
Abstract
Immunoglobulin G (IgG) molecules are glycoproteins with dual functionality. While participating in the destruction of virally infected cells or healthy tissues during autoimmune disease, IgG antibodies are also used as a therapeutic agent to suppress IgG-triggered autoimmune disease and inflammation. Research of recent years has put the IgG-associated sugar moiety in the spotlight for regulating these opposing activities. This review will focus on how certain IgG glycovariants impact different IgG-dependent effector functions and how this knowledge might be used to further improve the therapeutic effectiveness of this class of molecules.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
- Sialic Acid
- Sugar Moiety
- Chronic Inflammatory Demyelinating Polyneuropathy
- Sialic Acid Residue
- Fucose Residue
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
10.1 Introduction
In humans and mice five different isotypes of immunoglobulins (IgA, IgD, IgE, IgM and IgG) exist. In addition, there are several subclasses of IgA (IgA1 and IgA2) and IgG (IgG1-4 in humans and IgG1, IgG2a, IgG2b, and IgG3 in mice) building up a complex repertoire of molecules for the defense against microbial pathogens [1]. Despite this array of antibody isotypes and subclasses, antibodies of the IgG isotype are most frequently used as a platform for immune-therapeutic approaches [2–4]. This does not only include full-length antibody molecules but also a great variety of proteins fused to the IgG fragment crystallisable (Fc-fragment) to confer enhanced stability and serum half-life. Within the last 20 years, more than 20 monoclonal antibodies were approved for the use in human therapy of cancer and autoimmune disease. The success story of the human CD20-specific antibody Rituximab and the Her2/neu specific antibody Trastuzumab has fueled the interest in therapeutic IgG antibodies and several strategies are employed to further enhance the activity of this class of molecules. Besides improving the affinity of therapeutic IgGs for their cognate antigen or the generation of IgG molecules for novel target antigens, many strategies focus on enhancing IgG dependent effector functions such as release of pro-inflammatory mediators, antibody-dependent cellular phagocytosis (ADCP), antibody-dependent cellular cytotoxicity (ADCC), and complement-dependent cytotoxicity (CDC) [5–8]. Although CDC is a potent way to kill target cells in vitro, results by many groups obtained in different mouse in vivo model systems of autoimmune disease and antibody-dependent tumor immunotherapy argue for a limited involvement of this pathway in destruction of target cells [9–17]. In contrast, mice lacking functional Fc-receptors for IgG (Fcγ-receptors, FcγR) were protected from IgG-dependent destruction of healthy tissues during autoimmune disease or were unresponsive to anti-tumor antibodies in different tumor models [11, 13, 15, 16, 18]. There is clear evidence however, for a complement C5a mediated enhancement of FcγR activity through regulation of activating versus inhibitory FcγR expression [19, 20]. Therefore, many strategies to improve IgG activity are focused on the IgG-FcγR interaction. The family of FcγRs consists of several members with distinct features. There is one high affinity FcγR (FcγRI or CD64) which has the capacity to bind monomeric IgG similar to the high affinity receptor for IgE, FcεRI. All the other members have a much lower affinity for IgG and can only recognize antibodies in the form of multimeric immune complexes (Fig. 10.1). The second distinguishing feature is the signaling pathways initiated by the different family members. Thus, there are three activating (FcγRIA, IIA and IIIA in humans and FcγRI, III and IV in mice) and one inhibitory member (FcγRIIB) in mice and man. Whereas the activating FcγRs signal cell activation through immune-receptor tyrosine based activation motifs (ITAM) present in their intracellular domains or in signaling adaptor molecules such as the common FcR gamma chain (γ-chain), the inhibitory FcγRIIB contains and immune-receptor tyrosine based inhibitory motif (ITIM) and therefore initiates inhibitory signaling pathways [15, 16, 21, 22]. One exception to this rule is the GPI-linked FcγRIIIB, which is selectively expressed on human neutrophils and has no signaling capacity (Fig. 10.1). As activating and inhibitory FcγRs are co-expressed on the majority of innate immune effector cells, including basophils, eosinophil mast cells, neutrophils, monocytes, and macrophages immune complexes will trigger both, activating and inhibitory signaling pathways. Thus, co-expression of FcγRs sets a threshold for cell activation preventing unwanted activation of the powerful effector functions that can be unleashed by the innate immune system. On B cells, the inhibitory FcγRIIB is a crucial regulator of activating signaling pathways triggered by the B cell receptor, thereby preventing unwanted activation of B cells and generation of low affinity and potentially cross-reactive antibodies [15, 21, 22]. Mice deficient in FcγRIIB develop an autoimmune disease very similar to human systemic lupus erythematosus characterized by auto-antibodies specific for double stranded DNA, development of glomerulonephritis and a reduced life expectancy [23–26]. Another interesting result from these studies was that the low affinity FcγRs were the major contributors to IgG dependent effects in mice and humans [27]. Human lymphoma patients carrying allelic variants of FcγRIIA and FcγRIIIA with enhanced binding to human IgG1 showed improved anti-tumor responses under RituxiMab therapy or after vaccination against the lymphoma idiotype [28–30]. In a similar fashion, the mouse orthologous low affinity receptors FcγRIII and FcγRIV were dominantly involved in triggering IgG1, IgG2a and IgG2b dependent effects [5, 12, 14, 31–34]. One explanation for the relatively small contribution of the high affinity FcγRI to these antibody-dependent effects might be that due to the high affinity of this receptor immune complex, binding might be compromised in the presence of high levels of serum IgG. Indeed, a role for the high affinity receptor was detected in solid tumor models where the tumor is localized in tissues [35, 36].

The extended family of human and mouse Fcγ-receptors. Shown here are the members of the mouse (upper panel) and human (lower panel) FcγR family. Only the high affinity FcγRI has the capacity to interact with monomeric IgG, whereas all the other receptors can only bind to IgG in the form of an immune complex. There is one inhibitory Fc-receptor (FcγRIIB) which regulates activating signals initiated via FcγRIA, FcγRIIA, FcγRIIIA, and their respective mouse orthologous receptors. The GPI-linked human FcγRIIIB has not been identified in mice. In addition to the canonical FcγRs, mouse SIGN-R1 and human DC-SIGN have recently been shown to interact with IgG glycovariants rich in terminal sialic acid residues. See text for further details
Besides this pro-inflammatory activity, IgG is long known to have an active anti-inflammatory activity [37, 38]. The injection of high doses of pooled IgG fractions derived from thousands of donors (intravenous IgG or IVIg therapy) is an efficient means to suppress a variety of autoimmune diseases including immune-thrombocyotpenia (ITP), chronic inflammatory demyelinating polyneuropathy (CIDP), and rheumatoid arthritis [37, 38]. It is an unsolved mystery as to how IgG can mediate these opposing functions. A possible explanation for this conundrum was afforded by the finding that the IgG-associated sugar moiety is essential for both activities. Thus, IgG deglycosylation impairs both the pro and anti-inflammatory activity [39–42]. Using this Achilles heel of IgG, it was demonstrated that novel endoglycosidases that selectively remove the sugar moiety of IgG are a potent means to interfere with autoantibody induced inflammation in vivo [43–45]. In the following paragraphs we discuss which individual sugar residues participate in the modulation of IgG activity.
10.2 IgG Glycosylation is Essential for IgG Functionality
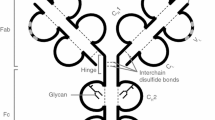
As is true for all antibody isotypes, IgG is a glycoprotein with a sugar moiety attached to each of the asparagin 297 (N297) residues in the CH2-domains of the two Fc-fragments. In contrast to other Ig isotypes, the IgG-associated sugar domain is not exposed on the IgG surface but rather buried within the hydrophobic core between the two Fc-fragments and impacts Fc-structure [1]. Removal of this sugar moiety impairs Fc-dependent effector functions such as binding to FcγRs and C1q [40, 42]. The core of this sugar moiety consists of a bi-antennary heptameric structure consisting of mannose and N-acetylglucosamine (GlcNAc), further decorated with terminal and branching residues including galactose, sialic acid, fucose, and N-acetylglucosamine (Fig. 10.2). Depending on the presence of terminal galactose residues, IgG glycovariants are termed IgG-G0 (no terminal galactose and sialic acid residues), IgG-G1 (one terminal galactose residue with or without an additional sialic acid residue), and IgG-G2 (two terminal galactose residues with or without sialic acid residues) [15, 46]. In contrast to IgG produced in tissue culture, serum IgG is heterogeneous with respect to the exact composition of these terminal and branching sugar residues [47]. Thus, between 30–40 different IgG glycovariants can be identified in the serum of healthy individuals [46]. Early studies in patients with arthritis, osteoarthritis and spondyloarthropathy demonstrated that the glycosylation pattern of serum IgG can be altered dramatically and correlate to disease activity [48–50]. Similar results were obtained in a variety of mouse strains naturally prone to autoimmune disease development or induced to develop autoimmune symptoms [39, 51, 52]. Apart from inflammatory diseases, changes in serum IgG glycosylation were noticed during pregnancy and aging, suggesting that an active process modulating the composition of the IgG linked sugar moiety might exist [53–55]. Interestingly a differential regulation of IgG glycoforms was noticed between pregnancy and autoimmune diseases. Thus, during autoimmune disease (representing a pro-inflammatory state), the level of IgG-G0 glycovariants lacking terminal galactose and sialic acid residues were increased, whereas during pregnancy (representing an anti-inflammatory state with suppression of certain autoimmune disease symptoms), the IgG-G0 forms were decreased [48, 49, 53, 54]. These observations support the hypothesis that some of these variable branching or terminal sugar residues might be involved in modulating antibody activity, which we will discuss in the next paragraphs.

The sugar moiety of IgG. Shown is the asparagine 297 (ASN 297) attached sugar moiety of IgG. Depending on the presence of none, one, or two terminal galactose residues IgG-G0, IgG-G1, and IgG-G2 glycovariants can be distinguished. All colored sugar residues including sialic acid (SA), galactose (Gal), fucose, and N-acetylglucosamine (GlcNAc) are variable, whereas the black colored residues of the heptameric core sugar structure consisting of mannose (Man) and GlcNAc are always present
10.3 The Role of Branching Fucose Residues
It is widely accepted that branching fucose residues are crucially involved in modulating the pro-inflammatory activity of IgG [56]. Several studies have shown that removal of fucose residues enhances the ADCC activity of therapeutic antibodies in vitro and in vivo [14, 56–59]. Although largely studied for the IgG1 subclass, which is most widely used in clinical applications, it was recently shown that all IgG subclasses show enhanced activity upon removal of fucose [60]. Of note, fucose removal seems to selectively enhance the affinity of IgG for human activating FcγRIIIA and its mouse orthologue FcγRIV. The binding to all other activating FcγRs was unchanged, regardless of the presence or absence of fucose. A possible explanation for this selective enhancement was afforded by a study that showed that yet another sugar side chain, this time attached to activating FcγRIIIA (and mouse FcγRIV), might explain this result [61]. All FcγRs are glycoproteins that contain multiple sugar domains. FcγRIIIA has five asparagine-linked glycosylation sites. Removal of the sugar moiety attached to the asparagine 162 (N162) residue of FcγRIIIA resulted in the inability to bind to IgG without fucose with enhanced affinity. Modeling the FcγR-associated sugar domain on the available co-crystal structure of FcγRIIIA bound to IgG1 suggested that the sugar domains of FcγRIIIA and the IgG molecule come in close contact if fucose residues are present, which might lead to a sterical hindrance effect [61]. In the absence of fucose, however, this inhibitory effect was removed, offering a possible explanation for the affinity data. A more recent study confirmed these results and showed that another sugar moiety attached to the asparagine 45 (N45) residue of FcγRIIIA further modulates the binding to IgG1. Thus, in the absence of the N45 linked sugar moiety, an increase of N162-dependent binding of FcγRIIIA to IgG without fucose could be observed [62]. It is currently unclear whether the level of FcγR glycosylation is stable or is subject to modulation during immune responses or cell activation. Regardless of these open questions, many companies are in the process of manufacturing and testing fucose-deficient glycovariants of antibodies already used successfully in clinical applications [56]. It should be kept in mind that in cases where FcγRIIA is the dominant triggering activating Fc-receptor, no increased antibody activity might be expected. In this scenario, the generation of optimized FcγRIIA binding antibodies generated through introduction of amino acid mutations into the IgG backbone that additionally enhance binding to this activating receptor might be the method of choice. Moreover, the combined engineering of both the sugar moiety and the amino acid backbone might be useful to generate more powerful antibody variants.
10.4 The Role of Branching N-Acetylglucosamine Residues
In contrast to the relative abundant presence of fucose in the sugar moiety of IgG, branching N-Acetylglucosamines (GlcNAc) are rather rare and even absent if the antibodies are produced in cell lines such as Chinese hamster ovarian (CHO) cells. In contrast, rat myeloma cell lines do add significant amounts of branching GlcNAcs, and it was observed that a humanized CAMPATH-1H (anti-CD52) antibody produced in this rat myeloma line had a higher ADCC activity [63]. Similar results were obtained by other studies that generated cell lines overexpressing the enzyme b [1, 4]-N-acetylglucosaminyltransferase III (GnTIII) which adds branching GlcNAc residues to the bi-antennary sugar moiety of IgG [64, 65]. One effect of GnTIII over-expression and addition of branching GlcNAcs is that other consecutive glycosylation enzymes such as Golgi-mannosidase II, galactosidase, and fucosyl transferases no longer recognize this sugar moiety efficently, resulting in lower levels of core fucosylation, which may explain the enhanced activity [65, 66]. Shinkawa and colleagues compared the effects of either enhanced levels of GlcNAcs or the lack of fucose, and found that compared to the more than 50-fold ADCC enhancement seen for IgG without fucose, the enhancement of activity for IgG with high levels of GlcNAcs was much smaller [58].
10.5 The Role of Terminal Galactose Residues
There are several conflicting reports about the role of terminal galactose residues in modulating IgG activity. Initial studies indicated that a highly galactosylated anti-D IgG1 antibody has a 2–3 fold higher ADCC activity [67, 68]. Others could not confirm these results when using anti-CD52, anti-IL5R and anti-CD20 antibodies [58, 69]. Complicating the situation further studies found IgG molecules lacking terminal galactose residues (the IgG-G0 glycovariant) are more active than their IgG-G1 or IgG-G2 counterparts [70]. These results were supported by the notion that patients with rheumatoid arthritis, primary osteoarthritis or spondyloarthrophathy and several autoimmune prone mouse strains showed an altered serum IgG glycosylation pattern with higher levels of the IgG-G0 glycoform lacking terminal sialic acid and galactose residues [48–50, 71, 72]. In vitro studies showed that a potential mechanism for this enhanced activity might be the exposure of the high mannose core sugar structure which could acquire the capacity to bind to mannan binding lectin (MBL), the first component of the lectin pathway of complement activation [73]. More recent studies in MBL knockout animals, however, argue against a significant involvement of this pathway and show that the activity of IgG-G0 glycovariants is still fully dependent on activating FcγRs and independent of the complement pathway [74]. Taken together, current evidence does not support an important role of terminal galactose residues in either enhancing or attenuating the activity of IgG in vivo.
10.6 The Role of Terminal Sialic Acid Residues
In addition to the absence of terminal galactose residues, IgG-G0 glycoforms also lack terminal sialic acid residues (Fig. 10.2). Whereas many of the previous studies had anticipated that the reduction of terminal galactose residues in patients with autoimmune disease would enhance their pro-inflammatory activity, an alternative explanation could be that IgG antibodies lacking these terminal sialic acid or galactose residues loose an active anti-inflammatory activity. There is long standing evidence that IgG molecules can have an anti-inflammatory activity. The infusion of large amounts of the pooled fraction of serum IgG obtained from thousands of donors is a well established and efficient treatment for many autoimmune diseases including immune-thrombocytopenia (ITP), chronic inflammatory demyelinating polyneuropathy (CIDP), and rheumatoid arthritis (RA) [37, 38]. Removing the sugar moiety from this pooled IgG fraction abolishes the anti-inflammatory activity [39]. A comparable level of reduction in IVIg activity was seen if terminal sialic acid residues were removed by treatment with neuraminidase. Consistently, enriching IVIg for terminal sialic acid residues increased its anti-inflammatory activity in models of arthritis and nephrotoxic nephritis [39]. Further support for this concept was provided by data showing that only 2,6-linked sialic acid residues (the predominant type of linkage for sialic acid in the sugar moiety of IgG) were responsible for the anti-inflammatory activity, enabling the generation of a recombinant IVIg product produced in tissue culture [75]. Importantly, the sugar structure itself was not sufficient to provide this anti-inflammatory activity, as other serum proteins containing the same sugar moiety with high levels of terminal sialic acid residues had no anti-inflammatory activity [39]. This indicates that both the IgG amino acid backbone and the sialic acid residues in the sugar moiety were essential for the anti-inflammatory activity. When terminal sialic acid residues come in close contact with the amino acid backbone, an altered tertiary structure of the IgG molecule imposed by the negatively charged acidic residues might be one mechanistic explanation. As human and mouse IgG glycovariants rich in terminal sialic acid residues show a reduced affinity for activating FcγRs, it seems clear that other receptors might be involved in recognizing sialic acid rich IgG [39, 76]. Indeed, a recent study showed that mouse SIGN-R1 or its human orthologue, DC-SIGN, have the capacity to recognize this IgG glycovariant [77]. Knock-out mice lacking SIGN-R1 expression showed an abrogation of IVIg activity in a model of serum transfer arthritis, strongly arguing for an important role of this receptor in the anti-inflammatory activity of IVIg. Nonetheless, it seems clear that the inhibitory FcγRIIB is also essential for IVIg activity. IVIg lost its therapeutic activity in FcγRIIB knock-out animals in models of ITP, nephrotoxic nephritis, and serum transfer arthritis [39, 78, 79]. A detailed analysis of FcγR expression on innate immune effector cells in the course of IVIg therapy showed that in mice and humans, IVIg induces an upregulation of the inhibitory FcγRIIB and a downregulation of activating FcγRs, resulting in an enhanced threshold for innate immune effector cell activation [78–80]. At present, it is unclear how this change in FcγR expression is achieved. It is tempting to speculate that anti-inflammatory cytokines might be involved in this pathway, although a recent study using several mouse strains deficient in a variety of cytokines could not detect a reduced anti-inflammatory activity [81]. Taken together, we begin to get a better picture of the mechanisms underlying the anti-inflammatory activity of IgG, although many open questions remain. Moreover, we have to point out that depending on the autoimmune disease, other anti-inflammatory pathways might exist. There are excellent reviews providing a more complete overview over this exciting field [38, 82].
10.7 Conclusion
Research during the last few years has provided convincing evidence that for understanding the many activities of IgG more than just protein-protein interactions have to be taken into consideration. The importance of certain sugar residues for the pro and anti-inflammatory functions of IgG have highlighted that the immunoglobulin attached sugar moiety is much more than just a scaffold for the correct three dimensional structure. The presence or absence of distinct sugar residues such as fucose or sialic acid can dramatically alter IgG activity and the change in serum IgG glycosylation during age, autoimmune disease, and pregnancy suggests that active regulatory mechanisms might exist that could be envisaged as a molecular switch keeping the humoral immune system in an active pro-inflammatory or a more anti-inflammatory state. We are clearly just at the beginning of understanding the mechanisms involved in fine-tuning IgG glycosylation and which functions the different glycoforms play during the steady state and infection.
References
Woof JM, Burton DR (2004) Human antibody-Fc receptor interactions illuminated by crystal structures. Nat Rev Immunol 4:89–99
von Mehren M, Adams GP, Weiner LM (2003) Monoclonal antibody therapy for cancer. Annu Rev Med 54:343–369, Epub 2001 Dec 2003
Waldmann TA (2003) Immunotherapy: past, present and future. Nat Med 9:269–277
Adams GP, Weiner LM (2005) Monoclonal antibody therapy of cancer. Nat Biotechnol 23:1147–1157
Nimmerjahn F, Ravetch JV (2007) Antibodies, Fc receptors and cancer. Curr Opin Immunol 19:239–245
Natsume A, Niwa R, Satoh M (2009) Improving effector functions of antibodies for cancer treatment: enhancing ADCC and CDC. Drug Des Dev Ther 3:7–16
Kubota T, Niwa R, Satoh M et al (2009) Engineered therapeutic antibodies with improved effector functions. Cancer Sci 100:1566–1572
Parren PW, Burton DR (2009) Immunology. Two-in-one designer antibodies. Science 323:1567–1568
Azeredo da Silveira S, Kikuchi S, Fossati-Jimack L et al (2002) Complement activation selectively potentiates the pathogenicity of the IgG2b and IgG3 isotypes of a high affinity anti-erythrocyte autoantibody. J Exp Med 195:665–672
Carroll MC (2004) The complement system in regulation of adaptive immunity. Nat Immunol 5:981–986
Clynes R, Ravetch JV (1995) Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity 3:21–26
Hamaguchi Y, Xiu Y, Komura K et al (2006) Antibody isotype-specific engagement of Fc gamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. J Exp Med 203:743–753
Hogarth PM (2002) Fc receptors are major mediators of antibody based inflammation in auto-immunity. Curr Opin Immunol 14:798–802
Nimmerjahn F, Ravetch JV (2005) Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science 310:1510–1512
Nimmerjahn F, Ravetch JV (2008) Fc gamma receptors as regulators of immune responses. Nat Rev Immunol 8:34–47
Takai T (2002) Roles of Fc receptors in autoimmunity. Nat Rev Immunol 2:580–592
Sylvestre D, Clynes R, Ma M et al (1996) Immunoglobulin G-mediated inflammatory responses develop normally in complement-deficient mice. J Exp Med 184:2385–2392
Takai T, Li M, Sylvestre D et al (1994) FcR gamma chain deletion results in pleiotropic effector cell defects. Cell 76:519–529
Schmidt RE, Gessner JE (2005) Fc receptors and their interaction with complement in auto-immunity. Immunol Lett 100:56–67
Shushakova N, Skokowa J, Schulman J et al (2002) C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J Clin Invest 110:1823–1830
Daeron M, Lesourne R (2006) Negative signaling in Fc receptor complexes. Adv Immunol 89:39–86
Bolland S, Ravetch JV (1999) Inhibitory pathways triggered by ITIM-containing receptors. Adv Immunol 72:149–177
Baerenwaldt A, Nimmerjahn F (2008) Immune regulation - Fc gamma RIIB - regulating the balance between protective and auto-reactive immune responses. Immunol Cell Biol 86:482–484
Bolland S, Ravetch JV (2000) Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 13:277–285
Bolland S, Yim YS, Tus K et al (2002) Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(−/−) mice. J Exp Med 195:1167–1174
Takai T, Ono M, Hikida M et al (1996) Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 379:346–349
Tan Sardjono C, Mottram PL, Hogarth PM (2003) The role of FcgammaRIIa as an inflammatory mediator in rheumatoid arthritis and systemic lupus erythematosus. Immunol Cell Biol 81:374–381
Cartron G, Dacheux L, Salles G et al (2002) Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99:754–758
Weng WK, Czerwinski D, Timmerman J et al (2004) Clinical outcome of lymphoma patients after idiotype vaccination is correlated with humoral immune response and immunoglobulin G Fc receptor genotype. J Clin Oncol 22:4717–4724, Epub 2004 Oct 4713
Weng WK, Levy R (2003) Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol 21:3940–3947, Epub 2003 Sep 3915
Baudino L, Nimmerjahn F, da Silveira SA et al (2008) Differential contribution of three activating IgG Fc receptors (Fc gamma RI, FcyRIII, and Fc gamma RIV) to IgG2a- and IgG2b-induced autoimmune hemolytic anemia in mice. J Immunol 180:1948–1953
Giorgini A, Brown HJ, Lock HR et al (2008) Fc gamma RIII and Fc gamma RIV are indispensable for acute glomerular inflammation induced by switch variant monoclonal antibodies. J Immunol 181:8745–8752
Hazenbos WL, Gessner JE, Hofhuis FM et al (1996) Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity 5:181–188
Syed SN, Konrad S, Wiege K et al (2009) Both FcgammaRIV and FcgammaRIII are essential receptors mediating type II and type III autoimmune responses via FcRgamma-LAT-dependent generation of C5a. Eur J Immunol 39:3343–3356
Otten MA, van der Bij GJ, Verbeek SJ et al (2008) Experimental antibody therapy of liver metastases reveals functional redundancy between Fc gamma RI and Fc gamma RIV. J Immunol 181:6829–6836
Bevaart L, Jansen MJ, van Vugt MJ et al (2006) The high-affinity IgG receptor, FcgammaRI, plays a central role in antibody therapy of experimental melanoma. Cancer Res 66:1261–1264
Nimmerjahn F, Ravetch JV (2008) Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol 26:513–533
Negi VS, Elluru S, Siberil S et al (2007) Intravenous immunoglobulin: an update on the clinical use and mechanisms of action. J Clin Immunol 27:233–245
Kaneko Y, Nimmerjahn F, Ravetch JV (2006) Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313:670–673
Duncan AR, Winter G (1988) The binding site for C1q on IgG. Nature 332:738–740
Nose M, Wigzell H (1983) Biological significance of carbohydrate chains on monoclonal antibodies. Proc Natl Acad Sci USA 80:6632–6636
Koide N, Nose M, Muramatsu T (1977) Recognition of IgG by Fc receptor and complement: effects of glycosidase digestion. Biochem Biophys Res Commun 75:838–844
Albert H, Collin M, Dudziak D et al (2008) In vivo enzymatic modulation of IgG glycosylation inhibits autoimmune disease in an IgG subclass-dependent manner. Proc Natl Acad Sci USA 105:15005–15009
Nandakumar KS, Collin M, Olsen A et al (2007) Endoglycosidase treatment abrogates IgG arthritogenicity: importance of IgG glycosylation in arthritis. Eur J Immunol 37:2973–2982
Collin M, Shannon O, Bjorck L (2008) IgG glycan hydrolysis by a bacterial enzyme as a therapy against autoimmune conditions. Proc Natl Acad Sci USA 105:4265–4270
Arnold JN, Wormald MR, Sim RB et al (2007) The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol 25:21–50
Kobata A (2008) The N-linked sugar chains of human immunoglobulin G: their unique pattern, and their functional roles. Biochim Biophys Acta 1780:472–478
Parekh RB, Dwek RA, Sutton BJ et al (1985) Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 316:452–457
Leirisalo-Repo M, Hernandez-Munoz HE, Rook GA (1999) Agalactosyl IgG is elevated in patients with active spondyloarthropathy. Rheumatol Int 18:171–176
Parekh RB, Roitt IM, Isenberg DA et al (1988) Galactosylation of IgG associated oligosaccharides: reduction in patients with adult and juvenile onset rheumatoid arthritis and relation to disease activity. Lancet 1:966–969
Mizuochi T, Hamako J, Nose M et al (1990) Structural changes in the oligosaccharide chains of IgG in autoimmune MRL/Mp-lpr/lpr mice. J Immunol 145:1794–1798
Bond A, Cooke A, Hay FC (1990) Glycosylation of IgG, immune complexes and IgG subclasses in the MRL-lpr/lpr mouse model of rheumatoid arthritis. Eur J Immunol 20:2229–2233
van de Geijn FE, Wuhrer M, Selman MH et al (2009) Immunoglobulin G galactosylation and sialylation are associated with pregnancy-induced improvement of rheumatoid arthritis and the postpartum flare: results from a large prospective cohort study. Arthritis Res Ther 11:R193
Rook GA, Steele J, Brealey R et al (1991) Changes in IgG glycoform levels are associated with remission of arthritis during pregnancy. J Autoimmun 4:779–794
Parekh R, Roitt I, Isenberg D et al (1988) Age-related galactosylation of the N-linked oligosaccharides of human serum IgG. J Exp Med 167:1731–1736
Satoh M, Iida S, Shitara K (2006) Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies. Expert Opin Biol Ther 6:1161–1173
Shields RL, Lai J, Keck R et al (2002) Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem 277:26733–26740, Epub 22002 May 26731
Shinkawa T, Nakamura K, Yamane N et al (2003) The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem 278:3466–3473, Epub 2002 Nov 3468
Niwa R, Sakurada M, Kobayashi Y et al (2005) Enhanced natural killer cell binding and activation by low-fucose IgG1 antibody results in potent antibody-dependent cellular cytotoxicity induction at lower antigen density. Clin Cancer Res 11:2327–2336
Niwa R, Natsume A, Uehara A et al (2005) IgG subclass-independent improvement of antibody-dependent cellular cytotoxicity by fucose removal from Asn297-linked oligosaccharides. J Immunol Meth 306:151–160
Ferrara C, Stuart F, Sondermann P et al (2006) The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem 281:5032–5036
Shibata-Koyama M, Iida S, Okazaki A et al (2009) The N-linked oligosaccharide at Fc gamma RIIIa Asn-45: an inhibitory element for high Fc gamma RIIIa binding affinity to IgG glycoforms lacking core fucosylation. Glycobiology 19:126–134
Lifely MR, Hale C, Boyce S et al (1995) Glycosylation and biological activity of CAMPATH-1H expressed in different cell lines and grown under different culture conditions. Glycobiology 5:813–822
Umana P, Jean-Mairet J, Moudry R et al (1999) Engineered glycoforms of an anti-neuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol 17:176–180
Schuster M, Umana P, Ferrara C et al (2005) Improved effector functions of a therapeutic monoclonal Lewis Y-specific antibody by glycoform engineering. Cancer Res 65:7934–7941
Schachter H (1986) Biosynthetic controls that determine the branching and micro-heterogeneity of protein-bound oligosaccharides. Adv Exp Med Biol 205:53–85
Kumpel BM, Rademacher TW, Rook GA et al (1994) Galactosylation of human IgG monoclonal anti-D produced by EBV-transformed B-lymphoblastoid cell lines is dependent on culture method and affects Fc receptor-mediated functional activity. Hum Antibodies Hybridomas 5:143–151
Kumpel BM, Wang Y, Griffiths HL et al (1995) The biological activity of human monoclonal IgG anti-D is reduced by beta-galactosidase treatment. Hum Antibodies Hybridomas 6:82–88
Boyd PN, Lines AC, Patel AK (1995) The effect of the removal of sialic acid, galactose and total carbohydrate on the functional activity of Campath-1H. Mol Immunol 32:1311–1318
Rademacher TW, Williams P, Dwek RA (1994) Agalactosyl glycoforms of IgG autoantibodies are pathogenic. Proc Natl Acad Sci USA 91:6123–6127
Kuroda Y, Nakata M, Hirose S et al (2001) Abnormal IgG galactosylation in MRL-lpr/lpr mice: pathogenic role in the development of arthritis. Pathol Int 51:909–915
Kuroda Y, Nakata M, Nose M et al (2001) Abnormal IgG galactosylation and arthritis in MRL-Fas(lpr) or MRL-FasL(gld) mice are under the control of the MRL genetic background. FEBS Lett 507:210–214
Malhotra R, Wormald MR, Rudd PM et al (1995) Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med 1:237–243
Nimmerjahn F, Anthony RM, Ravetch JV (2007) Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc Natl Acad Sci USA 104:8433–8437
Anthony RM, Nimmerjahn F, Ashline DJ et al (2008) Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 320:373–376
Scallon BJ, Tam SH, McCarthy SG et al (2007) Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol Immunol 44:1524–1534
Anthony RM, Wermeling F, Karlsson MC et al (2008) Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci USA 105:19571–19578
Kaneko Y, Nimmerjahn F, Madaio MP et al (2006) Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J Exp Med 203:789–797, Epub 2006 Mar 2006
Bruhns P, Samuelsson A, Pollard JW et al (2003) Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity 18:573–581
Tackenberg B, Jelcic I, Baerenwaldt A et al (2009) Impaired inhibitory Fc gamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci USA 106:4788–4792
Crow AR, Song S, Semple JW et al (2007) A role for IL-1 receptor antagonist or other cytokines in the acute therapeutic effects of IVIg? Blood 109:155–158
Baerenwaldt A, Biburger M, Nimmerjahn F (2010) Mechanisms of action of intravenous immunoglobulins. Expert Rev Clin Immunol 6:425–434
Acknowledgements
This work was supported by grants from the German Research Foundation (SFB 643, FOR832, GK1660, SPP1468) and the Bavarian Genome Research Network (BayGene) to F.N. We apologize to all colleagues whose important work could not be cited directly due to limitation in space. These references can be found in the review articles referred to in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Businees Media, LLC
About this paper
Cite this paper
Lux, A., Nimmerjahn, F. (2011). Impact of Differential Glycosylation on IgG Activity. In: Pulendran, B., Katsikis, P., Schoenberger, S. (eds) Crossroads between Innate and Adaptive Immunity III. Advances in Experimental Medicine and Biology, vol 780. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-5632-3_10
Download citation
DOI: https://doi.org/10.1007/978-1-4419-5632-3_10
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-5631-6
Online ISBN: 978-1-4419-5632-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)