
Abstract
Since its identification over a hundred years ago, the neurotransmitter acetylcholine (ACh) has proven to play an essential role in supporting many diverse functions. Some well-characterized functions include: chemical transmission at the neuromuscular junction; autonomic function in the peripheral nervous system; and, sustained attention, sleep/wake regulation, and learning and memory within the central nervous system. Within the brain, major cholinergic projection pathways from the basal forebrain and the brainstem support these centrally mediated processes, and dysregulation of the cholinergic system is implicated in cognitive decline associated with aging and dementias including Alzheimer’s disease. ACh exerts its effects by binding to two different membrane-bound receptor classes: (1) G‑protein coupled muscarinic acetylcholine receptors (mAChRs), and (2) ligand-gated nicotinic acetylcholine receptors (nAChRs). These receptor systems are described in detail within this chapter along with discussion on the successes and failures of synthetic ligands designed to selectively target receptor subtypes for treating brain disorders. New molecular approaches and advances in our understanding of the target biology combined with opportunities to re-purpose existing cholinergic drugs for new indications continue to highlight the exciting opportunities for modulating this system for therapeutic purposes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Cholinergic System Overview
1.1 Acetylcholine
The activity of a chemical substance that reduced heart rate frequency was first observed by the pharmacologist Otto Loewi in 1921 and was later identified as acetylcholine (ACh) by Henry Dale. Subsequently, ACh was shown to play a role in many essential functions including (1) chemical transmission at the neuromuscular junction, (2) autonomic function in the peripheral nervous system, and (3) centrally mediated cognitive processes such as attention, learning, and memory. ACh is synthesized from choline and acetyl-CoA through the enzyme choline acetyltransferase that occurs in different neurons as well as non-neuronal cells and is released locally (Wessler and Kirkpatrick 2008; Schubert et al. 2012; Beckmann and Lips 2013). ACh is hydrolyzed by acetylcholinesterase enzymes, which are abundant in the synaptic cleft, after its release from presynaptic neurons.
Two major cholinergic projection pathways occur in the brain (Fig. 1): (1) The magnocellular basal forebrain cholinergic system, which includes the nucleus basalis of Meynert, the medial septal nucleus, and the vertical and horizontal limbs of the diagonal band of Broca. The basal forebrain cholinergic system has extensive projections to neocortical regions, as well as to basolateral amygdala and olfactory bulb, hippocampus, and entorhinal cortices. (2) The brainstem cholinergic system which includes the pedunculopontine nucleus and the laterodorsal pontine tegmental nucleus and projects primarily to thalamic structures and to basal forebrain regions.

Schematic of major cholinergic projections in the human brain
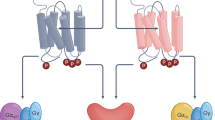
ACh exerts its effects by binding to two different membrane-bound receptor classes: (1) G protein-coupled muscarinic acetylcholine receptors (mAChRs), present in both the peripheral and central nervous systems, and (2) ligand-gated nicotinic acetylcholine receptors (nAChRs), which function in the peripheral and central nervous systems, in the neurons from the parasympathetic ganglia, at the neuromuscular junction, as well as in non-neuronal cells (Fig. 2). These receptor systems will be described in more detail in the following text.

Illustration of mAChR subtypes and a typical structure of a heteromeric nAChR. Schematic representation of the muscarinic and nicotinic receptors inserted in the membrane are represented in panels (a) and (c). Crystal structures of the muscarinic and nicotinic receptors in a side view are shown for comparison in panels (b) and (d). Structures correspond to publications Maeda et al. (2019) and Morales-Perez et al. (2016)
1.2 Muscarinic ACh Receptors (mAChRs)
Muscarinic acetylcholine receptors (mAChRs) are class A G protein-coupled receptors (GPCRs) and exist as five distinct subtypes (M1–M5) expressed in different brain regions and the periphery (Kruse et al. 2014). M1, M2, M3, M4, and M5 mAChR subtypes are encoded by separate genes (CHRM1–CHRM5) and are classified based on their tissue localization, molecular conformation, and activation of different intracellular signaling pathways. The M1, M3, and M5 mAChRs are excitatory and couple to Gq proteins, activating phospholipase C and subsequently mobilizing intracellular calcium, whereas M2 and M4 mAChRs act in an inhibitory manner by coupling to Gi/o proteins and inhibiting adenylate cyclase. M1–M4 mAChR subtypes have all been crystallized in an inactive state (Haga et al. 2012; Kruse et al. 2012; Thal et al. 2016). The M5 mAChR subtype is the most recent mAChR to be cloned (Bender et al. 2019).
M1–M5 mAChRs are integral membrane proteins with seven transmembrane segments that form a pocket in which ACh can penetrate from the extracellular space, bind with at high affinity, and activate intracellular GTP binding regulatory proteins (G proteins). Heteromeric G proteins consist of an α subunit, a β subunit, and a γ subunit, and when ACh or other agonists bind to the extracellular mAChR binding site, it causes a conformational change in the receptor that promotes the α subunit to separate from the βγ moiety which then binds to effector proteins engaging multiple signaling cascades that amplify the initial ligand-receptor interaction. Additionally, following the ligand-receptor interaction, internalization of the receptor can occur through phosphorylation of the G proteins by intracellular kinases that lead to their uncoupling from the receptor and aim to regulate the ensuing biological response.
Additionally, elegant cryo-electron microscopy of the M1 and M2 mAChRs by Maeda and colleagues has demonstrated unique features of the receptors and their interactions with G proteins that provide a provocative basis for how they may impact their signaling properties (Maeda et al. 2019). Whereas it was originally thoughts that GPCRs are formed by a single protein which spans the membrane seven times, advances in the field of GABAB receptors have highlighted that functional receptors can result from the dimerization of two subunits (Kuner et al. 1999). Although evidence of heterodimers between the M2 and M3 proteins were identified, several outstanding questions about the quaternary structure of the mAChRs still remain (Marsango et al. 2018).
1.3 Nicotinic ACh Receptors (nAChRs)
Neuronal nicotinic acetylcholine receptors (nAChRs) belong to the superfamily of ligand-gated ion channels (LGIC). These highly specialized membrane proteins consist of a high-affinity binding site for a given ligand (e.g., ACh) and a pore-forming domain which is normally closed and opens upon binding of the ligand. Multiple forms of LGICs can be identified and classified according to their structural determinant, pharmacology, or ionic selectivity. Work conducted initially at the neuromuscular junction revealed that nAChRs are formed by the assembly of five subunits around a central ionic pore, and each subunit spans the membrane four times with N- and C-terminal ends lying in the extracellular space (Bertrand et al. 2015). Maintained throughout evolution, these structural features can already be identified in LGICs expressed by bacteria which present a striking similarity with mammalian nAChRs (Hilf and Dutzler 2009; Nemecz et al. 2016). Today, 16 genes (α1–α10, β1–β4, γ, δ, ε; note that α8 was identified only in chicken) encoding nAChR subunits have been identified in the mammalian genome (Schaaf 2014; Bertrand et al. 2015). Although the nAChR genes show a high degree of conservation, variations exist among species, and functional differences have already been identified between rodents and humans, for example (Paradiso et al. 2001; Curtis et al. 2002; Shorey-Kendrick et al. 2015).
While crystallography and high-resolution electron microscopy brought our understanding of the structural features of LGIC to an entirely new level, these studies have also highlighted the underlying complexity of nAChRs (Morales-Perez et al. 2016; Walsh et al. 2018). One complexity is that a single receptor results from the assembly of 5 subunits, and given the possibility offered by the combinations of the 16 subunits, multiple forms of receptors have been identified (Bertrand et al. 2015). In their simplest form, nAChRs are homomeric or comprised of five identical subunits, such as the α7 nAChRs; however, most generally, nAChRs are composed of subunits of at least two or more forms such as the α4β2 which is the major brain nAChR subtype (Fig. 2). Moreover, even in a receptor containing two types of subunits (e.g., α and β), the α versus β ratio has been shown to yield structural differences in the protein interfaces and also to modify the receptor properties (Zwart and Vijverberg 1998; Nelson et al. 2003; Zhou et al. 2003; Tapia et al. 2007; Walsh et al. 2018). The α4β2 nAChR demonstrates high (nM) binding affinity to nicotine and ACh, whereas the α7 nAChR shows lower (μM) sensitivity to these same ligands. The α4β2nAChR can occur with two distinct stoichiometries: (α4)2 and (β2)3 subunits (high sensitivity state) or (α4)3 and (β2)2 subunits (low sensitivity state) that are characterized by their binding affinity for ACh (Zwart and Vijverberg 1998; Zhou et al. 2003). Interestingly, functional properties associated with differences in stoichiometric ratio are not just an in vitro phenomenon, but also have been observed in vivo (Lamotte d’Incamps et al. 2018).
To understand the role and contribution of nAChRs in brain function, it is therefore indispensable to know their precise brain localization as well as their structural arrangement. For example, while some receptors will be composed of α4 and β2, the introduction of an additional subunit in the receptor complex, such as α5 or other, will be accompanied by modifications in the functional and pharmacological properties of the receptors (Brown et al. 2007; Kuryatov et al. 2008; Grady et al. 2010; Besson et al. 2016). Moreover, as multiple studies have already highlighted, a single cell can express more than one receptor subtype, and therefore it is necessary to understand the precise receptor distribution to be able to evaluate their functional outcome (Klink et al. 2001).
2 Localization of AChRs in the Central Nervous System
2.1 mAChRs
The mAChR system is widespread throughout the brain and periphery; however, for the purposes of this chapter, we will focus on the mAChR brain expression and its related functions (Table 1). mAChRs show the densest expression within the caudate nucleus and putamen regions. The M1 and M2 subtypes are the most abundant mAChR subtypes in the brain; however, the M1, M2, and M4 subtypes have all received a great deal of attention as drug targets for neuropsychiatric and neurological disorders.
The M1 mAChR represents approximately 35–60% of the total mAChRs in the human brain (Volpicelli and Levey 2004). It is localized postsynaptically and is prominently expressed in the cerebral cortex, including frontal, temporal, parietal, and occipital cortices, and also is abundant in the hippocampus, striatum, amygdala, and thalamic brain regions (Levey et al. 1991; Crook et al. 2001). The M1 mAChR subtype is involved in learning and memory functions, and selective activation of the M1 mAChR has been investigated for its therapeutic potential as a cognitive-enhancing agent in disorders such as Alzheimer’s disease in which there is associated decline of these processes (Wess et al. 2007; Scarr 2012). Initial attempts to develop selective agonists to the M1 mAChR highlighted the challenge with the highly conserved homology that exists within the orthosteric binding site of mAChRs making it difficult to design selective subtype-specific ligands. Compounds such as xanomeline with M1- and M4-preferring agonist activity were tested for cognitive-enhancing potential, but dose-limiting side effects attributed to “off-target” parasympathomimetic activity at peripheral M2 and M3 mAChR subtypes were considered a limitation to their investigation (Bymaster et al. 2003; Wess et al. 2007).
The M2 mAChR is a cholinergic inhibitory autoreceptor localized on presynaptic terminals in many regions throughout the brain. M2 mAChRs are present on large cholinergic interneurons in the striatum and have high expression in cerebellum, thalamus, and nucleus basalis of Meynert along with some limbic structures, e.g., amygdala and hippocampus. Stimulating M2 mAChRs decreases cholinergic neurotransmission and impairs memory. This has led to investigation of M2 mAChR antagonists as an approach to restore ACh release and improve learning and memory for cognitive-impairing diseases in which the cholinergic system is compromised (Billard et al. 1995; Langmead et al. 2008). The limitations to this approach thus far have included the challenges associated with developing M2 mAChR selective antagonists that do not have off-target activity at other mAChRs (as described above for M1) but, also importantly, that do not engender liabilities in peripheral organs in which high levels of expression of M2 mAChRs have been shown (e.g., from M2 mAChR expression in the heart and consequently cardiovascular complications).
Relative to M1 and M2, both M3 and M4 mAChR subtypes show much lower expression in the brain. For example, the M3 subtype is estimated to constitute only 5–10% of all mAChRs (Levey et al. 1994). Both M3 and M4 mAChRs are involved in neurotransmitter regulation and are prominent in hippocampal subregions, cerebral cortex, and striatum. In the brain, the M3 subtype has a functional role in regulating insulin secretion making it an interesting target to investigate for its role in type 2 diabetes mellitus (Gautam et al. 2006). However, the primary functions attributed to the M3 subtype are peripheral, e.g., smooth muscle contraction, exocrine secretion (e.g., saliva), and endocrine function (Matsui et al. 2000).
The distribution of the M4 subtype largely overlaps with that of the M1 and M3 subtypes, and it functions primarily as an inhibitory autoreceptor that decreases ACh release. Additionally, with its expression in striatum, the M4 subtype has demonstrated a regulatory control on dopamine-mediated functions in this region. Genetic deletion of the M4 subtype receptor in mice results in increased locomotor stimulation in response to dopamine agonists (e.g., amphetamine, cocaine) (Wess et al. 2007). An exciting area of ongoing research is to target the M4 subunit as a therapeutic approach for movement disorders such as Parkinson’s disease (Langmead et al. 2008).
The M5 mAChR shows low expression in the brain, but is localized in dopamine-rich areas such as the ventral tegmental area and the substantia nigra suggesting that it could play a role in reward processing and movement, respectively. It also has been identified in the hippocampus, outermost layers of the cerebral cortex, and caudate putamen areas.
Developing a better understanding of the receptor distribution with the highest possible granularity including the homomer and/or heterodimer expression is expected to offer new alternatives to develop molecules displaying enough specificity to target a precise and well-localized subtype.
2.2 nAChRs
The homomeric α7 and heteromeric α4β2 nAChRs are the most prominent nAChRs in the mammalian brain, and both are involved in diverse functions. The α4β2 nAChRs have been identified in all layers of the cerebral cortex, hippocampal subregions, substantia nigra, and ventral tegmental area (Gotti et al. 2007). With high expression in dopamine-rich regions, the α4β2 nAChR has been studied for its involvement in hedonic processes and addiction particularly with regard to tobacco smoking. To this end, the α4β2 nAChR partial agonist, varenicline, has been brought to the market as a smoking cessation product.
High expression of the α7 nAChR in hippocampal subregions (CA1, CA3, dentate gyrus), the prefrontal cortex (layers I–VI), and also subcortical structures has directed attention to its role in cognitive processes (e.g., long-term memory, working memory, sensory gating). The α7 nAChR is localized presynaptically, postsynaptically, and perisynaptically contributing to its wide-ranging effects on neurotransmission (Jones and Wonnacott 2004). In the hippocampus, it has been identified postsynaptically on GABAergic neurons, whereas in brainstem nuclei such as the ventral tegmental area and the substantia nigra, the α7 nAChR localized presynaptically and is important in regulating excitatory neurotransmission (McGehee et al. 1995; Cheng and Yakel 2014).
3 nAChR Neuronal Expression and Neurotransmission
3.1 Ionic Selectivity and Ca2+ Permeability
Agonist stimulation of neuronal nAChRs causes the rapid opening of the ion channel and, in most cases, a depolarization of the cell in which these receptors are expressed. Structurally similar to the nAChRs expressed at the neuromuscular junction, the neuronal nAChRs are permeable to cations (Ballivet et al. 1988). Determination of the ionic selectivity using electrophysiological methods and ionic substitutions revealed that these receptors are permeable to sodium, potassium, and calcium (Vernino et al. 1992; Bertrand et al. 1993a, 1993b; Castro and Albuquerque 1995; Fucile et al. 2004). The calcium permeability was shown to vary as a function of the nAChR composition and is the highest for α7 and α9 nAChRs which are comparable or superior to the ionic permeability of the N-methyl-D-aspartic-acid (NMDA) receptor (Bertrand et al. 1993a, b; Séguéla et al. 1993; Elgoyhen et al. 1994; Castro and Albuquerque 1995; Sgard et al. 2002; Katz et al. 2000; Fucile et al. 2005; Uteshev 2012). Opening of divalent permeable channels can increase the intracellular calcium concentration in the proximity of the membrane and trigger the opening of the other ionic pore. For example, in the case of the α9 α10 receptors expressed in the outer hair cells of the inner ear, it was shown that activation of these receptors can activate potassium channels and indirectly cause the hyperpolarization of the cell (Janssen et al. 2004; Nie et al. 2004; Dani and Bertrand 2007; Roux et al. 2011).
Experiments conducted using site-directed mutagenesis at the homomeric α7 nAChRs revealed the presence of two binding sites, located at the inner mouth and in the upper part of the pore, that determine the level of calcium permeability (Bertrand et al. 1993a, b). The high degree of conservation of the second transmembrane segment (TM2) and, consequently, the cationic permeability in addition to the calcium permeability of these homomeric receptors suggest an important overall physiological role (Devillers-Thiery et al. 1992). Intracellular calcium homeostasis is an important physiological mechanism regulated, for example, by the calcium influx through receptors such as the N-methyl-D-aspartic acid (NMDA) or by the α7 nAChRs and by its sequestration in intracellular compartments such as the endoplasmic reticulum (ER). The intracellular calcium concentration is a ubiquitous second messenger regulating the activity of several membrane proteins including the calcium-activated potassium channels up to the control of many enzymatic pathways. Disruption of the intracellular calcium concentration is thought to be associated with several neurological disorders (Glaser et al. 2018).
Taking advantage of the homomeric nature of the α7 nAChRs, site-directed mutagenesis further allowed a more detailed characterization of the ionic selectivity filter of these cationic channels. It was shown that amino acids at the inner mouth of the channel determine the cation or anion selectivity, and interestingly, the introduction of a single proline amino acid is sufficient to switch the ionic selectivity (Galzi et al. 1992; Corringer et al. 1999). These observations were confirmed subsequently in invertebrates which have nicotinic-like channels that are natively permeable to anions and possess the critical amino acids earlier identified by mutagenesis (van Nierop et al. 2006; Juneja et al. 2014). Moreover, the same biophysical rules were found to apply for naturally occurring anion-permeable channels that were successfully switched to cation permeable by the introduction of the corresponding amino acids (Keramidas et al. 2000).
3.2 Voltage Dependence
Early characterization of the functional properties of nAChRs revealed that these receptors display a peculiar voltage sensitivity. While the nAChRs at the neuromuscular junction present an essentially Ohmic behavior, neuronal nAChRs display a strong inward rectification (Bertrand et al. 1991). Activation of the neuronal nAChRs causes the opening of the channels only when the cell membrane potential is negatively charged but the ionic pore becomes shut for resting potential smaller than −40 mV. As single channel conductance measurements revealed a resistive Ohmic behavior, it was concluded that rectification probably occurs by intracellular blockade (Bertrand et al. 1991; Haghighi and Cooper 2000). The mechanism responsible for this channel closure is thought to be caused by the blockade at the intracellular mouth by magnesium ions and/or intracellular polyamines (Forster and Bertrand 1995; Bonfante-Cabarcas et al. 1996; Stauderman et al. 1998). Inward rectification was reported for the different nAChR subtypes including the homomeric α7 and the heteromeric α4β2, α4α∗β∗ receptors and was observed for both recombinant and native nAChRs (Buisson et al. 1996; Gerzanich et al. 1997; Stauderman et al. 1998; Zaninetti et al. 1999; Alkondon et al. 2000; Nelson et al. 2001). Highly conserved across species, rectification is another hallmark of neuronal nAChRs. To understand the relevance of this mechanism, consider that release of ACh in the vicinity of the receptors will cause a physiological effect only when the cell is near its resting potential. On the contrary, if the cell is depolarized by any other activity, the ACh release will not provoke further signaling. This can also be summarized as a mechanism of coincidence detection in which the sequence of events is determinant for the functional outcome.
4 Synaptic Plasticity
A prominent role of the cholinergic system is its involvement in attention, learning, and memory processes, which has been established over decades of research using animal models (cholinergic lesions, receptor pharmacology, genetic manipulations), as well as in humans with clinically effective therapies that are prescribed for patients with cognitive dysfunction (e.g., cholinesterase inhibitors for Alzheimer’s disease). Underlying these in vivo studies and contributing to our mechanistic understanding of the cognitive involvement of the cholinergic system are in vitro models of synaptic plasticity (long-term potentiation and long-term depression).
The mAChR-dependent modulation of synaptic plasticity has been demonstrated within hippocampal neurocircuits, and this is perhaps best represented by studies investigating the M1 mAChR. M1 mAChRs localized on glutamatergic pyramidal neurons of the CA1 subregion of the hippocampus provide a direct excitatory outlet for cholinergic basal forebrain afferents and are believed to underlie the cholinergic potentiation of glutamate-mediated neurotransmission that results in a robust strengthening of glutamatergic synapses in pyramidal neurons in this region (Dennis et al. 2016). Studies conducted in the CA1 region of the mouse hippocampus have shown that mAChR agonists applied at low concentrations modulate plasticity of glutamatergic synapses in this region (Shinoe et al. 2005). It remains to be determined if the enhanced synaptic plasticity with M1 mAChR agonists extends to other brain regions such as the neocortex.
The characteristic high Ca2+permeability of the α7 nAChR combined with its localization on glutamatergic axon terminals can lead to enhanced synaptic plasticity following stimulation (e.g., with nicotine or with selective α7 nAChR agonists) as has been shown using the long-term potentiation (LTP), an in vitro model of learning and memory in the dentate gyrus region of the hippocampus. In addition, genetic deletion of the α7 nAChR in mice produces deficits in LTP following nicotine administration (Criscuolo et al. 2015).
5 Intracellular Signaling
mAChR and nAChR subtypes are all activated by acetylcholine, but as will be discussed within the current section, each receptor system is coupled to different second messenger pathways that can yield divergent signaling effects across cell types. Interestingly, activation of different classes of mAChRs and nAChRs, distinguished both by their location on the neurons and by their subunit composition within a single cell, can regulate differences in the sources of calcium mobilized (e.g., extracellular, intracellular stores) and result in altered physiological and dynamic intracellular responses that ultimately control cellular function at an individual neuron level (Rathouz et al. 1995), as will be highlighted more below.
5.1 mAChRs
As introduced earlier in the chapter, the M1, M3, and M5 mAChRs couple with Gq-type G proteins and mediate activation of phospholipase C (PLC) and inositol triphosphate/calcium signaling via pertussis toxin-insensitive G proteins of the Gq family. M1 receptors are the most abundant mAChR in the brain and have the richest biology at this point with which to focus our attention.
M1 mAChRs localized postsynaptically on pyramidal neurons in the cerebral cortex and hippocampus receive innervation from cholinergic projections from basal forebrain. Activation of the M1 receptor causes calcium release from intracellular stores via IP3-dependent calcium release that subsequently causes a transient inhibition driven by calcium-dependent small conductance potassium channels. In addition to the Gq-coupled activity, activation of M1 mAChRs in cortical pyramidal neurons produces a longer-lasting and voltage-dependent excitation that involves additional cation channels that have not been fully characterized to date (Dasari et al. 2017). Together, this signaling is hypothesized to underlie some of the functional effects of M1 activation on attention, learning, and memory.
M2 and M4 receptors signal through the pertussis-sensitive Gi/Go subfamily of G proteins and mediate inhibition of cAMP production. Targeting the M2 autoreceptor with selective antagonist ligands has been hypothesized to be a therapeutic strategy for treating Alzheimer’s disease through increasing ACh neurotransmission by blocking its Gi-coupled inhibitory actions on cholinergic neurons. This has been supported by animal studies demonstrating that antagonism of the M2 mAChR elevates extracellular ACh concentrations (Billard et al. 1995). However, the limitations with this approach have proved to be several-fold and include that M2 receptors are localized on both cholinergic and non-cholinergic terminals in the cortex and hippocampus; therefore, increasing cholinergic tone with an M2 antagonist yields a more complicated pharmacology that is not necessarily therapeutic (Levey 1996). In addition and perhaps a more prominent restraint is that activation of the M2 receptor plays an important role in the physiological regulation of cardiac function through its inhibitory actions on cAMP, as well as through its modulation of muscarinic potassium channels. Upon stimulation of the M2 receptor by ACh or other agonists, the α subunit separates from the βγ moiety, causing a decrease in adenylate cyclase, cAMP activity, and deceasing downstream signaling cascades. Activation of M2 also causes the βγ moiety to act on potassium channels. In the heart, M2 activates potassium channel and decreases heart rate (Krejci et al. 2004). Due to its prominent effects on cardiac function, it remains challenging to target the M2 receptor due to dose-limiting side effects that could prevent the therapeutic benefit from being achieved.
Much of the work investigating M4 signaling and function has been based on its abundant expression in the striatum, specifically in the dopamine D1 receptor containing spiny projections neurons of the direct output pathway. In this region where cholinergic projections densely innervate the dopamine pathways, the M4 subunit has been shown to suppress dopamine D1 receptor signaling through its inhibitory actions on the adenylate cyclase and cAMP pathway and diminish regulator of G protein signaling type 4 (RGS4) at corticostriatal glutamatergic neuronal synapses (Shen et al. 2015). This combination of activities is hypothesized to have therapeutic potential as it may be a mechanism to regulate aberrant corticostriatal synaptic plasticity that is involved in symptoms such as L-Dopa therapy-induced dyskinesias observed in patients with Parkinson’s disease who have been on chronic dopamine agonist-based therapies, and early evidence in animal models has supported this approach (Shen et al. 2015).
5.2 nAChRs
Signal transduction does not occur exclusively at the cell membrane, but also intracellularly. The high calcium permeability of the α7 or α9 containing nAChRs induces changes in the concentration of these divalent ions in the close vicinity of the membrane which, in turn, can trigger downstream signaling cascades and alteration in gene transcription. For the highly permeable α9 containing receptor, this can be best exemplified in the outer hair cells from the inner ear, wherein release of ACh triggers a hyperpolarization of the cells. Detailed analysis of these mechanisms revealed that the primary mechanism is the activation of α9α10 nAChRs which increases the intracellular calcium and, indirectly, triggers the opening of small conductance (SK) potassium channels that are at the origin of the cell hyperpolarization (Nie et al. 2004; Roux et al. 2011; Katz et al. 2000).
Similarly, activation of α7 containing nAChRs is known to increase the cytosolic calcium concentration, the outcome of which directly depends on other proteins through which this intracellular signaling will manifest. The physiological relevance of such mechanisms is readily understood in light of examples such as data from cortical pyramidal neurons. Accordingly, increased intracellular calcium in this cell segment activates potassium currents yielding modification of the signal processing (Berger and Lüscher 2003). An increase in the intracellular calcium concentration can yield multiple downstream cellular effects ranging from changes in the microtubules to alteration of the growth cone (King and Kabbani 2018).
Moreover, stimulation of α7 nAChRs triggers multiple intracellular cascades such as the neuroprotective Janus Kinase 2 (JAK2/STAT3/NF-kB (Marrero and Bencherif 2009)), extracellular related kinase 1 (ERK1) (Bencherif and Lippiello 2009), or mitogen-activated protein (MAP) kinase pathways (Gubbins et al. 2010). Activation of α7 nAChRs and subsequent engagement of intracellular signaling pathways can exert neurotrophic effects and has been suggested as a potential mechanism of neuroprotection in degenerative diseases such as Alzheimer’s disease (Ma and Qian 2019). Additionally, stimulation with the α7 nAChR agonist PNU-282987 and activation of key intracellular pathways exert a protective effect against myocardial reperfusion injury (Hou et al. 2018).
6 Receptor Desensitization
The response of receptors to a sustained exposure of agonist is generally characterized by first a rapid reaction which progressively declines over time. Known as desensitization, it is an active physiological mechanism aimed to return the system to a homeostatic state following continued exposure to an external stimulus. A simple example of desensitization occurs with the application of perfume; while it is easy to recognize its scent when first applied, after a few minutes, it is difficult to recognize whether it is on or not. While both mAChRs and nAChRs show desensitization properties, the magnitude of their effects and their mechanisms are clearly distinct, as it will be described below.
6.1 mAChRs
The desensitization mechanisms of G-coupled proteins are mainly due to internalization of these integral membrane proteins. Namely, stimulation of the receptors by the agonist causes changes in the receptor conformation and its interaction with the G proteins and, indirectly, triggers its internalization (van Koppen and Kaiser 2003). The progressive decline of the receptors at the cell surface causes a reduction in sensitivity to the agonist. The mAChR desensitization is a function of the receptor subtype and conditions. Recovery requires the incorporation of new receptors which are translocated from the intracellular pool into the plasma membrane. Given the complex cellular mechanisms involve in desensitization and recovery, the timing for these processes is rather slow.
6.2 nAChRs
Nicotinic acetylcholine receptors display desensitization and recovery mechanisms that are quite distinct from the mAChRs. In contrast to the G-coupled proteins, ligand-gated channels are fast responders and do not internalize during desensitization. Exposing nAChRs to brief pulses of agonist causes a brisk activation of the receptors, but in contrast, prolonged agonist application causes a desensitization of the receptors leading to a response with a peak and a plateau. The ratio between peak and plateau markedly differs with the receptor subtype with the fastest desensitization being observed with the α7 nAChRs and the slowest desensitization being observed at the nAChRs from the neuromuscular junction receptors. Major brain α4β2 nAChRs display a clear peak and plateau response during agonist exposure indicative of multiple phases of desensitization (Hogg and Bertrand 2007). The ability of the receptors to maintain a response during agonist exposure is thought to relate to their physiological function. Insertion in the plasma membrane of fast desensitizing receptors, such as the α7 nAChRs, is expected to cause only a transient activation of the cell, whereas a more sustained depolarization and, in consequence, physiological outcome will be observed in cells expressing more slowly desensitizing receptors such as the α4β2 nAChR or even further for the α3β4 nAChR. Given the specificity of cellular expression of some receptors in a particular neuronal pathway, such as in the interpeduncular nucleus and the fasciculus retroflexus (Perry et al. 2002), suggests that these brain areas will be more susceptible to prolonged stimulations by nicotinic agonists. The transition from peak to plateau characterizes the desensitization occurring during agonist exposure ranging from milliseconds to a few seconds, as its recovery is rather fast.
Desensitization is often subdivided into short- and long-term depending upon the exposure time of the receptor to the agonist and the persistence of the effects. For compound exposures of several minutes or hours, such as that caused by nicotine intake during smoking, a sustained inhibition of the receptors is observed. Various sets of experiments have shown that sustained exposure to 100 nM nicotine, which corresponds to the brain concentration observed after smoking a cigarette, is sufficient to cause a long-lasting receptor desensitization at the α4β2 nAChR (Ochoa et al. 1989; Lester and Dani 1995; Fenster et al. 1997; Paradiso and Brehm 1998; Dani et al. 2000; Besson et al. 2007). Long-term desensitization is closely related to the receptor composition (Vibat et al. 1995; Gerzanich et al. 1998; Dani and Bertrand 2007; Rollema et al. 2015). Desensitization to low concentrations of agonist can be observed by monitoring the amplitude of the response to a brief pulse of ACh, and it was shown that natural variants in the α4, β2, or α5 subunits influence the profile of desensitization and its recovery (Hoda et al. 2008; Improgo et al. 2010; Tammimäki et al. 2012).
7 Areas of Continued Investigation
A general division in neurotransmission is often made between ligand-gated ion channels which encompasses all the fast transmission mediated by ionotropic receptors and the opening of ion channels in the cell membrane, versus metabotropic receptors which are coupled to intracellular mechanisms such as the G proteins. This division was initially defined in terms of the pharmacology with nAChRs and their high sensitivity to the alkaloid nicotine found in the tobacco plant Nicotiana and for mAChRs and their sensitivity to alkaloids found in certain mushrooms such as the Amanita muscaria or the deadly poisoning Clitocybe dealbata. Based on the selectivity of these two substances, it was subsequently found that nicotine activates receptors that are ionotropic, which acts by the opening of the ionic pore (nAChRs), whereas muscarine indirectly modifies level of second messengers by its interaction with receptors (mAChRs) belonging to the family of GPCRs or seven transmembrane proteins.
Although these definitions largely hold true for most of the nAChR subtypes, their limit became obvious with the cloning and characterization of the pharmacological properties of the α9 nAChRs which displays a mixed nicotinic/muscarinic profile (Elgoyhen et al. 1994; Verbitsky et al. 2000). Nonetheless, the ionotropic properties of these receptors continued to fulfill the original distinction between fast-acting channels and receptors acting on a slower and prolonged time scale.
As a number of studies expanded our understanding of nAChR biology, especially within the homomeric α7 or heteromeric α9α10 receptors, several peculiarities also emerged, for example, recognition that the receptor expression occurred in many areas throughout the brain and periphery, as well as in the immune system (Wang et al. 2003; Peng et al. 2004; Razani-Boroujerdi et al. 2007). These results cast doubt about the sole ionotropic activity of these receptors with effects that could be associated only with ion fluxes. Follow-up investigation by different groups led to the proposition of a direct interaction between α7 nAChRs and metabotropic receptors that is reviewed in Kabbani and Nichols (2018). Evidence for the interaction between the α7 and metabotropic receptors has pointed to the specific intracellular sequence of the α7 protein which presents a unique amino acid sequence “LRMKRP” that is highly conserved throughout species (King et al. 2015). This observation might reconcile previous observations suggesting a mixed effect of α7 nAChRs on phosphorylation of multiple intracellular pathways (de Jonge and Ulloa 2007; Bencherif and Lippiello 2009; Maanen et al. 2009; Gubbins et al. 2010; Dhawan et al. 2012).
Genomic analysis of CHRNA7 in multiple species revealed a further complexity specific to this particular nAChR gene with a duplication of exons 6–10 that is observed only in human. Leading to a form that was subsequently termed CHRFAM7A, this genomic duplication encodes for a protein that closely resembles α7, but which is missing the N-terminal domain (Gault et al. 1998; Sinkus et al. 2015). Several studies have replicated this initial observation, and the key questions that were opened by this initial discovery included (a) “Is the dupα7 able to form functional receptors?” and (b) “Does the dupα7 assemble with α7 itself to form a hetero-oligomer?”. Experiments conducted by many different laboratories have concluded so far that while widely expressed, the dupα7 does not yield functional receptors. However, it was shown that α7 and the dupα7 can assemble in heteromers to form functional channels (Wang et al. 2010, Araud et al. 2011, de Lucas-Cerrillo et al. 2011,Lasala et al. 2019). Given the homologies between CHRNA7 and CHRFAM7A, this indicates that heteromeric receptors containing α7 and the dupα7 can equally interact with GPCRs and could participate to modulation of cellular functions.
Genetic studies conducted in humans have correlated variations in CHRFAM7A and neurological pathologies (Flomen et al. 2012; Williams et al. 2012; Rozycka et al. 2013; Kunii et al. 2015; Sinkus et al. 2015). Moreover, the potential role of the dupα7 was underlined by its high degree of expression in the immune system (Villiger et al. 2002; de Lucas-Cerrillo et al. 2011; Costantini et al. 2015; Dang et al. 2015; Baird et al. 2016). Studies conducted in human-induced pluripotent stem cells (iPSCs) might provide a model to get a better understanding of the physiological role of CHRFAM7A (Ihnatovych et al. 2019).
8 Cholinergic Receptors as Therapeutic Targets
8.1 mAChRs
Of the mAChR subtypes that have been investigated clinically for brain disorders, the focus largely has been on M1, M2, and M4 selective molecules. The non-selective M1 agonist xanomeline was taken into patients with Alzheimer’s disease, but was stopped due to an undesirable safety profile combined with minimal therapeutic benefit. More recently this molecule has been brought back into clinical development combined with the peripherally active non-selective mAChR antagonist, trospium chloride, that is approved for patients with overactive bladder. The hypothesis is that the combined therapy should allow engagement of centrally active mAChRs and antagonize peripheral mAChRs associated with adverse effects and medication discontinuation. The xanomeline/trospium chloride combined therapy is being investigated in patients with schizophrenia (ClinicalTrials.gov Identifier: NCT03697252).
An alternative approach to targeting mAChR subtype-specific molecules that bind to the orthosteric site is to identify molecules that can selectively modulate the mAChR subtype by targeting the allosteric site. By binding at a site distinct from the agonist (orthosteric site), these molecules bind at a distinct location on the receptor and are therefore more specific for a given amino acid sequence and tri-dimensional organization. Most recent advances in the optimization of allosteric modulators, which alter the affinity or efficacy of orthosteric ligands and offer selective and localized receptor modulation, have invigorated drug discovery efforts and resulted in several highly selective tool compounds that have demonstrated potential in preclinical studies and progressed to early clinical development (Felder et al. 2018). All of the mAChRs have at least one allosteric site that has been identified and provide an approach to selectively modulate mAChR subtypes independent of one another (Bock et al. 2018). In particular, positive allosteric modulators of the M1 and M4 mAChRs have been developed and are an exciting area of research that is enabling the biology of these receptors to be selectively interrogated (Kruse et al. 2012).
8.2 nAChRs
Development of nAChR subtype-specific compounds led to the discovery of several molecules which showed sufficient selectivity and robust pharmacological characteristics to be supported for testing in human clinical trials. The best examples can be shown for the α7 nAChRs, with the discovery of quinuclidine-based molecules such as the PNU-282987, TC-5619, MEM-3454/RG3487, or encenicline. Today, no less than 12 molecules showing specificity for the α7 nAChRs were brought into clinical trials (reviewed in Rollema et al. 2014)). However, despite these efforts, either beneficial effects were insufficient or side effects were not tolerable. Similar results were, unfortunately, observed with α4β2 nAChR-specific agonists such as ABT-418, sofinicline, pozanicline, ispronicline, or rivanicline (reviewed in Rollema et al. 2014).
In view of these efforts and failure to achieve the desired therapeutic effects, research of agonists specific to a given nAChR subtype and subsequent investments were slowed bringing this field almost to a complete halt in recent years. Nonetheless, it is important to underline that the development of an α4β2 nAChR partial agonist, varenicline, made a significant impact in the field of smoking cessation and was successfully introduced more than a decade ago (Rollema and Hurst 2018).
Although multiple brain diseases, such as Alzheimer’s, schizophrenia, or Parkinson’s, were investigated in clinical trials with selective nAChR ligands and highlight the diverse potential of these molecules, no nAChR drugs, except for varenicline, have made it as to the market as therapeutics (Terry 2008; Wallace and Bertrand 2013a, b; Perez-Lloret and Barrantes 2016; Hampel et al. 2019). Information about failures of clinical trials are limited, and it is often not possible to know if a compound was abandoned because of its side effects or lack of efficacy; however, it is possible to speculate about the encountered difficulties.
A first and an obvious difficulty are concerns around insufficient selectivity across receptor systems. For example, as in the case of encenicline, it was shown that this compound was active at the α7 nAChRs and was equipotent at the 5HT3 receptors. This cross interaction might be at the origin of the gastrointestinal difficulties that were reported in the clinical Phase 3 trial for this compound (Barbier et al. 2015; Keefe et al. 2015; Hayward et al. 2017; Godyń et al. 2016). A second and more difficult point concerns the desired mechanism of action in vivo. While it is easy to assess the effects of an agonist in vitro under controlled experimental conditions, the beneficial outcomes of such molecules in vivo are more difficult to comprehend. Especially when considering the desensitization properties of the nAChRs, it has remained a challenge how to effectively anticipate the outcome of sustained exposure to a low concentration of agonist under various disease conditions in which cholinergic tone and/or receptor expression may be different than in the healthy condition. Experiments conducted in vitro and in vivo with the α7 nAChRs and agonists selective for this subtype have highlighted that low concentrations of agonists can enhance the response to ACh by a mechanism described as priming (Prickaerts et al. 2012; Stoiljkovic et al. 2015). Similarly, priming was observed with the nicotine metabolite cotinine or the 5HT3 receptor antagonist tropisetron (Terry et al. 2015; Callahan et al. 2017). This mechanism would elegantly reconcile the enhancement of the cognitive performances observed with nicotinic derived compounds as well as the inverted U-shape observed between the compound concentration and performance increases (Wallace and Bertrand 2013a, b).
In view of a large body of evidence correlating the cholinergic system with cognitive performances, it is tempting to speculate that development of nAChR-specific ligands still has a promising future. In addition, such compounds might find additional and unexpected benefits by acting outside the CNS, such as the current repurposing of varenicline to treat dry eye afflictions.
References
Ahmed T, Zahid S, Mahboob A, Farhat SM (2017) Cholinergic system and post-translational modifications: an insight on the role in Alzheimer’s disease. Curr Neuropharmacol 15(4):480–494
Alkondon M, Braga MF, Pereira EF, Maelicke A, Albuquerque EX (2000) alpha7 nicotinic acetylcholine receptors and modulation of gabaergic synaptic transmission in the hippocampus. Eur J Pharmacol 393(1–3):59–67
Araud T, Graw S, Berger R, Lee M, Neveu E, Bertrand D, Leonard S (2011) The chimeric gene CHRFAM7A, a partial duplication of the CHRNA7 gene, is a dominant negative regulator of alpha7∗nAChR function. Biochem Pharmacol 82(8):904–914
Baird A, Coimbra R, Dang X, Eliceiri BP, Costantini TW (2016) Up-regulation of the human-specific CHRFAM7A gene in inflammatory bowel disease. BBA Clin 5:66–71
Ballivet M, Nef P, Couturier S, Rungger D, Bader CR, Bertrand D, Cooper E (1988) Electrophysiology of a chick neuronal nicotinic acetylcholine receptor expressed in Xenopus oocytes after cDNA injection. Neuron 1(9):847–852
Barbier AJ, Hilhorst M, Van Vliet A, Snyder P, Palfreyman MG, Gawryl M, Dgetluck N, Massaro M, Tiessen R, Timmerman W, Hilt DC (2015) Pharmacodynamics, pharmacokinetics, safety, and tolerability of encenicline, a selective α7 nicotinic receptor partial agonist, in single ascending-dose and bioavailability studies. Clin Ther 37(2):311–324
Beckmann J, Lips KS (2013) The non-neuronal cholinergic system in health and disease. Pharmacology 92(5–6):286–302
Bencherif M, Lippiello PM (2009) Alpha7 neuronal nicotinic receptors: the missing link to understanding Alzheimer's etiopathology? Med Hypotheses 74(2):281–285
Bender AM, Garrison AT, Lindsley CW (2019) The muscarinic acetylcholine receptor M5: therapeutic implications and allosteric modulation. ACS Chem Neurosci 10(3):1025–1034
Berger T, Lüscher H-R (2003) Timing and precision of spike initiation in layer V pyramidal cells of the rat somatosensory cortex. Cereb Cortex 13(3):274–281
Bertrand D, Cooper E, Valera S, Rungger D, Ballivet M (1991) Electrophysiology of neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes following nuclear injection of genes or cDNA. In: Conn M (ed) Methods in neuroscience, vol 4. Academic Press, New York, pp 174–193
Bertrand D, Galzi JL, Devillers-Thiery A, Bertrand S, Changeux JP (1993a) Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci U S A 90(15):6971–6975
Bertrand D, Galzi JL, Devillers-Thiery A, Bertrand S, Changeux JP (1993b) Stratification of the channel domain in neurotransmitter receptors. Curr Opin Cell Biol 5(4):688–693
Bertrand D, Lee CH, Flood D, Marger F, Donnelly-Roberts D (2015) Therapeutic potential of alpha7 nicotinic acetylcholine receptors. Pharmacol Rev 67(4):1025–1073
Besson M, Granon S, Mameli-Engvall M, Cloëz-Tayarani I, Maubourguet N, Cormier A, Cazala P, David V, Changeux J-P, Faure P (2007) Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc Natl Acad Sci U S A 104(19):8155–8160
Besson M, Guiducci S, Granon S, Guilloux J-P, Guiard B, Repérant C, Faure P, Pons S, Cannazza G, Zoli M, Gardier AM, Maskos U (2016) Alterations in alpha5∗ nicotinic acetylcholine receptors result in midbrain- and hippocampus-dependent behavioural and neural impairments. Psychopharmacology (Berl) 233(18):3297–3314
Billard W, Binch H 3rd, Crosby G, McQuade RD (1995) Identification of the primary muscarinic autoreceptor subtype in rat striatum as m2 through a correlation of in vivo microdialysis and in vitro receptor binding data. J Pharmacol Exp Ther 273(1):273–279
Bock A, Schrage R, Mohr K (2018) Allosteric modulators targeting CNS muscarinic receptors. Neuropharmacology 136(Pt C):427–437
Bonfante-Cabarcas R, Swanson KL, Alkondon M, Albuquerque EX (1996) Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. IV. Regulation by external Ca++ of alpha-bungarotoxin-sensitive receptor function and of rectification induced by internal Mg++. J Pharmacol Exp Ther 277(1):432–444
Brown RWB, Collins AC, Lindstrom JM, Whiteaker P (2007) Nicotinic alpha5 subunit deletion locally reduces high-affinity agonist activation without altering nicotinic receptor numbers. J Neurochem 103(1):204–215
Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D (1996) Human alpha4beta2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci 16(24):7880–7891
Bymaster FP, Carter PA, Yamada M, Gomeza J, Wess J, Hamilton SE, Nathanson NM, McKinzie DL, Felder CC (2003) Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur J Neurosci 17(7):1403–1410
Callahan PM, Bertrand D, Bertrand S, Plagenhoef MR, Terry AV Jr (2017) Tropisetron sensitizes alpha7 containing nicotinic receptors to low levels of acetylcholine in vitro and improves memory-related task performance in young and aged animals. Neuropharmacology 117:422–433
Castro NG, Albuquerque EX (1995) Alpha-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys J 68(2):516–524
Cheng Q, Yakel JL (2014) Presynaptic α7 nicotinic acetylcholine receptors enhance hippocampal mossy Fiber glutamatergic transmission via PKA activation. J Neurosci 34(1):124–133
Corringer PJ, Bertrand S, Galzi JL, Devillers-Thiery A, Changeux JP, Bertrand D (1999) Mutational analysis of the charge selectivity filter of the alpha 7 nicotinic acetylcholine receptor. Neuron 22(4):831–843
Costantini TW, Dang X, Yurchyshyna MV, Coimbra R, Eliceiri BP, Baird A (2015) A human-specific α7-nicotinic acetylcholine receptor gene in human leukocytes: identification, regulation and the consequences of CHRFAM7A expression. Mol Med 3(21):323–336
Criscuolo C, Accorroni A, Domenici L, Origlia N (2015) Impaired synaptic plasticity in the visual cortex of mice lacking alpha7-nicotinic receptor subunit. Neuroscience 294:166–171
Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B (2001) Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia: a study of Brodmann's areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. Am J Psychiatry 158(6):918–925
Curtis L, Buisson B, Bertrand S, Bertrand D (2002) Potentiation of human alpha4beta2 neuronal nicotinic acetylcholine receptor by estradiol. Mol Pharmacol 61(1):127–135
Dang X, Eliceiri BP, Baird A, Costantini TW (2015) CHRFAM7A: a human-specific α7-nicotinic acetylcholine receptor gene shows differential responsiveness of human intestinal epithelial cells to LPS. FASEB J 29(6):2292–2302
Dani JA, Bertrand D (2007) Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol 47:699–729
Dani JA, Radcliffe KA, Pidoplichko VI (2000) Variations in desensitization of nicotinic acetylcholine receptors from hippocampus and midbrain dopamine areas. Eur J Pharmacol 393(1–3):31–38
Dasari S, Hill C, Gulledge AT (2017) A unifying hypothesis for M1 muscarinic receptor signalling in pyramidal neurons. J Physiol 595(5):1711–1723
de Jonge WJ, Ulloa L (2007) The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151(7):915–929
de Lucas-Cerrillo AM, Maldifassi MC, Arnalich F, Renart J, Atienza G, Serantes R, Cruces J, Sánchez-Pacheco A, Andrés-Mateos E, Montiel C (2011) Function of partially duplicated human α77 nicotinic receptor subunit CHRFAM7A gene: potential implications for the cholinergic anti-inflammatory response. J Biol Chem 286(1):594–606
Dennis SH, Pasqui F, Colvin EM, Sanger H, Mogg AJ, Felder CC, Broad LM, Fitzjohn SM, Isaac JT, Mellor JR (2016) Activation of muscarinic M1 acetylcholine receptors induces long-term potentiation in the Hippocampus. Cereb Cortex 26(1):414–426
Devillers-Thiery A, Galzi JL, Bertrand S, Changeux JP, Bertrand D (1992) Stratified organization of the nicotinic acetylcholine receptor channel. Neuroreport 3(11):1001–1004
Dhawan S, Cailotto C, Harthoorn LF, de Jonge WJ (2012) Cholinergic signalling in gut immunity. Life Sci 91(21–22):1038–1042
Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S (1994) Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79(4):705–715
Felder CC, Goldsmith PJ, Jackson K, Sanger HE, Evans DA, Mogg AJ, Broad LM (2018) Current status of muscarinic M1 and M4 receptors as drug targets for neurodegenerative diseases. Neuropharmacology 136(Pt C):449–458
Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA (1997) Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci 17(15):5747–5759
Flomen RH, Shaikh M, Walshe M, Schulze K, Hall M-H, Picchioni M, Rijsdijk F, Toulopoulou T, Kravariti E, Murray RM, Asherson P, Makoff AJ, Bramon E (2012) Association between the 2-bp deletion polymorphism in the duplicated version of the alpha7 nicotinic receptor gene and P 50 sensory gating. Eur J Hum Genet 21(1):76–81
Forster I, Bertrand D (1995) Inward rectification of neuronal nicotinic acetylcholine receptors investigated by using the homomeric alpha 7 receptor. Proc Biol Sci 260(1358):139–148
Fucile S, Renzi M, Lauro C, Limatola C, Ciotti T, Eusebi F (2004) Nicotinic cholinergic stimulation promotes survival and reduces motility of cultured rat cerebellar granule cells. Neuroscience 127(1):53–61
Fucile S, Sucapane A, Eusebi F (2005) Ca2+ permeability of nicotinic acetylcholine receptors from rat dorsal root ganglion neurones. J Physiol 565(Pt 1):219–228
Galzi JL, Devillers-Thiery A, Hussy N, Bertrand S, Changeux JP, Bertrand D (1992) Mutations in the channel domain of a neuronal nicotinic receptor convert ion selectivity from cationic to anionic. Nature 359(6395):500–505
Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, Moore T, Jacobs S, Meriwether J, Choi MJ, Kim EJ, Walton K, Buiting K, Davis A, Breese C, Freedman R, Leonard S (1998) Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7). Genomics 52(2):173–185
Gautam D, Han SJ, Hamdan FF, Jeon J, Li B, Li JH, Cui Y, Mears D, Lu H, Deng C, Heard T, Wess J (2006) A critical role for beta cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo. Cell Metab 3(6):449–461
Gerzanich V, Kuryatov A, Anand R, Lindstrom J (1997) "Orphan" alpha 6 nicotinic AChR subunit can form a functional heteromeric acetylcholine receptor. Mol Pharmacol 51(2):320–327
Gerzanich V, Wang F, Kuryatov A, Lindstrom J (1998) Alpha 5 subunit alters desensitization, pharmacology, Ca++ permeability and Ca++ modulation of human neuronal alpha 3 nicotinic receptors. J Pharmacol Exp Ther 286(1):311–320
Glaser T, Arnaud Sampaio VF, Lameu C, Ulrich H (2018) Calcium signalling: a common target in neurological disorders and neurogenesis. Semin Cell Dev Biol S1084-9521(18):30068–30065
Godyń J, Jończyk J, Panek D, Malawska B (2016) Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep 68(1):127–138
Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M (2007) Heterogeneity and complexity of native brain nicotinic receptors. Biochem Pharmacol 74(8):1102–1111
Grady SR, Salminen O, McIntosh JM, Marks MJ, Collins AC (2010) Mouse striatal dopamine nerve terminals express alpha4alpha5beta2 and two stoichiometric forms of alpha4beta2∗-nicotinic acetylcholine receptors. J Mol Neurosci 40(1–2):91–95
Gubbins EJ, Gopalakrishnan M, Li J (2010) Alpha7 nAChR-mediated activation of MAP kinase pathways in PC12 cells. Brain Res 1328:1–11
Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482(7386):547–551
Haghighi AP, Cooper E (2000) A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine alpha3beta4 and alpha4beta2 receptors. J Neurosci 20(2):529–541
Hampel H, Mesulam MM, Cuello AC, Khachaturian AS, Vergallo A, Farlow MR, Snyder PJ, Giacobini E, Khachaturian ZS (2019) Revisiting the cholinergic hypothesis in Alzheimer’s disease: emerging evidence from translational and clinical research. J Prev Alzheimers Dis 6(1):2–15
Hayward A, Adamson L, Neill JC (2017) Partial agonism at the α7 nicotinic acetylcholine receptor improves attention, impulsive action and vigilance in low attentive rats. Eur Neuropsychopharmacol 7(4):325–335
Hilf RJC, Dutzler R (2009) Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457(7225):115–118
Hoda JC, Gu W, Friedli M, Phillips HA, Bertrand S, Antonarakis SE, Goudie D, Roberts R, Scheffer IE, Marini C, Patel J, Berkovic SF, Mulley JC, Steinlein OK, Bertrand D (2008) Human nocturnal frontal lobe epilepsy: pharmocogenomic profiles of pathogenic nicotinic acetylcholine receptor beta-subunit mutations outside the ion channel pore. Mol Pharmacol 74(2):379–391
Hogg RC, Bertrand D (2007) Partial agonists as therapeutic agents at neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 73(4):459–468
Hou Z, Zhou Y, Yang H, Liu Y, Mao X, Qin X, Li X, Zhang X, Hu Y (2018) Alpha7 nicotinic acetylcholine receptor activation protects against myocardial reperfusion injury through modulation of autophagy. Biochem Biophys Res Commun 500(2):357–364
Ihnatovych I, Nayak TK, Ouf A, Sule N, Birkaya B, Chaves L, Auerbach A, Szigeti K (2019) iPSC model of CHRFAM7A effect on α7 nicotinic acetylcholine receptor function in the human context. Transl Psychiatry 9(1):59
Improgo MRD, Scofield MD, Tapper AR, Gardner PD (2010) The nicotinic acetylcholine receptor CHRNA5/A3/B4 gene cluster: dual role in nicotine addiction and lung cancer. Prog Neurobiol 92:212–226
Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R (2004) The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc 52(1):80–85
Jones IW, Wonnacott S (2004) Precise localization of alpha7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci 24(50):11244–11252
Juneja P, Horlacher R, Bertrand D, Krause R, Marger F, Welte W (2014) An internally modulated, thermostable, pH-sensitive Cys loop receptor from the hydrothermal vent worm Alvinella pompejana. J Biol Chem 289(21):15130–15140
Kabbani N, Nichols RA (2018) Beyond the channel: metabotropic signaling by nicotinic receptors. Trends Pharmacol Sci 39(4):354–366
Katz E, Verbitsky M, Rothlin CV, Vetter DE, Heinemann SF, Elgoyhen AB (2000) High calcium permeability and calcium block of the alpha 9 nicotinic acetylcholine receptor. Hear Res 141(1–2):117–128
Keefe RSE, Meltzer HA, Dgetluck N, Gawryl M, Koenig G, Moebius HJ, Lombardo I, Hilt DC (2015) Randomized, double-blind, placebo-controlled study of Encenicline, an Alpha-7 nicotinic acetylcholine receptor agonist as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology 40(13):3053–3060
Keramidas A, Moorhouse AJ, French CR, Schofield PR, Barry PH (2000) M2 pore mutations convert the glycine receptor channel from being anion- to cation-selective. Biophys J 79(1):247–259
King JR, Kabbani N (2018) Alpha 7 nicotinic receptors attenuate neurite development through calcium activation of calpain at the growth cone. PLoS One 13(5):e0197247
King JR, Nordman JC, Bridges SP, Lin M-K, Kabbani N (2015) Identification and characterization of a G protein-binding cluster in α7 nicotinic acetylcholine receptors. J Biol Chem 290(33):20060–20070
Klink R, de Kerchove d’Exaerde A, Zoli M, Changeux JP (2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21(5):1452–1463
Krejci A, Michal P, Jakubik J, Ricny J, Dolezal V (2004) Regulation of signal transduction at M2 muscarinic receptor. Physiol Res 53(Suppl 1):S131–S140
Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482(7386):552–556
Kruse AC, Hu J, Kobilka BK, Wess J (2014) Muscarinic acetylcholine receptor X-ray structures: potential implications for drug development. Curr Opin Pharmacol 16:24–30
Kuner R, Köhr G, Grünewald S, Eisenhardt G, Bach A, Kornau H-C (1999) Role of Heteromer formation in GABAB receptor function. Science 283:74–77
Kunii Y, Zhang W, Xu Q, Hyde TM, McFadden W, Shin JH, Deep-Soboslay A, Ye T, Li C, Kleinman JE, Wang KH, Lipska BK (2015) CHRNA7 and CHRFAM7A mRNAs: co-localized and their expression levels altered in the postmortem dorsolateral prefrontal cortex in major psychiatric disorders. Am J Psychiatry 172(11):1122–1130
Kuryatov A, Onksen J, Lindstrom J (2008) Roles of accessory subunits in alpha4beta2(∗) nicotinic receptors. Mol Pharmacol 74(1):132–143
Lamotte d’Incamps B, Zorbaz T, Dingova D, Krejci E, Ascher P (2018) Stoichiometry of the Heteromeric nicotinic receptors of the Renshaw cell. J Neurosci 38(21):4943–4956
Langmead CJ, Watson J, Reavill C (2008) Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther 117(2):232–243
Lasala M, Fabiani C, Corradi J, Antollini S, Bouzat C (2019) Molecular modulation of human alpha7 nicotinic receptor by amyloid-beta peptides. Front Cell Neurosci 13:37
Lester RA, Dani JA (1995) Acetylcholine receptor desensitization induced by nicotine in rat medial habenula neurons. J Neurophysiol 74(1):195–206
Levey AI (1996) Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci U S A 93(24):13541–13546
Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR (1991) Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neuroscience 11:3218–3226
Levey AI, Edmunds SM, Heilman CJ, Desmond TJ, Frey KA (1994) Localization of muscarinic m3 receptor protein and M3 receptor binding in rat brain. Neuroscience 63(1):207–221
Ma K-G, Qian Y-H (2019) Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer's disease. Neuropeptides 73:96–106
Maanen MAV, Stoof SP, Zanden EPVD, Jonge WJD, Janssen RA, Fischer DF, Vandeghinste N, Brys R, Vervoordeldonk MJ, Tak PP (2009) The alpha7 nicotinic acetylcholine receptor on fibroblast-like synoviocytes and in synovial tissue from rheumatoid arthritis patients: a possible role for a key neurotransmitter in synovial inflammation. Arthritis Rheum 60(5):1272–1281
Maeda S, Qu Q, Robertson MJ, Skiniotis G, Kobilka BK (2019) Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364(6440):552–557
Marrero MB, Bencherif M (2009) Convergence of alpha 7 nicotinic acetylcholine receptor-activated pathways for anti-apoptosis and anti-inflammation: central role for JAK2 activation of STAT3 and NF-kappa B. Brain Res 1256:1–7
Marsango S, Ward R, Alvarez-Curto E, Milligan G (2018) Muscarinic receptor oligomerization. Neuropharmacology 136:401–410
Matsui M, Motomura D, Karasawa H, Fujikawa T, Jiang J, Komiya Y, Takahashi S, Taketo MM (2000) Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc Natl Acad Sci U S A 97(17):9579–9584
McGehee DS, Heath MJ, Gelber S, Devay P, Role LW (1995) Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269(5231):1692–1696
Morales-Perez CL, Noviello CM, Hibbs RE (2016) X-ray structure of the human α4β2 nicotinic receptor. Nature 538(7625):411–415
Nelson ME, Wang F, Kuryatov A, Choi CH, Gerzanich V, Lindstrom J (2001) Functional properties of human nicotinic AChRs expressed by IMR-32 neuroblastoma cells resemble those of alpha3beta4 AChRs expressed in permanently transfected HEK cells. J Gen Physiol 118(5):563–582
Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J (2003) Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol 63(2):332–341
Nemecz Á, Prevost MS, Menny A, Corringer P-J (2016) Emerging molecular mechanisms of signal transduction in Pentameric ligand-gated ion channels. Neuron 90(3):452–470
Nie L, Song H, Chen M-F, Chiamvimonvat N, Beisel KW, Yamoah EN, Vázquez AE (2004) Cloning and expression of a small-conductance Ca (2+)-activated K+ channel from the mouse cochlea: coexpression with alpha9/alpha10 acetylcholine receptors. J Neurophysiol 91(4):1536–1544
Ochoa EL, Chattopadhyay A, McNamee MG (1989) Desensitization of the nicotinic acetylcholine receptor: molecular mechanisms and effect of modulators. Cell Mol Neurobiol 9(2):141–178
Paradiso K, Brehm P (1998) Long-term desensitization of nicotinic acetylcholine receptors is regulated via protein kinase A-mediated phosphorylation. J Neurosci 18(22):9227–9237
Paradiso K, Zhang J, Steinbach JH (2001) The C terminus of the human nicotinic alpha4beta2 receptor forms a binding site required for potentiation by an estrogenic steroid. J Neurosci 21(17):6561–6568
Peng H, Ferris RL, Matthews T, Hiel H, Lopez-Albaitero A, Lustig LR (2004) Characterization of the human nicotinic acetylcholine receptor subunit alpha (alpha) 9 (CHRNA9) and alpha (alpha) 10 (CHRNA10) in lymphocytes. Life Sci 76(3):263–280
Perez-Lloret S, Barrantes FJ (2016) Deficits in cholinergic neurotransmission and their clinical correlates in Parkinson’s disease. NPJ Parkinsons Dis 2:16001
Perry DC, Xiao Y, Nguyen HN, Musachio JL, Dávila-García MI, Kellar KJ (2002) Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J Neurochem 82(3):468–481
Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, Methfessel C, Reneerkens OA, Flood DG, Hilt D, Gawryl M, Bertrand S, Bertrand D, Konig G (2012) EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology 62(2):1099–1110
Rathouz MM, Vijayaraghavan S, Berg DK (1995) Acetylcholine differentially affects intracellular calcium via nicotinic and muscarinic receptors on the same population of neurons. J Biol Chem 270(24):14366–14375
Razani-Boroujerdi S, Boyd RT, Dávila-García MI, Nandi JS, Mishra NC, Singh SP, Pena-Philippides JC, Langley R, Sopori ML (2007) T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol 179(5):2889–2898
Rollema H, Hurst RS (2018) The contribution of agonist and antagonist activities of α4β2∗ nAChR ligands to smoking cessation efficacy: a quantitative analysis of literature data. Psychopharmacology (Berl) 235(9):2479–2505
Rollema H, Bertrand D Hurst R (2014) Nicotinic agonists and antagonists. Encyclopedia of psychopharmacology, vol 16(6), Springer, Heidelberg, pp 733–742
Rollema H, Bertrand D, Hurst R (2015) Nicotinic agonists and antagonists. In: Stolerman IP, Price LH (eds) Encyclopedia of psychopharmacology. Springer, Berlin
Roux I, Wersinger E, McIntosh JM, Fuchs PA, Glowatzki E (2011) Onset of cholinergic efferent synaptic function in sensory hair cells of the rat cochlea. J Neurosci 31(42):15092–15101
Rozycka A, Dorszewska J, Steinborn B, Kempisty B, Lianeri M, Wisniewska K, Jagodzinski PP (2013) A transcript coding for a partially duplicated form of α7 nicotinic acetylcholine receptor is absent from the CD4+ T-lymphocytes of patients with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE). Folia Neuropathol 51(1):65–75
Scarr E (2012) Muscarinic receptors: their roles in disorders of the central nervous system and potential as therapeutic targets. CNS Neurosci Ther 18(5):369–379
Schaaf CP (2014) Nicotinic acetylcholine receptors in human genetic disease. Genet Med 16(9):649–656
Schubert J, Beckmann J, Hartmann S, Morhenn H-G, Szalay G, Heiss C, Schnettler R, Lips KS (2012) Expression of the non-neuronal cholinergic system in human knee synovial tissue from patients with rheumatoid arthritis and osteoarthritis. Life Sci 91(21–22):1048–1052
Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW (1993) Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 13(2):596–604
Sgard F, Charpantier E, Bertrand S, Walker N, Caput D, Graham D, Bertrand D, Besnard F (2002) A novel human nicotinic receptor subunit, alpha10, that confers functionality to the alpha9-subunit. Mol Pharmacol 61(1):150–159
Shen W, Plotkin JL, Francardo V, Ko WK, Xie Z, Li Q, Fieblinger T, Wess J, Neubig RR, Lindsley CW, Conn PJ, Greengard P, Bezard E, Cenci MA, Surmeier DJ (2015) M4 muscarinic receptor signaling ameliorates striatal plasticity deficits in models of L-DOPA-induced dyskinesia. Neuron 88(4):762–773
Shinoe T, Matsui M, Taketo MM, Manabe T (2005) Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J Neurosci 25(48):11194–11200
Shorey-Kendrick LE, Ford MM, Allen DC, Kuryatov A, Lindstrom J, Wilhelm L, Grant KA, Spindel ER (2015) Nicotinic receptors in non-human primates: analysis of genetic and functional conservation with humans. Neuropharmacology 96(Pt B):263–273
Sinkus ML, Graw S, Freedman R, Ross RG, Lester HA, Leonard S (2015) The human CHRNA7 and CHRFAM7A genes: a review of the genetics, regulation, and function. Neuropharmacology 96(Pt B):274–288
Stauderman KA, Mahaffy LS, Akong M, Veliçelebi G, Chavez-Noriega LE, Crona JH, Johnson EC, Elliott KJ, Gillespie A, Reid RT, Adams P, Harpold MM, Corey-Naeve J (1998) Characterization of human recombinant neuronal nicotinic acetylcholine receptor subunit combinations alpha2beta4, alpha3beta4 and alpha4beta4 stably expressed in HEK293 cells. J Pharmacol Exp Ther 284(2):777–789
Stoiljkovic M, Leventhal L, Chen A, Chen T, Driscoll R, Flood D, Hodgdon H, Hurst R, Nagy D, Piser T, Tang C, Townsend M, Tu Z, Bertrand D, Koenig G, Hajos M (2015) Concentration-response relationship of the alpha7 nicotinic acetylcholine receptor agonist FRM-17874 across multiple in vitro and in vivo assays. Biochem Pharmacol 97(4):576–589
Tammimäki A, Herder P, Li P, Esch C, Laughlin JR, Akk G, Stitzel JA (2012) Impact of human D398N single nucleotide polymorphism on intracellular calcium response mediated by α3β4α5 nicotinic acetylcholine receptors. Neuropharmacology 63(6):1002–1011
Tapia L, Kuryatov A, Lindstrom J (2007) Ca2+ permeability of the (alpha4)3(beta2)2 stoichiometry greatly exceeds that of (alpha4)2(beta2)3 human acetylcholine receptors. Mol Pharmacol 71(3):769–776
Terry AV (2008) Role of the central cholinergic system in the therapeutics of schizophrenia. Curr Neuropharmacol 6(3):286–292
Terry AV Jr, Callahan PM, Bertrand D (2015) R-(+) and S-(−) isomers of cotinine augment cholinergic responses in vitro and in vivo. J Pharmacol Exp Ther 352(2):405–418
Thal DM, Sun B, Feng D, Nawaratne V, Leach K, Felder CC, Bures MG, Evans DA, Weis WI, Bachhawat P, Kobilka TS, Sexton PM, Kobilka BK, Christopoulos A (2016) Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 531(7594):335–340
Uteshev VV (2012) α7 nicotinic ACh receptors as a ligand-gated source of Ca(2+) ions: the search for a Ca(2+) optimum. Adv Exp Med Biol 740:603–638
van Koppen CJ, Kaiser B (2003) Regulation of muscarinic acetylcholine receptor signaling. Pharmacol Ther 98(2):197–220
van Nierop P, Bertrand S, Munno DW, Gouwenberg Y, van Minnen J, Spafford JD, Syed NI, Bertrand D, Smit AB (2006) Identification and functional expression of a family of nicotinic acetylcholine receptor subunits in the central nervous system of the mollusc Lymnaea stagnalis. J Biol Chem 281(3):1680–1691
Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB (2000) Mixed nicotinic-muscarinic properties of the alpha9 nicotinic cholinergic receptor. Neuropharmacology 39(13):2515–2524
Vernino S, Amador M, Luetje CW, Patrick J, Dani JA (1992) Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron 8(1):127–134
Vibat CR, Lasalde JA, McNamee MG, Ochoa EL (1995) Differential desensitization properties of rat neuronal nicotinic acetylcholine receptor subunit combinations expressed in Xenopus laevis oocytes. Cell Mol Neurobiol 15(4):411–425
Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH, Bertrand D, Romand JA (2002) Expression of an alpha7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J Neuroimmunol 126(1–2):86–98
Volpicelli LA, Levey AI (2004) Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. In: Acetylcholine in the cerebral cortex. Elsevier, Amsterdam, pp 59–66
Wallace TL, Bertrand D (2013a) Alpha7 neuronal nicotinic receptors as a drug target in schizophrenia. Expert Opin Ther Targets 17(2):139–155
Wallace TL, Bertrand D (2013b) Importance of the nicotinic acetylcholine receptor system in the prefrontal cortex. Biochem Pharmacol 85(12):1713–1720
Walsh RM Jr, Roh SH, Gharpure A, Morales-Perez CL, Teng J, Hibbs RE (2018) Structural principles of distinct assemblies of the human alpha4beta2 nicotinic receptor. Nature 557(7704):261–265
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421(6921):384–388
Wang X-J, Liu Y-F, Wang Q-Y, Tsuruoka M, Ohta K, Wu S-X, Yakushiji M, Inoue T (2010) Functional expression of alpha 7 nicotinic acetylcholine receptors in human periodontal ligament fibroblasts and rat periodontal tissues. Cell Tissue Res 340(2):347–355
Wess J, Eglen RM, Gautam D (2007) Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov 6(9):721–733
Wessler I, Kirkpatrick CJ (2008) Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 154(8):1558–1571
Williams NM, Franke B, Mick E, Anney RJL, Freitag CM, Gill M, Thapar A, O'Donovan MC, Owen MJ, Holmans P, Kent L, Middleton F, Zhang-James Y, Liu L, Meyer J, Nguyen TT, Romanos J, Romanos M, Seitz C, Renner TJ, Walitza S, Warnke A, Palmason H, Buitelaar J, Rommelse N, Vasquez AA, Hawi Z, Langley K, Sergeant J, Steinhausen H-C, Roeyers H, Biederman J, Zaharieva I, Hakonarson H, Elia J, Lionel AC, Crosbie J, Marshall CR, Schachar R, Scherer SW, Todorov A, Smalley SL, Loo S, Nelson S, Shtir C, Asherson P, Reif A, Lesch K-P, Faraone SV (2012) Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: the role of rare variants and duplications at 15q13.3. Am J Psychiatry 169(2):195–204
Zaninetti M, Tribollet E, Bertrand D, Raggenbass M (1999) Presence of functional neuronal nicotinic acetylcholine receptors in brainstem motoneurons of the rat. Eur J Neurosci 11(8):2737–2748
Zhou Y, Nelson ME, Kuryatov A, Choi C, Cooper J, Lindstrom J (2003) Human alpha4beta2 acetylcholine receptors formed from linked subunits. J Neurosci 23(27):9004–9015
Zwart R, Vijverberg HP (1998) Four pharmacologically distinct subtypes of alpha4beta2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol Pharmacol 54(6):1124–1131
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bertrand, D., Wallace, T.L. (2020). A Review of the Cholinergic System and Therapeutic Approaches to Treat Brain Disorders. In: Shoaib, M., Wallace, T. (eds) Behavioral Pharmacology of the Cholinergic System. Current Topics in Behavioral Neurosciences, vol 45. Springer, Cham. https://doi.org/10.1007/7854_2020_141
Download citation
DOI: https://doi.org/10.1007/7854_2020_141
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-56012-6
Online ISBN: 978-3-030-56013-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)