(\frac{2}{{80}} = 0.025\). Therefore \(0.025 = 0.7{y_{\textrm{A}}} + 0.5y_{\textrm{A}}^2 + 3y_{\textrm{A}}^3\) which must be solved by trial-and-error to give \(y_{A} = 0.034\) and therefore \(K = 0.735\). Hence
or
The construction gives nine ideal stages and therefore twelve actual stages.
15.7 Supercritical Fluid Extraction
15.7.1 Introduction
In conventional extraction the relative solubility of the solute in the respective solvents is a function of the chemistry of the system and may be influenced to a small extent by changes in temperature. In contrast, supercritical fluid extraction (SCFE) employs a gas, such as carbon dioxide, at or near the supercritical state, as the second solvent. The use of such solvents has the particular advantages of increasing selectivity and of being able to control solubility by changes in pressure and temperature. Thus the extracted solute can be separated from the supercritical fluid by changing the process conditions. The SCFE solvent is then condensed and recycled, avoiding the need for distillation.
15.7.2 The Supercritical State
The definitions of gas and vapour; the transition between the liquid and the gaseous states; and the definition of the critical point are covered in Chapter 3. At the critical point (i.e. at the critical temperature and critical pressure of a substance) there is no distinction between the gaseous and liquid states and the physico-chemical properties of the substance lie between those of a gas and a liquid. A substance which is at a temperature above the critical temperature is said to be in a ‘fluid’ state. The phase diagram in Fig. 3.6 is presented as a plot of pressure against volume but this information can also be represented by a temperature-volume diagram as in Fig. 15.15. There is no discontinuity of properties in moving across the boundary between a vapour and a permanent gas (the line YZ in Fig. 15.15) nor, if the pressure is above the critical pressure, between a liquid and a gas (the line XY in Fig. 15.15). The densities, viscosities and diffusivities of supercritical fluids are intermediate between those of liquids and gases. Thus density lies in the range 100–1000 kg m–3; viscosity in the range 10–4–10–3 Pa s and diffusivity in the range \(10 ^{- 8} - {10^{ - 7}}{{\textrm{m}}^{ - 2}}{{\textrm{s}}^{ - 1}}\).

Temperature–volume diagram: supercritical fluid
15.7.3 Process Description
For extraction processes involving foods, carbon dioxide is commonly used as the supercritical fluid because it is inert, non-toxic, non-flammable and is readily available and inexpensive. The critical temperature of carbon dioxide is 31°C and above this temperature it enters the supercritical fluid (SCF) state with a critical density of 460 kg m–3. However, because of the lack of a discontinuity in physical properties in moving across the boundary between a vapour and a permanent gas, carbon dioxide and other potential fluids are used both in a supercritical state and in the form of a compressed gas immediately below the critical temperature. Substances under these conditions are therefore sometimes known as near critical fluids (NCF).
Figure 15.16 is a much simplified schematic diagram of a batch SCFE plant for the extraction of solutes from solid materials. It consists of an extraction vessel and an expansion (or separation) vessel. Carbon dioxide is stored as a liquid and is pumped into the extraction vessel via a heat exchanger. In the extraction vessel conditions are such that carbon dioxide exists as a SCF and its ability to extract the solute is at a maximum; the material to be extracted is in the form of a particulate solid. The residence time in the extraction vessel must be sufficient to allow adequate contact between the phases for diffusion of the solute from the solid phase into the SCF to take place and for equilibrium to be attained. The SCF, containing the dissolved solute, is then throttled through an expansion valve and fed into the expansion tank. The reduction in pressure converts the carbon dioxide to a vapour. The solubility is therefore much reduced and the solute is precipitated and removed. The carbon dioxide is recompressed to its supercritical state and recycled.

Schematic diagram of a batch SCFE process
15.7.4 Advantages of SCFE
SCFE has a number of advantages over more conventional extraction techniques. First, whilst the solubility of a solute in a supercritical fluid approaches that of the solubility in a liquid, the major advantage of SCFE is the improved selectivity of solvent rather than the degree of solubility. Second, supercritical fluids have relatively low viscosities and high diffusivities both of which result in increased mass transfer of solute into the fluid. However, despite this, the rate of mass transfer of solute into the supercritical fluid is usually the rate determining step in a SCFE process. Third, there is no requirement for a distillation stage to separate the preferential solvent and solute. Fourth, supercritical fluids are volatile and the recycling of solvent takes place at low temperatures (rather than at the high temperatures required in distillation) and therefore the solute, or extract, is not subjected to thermal damage; this is of particular relevance to temperature-sensitive foods. Finally, the process is inherently safer because of the use of non-toxic and non-inflammable solvents. Set against these advantages are two disadvantages: the need for high pressure equipment with the consequent high capital costs; and that SCFE is a batch process with all the disadvantages that this entails.
15.7.5 Food Applications of SCFE
The possible applications of supercritical fluid extraction in the food industry, both those which have been exploited commercially and those at the development stage, include the removal of flavours and colours from fruits, herbs and spices (e.g. ginger, coriander); the extraction and removal of fats and oils; the removal of hop extracts; and the extraction of egg phospholipids from dried egg yolk. SCFE of rapeseed oil and soya oil from seeds is a potential replacement for the conventional leaching process using hexane. In the decaffeination of coffee beans, supercritical carbon dioxide is a particularly effective selective solvent for caffeine and avoids the co-extraction of other flavour compounds.
Problems
-
15.1
A binary mixture is to be separated by differential batch distillation. 60 kmol of feed, containing 55 mol% ethanol in an second organic liquid, is charged to the still. The distillation is stopped when the residue composition reaches 20 mol% ethanol. Estimate the quantity of distillate produced and the mean mole fraction of ethanol in the distillate.
The equilibrium data are as follows:
$$\begin{array}{l} x\,\,\,\,\,0.20\,\,\,\,\,0.25\,\,\,\,\,0.30\,\,\,\,\,0.35\,\,\,\,\,0.40\,\,\,\,\,0.45\,\,\,\,\,0.50\,\,\,\,\,0.55 \\ y\,\,\,\,\,0.63\,\,\,\,\,0.68\,\,\,\,\,0.72\,\,\,\,\,0.75\,\,\,\,\,0.77\,\,\,\,\,0.80\,\,\,\,\,0.82\,\,\,\,\,0.84 \\ \end{array}$$ -
15.2
An equimolar mixture of two organic liquids A and B is to be distilled until the liquid remaining in the still contains 90 mol% B. If the mole fraction of A in the vapour phasey in equilibrium with the liquid composition x is given by \(y = 1.5x + 0.05\), determine the percentage of the feed that must be removed and the average composition of the distillate.
-
15.3
A solution containing 30% ethanol by mass in water is fed to a continuous fractionation column. Distillate and residue streams with composition of 88% ethanol by mass and 1% ethanol by mass, respectively, are required. The vapour–liquid equilibrium data (in terms of mole fraction of ethanol) are as follows:
$$\begin{array}{l} x\,\,\,\,\,0.05\,\,\,\,\,0.10\,\,\,\,\,0.20\,\,\,\,\,0.30\,\,\,\,\,0.40\,\,\,\,\,0.50\,\,\,\,\,0.60\,\,\,\,\,0.70\,\,\,\,\,0.80\,\,\,\,\,0.90 \\ y\,\,\,\,\,0.34\,\,\,\,\,0.42\,\,\,\,\,0.53\,\,\,\,\,0.58\,\,\,\,\,0.62\,\,\,\,\,0.66\,\,\,\,\,0.70\,\,\,\,\,0.76\,\,\,\,\,0.83\,\,\,\,\,0.90 \\ \end{array}$$Determine the number of ideal stages required assuming that the feed and product streams are at their respective boiling points. The column is operated at a reflux ratio of 3.
-
15.4
Calculate the minimum reflux ratio for the column in Problem 15.3.
-
15.5
A 60 mol% mixture of A in B is to be continuously fractionated to produce a distillate of 97 mol% A and a residue of 3 mol% A. The feed to the column is a saturated vapour. Determine the minimum reflux ratio and the number of ideal stages required at a reflux ratio of twice the minimum. The relative volatility of A to B is 2.3.
-
15.6
For the process in Example 15.6, determine the number of actual stages required if the stage efficiency is 80%.
-
15.7
Solute A is to be leached, using pure water, from a raw material containing 35% A and 65% insoluble solids. A flow rate of 1200 kg h–1 of water is required to process 3000 kg h–1 of feed. The underflow from each stage contains 0.5 kg of solution per kg of dry inert solids. Determine the composition and flow rate of the overflow product and estimate the number of stages required to recover 97% of the solute if the stage efficiency is 75%.
Abbreviations
- a :
-
Coefficient
- b :
-
Constant
- D :
-
Molar flow rate (molar mass) of distillate
- F :
-
Molar flow rate (molar mass) of feed
- h f :
-
Enthalpy per kmol of feed at its boiling point
- h fg :
-
Molar latent heat of vaporisation
- h L :
-
Molar specific enthalpy of liquid stream
- h V :
-
Molar specific enthalpy of vapour stream
- hʹ :
-
Enthalpy per kmol of feed
- H :
-
Mass flow rate of ‘difference’ stream
- K :
-
Mass of solution per unit mass of inert solids in the underflow
- L :
-
Molar flow rate of liquid
- LVC:
-
Less volatile component
- MVC:
-
More volatile component
- p :
-
Partial pressure
- pʹ :
-
Pure component vapour pressure
- P :
-
System pressure
- q :
-
Heat to vaporise 1 kmol of feed entering the column per unit molar latent heat of feed
- R :
-
Reflux ratio
- \({R_{\min }}\) :
-
Minimum reflux ratio
- S :
-
Molar flow rate of steam
- V :
-
Molar flow rate of vapour
- w :
-
Mass flow rate of overflow stream
- W :
-
Molar flow rate (molar mass) of residue; mass flow rate of the underflow stream
- x :
-
Mole fraction of MVC in liquid phase; mass fraction of solute in underflow
- x D :
-
Mole fraction of MVC in distillate
- x F :
-
Mole fraction of MVC in feed
- x q :
-
Co-ordinate of point of intersection of operating lines
- x W :
-
Mole fraction of MVC in residue
- y :
-
Mole fraction of MVC in vapour phase; mass fraction of solute in overflow
- y q :
-
Co-ordinate of point of intersection of operating lines
- A:
-
Component A; solute
- B:
-
Component B; inert solids
- S:
-
Solvent
- n :
-
Plate in rectifying section; leaching stage
- m :
-
Plate in stripping section
- L:
-
Liquid
- V:
-
Vapour
- α :
-
Relative volatility
Further Reading
Grandison, A. S. and Lewis, M. J. 1996. Separation processes in the food and biotechnology industries. Cambridge: Woodhead.
Treybal, R. E. 1980. Mass transfer operations. New York, NY: McGraw-Hill.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Smith, P. (2011). Mass Transfer Operations. In: Introduction to Food Process Engineering. Food Science Text Series. Springer, Boston, MA. https://doi.org/10.1007/978-1-4419-7662-8_15
Download citation
DOI: https://doi.org/10.1007/978-1-4419-7662-8_15
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4419-7661-1
Online ISBN: 978-1-4419-7662-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)