
Abstract
The mucosal epithelium is in direct contact with symbiotic and pathogenic microorganisms. Therefore, the mucosal surface is the principal portal of entry for invading pathogens and immune cells accumulated in the intestine to prevent infections. In addition to these conventional immune system functions, it has become clear that immune cells during steady-state continuously integrate microbial and nutrient-derived signals from the environment to support organ homeostasis. A major role in both processes is played by a recently discovered group of lymphocytes referred to as innate lymphoid cells (ILCs) that are specifically enriched at mucosal surfaces but are rather rare in secondary lymphoid organs. In analogy to the dichotomy between CD8 and CD4 T cells, we propose to classify ILCs into interleukin-7 receptor α-negative cytotoxic ILCs and IL-7Rα+ helper-like ILCs. Dysregulated immune responses triggered by the various ILC subsets have been linked to inflammatory diseases such as inflammatory bowel disease, atopic dermatitis and airway hyperresponsiveness. Here, we will review recent progress in determining the transcriptional and developmental programs that control ILC fate decisions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Aryl Hydrocarbon Receptor
- Secondary Lymphoid Organ
- Aryl Hydrocarbon Receptor Ligand
- Promyelocytic Leukemia Zinc Finger
- Innate Lymphoid Cell
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Innate Lymphoid Cells
Like B and T cells, innate lymphoid cells (ILCs) belong to the lymphoid lineage of hematopoietic cells. However, in contrast to B and T cells, ILCs lack the expression of rearranged antigen receptors and, therefore, are considered to be part of the innate immune system. Their existence has not been uncovered until recently, because ILCs are mainly localized at mucosal surfaces and, with the exception of conventional NK (cNK) cells, ILCs are very rare in secondary lymphoid organs. Although ILCs lack antigen receptor rearrangement, the basic transcriptional programs, guiding their fate decisions, are to a large degree evolutionary conserved (Tanriver and Diefenbach 2014) and they have often been compared to the one of T helper (Th) cell subsets (Spits et al. 2013; Walker et al. 2013).
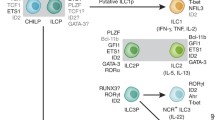
Based on three lines of evidence which are described in more detail in the following chapters, we have recently proposed to subdivide ILCs into cytotoxic or killer ILCs (i.e., cNK cells) and into helper-like ILCs such as ILC1s, ILC2s, and ILC3s (Fig. 1) (Klose et al. 2014), thereby providing a mirror image among innate lymphocytes of the dichotomy between cytotoxic CD8 versus helper CD4 T cells. (1) A recently described progenitor to ILCs gives rise to all helper-like ILCs but not to cNK cells (Klose et al. 2014; Constantinides et al. 2014). (2) Helper-like ILCs but not cNK cells express the interleukin 7 receptor α chain (IL-7Rα or CD127) and depend on the transcription factor GATA-3 for development (Klose et al. 2014; Yagi et al. 2014; Samson et al. 2003; Hoyler et al. 2012; Serafini et al. 2014). (3) The expression pattern of the related T-box transcription factors Eomes and T-bet among helper-like ILCs (T-bet in ILC1s) and cNK cells (co-expression of Eomes and T-bet) (Klose et al. 2014; Daussy et al. 2014; Gordon et al. 2012) parallels the pattern found in helper (T-bet+ Eomes−) versus cytotoxic T cells (T-bet+ Eomes+) (Intlekofer et al. 2005; Pearce et al. 2003). Conventional NK cells are the only known member of the group of cytotoxic ILCs . Their transcriptional regulation is described in detail in Chap. “Transcriptional control of NK cell development and function” of this book and is, therefore, not discussed here.

ILC differentiation
The classification of helper-like ILCs into three groups is based on their cytokine expression profile that is controlled by key transcription factors (Fig. 1) (Spits et al. 2013). ILC1s similar to Th1 cells express the transcription factor T-bet and secrete IFN-γ. In contrast, and in parallel to Th2 cells, ILC2s express high levels of GATA-3 which drives the secretion of cytokines such as IL-5, IL-9, and IL-13. ILC3s depend on the transcription factor RORγt. Two different ILC3 group members have been identified based on the expression of CCR6, both of which express RORγt (Klose et al. 2013; Sawa et al. 2010). CCR6+ ILC3s include the previously described lymphoid tissue inducer (LTi) cells (Mebius et al. 1997). LTi cells are essential for the generation of lymph nodes and Peyer’s patches and are localized in adult mice preferentially in lymphoid structures in the gut called cryptopatches (CP) and isolated lymphoid follicles (ILFs) (Eberl et al. 2004). CCR6+ ILC3s secrete cytokines such as IL-17A and IL-22 but do not express activating NK cell receptors such as NKp46, NKG2D, or NK1.1 (Takatori et al. 2009; Zenewicz et al. 2008; Cupedo et al. 2009). In sharp contrast, CCR6−/low ILC3s co-express T-bet in addition to RORγt. T-bet expression is required for the differentiation of CCR6−/low ILC3s into NK cell receptor-expressing (i.e., NKp46, NKG2D) ILC3s and for their production of IFN-γ (Klose et al. 2013; Rankin et al. 2013; Sciume et al. 2012a; Bernink et al. 2013). During these differentiation steps, T-bet expression increases and guides a transcriptional program which downregulates RORγt and IL-22 expression allowing for functional plasticity. This enigmatic conversion of ILC3s and the transcriptional regulation of other ILC fates is discussed in the following chapters (Vonarbourg et al. 2010; Bernink et al. 2013).
2 Id2 and GATA-3 Are Central Transcriptional Hubs for the Early Commitment to the Helper-Like ILC Fate
2.1 Regulation of Early Lymphocyte Differentiation by Id2
Hematopoietic cell development from a hematopoietic stem cell (HSC) to immune effector cells is characterized by progressive loss of proliferative and differentiation potential and a gain in effector functions. Irreversible commitment to a certain hematopoietic cell lineage is controlled by the action of one or more transcription factors which determine cell fate by direct or indirect regulation of target gene expression and by coordinating epigenetic changes.
A crucial branchpoint in hematopoiesis is the commitment to either the lymphoid or myeloid lineage represented by the differentiation of HSC into a common lymphoid precursor (CLP) (Kondo et al. 1997) or the common myeloid precursor (CMP) (Akashi et al. 2000), respectively. The CLP has the potential to differentiate into all known lymphoid lineages (i.e., T and B cells of the adaptive immune system and cytotoxic and helper-like ILCs) (Moro et al. 2010; Yang et al. 2011; Klose et al. 2014). The further separation between adaptive and innate lymphocytes is regulated by the E-family of transcription factors and their repressors, inhibitors of DNA binding (Id) (Murre 2005; Kee 2009; de Pooter and Kee 2010). E-proteins possess a helix-loop-helix (HLH) domain, allowing for dimerization of E proteins. A basic HLH DNA binding domain and two transcriptional regulation domains allow binding to chromatin and regulate transcription of target genes. E-proteins bind as dimers to the respective E-box sequence within their promoter or enhancer regions of genes. Id proteins lack the basic HLH domain, which allows DNA-binding but still possess a HLH domain for dimerization. Id proteins heterodimerize with E-proteins impairing their DNA binding and E-protein-mediated gene regulation (Murre 2005; Kee 2009). Expression of the E-protein family member E2A instructs specification of lymphoid progenitors to the B cell fate. E2A together with IL-7R signaling induces the expression of EBF-1 (early B cell factor-1), a pioneer transcription factor and a potent repressor of Id2 allowing for increased expression of E2A (Lin and Grosschedl 1995; Treiber et al. 2010). While repressing Id2, EBF-1 promotes expression of Pax5 widely considered as the “master regulator” of B cell fate (Nutt et al. 1999). Interestingly, deletion of EBF-1 in late pro-B cells reverted B cell commitment and reprogrammed EBF-1-deficient pro-B cells into T cells and helper-like ILCs (Nechanitzky et al. 2013). These data assign an important function to EBF-1 as a guardian of the B cell fate, which is at least in part mediated by the repression of Id2.
Another line of evidence supporting the notion that Id2 is a central hub for ILC fate came from the analysis of Id2-deficient mice. Id2 −/− mice could not generate ILCs including cNK cells and helper-like ILCs (i.e., ILC1s, ILC2s, and ILC3s), whereas the differentiation of B and T cells was largely unperturbed (Table 1) (Yokota et al. 1999; Moro et al. 2010; Hoyler et al. 2012). Therefore, it has been proposed that ILCs may develop from a common precursor which expresses Id2 and is developmentally dependent on Id2 (Sanos et al. 2011; Spits et al. 2013; Spits and Di Santo 2011). Indeed, investigation of Id2 expression in ILCs using Id2 reporter mice revealed that Id2 is indeed expressed in all ILCs and in lineage-specified ILC progenitors (or immature ILCs), whereas B cells and T cells have no or very low level Id2 expression, respectively (Hoyler et al. 2012; Boos et al. 2007; Carotta et al. 2011; Tachibana et al. 2011; Cherrier et al. 2012). Interestingly, cNK cells depicted lower expression of Id2 compared to ILC2s or ILC3s (Hoyler et al. 2012) and the refined NK cell precursor (rNKP) was largely Id2-negative (Klose et al. 2014) suggesting that Id2 expression is engaged after commitment to the cNK cell lineage. These data provide additional evidence for an early bifurcation between the cytotoxic arm of ILCs (i.e., cNK cells) and helper-like ILCs (IL-7Rα-expressing ILC1s, ILC2s, and ILC3s).
2.2 A Common Progenitor to All Helper-Like ILCs
As all ILCs express Id2 and are developmentally dependent on it, the existence of a common ILC precursor (variably called CILP or ILCP) has been proposed (Sanos et al. 2011; Spits et al. 2013; Spits and Di Santo 2011; Mortha and Diefenbach 2011). Recently, two groups independently reported two populations of Id2-expressing lymphoid-restricted progenitors in the bone marrow and in the fetal liver that were committed to the ILC fate (i.e., could not differentiate into B cells, T cells, or myeloid cells) but, as a population in vivo and on a single cell level in vitro, had the potential to differentiate into various helper-like ILC lineages (Klose et al. 2014; Constantinides et al. 2014). These populations carried markers of early hematopoietic development such as IL-7Rα (CD127), Kit (CD117), 2B4 (CD244) and CD27 but lacked expression of Flt3 and CD93, markers expressed by the CLP (Karsunky et al. 2003, 2008). In contrast to CLPs, this lymphoid progenitor population expressed Id2 and integrin α4β7, an integrin involved in the migration to mucosal surfaces (Holzmann et al. 1989; Wagner et al. 1996) and also found on ILC2s (Hoyler et al. 2012) and ILC3 progenitors (Possot et al. 2011; Cherrier et al. 2012). Lineage− Id2+ IL-7Rα+ α4β +7 Flt3− CD25− cells lacked expression of ILC lineage-specifying transcription factors such as RORγt, T-bet, Eomes and had low expression of GATA-3 arguing that these cells are not yet committed to a distinct ILC lineage (Klose et al. 2014; Constantinides et al. 2014).
Upon adoptive transfer of Lin− Id2+ IL-7Rα+ α4β +7 Flt3− CD25− cells into alymphoid mice, this population had no appreciable T, B, or myeloid cell potential but differentiated into functional ILC2s, ILC3s, and into a peculiar, intestinal mucosa and liver-resident Eomes− T-bet+ NKp46+ NK1.1+ ILC lineage, distinct of cNK cells (Klose et al. 2014). These data indicate that another branching point upstream of this progenitor may exist that is marked by a progenitor still having cNK cell potential, the common ILC progenitor (CILP) (Fig. 1). These data also provide evidence for partially distinct pathways of cNK cell and helper-like ILC development. Limiting dilution and single cell assays in vitro demonstrated that a single Lin− Id2+ IL-7Rα+ α4β +7 Flt3− CD25− cell had the potential to give rise to all three ILC lineages. Hence, Lin− Id2+ IL-7Rα+ α4β +7 Flt3− CD25− cells are now referred to as the common helper-like ILC progenitor (CHILP) (Klose et al. 2014; Constantinides et al. 2014). These data now create new opportunities to scrutinize the molecular signals required for the specification of the various helper-like ILC lineages from the CHILP .
Interestingly, the CHILP population contained a promyelocytic leukemia zinc finger protein (PLZF)− and a PLZF+ subset (Klose et al. 2014). PLZF (also known as ZBTB16) is a transcription factor of the BTB-POZ zinc finger family of transcription factors known to be important for NKT cell development and for the innate behavior of CD4 T cells (Kovalovsky et al. 2008; Savage et al. 2008). Lineage tracing experiments revealed that PLZF is not expressed by mature ILC populations but that it is transiently expressed during ILC development (Constantinides et al. 2014). Using a PLZF reporter allele, a bone marrow and a liver-resident PLZF+ lymphoid progenitor was identified which, in adoptive transfer experiments and in clonal differentiation assays in vitro, gave rise to non-NK ILC1-like cells, ILC2s and CCR6−/low ILC3s. Interestingly, PLZF+ progenitors had lost the potential to generate LTi cells (i.e., CCR6+ CD4+ ILC3s) (Constantinides et al. 2014), whereas PLZF− CHILPs still had LTi cell potential (Klose et al. 2014). Id2 is a regulator of PLZF expression and it is likely that PLZF− CHILPs are progenitors of PLZF+ CHILPs although this remains to be experimentally addressed (Verykokakis et al. 2013).
2.3 Development of All Helper-Like ILCs Depends on GATA-3
The six mammalian GATA-binding proteins (GATA-1-6) possess two zinc finger motifs, which have been proposed to originate from gene duplication events (Ho et al. 2009; Tanriver and Diefenbach 2014). GATA-1-3 are involved in the development of hematopoietic cells whereas GATA-4-6 regulate development of endodermal tissues. The two zinc fingers within the GATA proteins bind to a consensus sequence WGATAR (W: A/T, R: A/G) in the regulatory regions of multiple genes. In addition to the two zinc finger motifs, GATA-3 contains two transactivation domains and basic regions. The two zinc finger motifs allow GATA proteins to bind to two distant consensus sequences and mediate long-range looping of DNA (Hosoya et al. 2010; Chen et al. 2012). GATA-3 is already expressed early during hematopoietic development in hematopoietic stem cells and then further modulated, for example, during T cell development (Ku et al. 2012; Frelin et al. 2013; Zhong et al. 2005). This would suggest that GATA-3 may already play an important role early during commitment to different lymphoid lineages. Indeed, mice-derived from Rag-deficient blastocysts complemented with germline deleted GATA-3 stem cells, did not develop any T cells (Ting et al. 1996). It is now becoming apparent that GATA-3 plays important and stage-specifc roles for the development and function of helper-like ILCs . Early data showed that ILC2s (similar to Th2 cells) express high levels of GATA-3 and that deletion of GATA-3 in all Id2+ cells specifically affected the maintenance of ILC2s and ILC2Ps whereas ILC3s, which express intermediate GATA-3 levels, were not affected (Hoyler et al. 2012; Mjosberg et al. 2012; Yagi et al. 2014). Deletion of GATA-3 prior to the engagement of Id2 expression either by using Cre-mediated deletion in all hematopoietic cells (using Vav-Cre) or in fetal liver chimeras revealed that GATA-3 is essential for the development of all helper-like ILC lineages whereas differentiation of cNK cells was unaffected (Samson et al. 2003; Serafini et al. 2014; Yagi et al. 2014). Thus, similar to its role for helper versus cytotoxic T cell determination (Zhu et al. 2004), GATA-3 is an important transcriptional regulator required for the differentiation of all helper-like ILCs but not for the differentiation of killer ILCs (Table 1).
3 Group 1 ILCs Depend on T-bet for Development and Secrete IFN-γ
While group 2 and group 3 ILCs have been well-defined, group 1 ILCs have been not well characterized. It has been proposed that ILC1s may consist of T-bet-expressing “ex-RORγt+” ILC3s and cNK cells (Spits and Cupedo 2012; Spits and Di Santo 2011; Spits et al. 2013; Walker et al. 2013). For a detailed review on transcriptional regulation of cNK cell development the reader is kindly referred to Chap. “Transcriptional control of NK cell development and function” of this book. NKp46+ cells with a history of RORγt expression are of the ILC3 lineage and are further discussed in the section dealing with ILC3s below. However, recent data may have identified bona fide, non-NK cell ILC1 populations (Fuchs et al. 2013; Klose et al. 2014).
3.1 The Population of NKp46+ Cells Contains Various ILC Lineages
The identification of bona fide members of group 1 ILCs has proven difficult because the expression of T-bet and of NKp46 and other activating NK cell receptors (such as NKG2D and NK1.1) are not specific for one ILC lineage. NKp46 and NKG2D are expressed by cNK cells and ILC3 subsets (Cella et al. 2009; Cupedo et al. 2009; Sanos et al. 2009; Luci et al. 2009; Satoh-Takayama et al. 2008). In addition, T-bet is expressed by cNK cells (Townsend et al. 2004; Gordon et al. 2012) and by the CCR6−/low subset of RORγt+ ILC3s (Klose et al. 2013; Rankin et al. 2013; Sciume et al. 2012a; Powell et al. 2012). The fate of all these NKp46+ lymphocytes is, at least in part, mediated by the related T-box transcription factors T-bet and Eomes which control production of IFN-γ and expression of cytokine receptors (e.g., IL-2Rβ) or of the immunoreceptors NKp46, NKG2D or NK1.1 (Lazarevic and Glimcher 2011; Gordon et al. 2012). Given the promiscuous expression of T-bet in various ILC subsets and the broad expression of activating NK cell receptors, it was not obvious if a non-NK ILC1 population existed. For various reasons, it was somewhat unsatisfying that cNK cells would be bona fide ILC1s because they do not express IL-7Rα (as all other known ILC subsets) and, in striking contrast to Th1 cells, cNK cells develop and are maintained independently of T-bet (Townsend et al. 2004; Jenne et al. 2009; Gordon et al. 2012). Conventional NK cells co-express both T-box transcription factors and Eomes but not T-bet seemed to be required for cNK cell differentiation. In fact, Eomes-deficient mice have virtually no cNK cells (Fig. 1 and Table 1) (Gordon et al. 2012).
The analysis of small intestinal lamina propria-resident NKp46+ NK1.1+ cells, an operative definition of NK cells, using an Eomes reporter allele (Eomes Gfp/+ mice) in combination with a reporter allele allowing for lineage tracing of RORγt-expressing cells (RORγt-fate map (fm) mice) discerned three distinct populations of NKp46+ NK1.1+ cells, all of which expressed T-bet (Fig. 1) (Klose et al. 2014): (1) Eomes-positive RORγt-fm− cells represented cNK cells expressing class I MHC-specific inhibitory and activating Ly49 receptors. (2) Eomes-negative NKp46+ NK1.1+ RORγt-fm+ cells were part of the ILC3 lineage, enriched in plastic ILC3s which have downregulated RORγt and upregulated T-bet expression (Vonarbourg et al. 2010; Klose et al. 2013). (3) The third population expressed T-bet and IL-7Rα (consistent with an ILC phenotype) but was Eomes-negative and did not express RORγt and had not expressed this transcription factor during lineage differentiation (RORγt-fm-negative) (Klose et al. 2014). We refer to this non-NK, non-ILC3 population as ILC1s (Klose et al. 2014). Interestingly, all three subsets of NKp46+ NK1.1+ cells were represented in almost every organ investigated but the composition of the NKp46+ NK1.1+ population differed to a great extent. In secondary lymphoid organs, cNK cells represented the major population whereas ILC1s and ILC3s dominated in mucosal organs (Klose et al. 2014).
The analysis of these double reporter mice identified an ILC1 subset (i.e., T-bet+ Eomes− RORγt-fm− cells) in the bone marrow. In the past, a population of cells with a similar cell surface marker expression profile (IL-7Rα+ NK1.1+) was termed immature (i) NK cells (Huntington et al. 2007; Vosshenrich and Di Santo 2013), despite the fact, that they expressed markers such as CD51, CD69, and TRAIL which were not expressed by cNK cells before and after this developmental stage. Analysis of Eomes Gfp/+ x RORγt-fm reporter mice now revealed that bone marrow IL-7Rα+ NK1.1+ cells contain an Eomes+ CD49a− and an Eomes− CD49a+ population. Upon adoptive transfer into lymphopenic mice, Eomes+ CD49a− cells differentiated into cNK cells whereas Eomes− CD49a+ cells stably displayed an Eomes− RORγt-fm− phenotype. Thus, bone marrow Eomes− CD49a+ IL-7Rα+ NK1.1+ cells do not upregulate Eomes in vivo or in vitro and, therefore, do not belong to the cNK cell but rather to the ILC1 lineage (Klose et al. 2014).
Genome-wide gene expression profiling was quite informative in that it revealed that ILC1s were more closely related to NKp46+ NK1.1+ RORγt-fm+ cells than to cNK cells suggesting a core transcriptional program common to ILCs . When compared to cNK cells, ILC1s showed striking differences in the expression of chemokines and cytokines including their receptors and in the expression of integrins (Klose et al. 2014). Interestingly, the expression of various cell surface markers and transcription factors by ILCs (CD62L, CD69, CCR7, CD27, CXCR3, Id2, Eomes, T-bet) resembled the one described for central memory and tissue-resident memory T cells (Mackay et al. 2013; Kaech and Cui 2012; Hofmann and Pircher 2011). Therefore, ILCs seem to be programmed for tissue residency.
3.2 Transcriptional Regulation of ILC1 Development by T-bet, Nfil3 and GATA-3
In further support of the notion that ILC1s constitute a lineage independent of cNK cells or other defined ILC subsets, ILC1s have unique transcriptional requirements for differentiation and/or maintenance that sets them apart from cNK cells and from other ILC lineages (Table 1).
3.2.1 T-bet
As the various helper-like ILCs share transcriptional circuitry with Th cells, an important role of T-bet for the differentiation and/or maintenance of ILC1s was expected. Indeed, ILC1s expressed only T-bet but not the related T-box transcription factor Eomes (Klose et al. 2014). In sharp contrast to the mild phenotype of T-bet deficiency on cNK cell development (Townsend et al. 2004; Jenne et al. 2009), T-bet was essential for the ILC1 fate (Klose et al. 2014). Indeed, Tbx21 −/− mice had virtually no intestinal ILC1s whereas cNK cells and ILC3s were normally represented. Interestingly, T-bet was already required for the early commitment to the ILC1 lineage because iILC1s (Eomes− CD49a+ IL-7Rα+ NK1.1+ cells) in the bone marrow were affected as well. This is an interesting finding because cNK cells are overrepresented in the bone marrow of T-bet-deficient mice because T-bet directly regulates S1PR5 (sphingosine-1-phosphate receptor 5) expression required for cNK cell egress from the bone marrow (Jenne et al. 2009). The exact mechanism of how T-bet regulates ILC1 fate remains elusive. Overexpression of Bcl-2 in T-bet-deficient mice did not rescue ILC1 differentiation arguing for a role of T-bet in ILC fate decision rather than for their maintenance (C.S.N.K and A.D., unpublished data).
T-bet is required for the regulation of IL-2Rβ chain (CD122) expression, a component of the IL-15 receptor (Lazarevic and Glimcher 2011). Although ILC1s expressed IL-7Rα similar to the other, helper-like ILC subsets, but their development and/or maintenance was not affected in mice lacking the IL-7 receptor (Klose et al. 2014). Interestingly, ILC1s co-expressed IL-2Rβ and IL-7Rα but ILC1 numbers were significantly reduced in mice lacking IL-15. IL-15 dependency of ILC1s was not absolute, indicating redundant control of ILC1 maintenance by various factors (Klose et al. 2014). These data raise interesting questions about regional niches in the intestine that can be distinguished into high-IL-7 and high-IL-15 environments. It will be interesting to see if ILC1s can depend on IL-7 in organs with low IL-15 expression.
3.2.2 Nfil3 or E4BP4
Another transcription factor which might be involved in the IL-15 signaling cascade is Nfil3 (also known as E4BP4) (Gascoyne et al. 2009; Kamizono et al. 2009). Nfil3 is a basic leucine zipper transcription factor, essential for the development of cNK cells (Gascoyne et al. 2009; Kamizono et al. 2009). Recent data indicated that Nfil3 is already controlling development of the rNKP in the bone marrow (i.e., Lin− Kit+ IL-7Rα+ IL-2Rβ (CD122)+ 2B4+ CD27+ Flt3− cells) indicating an important role of Nfil3 for the early stages of NK cell development (Male et al. 2014). In contrast, T-bet or Eomes deficiency did not affect specification toward the rNKP. Of note a population referred to as the pre-NKP (i.e., Lin− Kit+ IL-7Rα+ IL-2Rβ (CD122)− 2B4+ CD27+ Flt3−) was also reduced in Nfil3 −/− mice. As this population is largely Id2-positive (M. Flach and A.D., unpublished data) and contains the CHILP , the question arises if Nfil3 may not play broader roles for the development of various ILC lineages. This is supported by our data showing that in mice with a deletion of Nfil3 in all hematopoietic cells, both mature intestinal and immature bone marrow ILC1s as well as NKp46+ NK1.1+ ILC3s were lacking (Klose et al. 2014). The broader roles of Nfil3 for ILC development and a careful analysis of Nfil3 target genes need to be addressed in future studies.
3.2.3 GATA-3
ILC1s express intermediate levels of GATA-3 (Klose et al. 2014). As alluded to above, deletion of GATA-3 in all hematopoietic cells cripples the development of all helper-like (IL-7Rα+) ILCs but leaves cNK cell differentiation intact (Yagi et al. 2014; Serafini et al. 2014; Samson et al. 2003). Conditional deletion of Gata3 by using NKp46-Cre or Id2-CreERt2 had no impact on the representation of cNK cells or ILC3s including NKp46+ ILC3s. However, deletion of Gata3 in all NKp46-expressing cells led to a dramatic reduction of ILC1s (Klose et al. 2014; Hoyler et al. 2012). Thus, GATA-3 plays multiple important roles for the differentiation of ILCs. (1) Early expression (before or at the Id2-positive stage) is required for specification of the CILP or CHILP to the helper-like ILC lineages. GATA-3 marks an important bifurcation between helper-like (GATA-3-dependent) and cytotoxic ILCs (GATA-3-independent). Future studies need to address at which progenitor stage GATA-3 is required and which GATA-3-controlled genes determine commitment to helper-like ILCs (Yagi et al. 2014). (2) Deletion of GATA-3 in all Id2-expressing cells did not affect cNK cell or ILC3 differentiation or maintenance. However, both GATA-3high ILC2s and GATA-3int ILC1s including their bone marrow-resident progenitors (ILC2Ps and iILC1s) required GATA-3 for maintenance (Klose et al. 2014; Hoyler et al. 2012). (3) Deletion of Gata3 in all NKp46+ cells led to reduced numbers of ILC1s whereas both other NKp46+ ILC subsets, cNK cells and ILC3s were normally represented (Klose et al. 2014).
3.3 Intraepithelial ILC1-Like Cells and Unusual NKp46+ ILC Subsets
Apart from ILC1s in the small intestine, other NKp46+ ILC subsets were described which differed in important aspects from cNK cells. These NKp46+ ILC subsets are found within specific tissue niches such as the epithelium of the small intestine, the thymus, or the liver.
3.3.1 Intraepithelial ILC1s
Intraepithelial ILC1s were mainly investigated in human tonsils and the epithelium of the intestine. These cells were found within the NKp44+ CD103+ and NKp44− CD103− compartment of CD56+ non-T cells and were potent producers of IFN-γ (Fuchs et al. 2013). NKp44+ CD103+ cells expressed molecules indicating TGF-β imprinting and additional surface markers not found on cNK cells such as CD160, CD49a, CXCR6, CD69, and CD39. Interestingly, they expressed the IL-2Rβ chain but not the IL-7Rα chain, which suggested that their development might be dependent on IL-15 signaling. In mice, a possibly related CD160+ NKp46+ ILC subset residing within the epithelium of the intestine was identified that produced IFN-γ. Il15ra-deficient mice had only a very mild reduction of intraepithelial ILC1s arguing that IL-15 signaling may not be indispensable for their development. It is unknown if intraepithelial ILC1s require IL-7 receptor signaling for differentiation and/or maintenance. In contrast to cNK cells, intraepithelial ILC1s were developmentally dependent on the transcription factor T-bet. Similar to cNK cells and ILC1s of the lamina propria (Klose et al. 2014), intraepithelial ILC1 development was perturbed in Nfil3-deficient mice. Like cNK cells, intraepithelial ILC1s expressed Eomes which distinguishes them from ILC1s described in the lamina propria of the small intestine (Klose et al. 2014). Analysis of intraepithelial NKp46+ NK1.1+ ILCs (all of which express CD160) using Eomes Gfp/+ x RORγt-fm double reporter mice revealed that this population is diverse and contained ILC1s, NKp46+ NK1.1+ ILC3s and cNK cells (Klose et al. 2014). The functional importance of intraepithelial ILC1s was demonstrated in an αCD40-triggered innate colitis model (Uhlig et al. 2006; Buonocore et al. 2010). Intraepithelial ILC1s were potent IFN-γ producers upon αCD40 injection and depletion of NKp46+ lymphocytes by NK1.1 antibody depletion ameliorated colitis severity (Fuchs et al. 2013). This might also be relevant with regard to human disease because NKp44+ CD103+ intraepithelial ILC1s were increased in patients with Crohn’s disease (Fuchs et al. 2013).
3.3.2 Thymic “NK Cells”
The IL-7Rα chain is expressed by the CLP and NKP but quickly downregulated during NK cell development. Therefore, an NKp46+ innate lymphocyte subset that expressed the IL-7Rα chain and that was mainly located in the thymus of mice had attracted interest and was termed “thymic NK cells” because their development depended on the presence of the thymus (Vosshenrich et al. 2006). In addition, thymic NK cells depicted a very different phenotype compared to cNK cells as they lacked expression of Ly49s but had higher expression of Kit and CD94. In contrast to cNK cells and ILC1s they developmentally depended on IL-7 (Vosshenrich et al. 2006). Similar to ILC1s, the transcription factor GATA-3 was essential for them to develop and they were potent producers of IFN-γ and TNF. It was suggested that they might correspond to the human CD56bright NK cell subset which has high capacity to produce cytokines but low cytotoxic activity (Vosshenrich et al. 2006). Even though ILC1s and “thymic NK cells” differ in their dependency on IL-15 or IL-7, respectively, it is conceivable that they are of the same lineage because ILC1s express both the IL-15 and IL-7 receptors and their maintenance may depend on the availability of these cytokines in certain tissues. Future experiments will need to more firmly establish if ILC1s and “thymic NK cells” are separate or related ILC lineages.
3.3.3 Liver “NK Cells”
The liver is home to a special NK cell population called TRAIL (tumor necrosis factor-related apoptosis-inducing ligand)+ NK cells which are very prominent in neonatal mice but then decline with the age of mice (Takeda et al. 2005). Besides TRAIL, the surface receptor by which they were identified, they also express other molecules such as CD49a and CXCR6, a chemokine receptor which recognizes CXCL16 that is secreted by endothelial cells of the liver sinusoids (Shi et al. 2011; Peng et al. 2013). TRAIL+ NK cells are distinct from liver cNK cells in that they do not express DX5 (CD49b) and Eomes (Gordon et al. 2012). TRAIL+ NK cells were able to kill target cells but are less cytotoxic than cNK cells. In addition to IFN-γ, which is expressed by all known NKp46+ cells, TRAIL+ NK cells secrete TNF, IL-13 and GM-CSF (Takeda et al. 2005; Daussy et al. 2014; Sojka et al. 2014). Because of their high representation in the liver of neonatal mice and based on in vivo differentiation studies, a model was proposed in which TRAIL+ NK cells are immature progenitors of cNK cells. Indeed, TRAIL+ NK cells expressed several markers which define immature cNK cells such as CD27 but not CD11b, a NK cell maturation marker. In addition, transfer of TRAIL+ NK cells into alymphoid mice led to the development of TRAIL− DX5+ cNK cells (Gordon et al. 2012). Development of TRAIL+ NK cells depended on T-bet but not Eomes, whereas mature cNK cells strictly depended on Eomes but not on T-bet. This could be in line with a model in which T-bet is essential to maintain immature TRAIL+ NK cells and Eomes to maintain mature cNK cells and that deficiency for one of these transcription factors preferentially allows for outgrowth of the NK cell population independent of that factor (Takeda et al. 2005; Gordon et al. 2012). Nevertheless, it is difficult to appreciate why Eomes+ cNK cells are normally represented in T-bet-deficient mice that lack their progenitors (i.e., TRAIL+ NK cells). In fact, several recent studies have called this model into question because they provided evidence that the transcriptome of TRAIL+ NK cells and cNK cells is very different, making it unlikely that they represent various maturation stages of cNK cells. In further support of this view, these studies failed to differentiate cNK cells from TRAIL+ NK cells (Daussy et al. 2014; Peng et al. 2013; Sojka et al. 2014). Thus, TRAIL+ NK cells and cNK cells may constitute distinct ILC lineages.
Interestingly, studies concerning immunological memory of NK cells showed that the memory potential is contained within CXCR6+ liver NK cells which are most likely identical to TRAIL+ NK cells (Peng et al. 2013; O’Leary et al. 2006; Paust et al. 2010). These findings would be difficult to reconcile with a model in which TRAIL+ NK cells are immature progenitors of cNK cells and, therefore, argue in favor of a model that would assign TRAIL+ NK cells and cNK cells to different lymphocyte lineages.
Future research will need to answer the important question regarding the lineage relationship of the unusual NK cell populations. Although such unconventional NKp46+ ILCs share some common markers such as expression of CXCR6 and CD49a and limited expression of Ly49 receptors, the lineage relationship remains difficult to determine because they are present in different organs. Therefore, it is unclear if difference in gene expression or dependency on cytokines that may be differentially expressed in the various organs reflects organ-specific imprinting of gene expression or if it indicates that these NKp46+ ILC subsets constitute different lymphoid lineages.
3.4 ILC1s Provide Innate Protection Against Intracellular Pathogens
IFN-γ is indispensable for the control of intracellular infections (Suzuki et al. 1988; Flynn et al. 1993; Buchmeier and Schreiber 1985; Wang et al. 1994). A central role in this process plays the cytokine IL-12 which is produced by various subsets of mononuclear phagocytes. IL-12 was initially identified as the natural killer cell stimulating factor (NKSF) (Kobayashi et al. 1989) and induces IFN-γ production by NK cells but also instructs the differentiation of IFN-γ-producing Th1 cells. IFN-γ released by NK cells and Th1 cells allows for the activation of macrophages to an antimicrobial state effectively controlling various intracellular infections. Deficiencies in components of the IFN-γ and IL-12 signaling pathways lead to susceptibility to intracellular bacterial infections (Altare et al. 1998; de Jong et al. 1998). ILC1s and ILC1-like cells express T-bet and high levels of the components of the IL-12 receptor. Interestingly, ILC1s similar to “thymic NK cells” reacted to IL-12 stimulation with IFN-γ production (Vosshenrich et al. 2006; Fuchs et al. 2013; Klose et al. 2014). This is in striking contrast to naïve cNK cells that are remarkably poor producers of IFN-γ after stimulation with IL-12 (Lucas et al. 2007; Klose et al. 2014). Thus, ILC1s may be an important immediate source of IFN-γ during infection with intracellular pathogens. While cNK cells contain granula with proteins allowing for cytotoxic function, ILC1s were relatively poor killer cells.
High IL-12 secretion is triggered by infection with the obligate intracellular parasite Toxoplasma gondii (T. gondii). Resistance to T. gondii infection is mediated to a large degree by IFN-γ and inflammatory monocytes, recruited to the sites of infection (Suzuki et al. 1988; Dunay et al. 2008). Previously, intestinal IL-15-dependent NK1.1+ or NKp46+ cells (“NK cells”) were shown to be an important early source of this cytokine during oral T. gondii infection. Furthermore, NKp46+ ILCs produced CCL3, a chemokine leading to the recruitment of inflammatory monocytes (Schulthess et al. 2012). Future studies will need to dissect which of the three NK1.1+ NKp46+ ILC subsets are the source of CCL3. Recently however, it became clear that among all NK1.1+ ILCs, ILC1s were the main producers of IFN-γ and TNF during a sublethal T. gondii infection in the small intestine. ILC1s were especially potent in the co-production of IFN-γ and TNF. T-bet deficient mice (lacking ILC1s) showed increased T. gondii titers and reduced recruitment of inflammatory monocytes during the early phases of infection consistent with an important role of T-bet-driven innate cytokines for early parasite control. Rag2 −/− Il2rg −/− mice that lack all lymphoid cells but have a largely normal myeloid compartment, were unable to control T. gondii infection and showed only very inefficient recruitment of inflammatory monocytes to the lamina propria. Interestingly, transfer of ILC1s into alymphoid mice led to increased recruitment of inflammatory monocytes and innate parasite control (Klose et al. 2014).
Collectively, ILC1s and cNK cells are distinct ILC lineages that can be discriminated on the basis of different developmental requirements for transcription factors, distinct tissue residency, and a distinct gene expression program. ILC1s build a first line defense at the mucosal barrier to quickly react with cytokine production to invading intracellular pathogens. In contrast, cNK cells are located preferentially in secondary lymphoid organs, where they require priming by mononuclear phagocytes (Lucas et al. 2007) and are able to proliferate (Sun et al. 2009) before invading inflamed tissues (Lucas et al. 2007) to combat infections through cell-mediated cytotoxicity and cytokine production.
4 ILC2s Express High Levels of GATA-3 and Secrete IL-5 and IL-13
4.1 ILC2 Differentiation
ILC2s are very potent producers of effector cytokines such as IL-5, IL-9, IL-13, and amphiregulin, the latter is a member of the expanded epithelial growth factor (EGF) family (Wilhelm et al. 2011; Turner et al. 2013; Monticelli et al. 2011; Neill et al. 2010; Moro et al. 2010; Zaiss et al. 2006). It is still controversial if ILC2s also secrete IL-4. Secretion of IL-5 from ILC2s is continuous (i.e., occurs during steady-state) (Hoyler et al. 2012) and is controlled by circadian rhythm (Nussbaum et al. 2013). ILC2s are mainly localized in the gastrointestinal tract, the respiratory tract, the skin and fat-associated lymphoid clusters, but rare in secondary lymphoid organs at steady-state. Their localization at barrier surfaces allows them to quickly react to invading helminthes (Hoyler et al. 2012; Neill et al. 2010; Moro et al. 2010; Saenz et al. 2010; Price et al. 2010; Mjosberg et al. 2011; Liang et al. 2012). Helminth larvae invade the skin, enter the heart, then the lung via the blood vessels where they are coughed up, swallowed and finally reproduce in the gastrointestinal tract where they are expelled (Camberis et al. 2003). The crucial role of ILC2s for the resistance against the nematode Nippostrongylus brasilienses (N. brasiliensis) was demonstrated in several studies (Moro et al. 2010; Neill et al. 2010; Hoyler et al. 2012; Wong et al. 2012). Because of the therapeutic relevance, much research has focused on the pathophysiological reaction of ILC2s in airway hyperreactivity, allergic lung disease, atopic dermatitis, and liver injury (McHedlidze et al. 2013; Salimi et al. 2013; Roediger et al. 2013; Halim et al. 2012a; Monticelli et al. 2011; Chang et al. 2011).
Epithelial cell-derived cytokines such as IL-33, IL-25, and TSLP are strong activators of ILC2s (Van Dyken et al. 2014; Saenz et al. 2013; Neill et al. 2010; Neill and McKenzie 2011; Mjosberg et al. 2011). In fact, an innate source of IL-25-responsive cells has been documented in the past and eventually led to the discovery of ILC2s (Fort et al. 2001; Hurst et al. 2002; Fallon et al. 2006). In mice, ILC2s have been characterized by high expression of IL-7Rα, Sca-1 and CD25. However, these markers are not specific for the ILC2 lineage as IL-7Rα and CD25 are expressed by all helper-like ILCs and Sca-1 is found on a subset of ILC3s (Vonarbourg et al. 2010; Buonocore et al. 2010). ICOS, the IL-33Rα-chain, and the IL-17Rβ-chain are more specific markers of ILC2s (Moro et al. 2010; Price et al. 2010; Liang et al. 2012). KLRG1 and Kit were identified as maturation markers on ILC2s (Hoyler et al. 2012).
Precursors of ILC2s (ILC2Ps) in the bone marrow lack these markers whereas KLRG1 and Kit were upregulated in mature ILC2s in mucosal tissues of the gastrointestinal tract (Fig. 1). ILC2Ps were poor producers of ILC2-derived cytokines such as IL-5 and IL-13 despite sharing a core cluster of ILC2 lineage-specific gene sets (Hoyler et al. 2012). Consistent with the progenitor phenotype of ILC2Ps, their potential to proliferate and reconstitute the ILC2 compartment of alymphoid mice was more efficient when compared to adoptively transferred mature ILC2s (Hoyler et al. 2012; Halim et al. 2012b; Brickshawana et al. 2011). Interestingly, a lymphoid progenitor population that contains the ILC2P has been first described in 1998 and was referred to as LSK− (Lin− Sca-1+ Kit−) cells (Randall and Weissman 1998). Because of their phenotypic resemblance of hematopoietic stem cells (LSK cells), it was speculated that the LSK− population constitutes a hematopoietic precursor population (Randall and Weissman 1998). However, adoptive transfer of LSK− cells into lethally irradiated hosts did not reconstitute the hematopoietic system. Later studies identified additional markers such as CD25 and IL-7Rα indicative of lymphoid fate, on a fraction of LSK− cells but failed to recover progeny of adoptively transferred LSK− cells from the spleen of transplanted mice (Kumar et al. 2008). More recently bone marrow ILC2Ps were identified as Lin− Sca-1high Id2+ GATA-3+ CD25+ IL-7Rα+ cells that were largely identical to LSK− cells (Hoyler et al. 2012). In line with the above-mentioned previous data (Randall and Weissman 1998; Kumar et al. 2008), adoptive transfer of ILC2Ps into alymphoid mice did not generate any appreciable progeny in secondary lymphoid organs (Hoyler et al. 2012). Strikingly, ILC2Ps very efficiently homed to the small intestinal lamina propria where they differentiated into mature ILC2s (Hoyler et al. 2012). Homing to mucosal sites such as the small intestine was, in part, due to expression of the chemokine receptor CCR9 (Hoyler et al. 2012). In addition, the ILC2P expressed integrin α4β7 which is involved in homing of lymphocytes to the intestine (Holzmann et al. 1989).
4.2 GATA-3
In analogy to Th2 cells (Zheng and Flavell 1997), ILC2s have high expression of the transcription factor GATA-3 (Hoyler et al. 2012; Mjosberg et al. 2012). ILC2Ps already showed high GATA-3 expression indicating that ILC2Ps were already specified for the ILC2 lineage. The roles of GATA-3 within the ILC lineage are complex and have been discussed above (Table 1). While all ILCs express intermediate levels of GATA-3, high level GATA-3 expression is a unique characteristic of ILC2s driving a distinct gene expression program (Yagi et al. 2014; Hoyler et al. 2012). Conditional deletion of GATA-3 in ILCs and T cells using Id2-Cre led to complete absence of ILC2s and ILC2Ps, whereas ILC3s were normally maintained (Hoyler et al. 2012; Furusawa et al. 2013; Mjosberg et al. 2012). Cre-mediated deletion of Gata3 in vitro demonstrated that GATA-3 is not only important for differentiation but also for the maintenance of the ILC2 lineage (Hoyler et al. 2012; Yang et al. 2013; Yagi et al. 2014). Thus, the ILC2 lineage is characterized by high GATA-3 expression and GATA-3 is required for the maintenance of ILC2s.
4.3 RORα
RAR (retinoic acid receptor)-related orphan receptors α, β and γ form a family of steroid hormone receptors which recognize a specific DNA sequence RGGGTCA (R: A/G). They consist of an N-terminal domain, the DNA-binding domain, a hinge domain and a C-terminal DNA-binding domain. RORα is expressed in two isoforms in mice (RORα1 and RORα4) and this transcription factor is important in cerebellum and cone development (Jetten 2009). In addition, RORα is involved in regulation of circadian rhythm (McIntosh et al. 2010; Jetten 2009). In contrast to RORγ, RORα is essential for the development of ILC2s. RORα is already expressed at the ILC2P stage and is further upregulated in mature ILC2s (Hoyler et al. 2012; Wong et al. 2012; Halim et al. 2012b). Germline deletion of the Rora gene led to defective differentiation of ILC2s. Particularly, expansion of ILC2s upon IL-25 injection, N. brasiliensis infection or papain challenge was severely impaired in the absence of Rora (Wong et al. 2012; Halim et al. 2012b). As a consequence Rora-deficient mice could not control N. brasiliensis infection and were resistant to papain-induced airway inflammation, both processes have been shown to be ILC2-dependent (Halim et al. 2012b; Wong et al. 2012). RORα-dependent gene sets in ILC2 are not well investigated, which will be an important goal for the future. One RORα target gene in Purkinje cells of the cerebellum is sonic hedgehog (Shh), a fundamental gene in cell proliferation and differentiation (Wallace 1999; Dahmane and Ruiz i Altaba 1999; Jetten 2009). However, whether this or similar genes are also controlled by RORα in ILC2s remains to be tested.
4.4 TCF-1 Signaling
TCF-1 is a transcription factor involved in the early specification of T cell fate (Verbeek et al. 1995; Okamura et al. 1998). Recent data showed that TCF-1 is also essential for the development of the entire ILC2 lineage including the ILC2P in the bone marrow (Yang et al. 2013). Consequently, Tcf7 −/− mice (Tcf7 is the gene encoding TCF-1) had strongly reduced ILC2 numbers in many organs. As a result, mice were susceptible to N. brasiliensis infection and resistant to papain-induced asthma (Mielke et al. 2013; Yang et al. 2013). The Tcf7-gene is regulated by Notch signaling in ILC2s and itself controls expression of IL-7Rα chain, an essential cytokine receptor component for development of ILC2 and ILC3 lineages (Yang et al. 2013; Wong et al. 2012).
4.5 Gfi1
The transcription factor growth factor independent 1 (Gfi1) is broadly expressed in the hematopoietic system and has critical roles in various hematopoietic processes including T lymphopoiesis (Spooner et al. 2009; Yucel et al. 2003). Moreover, Gfi1 functions in type 2 immune responses by controlling the IL-2-dependent population expansion of Th2 cells (Zhu et al. 2006). In analogy to T helper cells, ILC2s expressed Gfi1 already at the ILC2P stage and Gfi1 was further upregulated upon ILC2 maturation (Spooner et al. 2013). Gfi1 was required for differentiation or maintenance of ILC2s because Gfi1 −/− mice had reduced numbers of ILC2s and ILC2Ps. As a result, Gfi1-deficient mice were susceptible to N. brasiliensis infection and resistant to papain-induced lung inflammation.
Microarray analysis revealed that Gfi1 regulates key genes of ILC2 function and development including the expression of Il1rl1 (IL-33R subunit), Il17rb (IL-25R subunit), Il5, Crlf2, and Gata3. In particular, Gfi1 seems to regulate the responsiveness to IL-33, one of the most important activation signals of ILC2s (Van Dyken et al. 2014; Hoyler et al. 2012). On the other hand, Gfi1 suppresses genes involved in ILC3 commitment and function such as Rorc(γt), Sox4, Il17a, Il17f, and Il1r1. Indeed, deletion of Gfi1 allowed expression of ILC3-specific genes in ILC2s (Spooner et al. 2013). Thus, Gfi1 is a transcription factor that reciprocally regulates the type 2 and type 17 effector states in lymphoid cells of the innate and adaptive immune systems (Spooner et al. 2013).
5 The ILC3 Family Developmentally Depends on RORγt and Secretes IL-22
5.1 Two ILC3 Lineages: CCR6+ and CCR6−/low ILC3s
LTi cells , the founding member of group 3 ILCs , were first described in the late 1990s as hematopoietic cells (CD45+) that express CD4 but lack expression of T cell markers such as CD3 (Mebius et al. 1997). Recent data support the view that LTi cells are CCR6+ Kithigh ILCs and CD4 is only expressed on a subset (Sawa et al. 2010; Klose et al. 2013). It is currently unclear if CD4+ and CD4− CCR6+ ILC3s differ in their functional or transcriptional properties. It also remains to be established if CD4 expression is stable or if CCR6+ ILC3s can downregulate CD4 expression (Vonarbourg et al. 2010; Sawa et al. 2010). The CC-chemokine receptor CCR6 recognizes the ligand CCL20 expressed by stroma cells in cryptopatches and the antimicrobial protein β-defensin 3 (Yang et al. 1999). Sensing of gram-negative commensal bacteria by NOD1 stimulates CCL20 expression which regulates isolated lymphoid follicle but not cryptopatch formation (Bouskra et al. 2008). The lineage-specifying transcription for LTis cells is RORγt, an immune cell-specific alternative transcript of the Rorc gene (Villey et al. 1999; He et al. 1998). Targeted deletion of the Rorc gene or of Rorc(γt)-specific exons led to disappearance of LTi cells and, consequently, to a failure of lymphoid organ development with absence of lymph nodes, Peyer’s patches, cryptopatches, and isolated lymphoid follicles (Eberl et al. 2004; Sun et al. 2000; Kurebayashi et al. 2000; Eberl and Littman 2004). An extensive description of how LTi cells interact with stroma cells or endothelial cells and other lymphoid cells to initiate the process of lymphorganogenesis can be found elsewhere (van de Pavert and Mebius 2010).
In adult mice, LTi cells are mainly localized in cryptopatches and isolated lymphoid follicles (ILFs), where they are in immediate contact with B cells and mononuclear phagocytes (Kanamori et al. 1996; Hamada et al. 2002; McDonald et al. 2010). ILFs are major sites for T cell-independent IgA production and LTi cells and mononuclear phagocytes supported class switch recombination and differentiation of IgA-producing plasma cells by controlling gene expression in stroma cells (Tsuji et al. 2008).
While the role of CCR6+ ILC3s in supporting lymphoid organogenesis is well supported, their broader role for immunity at mucosal surfaces has only been recognized very recently. CCR6+ ILC3s are an important source of the cytokines IL-22 and IL-17A (Klose et al. 2013; Takatori et al. 2009; Zenewicz et al. 2008). IL-17A expression by ILC3s in the upper respiratory tract protected mice against fungal infections with Candida albicans (C. albicans) (Gladiator et al. 2013). IL-22 production is essential for resistance against attaching-and-effacing types of intestinal infections such as those with Citrobacter rodentium (C. rodentium), a mouse model for enteropathogenic Escherichia coli (E. coli) infections (Zheng et al. 2008; Sonnenberg et al. 2011b). It has been documented that in the first week of C. rodentium infection the majority of IL-22 is secreted by ILC3s (Sawa et al. 2011; Kiss et al. 2011; Sonnenberg et al. 2011b). CD4+ ILC3s seemed to have an important role in immunity to C. rodentium infection (Sonnenberg et al. 2011b). IL-22 is an extraordinary cytokine because its receptor which consists of the IL-22Rα1 chain and the IL-10Rβ chain is almost exclusively expressed on nonhematopoietic cells (Wolk et al. 2004; Sanos et al. 2011). Hence, ILC3s-derived IL-22 directly interacts with the surrounding epithelium, regulating resistance, homeostasis, proliferation, and repair of epithelial cells as well as containment of bacteria (Sonnenberg et al. 2011a, 2012; Sanos et al. 2011; Hanash et al. 2012; Dudakov et al. 2012; Zenewicz et al. 2007, 2008; Zheng et al. 2008; Pickert et al. 2009).
In late 2008, five independent groups reported a cell type which, similar to LTi cells, expressed and developmentally depended on the transcription factor RORγt, and, in addition, also expressed activating immunoreceptors characteristic for NK cells such as the natural cytotoxicity receptor 1 (NCR1, also known as NKp46) and NKG2D (Satoh-Takayama et al. 2008; Luci et al. 2009; Sanos et al. 2009; Cella et al. 2009; Cupedo et al. 2009). NKp46 was considered to be a fairly specific marker of cNK cells (Walzer et al. 2007). Therefore, the expression of characteristic markers of two different ILC lineages (cNK cells versus ILC3/LTi cells ) provoked questions about the lineage relationship of this newly discovered lymphoid cell type. In principle, three models have been proposed. CCR6− ILC3s could be a differentiation stage (1) of cNK cells (Cella et al. 2009), (2) of ILC3s (Sanos et al. 2009; Cupedo et al. 2009; Luci et al. 2009) or, (3) they could constitute an independent lymphoid lineage (Satoh-Takayama et al. 2008; Sawa et al. 2010).
Based on transfer experiments and fate-labeling studies it became apparent that NKp46+ ILC3s constituted a lineage distinct from cNK cells (Vonarbourg et al. 2010; Satoh-Takayama et al. 2010; Sawa et al. 2010). NKp46+ ILC3s differed from CCR6+ ILC3s in that they were CCR6-negative and expressed low levels of Kit (Sawa et al. 2010; Klose et al. 2013). It also became clear that a population of CCR6−/low ILC3s existed that was NKp46-negative. Cell transfer experiments and in vitro differentiation assays documented that NKp46+ CCR6−/low ILC3s were the progeny of NKp46− CCR6−/low ILC3s (Fig. 1) (Vonarbourg et al. 2010; Klose et al. 2013; Rankin et al. 2013; Cupedo et al. 2009). The lineage relationship to LTi cells was more difficult to tackle but several arguments can be taken into account collectively contending that CCR6−/low and CCR6+ ILC3s are distinct RORγt-expressing and RORγt-dependent ILC lineages. First, CCR6+ ILC3s did not appreciably differentiate into CCR6−/low ILC3s or NKp46+ ILC3s in vitro or when adoptively transferred into alymphoid mice (Sawa et al. 2010; Klose et al. 2013; Rankin et al. 2013). Second, CCR6−/low ILC3s but not CCR6+ ILC3s expressed the transcription factor T-bet and its target genes (e.g. NK receptors, IFN-γ) (Klose et al. 2013; Sciume et al. 2012b; Bernink et al. 2013). Third, several knockout mice show deficiency of either CCR6+ ILC3s or CCR6−/low ILC3s (Table 1) (Klose et al. 2013; Aliahmad et al. 2010; Kiss et al. 2011; Mielke et al. 2013). Fourth, fate-labeling studies showed different labeling efficiencies for CCR6+ and CCR6−/low ILC3s (Sawa et al. 2010). Fifth, CD4 depletion only affects CCR6+ ILC3s but not CCR6−/low ILC3s (Sawa et al. 2010). Despite this evidence, it remains to be determined if CCR6+ and CCR6−/low ILC3s may originate from a putative unidentified common ILC3 precursor or may have distinct progenitors (Sawa et al. 2010).
While CCR6+ ILC3s were a phenotypically and functionally stable RORγt+ T-bet− ILC lineage, CCR6−/low ILC3s showed phenotypic and functional plasticity. CCR6−/low ILC3s underwent a process that led to the upregulation of T-bet and the downregulation of RORγt (Klose et al. 2013; Vonarbourg et al. 2010; Bernink et al. 2013; Rankin et al. 2013). The following stages could be distinguished that likely represent consecutive differentiation stages: (1) CCR6−/low ILC3s, which express high levels of RORγt and are T-bet negative. (2) CCR6−/low ILC3s which express high levels of RORγt and low levels of T-bet but not yet NKp46. (3) CCR6−/low ILC3s which have intermediate levels of T-bet and RORγt and express NKp46 (often referred to as NKp46+ ILC3s or “NK22” cells). (4) CCR6−/low ILC3s which have downregulated RORγt but now have high levels of T-bet, NKp46 and NK1.1 (Fig. 1). Despite lack of expression of RORγt in that stage, fate-labeling studies clearly demonstrated that these cells are progeny of cells with a history of RORγt expression (Vonarbourg and Diefenbach 2012; Vonarbourg et al. 2010). The co-expression of RORγt and T-bet is remarkable because they were believed to instruct opposing transcriptional programs. Interestingly, “ex-RORγt+” ILC3s still express the IL-23 receptor and are now able to produce IFN-γ. IL-23-dependent IFN-γ production is crucial for intestinal inflammation in the αCD40-mediated colitis model (Uhlig et al. 2006). Depletion of ILCs in the αCD40 colitis model led to a significant reduction in pathology (Vonarbourg et al. 2010; Buonocore et al. 2010), while adoptive transfer of RORγt− but RORγt fate-label+ ILC3s promoted pathology in the αCD40 colitis model (Vonarbourg et al. 2010).
5.2 ILC3 Precursors in the Fetal Liver
The fetal liver harbors Flt3+ IL-7Rαint Kitint CLPs (Mebius et al. 2001; Possot et al. 2011). Induction of integrin α4β7 expression on these cells correlated with a loss of B and T cell potential (Yoshida et al. 2001). Flt3− IL-7Rαint Kitint α4β +7 cells were RORγt− (see above) and contained the CHILP (Klose et al. 2014; Constantinides et al. 2014). Finally, commitment to the ILC3 lineage is established through expression of the lineage-defining transcription factor RORγt correlating with high level IL-7Rα (IL-7Rαhigh α4β7+ RORγt+) (Yoshida et al. 2001; Cherrier et al. 2012). Alternatively, CXCR6 has been shown to be a reliable marker for ILC3 committed progenitors in the fetal liver (Possot et al. 2011). Of note, differentiation of IL-7Rαhigh α4β7+ RORγt+ was perturbed in mice genetically lacking Id2 or Rorc(γt) while the population of IL-7Rαint Kitint α4β +7 ILC progenitors was largely normal (Tachibana et al. 2011). Both Runx1 and its partner Cbfβ2 were required for the differentiation of IL-7Rαint Kitint α4β +7 ILC progenitors. In addition, Runx1/Cbfβ2 complexes were required for the upregulation of RORγt expression in IL-7Rαint Kitint α4β +7 ILC3 progenitors (Tachibana et al. 2011).
5.3 RORγt
Retinoic acid-related orphan receptor γ belongs to the same family of steroid hormone receptors as RORα (Jetten 2009). Two isoforms RORγ1 and RORγ2 are transcribed (He et al. 1998; Villey et al. 1999). While RORγ1 is expressed in multiple tissues RORγ2 (also called RORγt) was found to be expressed in the thymus and later in Th17, LTi cells and all other ILC3 populations (Eberl et al. 2004; Ivanov et al. 2006b). Targeted deletion of Rorc or the first (RORγ2-specifc) exon of RORγt led to the disappearance of lymphoid structures such as lymph nodes, Peyer’s patches or postnatally forming cryptopatches and isolated lymphoid follicles (Eberl et al. 2004; Sun et al. 2000). Some specialized lymphoid structure such as NALT could still be found in RORγt-deficient mice arguing that LTi cells are not strictly required for their development (Ivanov et al. 2006a). Both CCR6+ and CCR6−/low ILC3s are absolutely dependent on RORγt. Which fundamental processes are controlled by RORγt in ILC3s so that it is essential for development of both subsets is not well understood. In T cells, RORγt deficiency could be rescued by overexpression of Bcl-xL which might argue that anti-apoptotic signals are regulated by RORγt (Sun et al. 2000). Lineage committed precursors of ILC3s were described in the fetal liver but not the bone marrow (Yoshida et al. 2001). RORγt is essential for transition to the IL-7Rαhigh stage of CCR6+ ILC3s (see above) (Tachibana et al. 2011). This leads to the question which signals may instruct lineage commitment to the ILC3 lineage through regulating the expression of RORγt. Most of our knowledge about regulation of RORγt expression is based on studies in Th17 cells in which inflammatory cytokines such as TGF-β, IL-6, IL-1, IL-23 and IL-21 induced RORγt expression in the context of T cell receptor activation (Ivanov et al. 2006b; Bettelli et al. 2006). In the fetal liver, Runx1/Cbfβ2 complexes regulate earlier steps of ILC3 development. Interestingly, deletion of Cbfβ2 but not of P1-Runx1 led to reduced RORγt expression (Tachibana et al. 2011). Therefore, Cbfβ2 is a strong candidate for regulating RORγt in ILC3 precursors which is also supported by data from Th cells where Runx1/Cbfβ2 complexes control RORγt expression (Lazarevic et al. 2011). In the bone marrow, Notch has been shown to regulate RORγt expression (Possot et al. 2011).
How RORγt is induced in ILC3s is poorly understood. Interestingly, CCR6− ILC3s downregulate RORγt and upregulate T-bet (Klose et al. 2013). Regulation of the RORγt locus by T-bet and Runx1 is described for T cells but has not been investigated in ILC3s (Lazarevic et al. 2011). IL-7 is a cytokine which favors expression of RORγt (Vonarbourg and Diefenbach 2012). Most of RORγt+ lymphocytes are found in the small intestine. In contrast, IL-12 promotes loss of RORγt. In many other organs such as the colon or secondary lymphoid organs, downregulation of RORγt is predominant and therefore ex-RORγt cells are found (Vonarbourg et al. 2010).
5.4 TOX
Together with TOX2, TOX3 and TX4, thymocyte selection associated HMG box protein (TOX) forms a subfamily within the large HMG-box superfamily of transcription factors (Aliahmad et al. 2012). The various TOX proteins share a well-conserved DNA-binding domain which is composed of three α-helices. Apart from the HMG-box, all family members possess a lysine-rich region which may represent a nuclear localization sequence and a transactivation domain but have a different C-terminus (Aliahmad et al. 2012).
TOX is highly expressed in double-positive thymocytes and required for proper development of CD4 T cells and for the upregulation of Id2 (Aliahmad and Kaye 2008). In addition, TOX is essential for the development of secondary lymphoid organs such as lymph nodes and Peyer’s patches (Aliahmad et al. 2010). TOX-deficient mice show a very strong decrease of ILC3s with LTi function which explains the lack of lymphoid structures. Interestingly, Tox −/− mice also lacked mature cNK cells. This raises the question if TOX plays a decisive role in early commitment to the ILC fate by regulating E-protein activity. However, overexpression of Id2 in lymphoid precursor did not rescue cNK development (Aliahmad et al. 2010). Therefore, the mechanism of how TOX regulates ILC fates and at which stage(s) TOX is required will need further investigation.
5.5 T-bet
Metazoans have five T-box transcription factor genes which are characterized by encoding a conserved DNA-binding domain called T-box (Tanriver and Diefenbach 2014). The T-box domain recognizes a palindromic DNA sequence and this is the only identified DNA-binding domain of the T-box transcription factors (Naiche et al. 2005). The Tbr1 subfamily comprises three members Tbr1, T-bet, and Eomes (Takashima and Suzuki 2013). The latter two possess a nonredundant role in various tissues including immune cells such as CD8+ T cells und Th1 cells (Intlekofer et al. 2005; Szabo et al. 2000). In innate lymphocytes , Eomes is specifically expressed by cNK cells and is absolutely requirement for their proper development (Gordon et al. 2012). In addition, cNK cells also express T-bet, but germline deletion of Tbx21 (the gene encoding T-bet) has only a mild effect on the distribution of NK cells and is not absolutely required for their development (Jenne et al. 2009; Townsend et al. 2004). Hence, in cells such as cNK which express two members of T-box transcription factors, Eomes could compensate for some of the functions fulfilled by T-bet. In contrast to cNK cells, helper-like ILCs (such as ILC1s and CCR6− ILC3s) only express T-bet but not Eomes and T-bet expression is, therefore, an obligate requirement for some biological processes (Fig. 1) (Klose et al. 2013). T-bet is absolutely essential for the development of ILC1s which has been discussed above (Klose et al. 2014). In contrast, ILC3s develop in the absence of T-bet despite the fact that CCR6−/low ILC3s express this transcription factor (Klose et al. 2013, 2014). T-bet deficient mice had normal numbers of CCR6−/low ILC3s but lacked NKp46+ ILC3s (Table 1). Indeed, T-bet-deficient CCR6−/low ILC3s failed to upregulate NK receptors such as NKp46, NK1.1 and NKG2D (Klose et al. 2013; Sciume et al. 2012b; Rankin et al. 2013). CCR6−/low ILC3s undergo a three step differentiation process in which they first upregulate T-bet, then upregulate the expression of activating NK cell receptors (such as NKp46, NKG2D and NK1.1) and IFN-γ and finally downregulate RORγt (Vonarbourg et al. 2010; Bernink et al. 2013). T-bet deficient ILC3s fail to secrete the proinflammatory cytokines IFN-γ and TNF (Klose et al. 2013). Activation of goblet cells to release mucus during infection with Salmonella typhimurium (S. typhimurium) is strictly dependent on IFN-γ (Songhet et al. 2011). Interestingly, T-bet-deficient mice or ILC-depleted Rag2 −/− mice failed to trigger mucus release from goblet cells (Klose et al. 2013). In addition, T-bet deficient or ILC-depleted Rag2 −/− depicted less immunopathology and inflammation of the cecum during S. typhimurium infection (Klose et al. 2013).
Intestinal inflammation was also reported from Tbx21 −/− mice on a Rag2 −/− background, a syndrome called TRUC (T-bet Rag ulcerative colitis) (Garrett et al. 2007). These mice develop spontaneous colitis which is triggered by Helicobacter typhlonius. IL-17-producing CCR6+ ILC3s were identified as pathogenic lymphocytes population in this model because neutralization of IL-17A or depletion of ILC3s with Thy1 or IL-7Rα deficiency reduced intestinal inflammation. ILC3 activation to produce IL-17A was triggered by IL-23 derived from dendritic cells (Powell et al. 2012). Thus, T-bet expression in DCs or CCR6−/low ILC3s is required to restrain IL-17A production by CCR6+ IL-17A-producing ILC3s. A role of IL-17A-producing ILC3s as driver of colitis has also been documented for Helicobater hepaticus (H. hepaticus)-induced colitis in 129Sv mice. Thy1+ IL-17A producing ILCs expressing RORγt and T-bet were shown to drive colitis in this model (Buonocore et al. 2010).
5.6 AhR
The aryl hydrocarbon receptor (AhR) is basic helix-loop-helix transcription factor which belongs to the Per-Arnt-Sim family (Klose et al. 2012; McIntosh et al. 2010). Upon ligand binding, it translocates to the nucleus where it dimerizes with Arnt which allows regulation of gene transcription. Cytochrom p450 monooxidases are classical target genes and their promoters contain xenobiotic response elements. The AhR binds numerous ligands, the most famous being the environmental toxin dioxin (Fernandez-Salguero et al. 1995). Apart from toxins, endogenous ligands (such as tryptophan metabolites) or dietary phytochemicals (such as flavonoids, polyphenols, and glucosinolates) can activate the AhR (Stevens et al. 2009; Rannug et al. 1987; Opitz et al. 2011).
Ahr −/− mice have a strongly diminished ILC3 compartment. As a consequence, AhR-deficient mice failed to develop cryptopatches and ILFs, whereas the development of fetal lymphoid organs such as Peyer’s patches and peripheral lymph nodes was normal (Kiss et al. 2011; Qiu et al. 2011; Lee et al. 2012). While Peyer’s patches form during fetal development, cryptopatches appeared 2 to 3 weeks after birth. Therefore, the question arose if AhR was involved in the differentiation of a specific ILC3 subset. Interestingly, CCR6+ ILC3s were only mildly affected in Ahr-deficient mice, whereas CCR6−/low ILC3s were dramatically reduced (Klose et al. 2013). Increased apoptosis has been proposed as a mechanism (Qiu et al. 2011) whereas others have found a failure of CCR6−/low ILC3s to proliferate (Kiss et al. 2011). On a molecular level, AhR binds to the Kit promoter and regulates Notch2 expression (Kiss et al. 2011; Lee et al. 2012). Interestingly, mice with an impairment of Kit signaling also have reduced numbers of intestinal patches (Kiss et al. 2011; Chappaz et al. 2011). In addition, conditional deletion of RBP-Jκ, an essential component of the Notch signaling pathway, did not lead to a reduction in intestinal patches, but to a decrease in NKp46+ ILC3s (Lee et al. 2012).
While the frequency of CCR6+ ILC3s was less affected by AhR deficiency and CCR6+ ILC3s were only mildly reduced (Klose et al. 2013), AhR regulated expression of IL-22 and probably other key molecules in CCR6+ ILC3s. AhR physically interacted with RORγt and bound to the Il22 locus (Qiu et al. 2011). Reduced IL-22 expression caused reduced resistance against C. rodentium infection (Kiss et al. 2011; Lee et al. 2012).
As AhR is a ligand-activated transcription factor, the question arose which AhR ligands lead to the postnatal expansion of CCR6−/low ILC3s. Interestingly, dietary AhR ligands influenced the ILC3 pool size (Kiss et al. 2011). Strikingly, mice fed with a synthetic diet which contained low amounts of dietary AhR ligands (phytochemicals such as glucosinolates) had a phenotype similar to AhR-deficient mice (i.e., reduced expansion of ILC3s and diminished formation of cryptopatches and ILFs). Interestingly, addition of the tryptophan-derived phytochemical indol-3-carbinol to synthetic phytochemical-deprived diets led to a normal development of ILC3 compartment and intestinal patches (Kiss et al. 2011; Klose et al. 2012). Dietary-derived AhR ligands have also been shown to promote maintenance of intraepithelial lymphocytes (IELs) and promote homeostasis in the gut which protects mice to recover from DSS-colitis (Li et al. 2011). Hence, dietary-derived metabolites such as the glucosinolate glucobrassicin, which is contained in cruciferous vegetables, are involved in the regulation of intestinal immune homeostasis by regulating the pool size of ILC3s and IELs.
5.7 Notch and TCF-1 Signaling
T-bet directly binds to the Notch locus in Th1 cells (Germar et al. 2011; Weber et al. 2011). This signaling pathway regulates differentiation of CCR6−/low ILC3s. Differentiation on OP9 stroma cells, which express the Notch ligand delta-like 1, promoted differentiation of NKp46+ ILC3s (Rankin et al. 2013; Possot et al. 2011). Overexpression of constitutively active intracellular Notch partially rescued differentiation of NKp46+ ILC3s in T-bet-deficient mice. Deletion of signaling mediator RBP-Jκ leads to an impaired differentiation to NKp46+ ILC3s. AhR could be a regulator of Notch signaling in ILC3s (Rankin et al. 2013; Lee et al. 2012).
Notch signaling also regulated development of CCR6+ ILC3s in the fetal liver. Notch signaling was required for commitment to the ILC3 lineage and for the upregulation of RORγt. However, at later developmental stages, Notch blocked the differentiation of ILC3s with LTi function (Cherrier et al. 2012).
A target gene of Notch signaling is the HMG-box transcription factor TCF-1 (encoded by the Tcf7 gene). TCF-1 is an effector protein of the Wnt signaling pathway. Although expressed by all ILC3s, Tcf7-deficient mice have a selective defect in NKp46+ ILC3s. Tcf7 −/− mice were susceptible to C. rodentium infection (Mielke et al. 2013; Malhotra et al. 2013).
5.8 Runx1
Vertebrates have three genes of Runt-related transcription factors (Runx1-3), which are transcribed from two promoters and have several isoforms. All Runx proteins build heterodimers with CBFβ and recognize a common DNA motif TGPyGGTPy (Py: pyrimidine). DNA binding and heterodimerization is mediated by the Runt domain (Cohen 2009). Apart from regulating the development of multiple hematopoietic cells, Runx1 controls the early differentiation of ILC3 progenitors in the fetal liver. Mice with specific deletion of Runx1 from the proximal promoter or of CBFβ2 have impaired development of Peyer’s patches (Tachibana et al. 2011). These mice had reduced numbers of α4β +7 IL-7Rαmid cells in the fetal liver and subsequent stages of ILC3 development. Id2 and RORγt acted later in development because α4β +7 IL-7rαhi cells were affected but not IL-7rαmid cells. Interestingly, RORγt expression was reduced in mice with impaired Runx1 signaling, arguing that commitment to ILC3s lineage through upregulation of RORγt is in part mediated by Runx1 (Tachibana et al. 2011).
6 Conclusions and Perspectives
Much progress has been made during the last years to understand the basic composition of ILC lineages and subsets as well as to identify the transcriptional programs guiding their development. However, the exact molecular mechanism of their action and precise target genes are in many cases poorly understood and require more investigation.
ILCs constitute a potent first line of defense at mucosal surfaces where many pathogens invade the host’s tissues. ILCs mediate protective immune responses against bacteria, parasites, and fungi (Klose et al. 2013, 2014; Diefenbach 2013; Gladiator et al. 2013; Sonnenberg et al. 2011b; Neill et al. 2010; Moro et al. 2010). However, it remains incompletely understood how ILCs sense invading pathogens. ILC2s strongly react to epithelial cell-derived cytokines such as IL-33 whereas ILC3s are activated by myeloid cells surrounding cryptopatches or ILFs that can produce IL-23 or IL-1β (Mjosberg et al. 2011; Satpathy et al. 2013; Van Dyken et al. 2014). While activation by cues from neighboring cells may be the major route to activate ILCs, precedent from cNK cells and T/B cells would indicate that lymphocytes express immune recognition receptors allowing them to discriminate between “healthy” and “pathologically altered” cells (Diefenbach and Raulet 2003). Clearly, ILC1s and ILC3s express activating immunoreceptors previously characterized as stimulatory NK cell receptors such as NKp46, NKp44, NKG2D, and NK1.1. Genetic deletion of Ncr1 (the gene encoding NKp46) did not lead to impaired development of ILC3s and did not diminish resistance to C. rodentium infection (Satoh-Takayama et al. 2009). Interestingly, it has been documented that triggering of NKp44 on human ILC3s activates TNF production, whereas IL-22 release was mainly affected by cytokine cues (Glatzer et al. 2013). Human ILC3s may also express TLR2 and could be activated by directly sensing bacterial cell wall components (Crellin et al. 2010). However, the functional significance of direct sensing of pathogen-associated molecular patterns by ILCs remains elusive (Crellin et al. 2010). For ILC2s, immune recognition receptors have yet to be characterized. In addition to providing protection against various infections, inappropriate stimulation of ILCs has been tied to various inflammatory diseases such as inflammatory bowel diseases, psoriasis, allergic diseases, and airway hyperresponsiveness (Buonocore et al. 2010; Powell et al. 2012; Chang et al. 2011; Monticelli et al. 2011; Salimi et al. 2013). Future research into the signals leading to improper activation of ILCs may provide insights into therapeutically relevant pathways.
Another important line of research is how ILCs contribute to organ homeostasis. As mentioned throughout this review, ILCs produce soluble factors (e.g., IL-22, amphiregulin and other EGF family members) that control epithelial cell function (Sanos et al. 2011). In addition, ILCs (in particular ILC3s) have been documented to modify the function of stroma and endothelial cells (Eisenring et al. 2010; Dudakov et al. 2012). However, it remains unclear which molecular programs within nonhematopoietic cells is controlled by ILCs. Such ILC-controlled molecular pathways may be harnessed to improve organ homeostasis and tissue repair in the context of chronic and degenerative diseases.
A further crucial question is how ILCs influence adaptive immune responses. A recent study showed that T cells obtain restraining signals from class II MHC expressed on ILC3s (Hepworth et al. 2013). Specific deletion of class II MHC expression by ILC3s resulted in an autoinflammatory syndrome caused by inappropriately activated T cells. It was also demonstrated that ILC3s are required for maintenance of CD4+ memory T cells (Withers et al. 2012). In what way ILCs can shape T cell responses and how they interact with T cells requires more investigation (Hepworth et al. 2013). Another line of research has documented that ILCs may shape T cell function by affecting mononuclear phagocytes. ILC3s produced GM-CSF that conditioned mononuclear phagocytes in the intestinal lamina propria for priming of regulatory T cells (Mortha et al. 2014). In the papain-induced asthma model, IL-13 derived from ILC2s instructed dendritic cells to migrate to the lymph nodes in order to stimulate naïve T cells (Halim et al. 2014). Future research needs to flesh out the molecular circuitry by which ILCs crosstalk with components of the adaptive immune system.
Transcriptional control of helper-like ILC lineage commitment has often been compared to the one of CD4+ T helper cells. Are ILCs providing help for B cells? It has been demonstrated that ILC3s in cryptopatches and ILFs helped B cells to produce IgA (Tsuji et al. 2008). In addition, ILC3s in the marginal zone of the spleen supported T cell-independent IgA production through diverse mechanisms one being the production of GM-CSF and recruitment of neutrophils (Magri et al. 2014).
References
Akashi K, Traver D, Miyamoto T, Weissman IL (2000) A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404(6774):193–197. doi:10.1038/35004599
Aliahmad P, de la Torre B, Kaye J (2010) Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat Immunol 11(10):945–952
Aliahmad P, Kaye J (2008) Development of all CD4 T lineages requires nuclear factor TOX. J Exp Med 205(1):245–256
Aliahmad P, Seksenyan A, Kaye J (2012) The many roles of TOX in the immune system. Curr Opin Immunol 24(2):173–177. doi:10.1016/j.coi.2011.12.001
Altare F, Durandy A, Lammas D, Emile J, Lamhamedi S, Le Deist F, Drysdale P, Jouanguy E, Doffinger R, Bernaudin F, Jeppsson O, Gollob J, Meinl E, Segal A, Fischer A, Kumararatne D, Casanova J (1998) Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science 280:1432–1435
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, Bemelman WA, Mjosberg JM, Spits H (2013) Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14(3):221–229. doi:10.1038/ni.2534
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441(7090):235–238. doi:10.1038/nature04753
Boos MD, Yokota Y, Eberl G, Kee BL (2007) Mature natural killer cell and lymphoid tissue-inducing cell development requires Id2-mediated suppression of E protein activity. J Exp Med 204(5):1119–1130
Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G (2008) Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456(7221):507–510
Brickshawana A, Shapiro VS, Kita H, Pease LR (2011) Lineage(-)Sca1+ c-Kit(-)CD25+ cells are IL-33-responsive type 2 innate cells in the mouse bone marrow. J Immunol 187(11):5795–5804. doi:10.4049/jimmunol.1102242
Buchmeier NA, Schreiber RD (1985) Requirement of endogenous interferon-gamma production for resolution of Listeria monocytogenes infection. Proc Nat Acad Sci USA 82(21):7404–7408
Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464(7293):1371–1375
Camberis M, Le Gros G, Urban J Jr (2003) Animal model of Nippostrongylus brasiliensis and Heligmosomoides polygyrus. In: Coligan JE et al (ed) Current protocols in immunology, chapter 19, unit 19 12. doi:10.1002/0471142735.im1912s55
Carotta S, Pang SH, Nutt SL, Belz GT (2011) Identification of the earliest NK-cell precursor in the mouse BM. Blood 117(20):5449–5452. doi:10.1182/blood-2010-11-318956
Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M (2009) A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457(7230):722–725
Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT (2011) Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol 12(7):631–638. doi:10.1038/ni.2045
Chappaz S, Gartner C, Rodewald HR, Finke D (2011) Kit ligand and Il7 differentially regulate Peyer’s patch and lymph node development. J Immunol 185(6):3514–3519
Chen Y, Bates DL, Dey R, Chen PH, Machado AC, Laird-Offringa IA, Rohs R, Chen L (2012) DNA binding by GATA transcription factor suggests mechanisms of DNA looping and long-range gene regulation. Cell Rep 2(5):1197–1206. doi:10.1016/j.celrep.2012.10.012
Cherrier M, Sawa S, Eberl G (2012) Notch, Id2, and RORgammat sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J Exp Med 209(4):729–740. doi:10.1084/jem.20111594
Cohen MM Jr (2009) Perspectives on RUNX genes: an update. Am J Med Genet Part A 149A(12):2629–2646. doi:10.1002/ajmg.a.33021
Constantinides MG, McDonald BD, Verhoef PA, Bendelac A (2014) A committed precursor to innate lymphoid cells. Nature 508(7496):397–401. doi:10.1038/nature13047
Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, Di Santo JP, Spits H (2010) Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 33(5):752–764. doi:10.1016/j.immuni.2010.10.012
Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H (2009) Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol 10(1):66–74
Dahmane N, Ruiz i Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126(14):3089–3100
Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, Marvel J, Yoh K, Takahashi S, Prinz I, de Bernard S, Buffat L, Walzer T (2014) T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med 211(3):563–577. doi:10.1084/jem.20131560
de Jong R, Altare F, Haagen I, Elferink D, de Boer T, Vriesman P, Kabel P, Draaisma J, Dissel J, Kroon F, Casanova J, Ottenhoff T (1998) Severe mycobacterial and salmonella infections in interleukin-12 receptor-deficient patients. Science 280:1435–1438
de Pooter RF, Kee BL (2010) E proteins and the regulation of early lymphocyte development. Immunol Rev 238(1):93–109
Diefenbach A (2013) Innate lymphoid cells in the defense against infections. Eur J Microbiol Immunol 3(3):143–151. doi:10.1556/EuJMI.3.2013.3.1
Diefenbach A, Raulet DH (2003) Innate immune recognition by stimulatory immunoreceptors. Curr Opin Immunol 15(1):37–44
Dudakov JA, Hanash AM, Jenq RR, Young LF, Ghosh A, Singer NV, West ML, Smith OM, Holland AM, Tsai JJ, Boyd RL, van den Brink MR (2012) Interleukin-22 drives endogenous thymic regeneration in mice. Science 336(6077):91–95. doi:10.1126/science.1218004
Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, Sibley LD (2008) Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29(2):306–317. doi:10.1016/j.immuni.2008.05.019
Eberl G, Littman DR (2004) Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science 305(5681):248–251
Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR (2004) An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol 5(1):64–73
Eisenring M, vom Berg J, Kristiansen G, Saller E, Becher B (2010) IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol 11(11):1030–1038. doi:10.1038/ni.1947
Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, McIlgorm A, Jolin HE, McKenzie AN (2006) Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med 203(4):1105–1116. doi:10.1084/jem.20051615
Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ (1995) Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268(5211):722–726
Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR (1993) An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med 178(6):2249–2254
Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R, Muchamuel T, Hurst SD, Zurawski G, Leach MW, Gorman DM, Rennick DM (2001) IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 15(6):985–995
Frelin C, Herrington R, Janmohamed S, Barbara M, Tran G, Paige CJ, Benveniste P, Zuniga-Pflucker JC, Souabni A, Busslinger M, Iscove NN (2013) GATA-3 regulates the self-renewal of long-term hematopoietic stem cells. Nat Immunol 14(10):1037–1044. doi:10.1038/ni.2692
Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M (2013) Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of IL-12- and IL-15-Responsive IFN-gamma-Producing Cells. Immunity 38(4):769–781. doi:10.1016/j.immuni.2013.02.010
Furusawa J, Moro K, Motomura Y, Okamoto K, Zhu J, Takayanagi H, Kubo M, Koyasu S (2013) Critical role of p38 and GATA3 in natural helper cell function. J Immunol 191(4):1818–1826. doi:10.4049/jimmunol.1300379
Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH (2007) Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131(1):33–45. doi:10.1016/j.cell.2007.08.017
Gascoyne DM, Long E, Veiga-Fernandes H, de Boer J, Williams O, Seddon B, Coles M, Kioussis D, Brady HJ (2009) The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol 10(10):1118–1124
Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, Arnett KL, Blacklow SC, Aifantis I, Aster JC, Gounari F (2011) T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc Nat Acad Sci USA 108(50):20060–20065. doi:10.1073/pnas.1110230108
Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S (2013) Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol 190(2):521–525. doi:10.4049/jimmunol.1202924
Glatzer T, Killig M, Meisig J, Ommert I, Luetke-Eversloh M, Babic M, Paclik D, Bluthgen N, Seidl R, Seifarth C, Grone J, Lenarz M, Stolzel K, Fugmann D, Porgador A, Hauser A, Karlas A, Romagnani C (2013) RORgammat(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity 38(6):1223–1235. doi:10.1016/j.immuni.2013.05.013
Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, Reiner SL (2012) The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36(1):55–67. doi:10.1016/j.immuni.2011.11.016
Halim TY, Krauss RH, Sun AC, Takei F (2012a) Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 36(3):451–463. doi:10.1016/j.immuni.2011.12.020
Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F (2012b) Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity 37(3):463–474. doi:10.1016/j.immuni.2012.06.012
Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, Takei F (2014) Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40(3):425–435. doi:10.1016/j.immuni.2014.01.011
Hamada H, Hiroi T, Nishiyama Y, Takahashi H, Masunaga Y, Hachimura S, Kaminogawa S, Takahashi-Iwanaga H, Iwanaga T, Kiyono H, Yamamoto H, Ishikawa H (2002) Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J Immunol 168(1):57–64
Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, West ML, Jenq RR, Holland AM, Kappel LW, Ghosh A, Tsai JJ, Rao UK, Yim NL, Smith OM, Velardi E, Hawryluk EB, Murphy GF, Liu C, Fouser LA, Kolesnick R, Blazar BR, van den Brink MR (2012) Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 37(2):339–350. doi:10.1016/j.immuni.2012.05.028
He YW, Deftos ML, Ojala EW, Bevan MJ (1998) RORgamma t, a novel isoform of an orphan receptor, negatively regulates Fas ligand expression and IL-2 production in T cells. Immunity 9(6):797–806
Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, Mantegazza AR, Ma HL, Crawford A, Angelosanto JM, Wherry EJ, Koni PA, Bushman FD, Elson CO, Eberl G, Artis D, Sonnenberg GF (2013) Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498(7452):113–117. doi:10.1038/nature12240
Ho IC, Tai TS, Pai SY (2009) GATA3 and the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat Rev Immunol 9(2):125–135. doi:10.1038/nri2476
Hofmann M, Pircher H (2011) E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Nat Acad Sci USA 108(40):16741–16746. doi:10.1073/pnas.1107200108
Holzmann B, McIntyre BW, Weissman IL (1989) Identification of a murine Peyer’s patch–specific lymphocyte homing receptor as an integrin molecule with an alpha chain homologous to human VLA-4 alpha. Cell 56(1):37–46
Hosoya T, Maillard I, Engel JD (2010) From the cradle to the grave: activities of GATA-3 throughout T-cell development and differentiation. Immunol Rev 238(1):110–125. doi:10.1111/j.1600-065X.2010.00954.x
Hoyler T, Connor CA, Kiss EA, Diefenbach A (2013) T-bet and Gata3 in controlling type 1 and type 2 immunity mediated by innate lymphoid cells. Curr Opin Immunol 25(2):139–147. doi:10.1016/j.coi.2013.02.007
Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, Voehringer D, Busslinger M, Diefenbach A (2012) The Transcription Factor GATA-3 Controls Cell Fate and Maintenance of Type 2 Innate Lymphoid Cells. Immunity 37(4):634–648. doi:10.1016/j.immuni.2012.06.020
Huntington ND, Vosshenrich CA, Di Santo JP (2007) Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol 7(9):703–714. doi:10.1038/nri2154
Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, Menon S, Seymour B, Jackson C, Kung TT, Brieland JK, Zurawski SM, Chapman RW, Zurawski G, Coffman RL (2002) New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol 169(1):443–453
Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, Reiner SL (2005) Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol 6(12):1236–1244. doi:10.1038/ni1268
Ivanov II, Diehl GE, Littman DR (2006a) Lymphoid tissue inducer cells in intestinal immunity. Curr Top Microbiol Immunol 308:59–82
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006b) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126(6):1121–1133
Jenne CN, Enders A, Rivera R, Watson SR, Bankovich AJ, Pereira JP, Xu Y, Roots CM, Beilke JN, Banerjee A, Reiner SL, Miller SA, Weinmann AS, Goodnow CC, Lanier LL, Cyster JG, Chun J (2009) T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med 206(11):2469–2481. doi:10.1084/jem.20090525
Jetten AM (2009) Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal 7:e003. doi:10.1621/nrs.07003
Kaech SM, Cui W (2012) Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12(11):749–761. doi:10.1038/nri3307
Kamizono S, Duncan GS, Seidel MG, Morimoto A, Hamada K, Grosveld G, Akashi K, Lind EF, Haight JP, Ohashi PS, Look AT, Mak TW (2009) Nfil3/E4bp4 is required for the development and maturation of NK cells in vivo. J Exp Med 206(13):2977–2986
Kanamori Y, Ishimaru K, Nanno M, Maki K, Ikuta K, Nariuchi H, Ishikawa H (1996) Identification of novel lymphoid tissues in murine intestinal mucosa where clusters of c-kit+ IL-7R+ Thy1+ lympho-hemopoietic progenitors develop. J Exp Med 184(4):1449–1459
Karsunky H, Inlay MA, Serwold T, Bhattacharya D, Weissman IL (2008) Flk2+ common lymphoid progenitors possess equivalent differentiation potential for the B and T lineages. Blood 111(12):5562–5570. doi:10.1182/blood-2007-11-126219
Karsunky H, Merad M, Cozzio A, Weissman IL, Manz MG (2003) Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med 198(2):305–313. doi:10.1084/jem.20030323
Kee BL (2009) E and ID proteins branch out. Nat Rev Immunol 9(3):175–184
Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, Diefenbach A (2011) Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334(6062):1561–1565. doi:10.1126/science.1214914
Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, Domingues RG, Veiga-Fernandes H, Arnold SJ, Busslinger M, Dunay IR, Tanriver Y, Diefenbach A (2014) Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell 157(2):340–356. doi:10.1016/j.cell.2014.03.030
Klose CS, Hoyler T, Kiss EA, Tanriver Y, Diefenbach A (2012) Transcriptional control of innate lymphocyte fate decisions. Curr Opin Immunol 24(3):290–296. doi:10.1016/j.coi.2012.04.004
Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, Goppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A (2013) A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 494(7436):261–265. doi:10.1038/nature11813
Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G (1989) Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med 170(3):827–845
Kondo M, Weissman IL, Akashi K (1997) Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 91(5):661–672
Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, Chua K, Eidson M, Kim HJ, Im JS, Pandolfi PP, Sant’Angelo DB (2008) The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol 9(9):1055–1064. doi:10.1038/ni.1641
Ku CJ, Hosoya T, Maillard I, Engel JD (2012) GATA-3 regulates hematopoietic stem cell maintenance and cell-cycle entry. Blood 119(10):2242–2251. doi:10.1182/blood-2011-07-366070
Kumar R, Fossati V, Israel M, Snoeck HW (2008) Lin-Sca1+ kit- bone marrow cells contain early lymphoid-committed precursors that are distinct from common lymphoid progenitors. J Immunol 181(11):7507–7513
Kurebayashi S, Ueda E, Sakaue M, Patel DD, Medvedev A, Zhang F, Jetten AM (2000) Retinoid-related orphan receptor gamma (RORgamma) is essential for lymphoid organogenesis and controls apoptosis during thymopoiesis. Proc Nat Acad Sci USA 97(18):10132–10137
Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH (2011) T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat Immunol 12(1):96–104. doi:10.1038/ni.1969
Lazarevic V, Glimcher LH (2011) T-bet in disease. Nat Immunol 12(7):597–606. doi:10.1038/ni.2059
Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, Colonna M (2012) AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol 13(2):144–151. doi:10.1038/ni.2187
Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, Veldhoen M (2011) Exogenous Stimuli Maintain Intraepithelial Lymphocytes via Aryl Hydrocarbon Receptor Activation. Cell 147(3):629–640. doi:10.1016/j.cell.2011.09.025
Liang HE, Reinhardt RL, Bando JK, Sullivan BM, Ho IC, Locksley RM (2012) Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat Immunol 13(1):58–66. doi:10.1038/ni.2182
Lin H, Grosschedl R (1995) Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature 376(6537):263–267. doi:10.1038/376263a0
Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A (2007) Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26(4):503–517
Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D, Dalod M, Littman DR, Vivier E, Tomasello E (2009) Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol 10(1):75–82
Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T (2013) The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14(12):1294–1301. doi:10.1038/ni.2744
Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, Barra CM, Comerma L, Chudnovskiy A, Gentile M, Llige D, Cols M, Serrano S, Arostegui JI, Juan M, Yague J, Merad M, Fagarasan S, Cerutti A (2014) Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol. doi:10.1038/ni.2830
Male V, Nisoli I, Kostrzewski T, Allan DS, Carlyle JR, Lord GM, Wack A, Brady HJ (2014) The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med 211(4):635–642. doi:10.1084/jem.20132398
Malhotra N, Narayan K, Cho OH, Sylvia KE, Yin C, Melichar H, Rashighi M, Lefebvre V, Harris JE, Berg LJ, Kang J (2013) A network of high-mobility group box transcription factors programs innate interleukin-17 production. Immunity 38(4):681–693. doi:10.1016/j.immuni.2013.01.010
McDonald KG, McDonough JS, Dieckgraefe BK, Newberry RD (2010) Dendritic cells produce CXCL13 and participate in the development of murine small intestine lymphoid tissues. Am J Pathol 176(5):2367–2377. doi:10.2353/ajpath.2010.090723
McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, Voehringer D, McKenzie AN, Neurath MF, Pflanz S, Wirtz S (2013) Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 39(2):357–371. doi:10.1016/j.immuni.2013.07.018
McIntosh BE, Hogenesch JB, Bradfield CA (2010) Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu Rev Physiol 72:625–645. doi:10.1146/annurev-physiol-021909-135922
Mebius RE, Miyamoto T, Christensen J, Domen J, Cupedo T, Weissman IL, Akashi K (2001) The fetal liver counterpart of adult common lymphoid progenitors gives rise to all lymphoid lineages, CD45+CD4+CD3- cells, as well as macrophages. J Immunol 166(11):6593–6601
Mebius RE, Rennert P, Weissman IL (1997) Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 7(4):493–504
Mielke LA, Groom JR, Rankin LC, Seillet C, Masson F, Putoczki T, Belz GT (2013) TCF-1 controls ILC2 and NKp46+RORgammat+ innate lymphocyte differentiation and protection in intestinal inflammation. J Immunol 191(8):4383–4391. doi:10.4049/jimmunol.1301228
Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, Te Velde AA, Fokkens WJ, van Drunen CM, Spits H (2012) The Transcription Factor GATA3 Is Essential for the Function of Human Type 2 Innate Lymphoid Cells. Immunity 37(4):649–659. doi:10.1016/j.immuni.2012.08.015
Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, Spits H (2011) Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol 12(11):1055–1062. doi:10.1038/ni.2104
Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12(11):1045–1054. doi:10.1031/ni.2131
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S (2010) Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463(7280):540–544
Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, Merad M (2014) Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343(6178):1249288. doi:10.1126/science.1249288
Mortha A, Diefenbach A (2011) Natural killer cell receptor-expressing innate lymphocytes: more than just NK cells. Cell Mol Life Sci: CMLS 68(21):3541–3555. doi:10.1007/s00018-011-0803-6
Murre C (2005) Helix-loop-helix proteins and lymphocyte development. Nat Immunol 6(11):1079–1086. doi:10.1038/ni1260
Naiche LA, Harrelson Z, Kelly RG, Papaioannou VE (2005) T-box genes in vertebrate development. Annu Rev Genet 39:219–239. doi:10.1146/annurev.genet.39.073003.105925
Nechanitzky R, Akbas D, Scherer S, Gyory I, Hoyler T, Ramamoorthy S, Diefenbach A, Grosschedl R (2013) Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nat Immunol 14(8):867–875. doi:10.1038/ni.2641
Neill DR, McKenzie AN (2011) Nuocytes and beyond: new insights into helminth expulsion. Trends Parasitol 27(5):214–221. doi:10.1016/j.pt.2011.01.001
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie AN (2010) Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464(7293):1367–1370
Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, Thornton EE, Krummel MF, Chawla A, Liang HE, Locksley RM (2013) Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502(7470):245–248. doi:10.1038/nature12526
Nutt SL, Heavey B, Rolink AG, Busslinger M (1999) Commitment to the B-lymphoid lineage depends on the transcription factor Pax5 [see comments]. Nature 401(6753):556–562
O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH (2006) T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol 7(5):507–516
Okamura RM, Sigvardsson M, Galceran J, Verbeek S, Clevers H, Grosschedl R (1998) Redundant regulation of T cell differentiation and TCRalpha gene expression by the transcription factors LEF-1 and TCF-1. Immunity 8(1):11–20
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478(7368):197–203. doi:10.1038/nature10491
Paust S, Gill HS, Wang BZ, Flynn MP, Moseman EA, Senman B, Szczepanik M, Telenti A, Askenase PW, Compans RW, von Andrian UH (2010) Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat Immunol 11(12):1127–1135. doi:10.1038/ni.1953
Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL (2003) Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302(5647):1041–1043. doi:10.1126/science.1090148
Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, Sun R, Yokoyama WM, Tian Z (2013) Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest 123(4):1444–1456. doi:10.1172/JCI66381
Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, Ouyang W, Neurath MF, Becker C (2009) STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 206(7):1465–1472
Possot C, Schmutz S, Chea S, Boucontet L, Louise A, Cumano A, Golub R (2011) Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nat Immunol 12(10):949–958. doi:10.1038/ni.2105
Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E, Jackson I, Hashim A, Curtis MA, Jenner RG, Howard JK, Parkhill J, Macdonald TT, Lord GM (2012) The Transcription Factor T-bet Regulates Intestinal Inflammation Mediated by Interleukin-7 Receptor(+) Innate Lymphoid Cells. Immunity 37(4):674–684. doi:10.1016/j.immuni.2012.09.008
Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM (2010) Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Nat Acad Sci USA 107(25):11489–11494
Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L (2011) The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 36:92–104. doi:10.1016/j.immuni.2011.11.011
Randall TD, Weissman IL (1998) Characterization of a population of cells in the bone marrow that phenotypically mimics hematopoietic stem cells: resting stem cells or mystery population? Stem Cells 16(1):38–48. doi:10.1002/stem.160038
Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, McKenzie AN, Carotta S, Nutt SL, Belz GT (2013) The transcription factor T-bet is essential for the development of NKp46(+) innate lymphocytes via the Notch pathway. Nat Immunol 14(4):389–395. doi:10.1038/ni.2545
Rannug A, Rannug U, Rosenkranz HS, Winqvist L, Westerholm R, Agurell E, Grafstrom AK (1987) Certain photooxidized derivatives of tryptophan bind with very high affinity to the Ah receptor and are likely to be endogenous signal substances. J Biol Chem 262(32):15422–15427
Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, Mitchell AJ, Tay SS, Jain R, Forbes-Blom E, Chen X, Tong PL, Bolton HA, Artis D, Paul WE, Fazekas de St Groth B, Grimbaldeston MA, Le Gros G, Weninger W (2013) Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol 14(6):564–573. doi:10.1038/ni.2584
Saenz SA, Siracusa MC, Monticelli LA, Ziegler CG, Kim BS, Brestoff JR, Peterson LW, Wherry EJ, Goldrath AW, Bhandoola A, Artis D (2013) IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J Exp Med 210(9):1823–1837. doi:10.1084/jem.20122332
Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF Jr, Tocker JE, Budelsky AL, Kleinschek MA, Kastelein RA, Kambayashi T, Bhandoola A, Artis D (2010) IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 464(7293):1362–1366. doi:10.1038/nature08901
Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST, McKenzie AN, Fallon PG, Ogg GS (2013) A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med 210(13):2939–2950. doi:10.1084/jem.20130351
Samson SI, Richard O, Tavian M, Ranson T, Vosshenrich CA, Colucci F, Buer J, Grosveld F, Godin I, Di Santo JP (2003) GATA-3 promotes maturation, IFN-gamma production, and liver-specific homing of NK cells. Immunity 19(5):701–711
Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, Diefenbach A (2009) RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol 10(1):83–91
Sanos SL, Vonarbourg C, Mortha A, Diefenbach A (2011) Control of epithelial cell function by interleukin-22-producing RORgammat+ innate lymphoid cells. Immunology 132(4):453–465. doi:10.1111/j.1365-2567.2011.03410.x
Satoh-Takayama N, Dumoutier L, Lesjean-Pottier S, Ribeiro VS, Mandelboim O, Renauld JC, Vosshenrich CA, Di Santo JP (2009) The natural cytotoxicity receptor NKp46 is dispensable for IL-22-mediated innate intestinal immune defense against Citrobacter rodentium. J Immunol 183(10):6579–6587
Satoh-Takayama N, Lesjean-Pottier S, Vieira P, Sawa S, Eberl G, Vosshenrich CA, Di Santo JP (2010) IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. J Exp Med 207(2):273–280
Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, Eberl G, Di Santo JP (2008) Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29(6):958–970
Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, Wu X, Thomas SR, Lee WL, Turkoz M, McDonald KG, Meredith MM, Song C, Guidos CJ, Newberry RD, Ouyang W, Murphy TL, Stappenbeck TS, Gommerman JL, Nussenzweig MC, Colonna M, Kopan R, Murphy KM (2013) Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol 14(9):937–948. doi:10.1038/ni.2679
Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, Lantz O, Bendelac A (2008) The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity 29(3):391–403. doi:10.1016/j.immuni.2008.07.011
Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP, Eberl G (2010) Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science 330(6004):665–669. doi:10.1126/science.1194597
Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, Cua D, Di Santo JP, Eberl G (2011) RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol 12(4):320–326. doi:10.1038/ni.2002
Schulthess J, Meresse B, Ramiro-Puig E, Montcuquet N, Darche S, Begue B, Ruemmele F, Combadiere C, Di Santo JP, Buzoni-Gatel D, Cerf-Bensussan N (2012) Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity 37(1):108–121. doi:10.1016/j.immuni.2012.05.013
Sciume G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, Spencer SP, Wilhelm C, Poholek AC, Vahedi G, Kanno Y, Belkaid Y, O’Shea JJ (2012a) Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med 209(13):2331–2338. doi:10.1084/jem.20122097
Sciume G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, Spencer SP, Wilhelm C, Poholek AC, Vahedi G, Kanno Y, Belkaid Y, O’Shea JJ (2012b) Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med 209(13):2331–2338. doi:10.1084/jem.20122097
Serafini N, Klein Wolterink RG, Satoh-Takayama N, Xu W, Vosshenrich CA, Hendriks RW, Di Santo JP (2014) Gata3 drives development of RORgammat+ group 3 innate lymphoid cells. J Exp Med 211(2):199–208. doi:10.1084/jem.20131038
Shi FD, Ljunggren HG, La Cava A, Van Kaer L (2011) Organ-specific features of natural killer cells. Nat Rev Immunol 11(10):658–671. doi:10.1038/nri3065
Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, Zhong C, Chase JM, Rothman PB, Yu J, Riley JK, Zhu J, Tian Z, Yokoyama WM (2014) Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife 3:e01659. doi:10.7554/eLife.01659
Songhet P, Barthel M, Stecher B, Muller AJ, Kremer M, Hansson GC, Hardt WD (2011) Stromal IFN-gammaR-signaling modulates goblet cell function during Salmonella Typhimurium infection. PLoS ONE 6(7):e22459. doi:10.1371/journal.pone.0022459
Sonnenberg GF, Fouser LA, Artis D (2011a) Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol 12(5):383–390. doi:10.1038/ni.2025
Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, Tardif MR, Sathaliyawala T, Kubota M, Farber DL, Collman RG, Shaked A, Fouser LA, Weiner DB, Tessier PA, Friedman JR, Kiyono H, Bushman FD, Chang KM, Artis D (2012) Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336(6086):1321–1325. doi:10.1126/science.1222551
Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D (2011b) CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34(1):122–134. doi:10.1016/j.immuni.2010.12.009
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E (2013) Innate lymphoid cells - a proposal for uniform nomenclature. Nat Rev Immunol 13(2):145–149. doi:10.1038/nri3365
Spits H, Cupedo T (2012) Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol 30:647–675. doi:10.1146/annurev-immunol-020711-075053
Spits H, Di Santo JP (2011) The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 12(1):21–27
Spooner CJ, Cheng JX, Pujadas E, Laslo P, Singh H (2009) A recurrent network involving the transcription factors PU.1 and Gfi1 orchestrates innate and adaptive immune cell fates. Immunity 31(4):576–586. doi:10.1016/j.immuni.2009.07.011
Spooner CJ, Lesch J, Yan D, Khan AA, Abbas A, Ramirez-Carrozzi V, Zhou M, Soriano R, Eastham-Anderson J, Diehl L, Lee WP, Modrusan Z, Pappu R, Xu M, DeVoss J, Singh H (2013) Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol 14(12):1229–1236. doi:10.1038/ni.2743
Stevens EA, Mezrich JD, Bradfield CA (2009) The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology 127(3):299–311. doi:10.1111/j.1365-2567.2009.03054.x
Sun JC, Beilke JN, Lanier LL (2009) Adaptive immune features of natural killer cells. Nature 457(7229):557–561
Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, Littman DR (2000) Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288(5475):2369–2373
Suzuki Y, Orellana MA, Schreiber RD, Remington JS (1988) Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240(4851):516–518
Szabo SJ, Kim ST, Costa GL, Zhang XK, Fathman CG, Glimcher LH (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100(6):655–669
Tachibana M, Tenno M, Tezuka C, Sugiyama M, Yoshida H, Taniuchi I (2011) Runx1/Cbfbeta2 complexes are required for lymphoid tissue inducer cell differentiation at two developmental stages. J Immunol 186(3):1450–1457. doi:10.4049/jimmunol.1000162
Takashima Y, Suzuki A (2013) Regulation of organogenesis and stem cell properties by T-box transcription factors. Cell Mol Life Sci : CMLS 70(20):3929–3945. doi:10.1007/s00018-013-1305-5
Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O’Shea JJ (2009) Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med 206(1):35–41
Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, Yagita H, Kinoshita K, Okumura K, Smyth MJ (2005) TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 105(5):2082–2089. doi:10.1182/blood-2004-08-3262
Tanriver Y, Diefenbach A (2014) Transcription factors controlling development and function of innate lymphoid cells. Int Immunol 26(3):119–128. doi:10.1093/intimm/dxt063
Ting CN, Olson MC, Barton KP, Leiden JM (1996) Transcription factor GATA-3 is required for development of the T-cell lineage. Nature 384(6608):474–478
Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, Gapin L, Glimcher LH (2004) T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity 20(4):477–494
Treiber T, Mandel EM, Pott S, Gyory I, Firner S, Liu ET, Grosschedl R (2010) Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription- independent poising of chromatin. Immunity 32(5):714–725. doi:10.1016/j.immuni.2010.04.013
Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, Itoh K, Littman DR, Fagarasan S (2008) Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29(2):261–271
Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, Panzer U, Helmby H, Stockinger B (2013) IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med 210(13):2951–2965. doi:10.1084/jem.20130071
Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F (2006) Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25(2):309–318
van de Pavert SA, Mebius RE (2010) New insights into the development of lymphoid tissues. Nat Rev Immunol 10(9):664–674. doi:10.1038/nri2832
Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, McKenzie AN, Krummel MF, Liang HE, Locksley RM (2014) Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity 40(3):414–424. doi:10.1016/j.immuni.2014.02.003
Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, Oosterwegel M, Wilson A, MacDonald HR, Clevers H (1995) An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature 374(6517):70–74
Verykokakis M, Krishnamoorthy V, Iavarone A, Lasorella A, Sigvardsson M, Kee BL (2013) Essential Functions for ID Proteins at Multiple Checkpoints in Invariant NKT Cell Development. J Immunol 191(12):5973–5983. doi:10.4049/jimmunol.1301521
Villey I, de Chasseval R, de Villartay JP (1999) RORgammaT, a thymus-specific isoform of the orphan nuclear receptor RORgamma / TOR, is up-regulated by signaling through the pre-T cell receptor and binds to the TEA promoter. Eur J Immunol 29(12):4072–4080
Vonarbourg C, Diefenbach A (2012) Multifaceted roles of interleukin-7 signaling for the development and function of innate lymphoid cells. Semin Immunol 24(3):165–174. doi:10.1016/j.smim.2012.03.002
Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, Honig M, Pannicke U, Schwarz K, Ware CF, Finke D, Diefenbach A (2010) Regulated Expression of Nuclear Receptor RORgammat Confers Distinct Functional Fates to NK Cell Receptor-Expressing RORgammat(+) Innate Lymphocytes. Immunity 33(5):736–751
Vosshenrich CA, Di Santo JP (2013) Developmental programming of natural killer and innate lymphoid cells. Curr Opin Immunol 25(2):130–138. doi:10.1016/j.coi.2013.02.002
Vosshenrich CA, Garcia-Ojeda ME, Samson-Villeger SI, Pasqualetto V, Enault L, Richard-Le Goff O, Corcuff E, Guy-Grand D, Rocha B, Cumano A, Rogge L, Ezine S, Di Santo JP (2006) A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol 7(11):1217–1224
Wagner N, Lohler J, Kunkel EJ, Ley K, Leung E, Krissansen G, Rajewsky K, Muller W (1996) Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature 382(6589):366–370. doi:10.1038/382366a0
Walker JA, Barlow JL, McKenzie AN (2013) Innate lymphoid cells - how did we miss them? Nat Rev Immunol 13(2):75–87. doi:10.1038/nri3349
Wallace VA (1999) Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol : CB 9(8):445–448
Walzer T, Blery M, Chaix J, Fuseri N, Chasson L, Robbins SH, Jaeger S, Andre P, Gauthier L, Daniel L, Chemin K, Morel Y, Dalod M, Imbert J, Pierres M, Moretta A, Romagne F, Vivier E (2007) Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Nat Acad Sci USA 104(9):3384–3389
Wang ZE, Reiner SL, Zheng S, Dalton DK, Locksley RM (1994) CD4+ effector cells default to the Th2 pathway in interferon gamma-deficient mice infected with Leishmania major. J Exp Med 179(4):1367–1371
Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, Bhandoola A (2011) A critical role for TCF-1 in T-lineage specification and differentiation. Nature 476(7358):63–68. doi:10.1038/nature10279
Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, Sparwasser T, Helmby H, Stockinger B (2011) An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol 12(11):1071–1077. doi:10.1038/ni.2133
Withers DR, Gaspal FM, Mackley EC, Marriott CL, Ross EA, Desanti GE, Roberts NA, White AJ, Flores-Langarica A, McConnell FM, Anderson G, Lane PJ (2012) Cutting edge: lymphoid tissue inducer cells maintain memory CD4 T cells within secondary lymphoid tissue. J Immunol 189(5):2094–2098. doi:10.4049/jimmunol.1201639
Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R (2004) IL-22 increases the innate immunity of tissues. Immunity 21(2):241–254
Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, Barlow JL, Neill DR, Panova V, Koch U, Radtke F, Hardman CS, Hwang YY, Fallon PG, McKenzie AN (2012) Transcription factor RORalpha is critical for nuocyte development. Nat Immunol 13:229–236. doi:10.1038/ni.2208
Yagi R, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, Hu G, Barron L, Sharma S, Nakayama T, Belkaid Y, Zhao K, Zhu J (2014) The Transcription Factor GATA3 Is Critical for the Development of All IL-7Ralpha-Expressing Innate Lymphoid Cells. Immunity 40(3):378–388. doi:10.1016/j.immuni.2014.01.012
Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, Oppenheim JJ (1999) Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286(5439):525–528
Yang Q, Monticelli LA, Saenz SA, Chi AW, Sonnenberg GF, Tang J, De Obaldia ME, Bailis W, Bryson JL, Toscano K, Huang J, Haczku A, Pear WS, Artis D, Bhandoola A (2013) T cell factor 1 is required for group 2 innate lymphoid cell generation. Immunity 38(4):694–704. doi:10.1016/j.immuni.2012.12.003
Yang Q, Saenz SA, Zlotoff DA, Artis D, Bhandoola A (2011) Cutting edge: Natural helper cells derive from lymphoid progenitors. J Immunol 187(11):5505–5509. doi:10.4049/jimmunol.1102039
Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, Gruss P (1999) Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature 397(6721):702–706
Yoshida H, Kawamoto H, Santee SM, Hashi H, Honda K, Nishikawa S, Ware CF, Katsura Y, Nishikawa SI (2001) Expression of alpha(4)beta(7) integrin defines a distinct pathway of lymphoid progenitors committed to T cells, fetal intestinal lymphotoxin producer, NK, and dendritic cells. J Immunol 167(5):2511–2521
Yucel R, Karsunky H, Klein-Hitpass L, Moroy T (2003) The transcriptional repressor Gfi1 affects development of early, uncommitted c-Kit+ T cell progenitors and CD4/CD8 lineage decision in the thymus. J Exp Med 197(7):831–844. doi:10.1084/jem.20021417
Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, Mosmann TR (2006) Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 314(5806):1746. doi:10.1126/science.1133715
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA (2007) Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27(4):647–659
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA (2008) Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29(6):947–957
Zheng W, Flavell RA (1997) The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89(4):587–596
Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W (2008) Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14(3):282–289
Zhong JF, Zhao Y, Sutton S, Su A, Zhan Y, Zhu L, Yan C, Gallaher T, Johnston PB, Anderson WF, Cooke MP (2005) Gene expression profile of murine long-term reconstituting vs. short-term reconstituting hematopoietic stem cells. Proc Nat Acad Sci USA 102(7):2448–2453. doi:10.1073/pnas.0409459102
Zhu J, Jankovic D, Grinberg A, Guo L, Paul WE (2006) Gfi-1 plays an important role in IL-2-mediated Th2 cell expansion. Proc Nat Acad Sci USA 103(48):18214–18219. doi:10.1073/pnas.0608981103
Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, Urban JF Jr, Guo L, Paul WE (2004) Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol 5(11):1157–1165. doi:10.1038/ni1128
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Klose, C.S.N., Diefenbach, A. (2014). Transcription Factors Controlling Innate Lymphoid Cell Fate Decisions. In: Ellmeier, W., Taniuchi, I. (eds) Transcriptional Control of Lineage Differentiation in Immune Cells. Current Topics in Microbiology and Immunology, vol 381. Springer, Cham. https://doi.org/10.1007/82_2014_381
Download citation
DOI: https://doi.org/10.1007/82_2014_381
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-07394-1
Online ISBN: 978-3-319-07395-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)