
Abstract
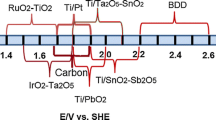
Electrochemical oxidation is a promising method for the treatment of wastewaters containing organic compounds. As a general rule, the electrochemical incineration of organics at a given electrode can take place at satisfactory rates and without electrode deactivation only at high anodic potentials in the region of the water discharge due to the participation of the intermediates of oxygen evolution. The nature of the electrode material strongly influences both the selectivity and the efficiency of the process. In particular, anodes with low oxygen evolution overpotential (i.e., good catalysts for oxygen evolution reactions), such as graphite, IrO2, RuO2, and Pt only permit the partial oxidation of organics, while anodes with high oxygen evolution overpotential (i.e., anodes that are poor catalysts for oxygen evolution reactions), such as SnO2, PbO2, and boron-doped diamond (BDD) favor the complete oxidation of organics to CO2 and so are ideal electrodes for wastewater treatment.However, the application of SnO2 and PbO2 anodes may be limited by their short service life and the risk of lead contamination, while BDD electrodes exhibit good chemical and electrochemical stability, a long life, and a wide potential window for water discharge, and are thus promising anodes for industrial-scale wastewater treatment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction
In recent decades, oxidative electrochemical technologies, providing versatility, energy efficiency, amenability to automation, environmental compatibility, and cost effectiveness have reached a promising stage of development and can now be effectively used for the destruction of toxic or biorefractory organics (Rajeshwar et al. 1994; Rajeshwar and Ibanez 1997; Chen 2004).
The overall performance of the electrochemical processes is determined by the complex interplay of parameters that may be optimized to obtain an effective and economical incineration of pollutants. The principal factors determining the electrolysis performance will be (Pletcher and Walsh 1982) as follows:
-
1.
Electrode potential and current density. Control which reaction should occur and its rate and commonly determine the efficiency of the process
-
2.
Current distribution. Determines the spatial distribution of the consumption of reactants and hence, it must be as homogeneous as possible
-
3.
Mass-transport regime. A high mass-transport coefficient that leads to a greater uniformity of pollutant concentration in the reaction layer near the electrode surface and to generally a higher efficiency
-
4.
Cell design. The cell dimension, the presence or the absence of a separator, the design of the electrode, etc. affect the figures of merit of the electrochemical process
-
5.
Electrolysis medium. The choice of electrolyte and its concentration, pH, temperature
-
6.
Electrode materials. The ideal electrode material for the degradation of organic pollutants should be totally stable in the electrolysis medium; cheap; and exhibit high activity toward organic oxidation and low activity toward secondary reactions (e.g., oxygen evolution).
Even if we still remain far from meeting all these requirements for an electrode, a significant step has been made toward the production of better electrode material. The present chapter reviews and discusses the crucial role of the electrode materials in the electrochemical treatment of wastewater containing organic pollutants.
2.2 Electrochemical Parameters
Before analyzing the influence of the electrode material on the electrooxidation of organic pollutants, it is necessary to review some global parameters commonly used to estimate the progress and the efficiency of electrochemical treatments.
The instantaneous current efficiency (ICE) of the electrooxidation is determined by chemical oxygen demand (COD) measurements, using the relationship (Comninellis and Pulgarin 1991):
where (COD) t and \({\left (\mathrm{COD}\right )}_{t+\Delta t}\) are the chemical oxygen demands at times t and t + Δt(in gO2 dm− 3), respectively; I is the current (A); F is Faraday’s constant (96,500C mol− 1); and V is the volume of the electrolyte (dm3).
The Electrochemical Oxidation Index (EOI), which is the average value of the current efficiency during the oxidation, is determined from the ICE using the relationship (Comninellis and Pulgarin 1991):
where τ is the time at which the ICE is almost zero.
The Electrochemical Oxygen Demand is calculated using the relationship (Comninellis and Pulgarin 1991):
where [organic] is the amount of the organic in the electrolyte (g) and EOD is expressed in gO2(gorganic)− 1.
The General Current Efficiency for the anodic oxidation is calculated from the COD values, using the following relationship (Martinez-Huitle et al. 2004b):
where (COD)0 and \({\left (\mathrm{COD}\right )}_{t+\Delta t}\) are the chemical oxygen demands (g dm− 3) at times t = 0 (initial) and t, respectively; I is the current (A), F is Faraday’s constant (96,500C mol− 1); V the volume of the electrolyte (dm3), and 8 is the oxygen equivalent mass (g eq− 1). This equation is similar to that proposed by Comninellis and Pulgarin (1991) for the determination of the ICE, although the expression used for the GCE represents an average value between the initial time t and t + Δt.
The overall capacity of the electrochemical oxidation is expressed in terms of space–time yield (YST) using the following relationship (Chen 2004):
where a is the specific electrode area (m− 1), defined as the ratio of the electrode area to the reactor volume; i is the current density (A m− 2); CE is the current efficiency; n is the number of electrons in the reaction involved; F is Faraday’s constant (96,500Cmol− 1); and M FW is the molar mass (g mol− 1).
2.3 Oxidation Mechanisms
Electrooxidation of organic pollutants can be performed in several different ways, including direct and indirect oxidation, which are schematized in Fig. 2.1.

Scheme of the electrochemical processes for the removal of organic compounds (R): (a) direct electrolysis; (b) via hydroxyl radicals produced by the discharge of the water; and (c) via inorganic mediators
It has been generally observed that the nature of the electrode material, the experimental conditions, and the electrolyte composition strongly influence the oxidation mechanism.
In direct electrolysis, the pollutants are oxidized after adsorption on the anode surface without the involvement of any substances other than the electron, which is a “clean reagent”:
Direct electrooxidation is theoretically possible at low potentials, before oxygen evolution, but the reaction rate usually has low kinetics that depends on the electrocatalytic activity of the anode. High electrochemical rates have been observed using noble metals such as Pt and Pd, and metal-oxide anodes such as iridium dioxide, ruthenium–titanium dioxide, and iridium–titanium dioxide (Foti et al. 1997).
However, the main problem of electrooxidation at a fixed anodic potential before oxygen evolution is a decrease in the catalytic activity, commonly called the poisoning effect, due to the formation of a polymer layer on the anode surface. This deactivation, which depends on the adsorption properties of the anode surface and the concentration and the nature of the organic compounds, is more accentuated in the presence of aromatic organic substrates such as phenol (Gattrell and Kirk 1993; Foti et al. 1997), chlorophenols (Rodgers et al. 1999; Rodrigo et al. 2001), naphthol (Panizza and Cerisola 2003b), and pyridine (Iniesta et al. 2001b).
In indirect oxidation, organic pollutants do not exchange electrons directly with the anode surface; but they exchange through the mediation of some electroactive species regenerated there, which act/acts as an intermediary for shuttling electrons between the electrode and the organics. Indirect electrolysis can be a reversible or an irreversible process.
In the reversible process, the redox reagents are turned over several times and recycled. The reversible mediators of oxidation can be a metallic redox couple, such as Ag+ ∕ Ag2 +(Farmer et al. 1992), Co2 + ∕ Co3 +(Leffrang et al. 1995), Ce3 + ∕ Ce4 +(Nelson 2002), Fe2 + ∕ Fe3 +(Dhooge and Park 1983), or inorganic ions such as Cl− ∕ ClO− (Comninellis and Nerini 1995; Szpyrkowicz et al. 1995; (Panizza and Cerisola 2003a) or Br− ∕ BrO− (Martinez-Huitle et al. 2005) added to or present in the electrolyte. The main drawback of the use of a solution redox couple is the need to subsequently separate the oxidation products from the mediator.
In the irreversible process, a strong oxidizing chemical (e.g., ozone (Foller and Tobias 1982; Tatapudi and Fenton 1993; Feng et al. 1994), hydrogen peroxide (Do and Chen 1993; Brillas et al. 1995, 2003; Alvarez-Gallegos and Pletcher 1998; Boye et al. 2002), etc.) is generated in situ to mineralize the organic pollutants.
Another mechanism for the indirect electrochemical oxidation of organics at high potential, proposed by Johnson et al. (Chang and Johnson 1990; Johnson et al. 1999), is based on intermediates of the oxygen evolution reaction. This process involves the transfer of anodic oxygen from H2O to the organics via adsorbed hydroxyl radicals generated by the water discharge:
where S represents the surface sites for adsorption of the OH⋅ species.
An inevitable but undesirable concomitant reaction is the evolution of oxygen by the oxidation of the water
Comninellis et al. (Comninellis 1994; Comninellis and De Battisti 1996; Simond et al. 1997; Foti et al. 1999) found that the nature of the electrode material strongly influences both the selectivity and the efficiency of the process and, in particular, several anodes favored the partial and selective oxidation of pollutants (i.e., conversion), while others favored complete combustion to CO2. In order to interpret these observations, they proposed a comprehensive model for the oxidation of organics at metal oxide electrodes with simultaneous oxygen evolution.
In a similar way to the mechanism proposed by Johnson, the first step in the oxygen transfer reaction is the discharge of water molecules to form adsorbed hydroxyl radicals
The following steps depend on the nature of the electrode materials, and make it possible to distinguish between two limiting classes of electrodes, defined as “active” and “nonactive” anodes:
-
(a)
At “active” electrodes, where higher oxidation states are available on the electrode surface, the adsorbed hydroxyl radicals may interact with the anode, forming the so-called higher oxide:
The surface redox couple MO x + 1 ∕ MO x , which is sometimes called chemisorbed “active oxygen,” can act as a mediator in the conversion or selective oxidation of organics on “active” electrodes:
-
(b)
At “nonactive” electrodes, where the formation of a higher oxide is excluded, hydroxyl radicals, called physisorbed “active oxygen,” may assist the nonselective oxidation of organics, which may result in complete combustion to CO2:
${ \mathrm{MO}}_{x}\left ({}^{\cdot }\mathrm{OH}\right ) + \mathrm{R} \rightarrow {\mathrm{MO}}_{ x} + m\,{\mathrm{CO}}_{2} + n\,{\mathrm{H}}_{2}\mathrm{O} +{ \mathrm{H}}^{+} +{ \mathrm{e}}^{-}$(2.13)
However, both the chemisorbed and the physisorbed “active oxygen,” also undergo a competitive side reaction, i.e., oxygen evolution, resulting in a decrease in the efficiency of the anodic process.
As a general rule, anodes with low oxygen evolution overpotential (i.e., anodes that are good catalysts for the oxygen evolution reaction), such as carbon, graphite, IrO2, RuO2, or platinum, have “active” behavior and only permit the partial oxidation of organics, while anodes with high oxygen evolution overpotential (i.e., anodes that are poor catalysts for the oxygen evolution reaction), such as antimony-doped tin oxide, lead dioxide, or boron-doped diamond (BDD), have “nonactive” behavior and favor the complete oxidation of the organics to CO2 and so are ideal electrodes for wastewater treatment. Moreover, radical trap experiments using N, N-dimethyl-p-nitrosoaniline (DMPO) as an OH⋅ scavenger have demonstrated that a larger concentration of OH⋅ is present on nonactive anodes than on active ones (Comninellis 1994; Marselli et al. 2003). The larger OH⋅ concentration has been suggested as the cause of the complete combustion of organics to CO2 on nonactive anodes.
2.4 Electrode Materials
As mentioned above, the nature of the electrode material influences the selectivity and the efficiency of an electrochemical process for the oxidation of organic compounds and for this reason, in literature, many anodic materials have been tested to find the optimum one. According to the model proposed by Comninellis (1994), anode materials are divided for simplicity into two classes as follows:
Class 1 anodes, or active anodes, have low oxygen evolution overpotential and consequently are good electrocatalysts for the oxygen evolution reaction:
-
–
Carbon and graphite
-
–
Platinum-based anodes
-
–
Iridium-based oxides
-
–
Ruthenium-based oxides
Class 2 anodes, or nonactive anodes, have high oxygen evolution overpotential and consequently are poor electrocatalysts for the oxygen evolution reaction:
-
–
Antimony-doped tin oxide
-
–
Lead dioxide
-
–
Boron-doped diamond
The oxygen evolution potentials in H2SO4 of the most extensively investigated anode materials are compared in Table 2.1.
2.4.1 Carbon and Graphite
Carbon and graphite electrodes are very cheap and have a large surface area and so they have been widely used for the removal of organics in electrochemical reactors with three-dimensional electrodes (e.g., packed bed, fluidized bed, carbon particles, porous electrode, etc.). However, with these materials the electrooxidation is generally accompanied by surface corrosion, especially at high current densities. Gattrell and Kirk (1990) used reticulated glassy carbon anodes in a flow-by cell for the oxidation of phenol. During the electrolysis there was a rapid decrease in the reaction rate due to the blocking of the electrode surface with insoluble polymeric products that were slow to oxidize or desorb. Moreover, increasing the phenol concentration from 0.005 to 0.02 M increased the current efficiency but also increased the fraction of phenol that reacted to form polymeric products. They observed that high-temperature (Fig. 2.2) and high-applied potentials (i.e., greater than 1.9 V vs. SCE) not only resulted in a more complete oxidation of the phenol, but also resulted in a decrease in the current efficiency and faster electrode corrosion.

The effect of temperature on the rate of phenol oxidation at a glassy carbon electrode (Gattrell and Kirk 1993)
The electrochemical 2-chlorophenol and 2,6-dichlorophenol removal from aqueous solutions using porous carbon felt (Polcaro and Palmas 1997) or a fixed bed of carbon pellets (Polcaro et al. 2000) as three-dimensional electrodes was investigated by Polcaro’s group. The group’s experimental setup consisted of a two-compartment electrochemical cell separated by an anionic membrane where the carbon felt or pellets could be lodged and the solution was recirculated by peristaltic pumps. Both carbon-based anodes effectively removed the chlorophenols as well as their reaction intermediates. The HPLC chromatographs showed that the main intermediates were aliphatic compounds such as oxalic and maleic acids, while phenol, cathecol, and benzoquinone were not observed; this may indicate that cleavage of chlorine occurs at the same time as the ring opening. They also demonstrated that the electrolyte velocity through the electrode did not affect the reaction behavior, while the most important parameter was the current density per unit electrolyte volume: Using an applied current density of 5 mA cm− 2 of electrode, average current efficiency values were from 25 to 30%. Moreover, under these conditions, they observed only low corrosion effects on the superficial characteristics of the anodes after they had been working for several hours.
The electrochemical treatment of phenol using graphite electrodes was recently investigated by (Awad and Abuzaid 1997, 1999, 2000) and the effect of residence time at different applied currents was elucidated. Phenol removal efficiency was found to increase with the increase in current and residence time and reached about 50% at a current of 2.0 A and a residence time of 35 min (Fig. 2.3).

Relationship between phenol removal efficiency and current, under different flow rates and residence time values, during phenol oxidation at porous graphite anodes (Awad and Abuzaid 2000)
Other types of carbon-based electrodes, such as activated carbon (Canizares et al. 1999), graphite particles (Piya-areetham et al. 2006), graphite Rashig rings (:̧def :̧def Ogutveren et al. 1999), and carbon black slurry (Boudenne et al. 1996; Boudenne and Cerclier 1999) have also been employed sometimes for the treatment of organic compounds.
Carbon-based materials have also been widely used as cathodes in indirect electrolyses of organics generating in situ hydrogen peroxide, by two-electron reduction of oxygen on the cathode surface:
In fact, carbon and graphite exhibit good electrochemical activities for oxygen reduction, high overpotential for hydrogen evolution, and low catalytic activity for hydrogen peroxide decomposition (Do and Chen 1994a, b; Ponce-de-Leon and Pletcher 1995).
It is well known that in acidic solutions, the addition of a small concentration of Fe(II) to the electrogenerated H2O2 enhances the rate and the efficiency of the oxidation of organics, due to the formation of highly oxidizing OH⋅ radicals, according to Fenton’s classical mechanism (Brillas et al. 1996):
This reaction is propagated by Fe2 + regeneration, which takes place through the reduction of Fe3 + with H2O2, the hydroperoxyl radical \({\mathrm{HO}}_{2}^{\cdot }\) previously formed, and the organic radical intermediates and by reduction at the cathode:
In this field, several authors have reported the complete removal of organic pollutants, such as formaldehyde (Do and Chen 1993), aniline (Brillas et al. 1996), phenol (Alvarez-Gallegos and Pletcher 1999), pesticides (Guivarch et al. 2003), herbicides (Boye et al. 2002), and industrial effluent containing naphthalene- and anthraquinone-sulfonic acids (Panizza and Cerisola 2001) by in situ electrogenerated hydrogen peroxide catalyzed by iron ions.
2.4.2 Platinum
The platinum electrode is one of the most commonly used anodes in both preparative electrolysis and synthesis because of its good chemical resistance to corrosion even in strongly aggressive media. The behavior of platinum electrodes in the electrochemical oxidation of organic pollutants has been widely reported in literature, showing a significant electrocatalytic activity (Soriaga and Hubbard 1982; Lamy et al. 1983; Foti et al. 1997; Rodgers et al. 1999).
For example, Lamy (1984) studied the oxidation of organic compounds (e.g., methanol, ethanol, butanol, ethylene-glycol, C2 oxygenated compounds, etc.) on noble metal electrodes (e.g., platinum, gold, rhodium, and palladium) in aqueous solutions and verified that platinum appears to be the best electrocatalyst, particularly in an acidic medium.
Gattrell and Kirk (Gattrell and Kirk 1993) investigated the oxidation of phenol at platinum and peroxidized platinum anodes using cyclic voltammetry and chronoamperometry. Their studies demonstrated that the phenol can be irreversibly adsorbed on metallic platinum, quickly passivating the electrode. However, the presence of a platinum oxide layer on the electrode surface slightly inhibited the formation of the passivating film due to the decreased adsorption strength of the reaction products at the oxide surface. In long-term electrolyses, the activity of metallic platinum and platinum oxide had the same behavior.
The electrochemical oxidation of phenol at platinum anodes was also studied in depth by Comninellis and Pulgarin (Comninellis and Pulgarin 1991) under different conditions for wastewater treatment. A yellow-brown polymeric product was formed during the oxidation of phenol, and this film formation strongly depended on the experimental conditions. They showed that the process was not limited by mass transfer at the anode and the oxidation occurred through an electrophilic attack on the aromatic ring by the OH⋅. The EOI and the EOD values were slightly influenced by the current density and temperature, but they increased with the pH (Table 2.2) and phenol concentration (Fig. 2.4). However, even at pH = 12. 5 the EOI obtained during the electrochemical oxidation of 0.01 M of phenol at T = 70°C and i = 57mA cm− 2 was only 0.143. Such a low value was due to the formation of reaction intermediates, mainly aliphatic acids (e.g., maleic, fumaric, and oxalic acid), which resisted further electrooxidation.

Influence of the initial phenol concentration on the EOI and EOD values obtained during electrochemical oxidation at platinum electrodes. I = 57 mA cm− 2, T = 70°C, pH 12. 5 (Comninellis and Pulgarin 1991)
The oxidation of a wide range of phenol and other biorefractory organic compounds (e.g., ethanol, aliphatic acids, naphthalene and anthraquinone sulfonic acids, aniline, nitrobenzene, etc.) on platinized titanium was also studied by Kotz et al. (Kotz et al. 1991; Stucki et al. 1991). The elimination of TOC was rather ineffective due to the leakage current for oxygen evolution, and the average EOI was about 0.05.
More recently, Bonfatti et al. (1999) have verified that the reactivity of glucose at Ti/Pt electrodes was acceptable in all current densities, slightly higher at 600A m− 2; however, the electrochemical mineralization was low, particularly over a long electrolysis time, due to the accumulation of intermediates, mainly glucaric acid, which resisted further attack at the platinum electrode. The situation improved by increasing the temperature to 56°C.
Similar results were also obtained by Socha et al. (2005) for the electrochemical treatment of 1-Aminonaphthalene-3,6-disulfonic acid (ANDS), a component of the wastewater produced during the synthesis of many synthetic dyes. An increase in the temperature caused a slight increase in the efficiency of the substrate oxidation (Table 2.3), but even at 80°C the removal of TOC and COD was only 18 and 36%, although the decolorization of the solution, i.e., decrease of absorbance, was achieved.
2.4.3 Dimensionally Stable Anodes
The dimensionally stable anodes (DSAⓇ) consist of a titanium base metal covered by a thin conducting layer of metal oxide or mixed metal-oxide oxides. Since their discovery by Beer (1966) in the late 1960s, a lot of work has been done on DSAⓇ and on finding and preparing new coating layers for many electrochemical applications. The development of anodes coated with a layer of RuO2 and TiO2 brought about significant improvements in the chlor-alkali industry (DSA − Cl2), while the anodes coated with IrO2 have been commercially used for oxygen evolution reactions (DSA − O2) in acidic media in several electrochemical processes, such as water electrolysis and metal electrowinning. Recently, DSAⓇ anodes with a different coating composition have been also studied for applications in the oxidation of organics (Bock and MacDougall 2000; Lanza and Bertazzoli 2002; Malpass et al. 2006).
Johnson et al. (Houk et al. 1998; Johnson et al. 1999) studied the incineration of 4-chlorophenol and benzoquinone using quaternary metal-oxide anodes (Ti, Ru, Sn, and Sb). They demonstrated that this type of electrode is stable and electrochemically active for the oxidation of organic compounds when it is used in the absence of a soluble supporting electrolyte, with a NafionⓇ membrane as solid-state electrolyte; however, the electrolysis time for complete COD and TOC removal was excessively long and current efficiency was low.
The electrocatalytic behavior of olefins was studied by Zanta et al. (2000) at thermally prepared ruthenium–titanium- and iridium–titanium-dioxide-coated anodes. The aliphatic olefins were shown to be inactive in the region before oxygen evolution, while aromatic ones showed one or two oxidation peaks, and the catalytic activity seemed to be the same for both substrates. However, as for platinum anodes, voltammetric studies and FTIR analyses have also shown the formation of a polymeric film that blocks the surface of the electrode and decreases its activity.
Many studies of the oxidation of organic compounds with Ti∕IrO2 electrodes have been carried out by Comninellis’ group (Pulgarin et al. 1994; Foti et al. 1997, 1999; Simond et al. 1997). In the region before oxygen evolution, Ti∕IrO2 did not show any electrocatalytic activity for the oxidation of alcohols (methanol, propanol, and butanol) and carboxylic acids (maleic, oxalic, and formic acid). Instead, in the presence of phenol, Ti∕IrO2 had high electrocatalytic activity, but this was quickly diminished because of the formation of a polymer film on the surface of the electrode.
Studies of the electrochemical detoxification of 1,4-benzoquinone (Pulgarin et al. 1994) showed that the primary oxidation, i.e., elimination of benzoquinone, was obtained with Ti∕IrO2 anodes, resulting in an accumulation of carboxylic acids: maleic, fumaric, mesoxalic, and oxalic acids that were only poorly degraded at the Ti∕IrO2 anode (Fig. 2.5).

Evolution of benzoquinone, intermediates and COD during the oxidation of benzoquinone at Ti/IrO2 anodes as a function of charge: open square with dot COD; filled circle benzoquinone; open diamond aliphatic acids, dashed line others; plus CO2. Reprinted from Pulgarin et al. (1994), Copyright (1994), with permission from Elsevier
Several authors have reported that DSA-type anodes coated with a layer of RuO2 or IrO2 and other oxides can be used efficiently for organic disposal by indirect electrolysis generating in situ active chlorine by the oxidation of chloride ions present in the solution, according to the following reaction:
As a function of pH, chlorine remains in the solution as aqueous chlorine (pH < 3) or disproportionates to hypochloric acid (pH < 7. 5) or hypochlorite ions (pH > 7. 5):
This process can effectively oxidize many pollutants, however, it has the drawback of permitting the formation of chlorinated organic compounds during the electrolysis.
Comninellis and Nerini (1995) studied the oxidation of phenol with Ti∕SnO2 and Ti∕IrO2 anodes in the presence of sodium chloride. They showed that the addition of 85 mM of NaCl to the solution catalyzed the oxidation of phenol at Ti∕IrO2 anodes due to the participation of electrogenerated ClO−, increasing the EOI from about 0.06 to 0.56. Surprisingly, the COD elimination was independent of the NaCl concentration and the applied current density. Unfortunately, in their experimental conditions, organochlorinated intermediates, which were further oxidized to volatile organics (CHCl3), were formed.
A systematic study of the kinetics and the influence of operating conditions on the anodic mineralization of formaldehyde with electrogenerated hypochlorite ions at a mixed-oxide anode of SnO2–PdO–RuO2–TiO2 (SPR) was undertaken by Do and Yeh (Do and Yeh 1995; Do et al. 1997). During the oxidation of the formaldehyde the current efficiency increased with the stirring of the solution, the concentration of the chloride ions, the pH, and the concentration of the formaldehyde, while it decreases with current density. The maximum degradation fraction during the electrolysis at i = 75mA cm− 2, at pH = 13, and in the presence of 1 M NaCl was about 90%.
The influence of the DSAⓇ-coating composition and experimental condition on the electrochemical treatment of disperse dyes mediated by chloride ions was investigated by Szpyrkowicz et al. (2000). The efficiency of the treatment depended on the nature of the supporting electrolyte and the bulk pH in the reactor and, to a lesser degree, on the type of the anode material. The best results were obtained in a chloride-rich medium under acidic pH using a Ti/Pt–Ir anode (Table 2.4). However, despite the excellent destruction of the synthetic dyes, the indirect electrooxidation resulted in the production of many chloroorganic compounds, which was a major disadvantage of this method (Naumczyk et al. 1996).
More recently, Panizza (Panizza and Cerisola 2003a) investigated the oxidation of 2-naphthol with in situ electrogenerated active chlorine using an undivided flow cell with a Ti–Ru–Sn ternary oxide anode and a stainless steel cathode. The degradation rate increased with the NaCl concentration and pH, but it was almost independent of the current density. A higher EOI value, about 0.302, was achieved with a concentration of NaCl of 7. 5 g dm− 3 at pH 12 (Fig. 2.6). A small quantity of organochlorinated compounds was detected in the solution during oxidation, but these compounds were then mineralized to CO2 or oxidized to volatile chlorinated compounds (i.e., chloroform).

3D representation of the combined effect of NaCl concentration and pH on the EOI values during the electrolyses of 2-naphthol on a Ti–Ru–Sn ternary oxide anode. Initial 2-naphthol concentration = 5mM; i = 75 mA cm− 2; flow rate = 180 l h− 1 (Panizza and Cerisola 2004b)
On the contrary, Bonfatti et al. (2000a, b) demonstrated that by choosing optimal experimental conditions, glucose can be incinerated by chlorine-mediated electrolysis without the formation of any chlorinated organics. In the presence of 2–5 g dm− 3 of NaCl, the formation of active chlorine makes the mineralization process substantially insensitive to the nature of the electrode surface, its rate being the same at Ti∕PbO2, Ti∕SnO2, and Ti/Pt electrodes. In particular, the removal of glucose was found to be faster at a chloride concentration of 5 g dm− 3, a lower temperature, and a higher current density.
Chlorine-mediated electrolysis has also been used efficiently for the treatment of real wastewater such as landfill leachate (Chiang et al. 1995; Vlyssides et al. 2003), textile effluents (Lin and Chen 1997; Vlyssides et al. 2000; Yang et al. 2000; Iniesta et al. 2002), olive oil wastewater (Israilides et al. 1997; Panizza and Cerisola 2006c), industrial effluent containing aromatic sulfonated acids (Panizza et al. 2000), and tannery wastewaters (Vlyssides and Israilides 1997; Szpyrkowicz et al. 2001; Panizza and Cerisola 2004a).
2.4.4 Tin Dioxide
During the last 10 years, many papers have demonstrated that conductive Sb-doped SnO2 anodes, which have an onset potential for O2 evolution of about 1.9 V vs. SHE, are highly effective for the electrooxidation of organics in wastewater treatment (Pulgarin et al. 1994; Cossu et al. 1998; Bock and MacDougall 1999; Grimm et al. 2000).
Kotz et al. (Kotz et al. 1991; Stucki et al. 1991) reported that anodic oxidation of a wide range of organic compounds at SnO2 was very much unselective, which means that the electrode can be applied to a multitude of different wastewater compositions, and proceeded with an average efficiency that was five times higher than with Pt anodes (Table 2.5).
The electrochemical oxidation of phenol on doped SnO2 and platinum anodes was studied by Comninellis and Pulgarin (1993). The rate of phenol removal was almost the same for both anodes but the rate of TOC elimination and the ICE were much higher for the SnO2 electrode (Fig. 2.7).

Evolution of the ICE with a specific charge during the oxidation of phenol at (a) Pt anode; (b) SnO2 anode. Conditions: T = 70°C, pH = 2, Initial phenol concentration: 21 mM, i = 50 mA cm− 2 (Comninellis and Pulgarin 1993)
The intermediate products of the oxidation of phenol, mainly aliphatic acids, were oxidized rapidly by the Ti∕SnO2–Sb anode, but on the contrary, they were practically unaffected by the Pt anodes.
Similar results were also obtained by Li et al. (2005) for the oxidation of phenol at Ti∕SnO2−Sb, Ti∕RuO2, and Pt anodes. The best result for phenol oxidation and TOC removal was obtained with the Ti∕SnO2 − Sb anode, followed by the Pt anode and then by the Ti∕RuO2 anode.
Polcaro et al. (1999) compared the performance of Ti/PbO2 and Ti∕SnO2 anodes for the electrochemical oxidation of 2-chlorophenol, showing that, although both electrodes gave similar Faradic yields, the Ti∕SnO2 anode was preferred because of its greater ability to oxidize toxic compounds. Stopping the electrolysis when only a small amount of easily biodegradable oxalic acid was present in the effluent (i.e., COD = 300 mg dm− 3), the current efficiency was 50%.
However, despite the high removal ability of organic pollutants, the SnO2 anodes have the major drawback of a short service life that limits their practical applications (Lipp and Pletcher 1997). Correa-Lozano et al. (1997) investigated the stability of the Ti∕Sb2O5 − SnO2 and found that the service life of those produced by spray pyrolysis can be improved by using a high electrode loading (about 100 g m− 2) and a preparation temperature of 550°C, but even under these conditions their life remained less than 12 h at 100 mA cm− 2 in 1 M H2SO4 at T = 25°C.
Ways of improving these anodes are now being investigated in many laboratories and it has been demonstrated that the service life of Sb-doped SnO2 electrodes can be increased by the incorporation of new dopants such as platinum (Vicent et al. 1998); by the introduction of interlayers between the titanium and the active oxide (Zanta et al. 2003), such as IrO2; or by improvements to the preparation method (Lipp and Pletcher 1997). However, the EOI values for the incineration of glucose at composite Pt − SnO2 electrodes were even lower than those at pure Pt electrodes (Bonfatti et al. 1999), while the onset potential for oxygen evolution of Ti∕IrO2 ∕ SnO2 decreased significantly up to 1.5 V vs. NHE (Chen et al. 2002).
2.4.5 Lead Dioxide
Lead dioxide anodes have a long history of use as electrode materials for the oxidation of organics because of their good conductivity and large overpotential for oxygen evolution in acidic media, enabling the production of hydroxyl radicals during water discharge (Cossu et al. 1998; Saracco et al. 2000; Gherardini et al. 2001; Keech and Bunce 2003). The possible release of toxic ions, especially in basic solutions, is the main drawback of these electrodes.
Early papers (Smith-de-Sucre and Watkinson 1981; Kirket et al. 1985; Sharifian and Kirk 1986) studied the oxidation of phenol and aniline using a packed-bed reactor of PbO2 pellets with a recirculating anolyte. The phenol and aniline in the solution were oxidized readily, but further oxidation of intermediates to carbon dioxide was more difficult. The extent of organic and TOC removal increased with the applied current density. Bonfatti et al. (1999), comparing the oxidation of glucose on different electrode materials such as Ti∕PbO2, Ti/Pt, and Ti∕Pt − SnO2, showed that the incineration of glucose and its oxidation intermediates (i.e., gluconic and glucaric acid) took place at a reasonable rate only at Ti∕PbO2 electrodes, and the highest Faradic yield was found at 300 A m− 2.
The electrochemical oxidation of phenol was thoroughly investigated under different experimental conditions Belhadj and Savall (Tahar and Savall 1998, 1999a, b) in a two-compartment cell. Phenol and its intermediates (benzoquinone, maleic, and fumaric acids) were completely eliminated at a pure Ta∕PbO2 anode through the intermediation of hydroxyl radicals adsorbed at the active site of the electrode (Fig. 2.8). The consumption rate of the phenol was mass-transport limited, and was favored by a high-temperature and low-current density. The mean Faradic yield reached 70% at the beginning of the electrolysis at T = 70°C for an anodic current density of 100 mA cm− 2. These authors also studied the effect of the type of substrate (Pb, Ti∕IrO2–Ta2O5, and Ta) or electrode formulation on the stability and efficiency of PbO2 deposits for phenol degradation. The efficiency of the electrodes for complete TOC removal decreased according to the type of substrate used, as follows: Ta > Ti ∕ (IrO2–Ta2O5) > Pb, andTa∕PbO2 was also more stable. Regarding the composition of the electrode, the pure PbO2 anode was more efficient for phenol degradation than Bi2O5–PbO2 or perchlorate-doped PbO2.

Variation of the concentration of (1) phenol, (2) 1,4-benzoquinone, and TOC during electrochemical oxidation of phenol on a Ta ∕ PbO2 anode at different temperatures. Initial phenol concentration 0. 021 mol dm− 3; i = 100 mA cm− 2 (Tahar and Savall 1998)
On the contrary, other studies by Johnson’s group (Chang and Johnson 1990; Kawagoe and Johnson 1994; Feng et al. 1995) showed that the electrocatalytic properties and fouling resistance of PbO2 film electrodes was enhanced by the incorporation of metallic species, such as Fe(III) or Bi(V), in the films. For example, the current efficiency for the electrochemical incineration of 10 mM of benzoquinone at 10 mA cm− 2 increased from 7.4 to 23.5% when the PbO2 anode was replaced with the Fe–PbO2 one. They also observed that a significant increase in the efficiency of the anodic degradation of benzoquinone was observed as a result of raising the temperature from 20 to 80°C.
The electrochemical degradation of phenol with several metal-oxide electrodes (i.e., PbO2, Ti∕Sb–Sn–RuO2, Ti∕Sb–Sn–RuO2–Gd, and Ti∕RuO2) and Pt anodes was carried out by Feng (Feng and Li 2003) and different TOC removal and phenol degradation rates were observed for different anodes (Fig. 2.9). Phenol was completely oxidized with all the electrodes, more rapidly at PbO2 and Pt anodes, however, complete removal of TOC only took place on PbO2-coated anodes.

Electrochemical degradation of 100-ppm phenol as a function of charge passed for different electrode materials, i = 10 mA cm− 2, (a) Phenol removal; (b) TOC removal; (filled square) Ti ∕ RuO2; (filled triangle) Ti ∕ Sb-Sn-RuO2; (filled circle) Ti ∕ Sb–Sn–RuO2–Gd; (open square) Ti ∕ PbO2; and (open circle) Pt. Reprinted from Feng and Li (2003), Copyright (2003), with permission from Elsevier
Martinez-Huitle et al. (2004a) studied the electrochemical oxidation of oxalic acid at Ti∕PbO2, highly BDD, Pt, Au, and Ti∕IrO2–Ta2O5 electrodes showing that oxalic acid was oxidized to CO2 with different results at several substrates. Higher current efficiencies were obtained at Ti∕PbO2 because the interaction of the oxalic acid with the PbO2 surface was particularly strong, and its anodic oxidation was only limited by mass transfer at higher current densities and lower substrate concentrations.
2.4.6 Boron-Doped Diamond
High-quality BDD electrodes possess several technologically important characteristics including an inert surface with low adsorption properties, remarkable corrosion stability even in strong acidic media, and extremely high oxygen evolution overpotential. Thanks to these properties. During electrolysis in the region of water discharge, a BDD anode produces a large quantity of the OH⋅ that is weakly adsorbed on its surface, and consequently it has high reactivity for organic oxidation, providing the possibility of efficient application to water treatment (Comninellis et al. 2005; Panizza and Cerisola 2005).
So far, many papers have demonstrated that BDD anodes allow complete mineralization of several types of organic compounds, such as carboxylic acids (Gandini et al. 2000; Canizares et al. 2003a), polyacrilates (Bellagamba et al. 2002), herbicides (Brillas et al. 2004), cyanides (Perret et al. 1999), wastewater from automotive industry (Troster et al. 2002), surfactants (Lissens et al. 2003; Panizza et al. 2005), benzoic acid (Montilla et al. 2002), industrial wastewaters (Kraft et al. 2003; Panizza and Cerisola 2006a), naphthol (Panizza et al. 2001a), phenol (Perret et al. 1999; Iniesta et al. 2001a; Canizares et al. 2002; Morao et al. 2004), chlorophenols (Gherardini et al. 2001; Rodrigo et al. 2001; Canizares et al. 2004a), nitrophenols (Canizares et al. 2004b), synthetic dyes (Hattori et al. 2003; Fernandes et al. 2004), and other pollutants, with high current efficiency.
It has been shown that the oxidation is controlled by the diffusion of the pollutants toward the electrode surface, where the hydroxyl radicals are produced, and the current efficiency is favored by high mass-transport coefficient, high organic concentration, and low current density. Performing electrolysis under optimum conditions, without diffusion limitation, the current efficiency approaches 100%.
Comninellis and coworkers (Foti et al. 1999; Gandini et al. 1999, 2000; Perret et al. 1999; Gherardini et al. 2001; Rodrigo et al. 2001) thoroughly investigated the behavior of Si/BDD anodes in an acidic solution for the oxidation of a wide range of pollutants, and they observed that complete mineralization was obtained with all the experimental conditions studied, and the current efficiency was influenced by the initial concentration and applied current. In particular, for high organic concentrations and low current densities, the COD decreased linearly and the ICE remained about 100%, indicating a kinetically controlled process, while for low organic concentrations or high current densities, the COD decreased exponentially and the ICE began to fall due to the mass-transport limitation and the side reactions of oxygen evolution. For example, Fig. 2.10 shows the trend of the COD and ICE during the electrochemical oxidation of different concentrations of 4-chlorophenol. In order to describe these results, the authors developed a comprehensive kinetic model that made it possible to predict the trend of the COD and current efficiency for the electrochemical combustion of the organic with BDD electrodes and estimate the energy consumption during the process (Panizza et al. 2001b).

Evolution of COD and ICE (inset) with the specific electrical charge passed during the oxidation of 4-chlorophenol (4-CP) on a boron-doped diamond anode. Electrolyte: sulfuric acid 1 M; T = 30°C; i = 30 mA cm− 2; initial 4-CP concentration: (open square) 3.9 mM; (cross) 7.8 mM; (filled circle) 15.6 mM (Rodrigo et al. 2001)
The oxidation of different phenolic compounds (phenol, chlorophenols, and nitrophenols) and carboxylic acids on BDD anodes was also studied by the group of Canizares et al. (2002, 2003b, 2004a, c). They reported that the organic compounds were completely mineralized regardless of the characteristics of the wastewater (initial concentration, pH, and supporting media) and operating conditions (temperature and current density) used. In particular, high concentration and low current density values increased the efficiency of the electrochemical oxidation of different chlorophenols (Table 2.6).
They also found that, depending on the electrolyte composition, the organics were oxidized on both the electrode surface by reaction with hydroxyl radicals and in the bulk of the solution by inorganic oxidants electrogenerated on the BDD anodes, such as peroxodisulfuric acid from sulfuric acid oxidation:
Polcaro et al. (2003, 2005) verified that during the oxidation of organic compounds, such as phenol, diuron, 3,4-dichloroaniline, and triazines, the crucial point to obtain high Faradic yields is the rate of mass transfer of the reactant toward the electrode surface (Fig. 2.11). Thus, they developed an impinging cell that enabled them to obtain high mass-transfer coefficients (e.g., 10− 4 m s− 1). With this cell, at a current density of 150 A m− 2, they achieved a Faradic yield of 100%, up to the almost complete disappearance of the organic load.

Trend of phenol concentration (solid symbols) and COD (hollow symbols) as function of electrolysis time during the oxidation of phenol at BDD: (open circle, filled circle) i = 305 A m− 2, Re = 13,250; (open triangle, filled triangle) i = 153 A m− 2, Re = 3,500; (open square, filled square) i = 153 A m− 2, Re = 13,250; and (open diamond, filled diamond) i = 305 A m− 2, Re = 3,500 (Polcaro et al. 2003)
Some investigations have also tried to compare the behavior of BDD with other electrodes, such as SnO2, PbO2, IrO2, for the oxidation of organic pollutants. Chen et al. (2003) reported that the current efficiency obtained with Ti/BDD in oxidizing acetic acid, maleic acid, phenol, and dyes was 1.6–4.3-fold higher than that obtained with the typical Ti∕Sb2O5–SnO2 electrode. Other papers have demonstrated that Si/BDD electrodes are able to achieve faster oxidation and better incineration efficiency than Ti∕PbO2 in the treatment of naphthol (Panizza and Cerisola 2004b) (Fig. 2.12), 4-chlorophenol (Gherardini et al. 2001), and chloranilic acid (Martinez-Huitle et al. 2004b).

Comparison of the trend of COD and ICE (inset) during the oxidation of 2-naphthol at the (open circle) Ti/PbO2 anode, i = 25 mA cm− 2; flow rate 180 dm3 h− 1; the (open square) BDD anode; i = 25 mA cm− 2; flow rate 180 dm3 h− 1; and the (open triangle) Ti–Ru–Sn ternary oxide anode, NaCl = 7. 5 g dm− 3; i = 50 mA cm− 2; flow rate 180 dm3 h− 1. (Panizza and Cerisola 2004b)
Diamond electrodes have also been studied with the aim of developing highly efficient electrochemical processes for water disinfection for domestic water treatment purposes or industrial water cooling systems. The good electrochemical stability and high overpotential for water electrolysis allows the production of a mixture of very strong oxidants under several disinfection mechanisms, without using any chemicals. In fact, besides hydroxyl radicals and hydrogen peroxide, directly produced by the water, the presence of chlorides, sulfates, and carbonates induces a very efficient generation of free chlorine, peroxodisulfate, and percarbonates, respectively (Rychen et al. 2003).
However, despite the numerous advantages of diamond electrodes, their high cost and the difficulties in finding an appropriate substrate on which to deposit the thin diamond layer are their major drawbacks. In fact, stable diamond films can really only be deposited on Silicon, Tantalum, Niobium, and Tungsten, but these materials are not suitable for large-scale use. In fact, a silicon substrate is very brittle and its conductivity is poor and Tantalum, Niobium, and Tungsten are too expensive. Titanium possesses good electrical conductivity, sufficient mechanical strength, electrochemical inertness, and is inexpensive. However, the stability of the diamond layer deposited on the Titanium substrate is still not satisfactory, because cracks may appear and may cause the detachment of the diamond film during long-term electrolysis.
2.5 Conclusions
This paper has presented and briefly discussed the performance of different electrode materials for the electrochemical oxidation of organic pollutants for wastewater treatment. Literature results have demonstrated that anodes with low oxygen evolution overpotential, such as graphite, IrO2, RuO2, or Pt only permit the primary oxidation of organics (i.e., conversion), but not the complete mineralization, due to the accumulation of oxidation intermediates, mainly aliphatic acids, which are quite stable against further attack at these electrodes.
The complete mineralization of the organics to CO2 and good Faradic efficiency can be obtained using high oxygen overpotential anodes, such as SnO2, PbO2, and BDD, because these electrodes involve the production of oxygen evolution intermediates, mainly hydroxyl radicals, that nonselectively oxidize the organic pollutants and their intermediates.
Despite their notable ability to remove organics, doped-SnO2 anodes have the major drawback of a short service life that limits their practical applications and, consequently, ways to improve the service life of these anodes is now under investigation.
Even the application of Ti∕PbO2 anodes to wastewater treatment may be limited by the possible release of toxic lead ions, due to their dissolution under specific anodic polarization and solution composition.
On the contrary, conducting diamonds offer significant advantages over other electrode materials in terms of current efficiency and stability for a variety of electrochemical processes. However, further improvements, such as finding an appropriate substrate on which to deposit the thin diamond layer and the reduction of production costs, are required before their wide industrial application.
References
Alvarez-Gallegos, A. and Pletcher, D. (1998) Removal of low level organics via hydrogen peroxide formed in a reticulated vitreous carbon cathode cell, Part 1. The electrosynthesis of hydrogen peroxide in aqueous acidic solutions. Electrochim. Acta 44, 853–861.
Alvarez-Gallegos, A. and Pletcher, D. (1999) The removal of low level organics via hydrogen peroxide formed in a reticulated vitreous carbon cathode cell, Part 2. The removal of phenols and related compounds from aqueous solutions. Electrochim. Acta 44, 2483–2492.
Awad, Y. M. and Abuzaid, N. S. (1997) Electrochemical treatment of phenolic wastewater: Efficiency, design considerations and economic evaluation. J. Environ. Sci. Health A 32, 1393–1414.
Awad, Y. M. and Abuzaid, N. S. (1999) Electrochemical oxidation of phenol using graphite anodes. Sep. Sci. Technol. 34, 699–708.
Awad, Y. M. and Abuzaid, N. S. (2000) Influence of residence time on the anodic oxidation of phenol. Sep. Purif. Technol. 18, 227–236.
Beer, H. B. (1966) US Patent Appl. 549 194.
Bellagamba, R., Michaud, P. A., Comninellis, C. and Vatistas, N. (2002) Electro-combustion of polyacrylates with boron-doped diamond anodes. Electrochem. Commun. 4, 171–176.
Bock, C. and MacDougall, B. (1999) Anodic oxidation of p-benzoquinone and maleic acid. J. Electrochem. Soc. 146, 2925–2932.
Bock, C. and MacDougall, B. (2000) Influence of metal oxide properties on the oxidation of organics. J. Electroanal. Chem. 491, 48–54.
Bonfatti, F., Ferro, S., Lavezzo, F., Malacarne, M., Lodi, G. and De Battisti, A. (1999) Electro-chemical incineration of glucose as a model organic substrate. I. Role of the electrode material. J. Electrochem. Soc. 146, 2175–2179.
Bonfatti, F., De Battisti, A., Ferro, S., Lodi, G. and Osti, S. (2000a) Anodic mineralization of organic substrates in chloride-containing aqueous media. Electrochim. Acta 46, 305–314.
Bonfatti, F., Ferro, S., Lavezzo, F., Malacarne, M., Lodi, G. and De Battisti, A. (2000b) Electro-chemical incineration of glucose as a model organic substrate. II. Role of active chlorine mediation. J. Electrochem. Soc. 147, 592–596.
Boudenne, J. L. and Cerclier, O. (1999) Performance of carbon black-slurry electrodes for 4-chlorophenol oxidation. Water Res. 33, 494–504.
Boudenne, J. L., Cerclier, O., Galea, J. and Vlist, E. V. D. (1996) Electrochemical oxidation of aqueous phenol at a carbon black slurry electrode. Appl. Catal. A: General 143, 185–202.
Boye, B., Dieng, M. M. and Brillas, E. (2002) Degradation of herbicide 4-chlorophenoxyacetic acid by advanced electrochemical oxidation methods. Environ. Sci. Technol. 36, 3030–3035.
Brillas, E., Bastida, R. M., Llosa, E. and Casado, J. (1995) Electrochemical destruction of aniline and chloroaniline for wastewater treatment using a carbon PTFE O2-fed cathode. J. Electrochem. Soc. 142, 1733–1741.
Brillas, E., Mur, E. and Casado, J. (1996) Iron(II) catalysis of the mineralization of aniline using a carbon-PTFE O2-fed cathode. J. Electrochem. Soc. 143, L49–L53.
Brillas, E., Boye, B. and Dieng, M. M. (2003) Peroxi-coagulation and photoperoxi-coagulation treatments of the herbicide 4-chlorophenoxyacetic acid in aqueous medium using an oxygen-diffusion cathode. J. Electrochem. Soc. 150, E148–E154.
Brillas, E., Boye, B., Sires, I., Garrido, J. A., Rodriguez, R. M., Arias, C., Cabot, P. L. and Comninellis, C. (2004) Electrochemical destruction of chlorophenoxy herbicides by anodic oxidation and electro-Fenton using a boron-doped diamond electrode. Electrochim. Acta 49, 4487–4496.
Canizares, P., Dominguez, J. A., Rodrigo, M. A., Villasenor, J. and Rodriguez, J. (1999) Effect of the current intensity in the electrochemical oxidation of aqueous phenol wastes at an activated carbon and steel anode. Ind. Eng. Chem. Res. 38, 3779–3785.
Canizares, P., Diaz, M., Dominguez, J. A., Garcia-Gomez, J. and Rodrigo, M. A. (2002) Electrochemical oxidation of aqueous phenol wastes on synthetic diamond thin-film electrodes. Ind. Eng. Chem. Res. 41, 4187–4194.
Canizares, P., Garcia-Gomez, J., Lobato, J. and Rodrigo, M. A. (2003a) Electrochemical oxidation of aqueous carboxylic acid wastes using diamond thin-film electrodes. Ind. Eng. Chem. Res. 42, 956–962.
Canizares, P., Garcia-Gomez, J., Saez, C. and Rodrigo, M. A. (2003b) Electrochemical oxida-tion of several chlorophenols on diamond electrodes: Part I. Reaction mechanism. J. Appl. Electrochem. 33, 917–927.
Canizares, P., Garcia-Gomez, J., Saez, C. and Rodrigo, M. A. (2004a) Electrochemical oxida-tion of several chlorophenols on diamond electrodes: Part II. Influence of waste character-istics and operating conditions. J. Appl. Electrochem. 34, 87–94.
Canizares, P., Saez, C., Lobato, J. and Rodrigo, M. A. (2004b) Electrochemical treatment of 2,4-dinitrophenol aqueous wastes using boron-doped diamond anodes. Electrochim. Acta 49, 4641–4650.
Canizares, P., Saez, C., Lobato, J. and Rodrigo, M. A. (2004c) Electrochemical treatment of 4-nitrophenol-containing aqueous wastes using boron-doped diamond anodes. Ind. Eng. Chem. Res. 43, 1944–1951.
Chang, H. and Johnson, D. C. (1990) Electrocatalysis of anodic oxygen-transfer reactions. J. Electrochem. Soc. 137, 2452–2457.
Chen, G. (2004) Electrochemical technologies in wastewater treatment. Sep. Purif. Technol. 38, 11–41.
Chen, G., Chen, X. and Yue, P. L. (2002) Electrochemical behavior of novel Ti ∕ IrOx − Sb2O5 − SnO2 anodes. J. Phys. Chem. B 106, 4364–4369.
Chen, X., Chen, G., Gao, F. and Yue, P. L. (2003) High-performance Ti/BDD electrodes for pollutant oxidation. Environ. Sci. Technol. 21, 5021–5026.
Chiang, L. C., Chang, J. E. and Wen, T. C. (1995) Indirect oxidation effect in electrochemical oxidation treatment of landfill leachate. Water Res. 29, 671–678.
Comninellis, C. (1994) Electrocatalysis in the electrochemical conversion/combustion of organic pollutants for waste water treatment. Electrochim. Acta 39, 1857–1862.
Comninellis, C. and De Battisti, A. (1996) Electrocatalysis in anodic oxidation of organics with simultaneous oxygen evolution. J. Chim. Phys. 93, 673–679.
Comninellis, C. and Nerini, A. (1995) Anodic oxidation of phenol in the presence of NaCl for wastewater treatment. J. Appl. Electrochem. 25, 23–28.
Comninellis, C. and Pulgarin, C. (1991) Anodic oxidation of phenol for wastewater treatment. J. Appl. Electrochem. 21, 703–708.
Comninellis, C. and Pulgarin, C. (1993) Electrochemical oxidation of phenol for wastewater treatment using SnO2 anodes. J. Appl. Electrochem. 23, 108–112.
Comninellis, C., Duo, I., Michaud, P. A., Marselli, B. and Park, S. M. (2005) Application of syhthetic boron-doped diamond electrodes in electrooxidation processes. In: A. Fujishima, Y. Einaga, T. N. Rao and D. A. Tryk (Eds.), Diamond Electrochemistry. Elsevier, Amsterdam, pp. 449–476.
Correa-Lozano, B., Comninellis, C. and De Battisti, A. (1997) Service life of Ti ∕ SnO2 − Sb2O5 anodes. J. Appl. Electrochem. 27, 970–974.
Cossu, R., Polcaro, A. M., Lavagnolo, M. C., Mascia, M., Palmas, S. and Renoldi, F. (1998) Electrochemical treatment of landfill leachate: Oxidation at Ti ∕ PbO2 and Ti ∕ SnO2 anodes. Ind. Eng. Chem. Res. 32, 3570–3573.
Dhooge, P. M. and Park, S. M. (1983) Electrochemistry of coal slurries - 2. Studies on various experimental parameters affecting oxidation of coal slurries. J. Electrochem. Soc. 130, 1029–1036.
Do, J. S. and Chen, C. P. (1993) In situ oxidative degradation of formaldehyde with electro-generated hydrogen peroxide. J. Electrochem. Soc. 140, 1632–1637.
Do, J. S. and Chen, C. P. (1994a) In situ oxidative degradation of formaldehyde with hydrogen peroxide electrogenerated on the modified graphite. J. Appl. Electrochem. 24, 936–942.
Do, J. S. and Chen, C. P. (1994b) Kinetics of in situ oxidative degradation of formaldehyde with electrogenerated hydrogen peroxide. Ind. Eng. Chem. Res. 33, 387–394.
Do, J. S. and Yeh, W. C. (1995) In situ degradation of formaldehyde with electrogenerated hypochlorite ion. J. Appl. Electrochem. 25, 483–489.
Do, J. S., Yeh, W. C. and Chao, I. Y. (1997) Kinetic of the oxidative degradation of formaldehyde with electrogen hypochlorite. Ind. Eng. Chem. Res. 36, 349–356.
Farmer, J. C., Wang, F. T., Hawley-Fedder, R. A., Lewis, P. R., Summers, L. J. and Foiles, L. (1992) Electrochemical treatment of mixed and hazardous wastes: Oxidation of ethylene glycole and benzene by silver(II). J. Electrochem. Soc. 139, 654–662.
Feng, Y. J. and Li, X. Y. (2003) Electro-catalytic oxidation of phenol on several metal-oxide electrodes in aqueous solution. Water Res. 37, 2399–2407.
Feng, J., Johnson, D. C., Lowery, S. N. and Carey, J. (1994) Electrocatalysis of anodic oxygen-transfer reactions evolution of ozone. J. Electrochem. Soc. 141, 2708–2711.
Feng, J., Houk, L. L., Johnson, D. C., Lowery, S. N. and Carey, J. J. (1995) Electrocatalysis of anodic oxygen-transfer reactions: The electrochemical incineration of benzoquinone. J. Electrochem. Soc. 142, 3626–3632.
Fernandes, A., Morao, A., Magrinho, M., Lopes, A. and Goncalves, I. (2004) Electrochemical degradation of C. I. Acid Orange 7. Dyes Pigm. 61, 287–296.
Foller, P. C. and Tobias, C. W. (1982) The anodic evolution of ozone. J. Electrochem. Soc. 129, 506–515.
Foti, G., Gandini, D. and Comninellis, C. (1997) Anodic oxidation of organics on thermally prepared oxide electrodes. Curr. Top. Electrochem. 5, 71–91.
Foti, G., Gandini, D., Comninellis, C., Perret, A. and Haenni, W. (1999) Oxidation of organics by intermediates of water discharge on IrO2 and synthetic diamond anodes. Electrochem. Solid State Lett. 2, 228–230.
Gandini, D., Comninellis, C., Perret, A. and Haenni, W. (1999) Anodic oxidation of organics on synthetic diamond thin-film electrodes. ICHEME Symp. Series 145, 181–190.
Gandini, D., Mahe, E., Michaud, P. A., Haenni, W., Perret, A. and Comninellis, C. (2000) Oxidation of carboxylic acids at boron-doped diamond electrodes for wastewater treatment. J. Appl. Electrochem. 30, 1345–1350.
Gattrell, M. and Kirk, D. (1990) The electrochemical oxidation of aqueous phenol at a glassy carbon electrode. Can. J. Chem. Eng. 68, 997–1003.
Gattrell, M. and Kirk, D. (1993) A study of the oxidation of phenol at platinum and preoxidized platinum surfaces. J. Electrochem. Soc. 140, 1534–1540.
Gherardini, L., Michaud, P. A., Panizza, M., Comninellis, C. and Vatistas, N. (2001) Electro-chemical oxidation of 4-chlorophenol for wastewater treatment. Definition of normalized current efficiency. J. Electrochem. Soc. 148, D78.
Grimm, J. H., Bessarabov, D. G., Simon, U. and Sanderson, R. D. (2000) Characterization of doped tin dioxide anodes prepared by a sol-gel technique and their application in an SPE-reactor. J. Appl. Electrochem. 30, 293–302.
Guivarch, E., Oturan, N. and Oturan, M. A. (2003) Removal of organophosphorus pesticides from water by electrogenerated Fenton’s reagent. Environ. Chem. Lett. 1, 165–168.
Hattori, S., Doi, M., Takahashi, E., Kurosu, T., Nara, M., Nakamatsu, S., Nishiki, Y., Furuta, T. and Iida, M. (2003) Electrolytic decomposition of amaranth dyestuff using diamond electrodes. J. Appl. Electrochem. 33, 85–91.
Houk, L. L., Johnson, S. K., Feng, J., Houk, R. S. and Johnson, D. C. (1998) Electrochemical incineration of benzoquinone in aqueous media using a quaternary metal oxide electrode in the absence of a soluble supporting electrolyte. J. Appl. Electrochem. 28, 1167–1177.
Iniesta, J., Michaud, P. A., Panizza, M., Cerisola, G., Aldaz, A. and Comninellis, C. (2001a) Electrochemical oxidation of phenol at boron-doped diamond electrode. Electrochim. Acta 46, 3573–3578.
Iniesta, J., Michaud, P. A., Panizza, M. and Comninellis, C. (2001b) Electrochemical oxidation of 3-methylpyridine at a boron-doped diamond electrode: Application to electroorganic synthesis and wastewater treatment. Electrochem. Commun. 3, 346–351.
Iniesta, J., Exposito, E., Gonzalez-Garcia, J., Montiel, V. and Aldaz, A. (2002) Electrochemical treatment of industrial wastewater containing phenols. J. Electrochem. Soc. 149, D57-D62.
Israilides, C. J., Vlyssides, A. G., Mourafeti, V. N. and Karvouni, G. (1997) Olive oil waste-water treatment with the use of an electrolysis system. Bioresource Technol. 61, 163–170.
Johnson, S. K., Houk, L. L., Feng, J., Houk, R. S. and Johnson, D. C. (1999) Electrochemical incineration of 4-chlorophenol and the identification of products and intermediates by mass spectrometry. Environ. Sci. Technol. 33, 2638–2644.
Kawagoe, K. T. and Johnson, D. C. (1994) Electrocatalysis of anodic oxygen-transfer reactions. Oxidation of phenol and benzene at bismuth-doped lead dioxide electrodes in acidic solutions. J. Electrochem. Soc. 141, 3404–3409.
Keech, P. G. and Bunce, N. J. (2003) Electrochemical oxidation of simple indoles at a PbO2 anode. J. Appl. Electrochem. 33, 79–83.
Kirk, D., Sharifian, H. and Foulkes, F. R. (1985) Anodic oxidation of aniline for waste water treatment. J. Appl. Electrochem. 15, 285–292.
Kotz, R., Stucki, S. and Carcer, B. (1991) Electrochemical wastewater treatment using high overvoltage anodes. Part I: physical and electrochemical properties of SnO2 anodes. J. Appl. Electrochem. 21, 14–20.
Kraft, A., Stadelmann, M. and Blaschke, M. (2003) Anodic oxidation with doped diamond electrodes: A new advanced oxidation process. J. Hazard. Mater. 103, 247–261.
Lamy, C. (1984) Electrocatalytic oxidation of organic compounds on noble metals in aqueous solution. Electrochim. Acta 29, 1581–1588.
Lamy, C., Leger, J. M., Clavilier, J. and Parsons, R. (1983) Structural effects in electrocatalysis: A comparative study of the oxidation of CO, HCOOH and CH3OH on single crystal Pt electrodes. J. Electroanal. Chem. 150, 71–77.
Lanza, M. R. V. and Bertazzoli, R. (2002) Cyanide oxidation from wastewater in a flow electrochemical reactor. Ind. Eng. Chem. Res. 41, 22–26.
Leffrang, U., Ebert, K., Flory, K., Galla, U. and Schmeider, H. (1995) Organic waste destruction by indirect electrooxidation. Sep. Purif. Technol. 30, 1883–1899.
Li, X.-y., Cui, Y.-h., Feng, Y.-j., Xie, Z.-m. and Gu, J.-D. (2005) Reaction pathways and mechanisms of the electrochemical degradation of phenol on different electrodes. Water Res. 39, 1972–1981.
Lin, S. H. and Chen, M. L. (1997) Treatment of textile wastewater by chemical methods for reuse. Water Res. 31, 868–876.
Lipp, L. and Pletcher, D. (1997) Preparation and characterization of tin dioxide coated titanium electrodes. Electrochim. Acta 42, 1091–1099.
Lissens, G., Pieters, J., Verhaege, M., Pinoy, L. and Verstraete, W. (2003) Electrochemical degradation of surfactants by intermediates of water discharge at carbon-based electrodes. Electrochim. Acta 48, 1655–1663.
Malpass, G. R. P., Neves, R. S. and Motheo, A. J. (2006) A comparative study of commercial and laboratory-made Ti ∕ Ru0. 3Ti0. 7O2 DSA electrodes: “In situ” and “ex situ” surface characterisation and organic oxidation activity. Electrochim. Acta 52, 936–944.
Marselli, B., Garcia-Gomez, J., Michaud, P. A., Rodrigo, M. A. and Comninellis, C. (2003) Electrogeneration of hydroxyl radicals on boron-doped diamond electrodes. J. Electrochem. Soc. 150, D79–D83.
Martinez-Huitle, C. A., Ferro, S. and De Battisti, A. (2004a) Electrochemical incineration of oxalic acid: Role of electrode material. Electrochim. Acta 49, 4027–4034.
Martinez-Huitle, C. A., Quiroz, M. A., Comninellis, C., Ferro, S. and De Battisti, A. (2004b) Electrochemical incineration of chloranilic acid using Ti ∕ IrO2, Pb ∕ PbO2 and Si/BDD electrodes. Electrochim. Acta 50, 949–956.
Martinez-Huitle, C. A., Ferro, S. and De Battisti, A. (2005) Electrochemical incineration in the presence of halides. Electrochem. Solid State Lett. 8, 35–39.
Montilla, F., Michaud, P. A., Morallon, E., Vazquez, J. L. and Comninellis, C. (2002) Electro-chemical oxidation of benzoic acid at boron-doped diamond electrodes. Electrochim. Acta 47, 3509–3513.
Morao, A., Lopes, A., Amorim, M. T. P. d. and Goncalves, I. C. (2004) Degradation of mixtures of phenols using boron doped diamond electrodes for wastewater treatment. Electrochim. Acta 49, 1587–1595.
Naumczyk, J., Szpyrkowicz, L. and Zillio-Grandi, F. (1996) Electrochemical treatment of textile wastewater. Water Sci. Technol. 34, 17–24.
Nelson, N. (2002) Electrochemical destruction of organic hazardous wastes. Plainum Met. Rev. 46, 18–23.
Ogutveren, U. B., Toru, E. and Koparal, S. (1999) Removal of cyanide by anodic oxidation for wastewater treatment. Water Res. 33, 1851–1856.
Panizza, M. and Cerisola, G. (2001) Removal of organic pollutants from industrial wastewater by electrogenerated Fenton’s reagent. Water Res. 35, 3987–3992.
Panizza, M. and Cerisola, G. (2003a) Electrochemical oxidation of 2-naphthol with in situ electrogenerated active chlorine. Electrochim. Acta 48, 1515–1519.
Panizza, M. and Cerisola, G. (2003b) Influence of anode material on the electrochemical oxidation of 2-naphthol. Part 1. Cyclic voltammetry and potential step experiments. Electrochim. Acta 48, 3491–3497.
Panizza, M. and Cerisola, G. (2004a) Electrochemical oxidation as final treatment of synthetic tannery wastewater. Environ. Sci. Technol. 38, 5470–5475.
Panizza, M. and Cerisola, G. (2004b) Influence of anode material on the electrochemical oxidation of 2-naphthol: Part 2. Bulk electrolysis experiments. Electrochim. Acta 49, 3221–3226.
Panizza, M. and Cerisola, G. (2005) Application of diamond electrodes to electrochemical processes. Electrochim. Acta 51, 191–199.
Panizza, M. and Cerisola, G. (2006a) Electrochemical oxidation of aromatic sulphonated acids on a boron-doped diamond electrode. Int. J. Environ. Pollut. 27, 64–74.
Panizza, M. and Cerisola, G. (2006b) Electrochemical processes for the treatment of organic pollutants. In: D. V. Zinger (Eds.), Advances in Chemistry Research, Vol. 2. Nova Science, New York, NY, pp. 1–38.
Panizza, M. and Cerisola, G. (2006c) Olive mill wastewater treatment by anodic oxidation with parallel plate electrodes. Water Res. 40, 1179–1184.
Panizza, M., Bocca, C. and Cerisola, G. (2000) Electrochemical treatment of wastewater containing poliaromatic organic pollutants. Water Res. 34, 2601–2605.
Panizza, M., Michaud, P. A., Cerisola, G. and Comninellis, C. (2001a) Anodic oxidation of 2-naphthol at boron-doped diamond electrodes. J. Electroanal. Chem. 507, 206.
Panizza, M., Michaud, P. A., Cerisola, G. and Comninellis, C. (2001b) Electrochemical treatment of wastewater containing organic pollutants on boron-doped diamond electrodes. Prediction of specific energy consumption and required electrode area. Electrochem. Commun. 3, 336.
Panizza, M., Delucchi, M. and Cerisola, G. (2005) Electrochemical degradation of anionic surfactants. J. Appl. Electrochem. 35, 357–361.
Perret, A., Haenni, W., Skinner, N., Tang, X. M., Gandini, D., Comninellis, C., Correa, B. and Foti, G. (1999) Electrochemical behavior of synthetic diamond thin film electrodes. Diam. Relat. Mater. 8, 820–823.
Piya-areetham, P., Shenchunthichai, K. and Hunsom, M. (2006) Application of electrooxidation process for treating concentrated wastewater from distillery industry with a voluminous electrode. Water Res. 40, 2857–2864.
Pletcher, D. and Walsh, F. C. (1982) Industrial Electrochemistry. Chapman and Hall, London.
Polcaro, A. M. and Palmas, S. (1997) Electrochemical oxidation of chlorophenols. Ind. Eng. Chem. Res., 1791–1798.
Polcaro, A. M., Palmas, S., Renoldi, F. and Mascia, M. (1999) On the performance of Ti∕SnO2 and Ti∕PbO2 anodes in electrochemical degradation of 2-chlorophenol for wastewater treatment. J. Appl. Electrochem. 29, 147–151.
Polcaro, A. M., Palmas, S., Renoldi, F. and Mascia, M. (2000) Three-dimensional electrodes for the electrochemical combustion of organic pollutants. Electrochim. Acta 46, 389–394.
Polcaro, A. M., Vacca, A., Palmas, S. and Mascia, M. (2003) Electrochemical treatment of wastewater containing phenolic compounds: Oxidation at boron-doped diamond electrodes. J. Appl. Electrochem. 33, 885–892.
Polcaro, A. M., Vacca, A., Mascia, M. and Palmas, S. (2005) Oxidation at boron doped diamond electrodes: An effective method to mineralise triazines. Electrochim. Acta 50, 1841–1847.
Ponce-de-Leon, C. and Pletcher, D. (1995) Removal of formaldehyde from aqueous solutions via oxygen reduction using a reticulated vitreous carbon cathode cell. J. Appl. Electrochem. 25, 307–314.
Pulgarin, C., Adler, N., Peringer, P. and Comninellis, C. (1994) Electrochemical detoxification of a 1,4-benzoquinone solution in wastewater treatment. Water Res. 28, 887–893.
Rajeshwar, K. and Ibanez, J. G. (1997) Environmental Electrochemistry. Fundamentals and Applications in Pollution Abatement. Academic, London.
Rajeshwar, K., Ibanez, J. G. and Swain, G. M. (1994) Electrochemistry and environment. J. Appl. Electrochem. 24, 1077–1091.
Rodgers, J. D., Jedral, W. and Bunce, N. J. (1999) Electrochemical oxidation of chlorinated phenols. Environ. Sci. Technol. 33, 1453–1457.
Rodrigo, M. A., Michaud, P. A., Duo, I., Panizza, M., Cerisola, G. and Comninellis, C. (2001) Oxidation of 4-Chlorophenol at boron-doped diamond electrodes for wastewater treatment. J. Electrochem. Soc. 148, D60–D64.
Rychen, P., Pupunat, L., Haenni, W. and Santoli, E. (2003) Water treatment applications with BDD electrodes and the DiaCell concept. New Diam. Front. Carbon Technol. 13, 109–117.
Saracco, G., Solarino, L., Aigotti, R., Specchia, V. and Maja, M. (2000) Electrochemical oxidation of organic pollutants at low electrolyte concentrations. Electrochim. Acta 46, 373–380.
Sharifian, H. and Kirk, D. (1986) Electrochemical oxidation of phenol. J. Electrochem. Soc. 113, 921–924.
Simond, O., Schaller, V. and Comninellis, C. (1997) Theoretical model for the anodic oxidation of organics on metal oxide electrodes. Electrochim. Acta 42, 2009–2012.
Smith-de-Sucre, V. and Watkinson, A. P. (1981) Anodic oxidation of phenol for wastewater treatment. Can. J. Chem. Eng. 59, 52–59.
Socha, A., Chrzescijanska, E. and Kusmierek, E. (2005) Electrochemical and photoelectro-chemical treatment of 1-aminonaphthalene-3,6-disulphonic acid. Dyes Pigm. 67, 71–75.
Soriaga, M. P. and Hubbard, A. T. (1982) Determination of the orientation of adsorbed molecules at solid–liquid interfaces by thin-layer electrochemistry: Aromatic compounds at platinum electrodes. J. Am. Chem. Soc. 104, 2735–2742.
Stucki, S., Kotz, R., Carcer, B. and Suter, W. (1991) Electrochemical wastewater treatment using high overvoltage anodes. Part II: Anode performance and applications. J. Appl. Electrochem. 21, 99–104.
Szpyrkowicz, L., Naumczyk, J. and Zilio-Grandi, F. (1995) Electrochemical treatment of tannery wastewater using Ti/Pt and Ti/Pt/Ir electrodes. Water Res. 29, 517–524.
Szpyrkowicz, L., Juzzolino, C., Kaul, S. N., Daniele, S. and DeFaveri, M. (2000) Electro-chemical oxidation of dyeing baths bearing disperse dyes. Ind. Eng. Chem. Res. 39, 3241–3248.
Szpyrkowicz, L., Kelsall, G. H., Kaul, S. N. and DeFaveri, M. (2001) Performance of electro-chemical reactor for treatment of tannery wastewaters. Chem. Eng. Sci. 56, 1579–1586.
Tahar, N. B. and Savall, A. (1998) Mechanistic aspects of phenol electrochemical degradation by oxidation on a Ta/PbO2 anode. J. Electrochem. Soc. 145, 3427–3434.
Tahar, N. B. and Savall, A. (1999a) A comparison of different lead dioxide coated electrodes for the electrochemical destruction of phenol. J. New Mat. Electr. Sys. 2, 19–26.
Tahar, N. B. and Savall, A. (1999b) Electrochemical degradation of phenol in aqueous solution on bismuth doped lead dioxide: A comparison of the activities of various electrode formulations. J. Appl. Electrochem. 29, 277–283.
Tatapudi, P. and Fenton, J. M. (1993) Synthesis of ozone in a proton exchange membrane electrochemical reactor. J. Electrochem. Soc. 140, 3527–3530.
Troster, I., Fryda, M., Herrmann, D., Schafer, L., Haenni, W., Perret, A., Blaschke, M., Kraft, A. and Stadelmann, M. (2002) Electrochemical advanced oxidation process for water treatment using DiaChem electrodes. Diam. Relat. Mater. 11, 640–645.
Vicent, F., Morallon, E., Quijada, C., Vazquez, J. L., Aldaz, A. and Cases, F. (1998) Characterization and stability of doped SnO2 anodes. J. Appl. Electrochem. 28, 607–612.
Vlyssides, A. G. and Israilides, C. J. (1997) Detoxification of tannery waste liquors with an electrolysis system. Environ. Pollut. 97, 147–152.
Vlyssides, A. G., Papaioannou, D., Loizidoy, M., Karlis, P. K. and Zorpas, A. A. (2000) Testing an electrochemical method for treatment of textile dye wastewater. Waste Manage. 20, 569–574.
Vlyssides, A. G., Karlis, P. K. and Mahnken, G. (2003) Influence of various parameters on the electrochemical treatment of landfill leachates. J. Appl. Electrochem. 33, 155–159.
Yang, C. H., Lee, C. C. and Wen, T. C. (2000) Hypochlorite generation on Ru–Ti binary oxide for the treatment of dye wastewater. J. Appl. Electrochem. 30, 1043–1051.
Zanta, C. L. P. S., Andrade, A. R. d. and Boodts, J. F. C. (2000) Electrochemical behaviour of olefins: Oxidation at ruthenium-titanium dioxide and iridium-titanium dioxide coated electrodes. J. Appl. Electrochem., 467–474.
Zanta, C. L. P. S., Michaud, P. A., Comninellis, C., Andrade, A. R. D. and Boodts, J. F. C. (2003) Electrochemical oxidation of p-chlorophenol on SnO2-Sb2O5 based anodes for wastewater treatment. J. Appl. Electrochem. 33, 1211–1215.
Acknowledgments
The author wishes to express his sincere thanks to Prof. Giacomo Cerisola for his helpful discussions during the preparation of this article and to the journals and all the authors who gave permission for the reproduction of figures and tables.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Panizza, M. (2010). Importance of Electrode Material in the Electrochemical Treatment of Wastewater Containing Organic Pollutants. In: Comninellis, C., Chen, G. (eds) Electrochemistry for the Environment. Springer, New York, NY. https://doi.org/10.1007/978-0-387-68318-8_2
Download citation
DOI: https://doi.org/10.1007/978-0-387-68318-8_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-0-387-36922-8
Online ISBN: 978-0-387-68318-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)