
Abstract
The spatiotemporal distribution of cytosolic Ca2+ ions is a key determinant of neuronal behavior and survival. Distinct sources of Ca2+ ions including ligand- and voltage-gated Ca2+ channels contribute to intracellular Ca2+ homeostasis. Many normal physiological and therapeutic neuronal functions are Ca2+-dependent, however an excess of cytosolic Ca2+ or a lack of the appropriate balance between Ca2+ entry and clearance may destroy cellular integrity and cause cellular death. Therefore, the existence of optimal spatiotemporal patterns of cytosolic Ca2+ elevations and thus, optimal activation of ligand- and voltage-gated Ca2+ ion channels are postulated to benefit neuronal function and survival. Alpha7 nicotinic acetylcholine receptors (nAChRs) are highly permeable to Ca2+ ions and play an important role in modulation of neurotransmitter release, gene expression and neuroprotection in a variety of neuronal and non-neuronal cells. In this review, the focus is placed on α7 nAChR-mediated currents and Ca2+ influx and how this source of Ca2+ entry compares to NMDA receptors in supporting cytosolic Ca2+ homeostasis, neuronal function and survival.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- α7 nAChR
- NMDA
- Ca2+
- Permeability
- Ion channel
- Receptor
- ACh
- Choline
- Nicotinic
- Positive allosteric modulator
- PNU-120596
- Neuroprotection
- Cognitive
- Cognition
- Dementia
- Neurotoxicity
- Synaptic
- Extrasynaptic
- Alzheimer
- Schizophrenia
- Trauma
- Auditory
- Gating
Ligand- and Voltage-Gated Sources of Ca2+ Ions
Background
Changes in cytosolic Ca2+ levels act as a messenger relaying information from the cellular membrane to the cellular cytoplasm and the nucleus. In neurons and other excitable cells, this message encodes the amplitude and duration of activation of voltage- and/or ligand-gated ion channels. The cellular response then includes a sequence of intracellular biochemical reactions that alter the expression and function of genes and proteins. In healthy neurons, the expression of different Ca2+ sources and the spatiotemporal patterns of Ca2+ entry are well-balanced and an adequate match between Ca2+ demand and supply is usually observed. However, when Ca2+ sources become dysfunctional due to age, disease, or trauma, persistent imbalance in Ca2+ entry and clearance destroys cellular integrity, leading to cellular damage, dysfunction, and excessive proliferation or death depending on the type of cells and the strength of the insult. Neuronal damage or loss may result in severe chronic neurodegenerative conditions including sensorimotor deficits and dementia. Therefore, a tight but subtle control of cytosolic Ca2+ levels is required for neuronal health, development and function. Understanding the pharmacology and mechanisms of cytosolic Ca2+ messaging is essential for developing successful preventative strategies and treatments for neurodegenerative conditions associated with aging, dementia and brain trauma.
Inadequate vs. Optimal Ca2+ Entries and Neuronal Fate
An important common motif in the livelihood of central neurons is the existence of an optimum in the cytosolic Ca2+ concentration ([Ca2+]i) and the spatiotemporal patterns of cytosolic Ca2+ elevations. This optimum promotes neuronal survival and delivers functional benefits to neurons. The farther [Ca2+]i is from its optimum, the greater is the likelihood of neuronal damage and death. Accordingly, excessive elevations in [Ca2+]i mediated by excessive activation of ligand- and/or voltage-gated Ca2+ ion channels have been associated with a loss of neuronal function and neuronal death (see, for instance, [1–11]). Moreover, in a number of in vivo and in vitro experimental models of normal aging and Alzheimer’s disease (AD), elevated levels of cytosolic Ca2+ have been linked to age- and disease-related dysregulations in the function of voltage-gated Ca2+ ion channels (VGCCs) and N-Methyl-D-Aspartate (NMDA) receptor-mediated ion channels [2, 3, 6, 7, 10–17]. Conversely, moderate elevations in [Ca2+]i, for example, via a K+-induced depolarization or weak persistent activation of highly Ca2+-permeable α7 nicotinic acetylcholine receptors (nAChRs) have been shown to protect neurons from death in a variety of toxicity models [18–28]. In addition, some biologically active compounds (e.g., estrogen, insulin-related growth factor 1 and positive allosteric modulators of α7 nAChRs) potentiate Ca2+ permeable voltage- or ligand-gated ion channels and increase Ca2+ influx [29–37] which can be neuroprotective and cognitively beneficial.
Originally, the concept of excitotoxicity linked neuronal injury to excessive elevations in [Ca2+]i which resulted from activation of a variety of Ca2+ sources including ligand- and voltage-gated Ca2+ ion channels [38]. As such, the “Ca2+ set-point” hypothesis was introduced, proposing four stages of neuronal responsiveness to elevation in [Ca2+]i elicited by K+-dependent depolarization or electrical stimulation [1, 22, 39]: (1) a lack of neuroprotection in the near absence of cytosolic Ca2+ regardless of neurotrophic support (stage 1); (2) neuronal survival in the presence of normal cytosolic Ca2+ (∼100 nM) with neurotrophic support (stage 2); (3) neuronal survival in the presence of moderate elevation in cytosolic Ca2+ (∼200 nM) regardless of neurotrophic support (stage 3) and (4) an excess (>1 μM) of Ca2+ and neuronal death (stage 4). Although the Ca2+ set-point hypothesis supported the concept of Ca2+ optimum for neuronal survival and function, it did not explain the role of specific pathways of Ca2+ entry leaving a key question unanswered: can an elevation in [Ca2+]i be optimal regardless of the pathway of Ca2+ entry?
Role of NMDARs
Further studies revealed that elevations in [Ca2+]i are derivatives of a more elementary chain of events consisting of Ca2+ entry and intracellular Ca2+ processing. According to this concept, neuronal fate (i.e., survival or death) is predominantly determined by the source of Ca2+ entry rather than [Ca2+]i [40]: i.e., Ca2+ ions entering the cell via NMDARs are much more likely to cause damage to the cell than similar amounts of Ca2+ ions entering the cell via VGCCs. In fact, VGCC-mediated elevations in [Ca2+]i are more likely to be neuroprotective than neurotoxic (see above and [1, 20, 22, 24, 39, 41]). However, moderate activation of NMDARs during preconditioning in low concentrations of glutamate (<50 μM) as well as activation of nAChRs by nicotine have also been found to promote neuronal survival (see below and [41–44]). In general, a proper investigation of neuroprotective and neurotoxic effects of individual Ca2+ sources requires selective pharmacological tools because multiple Ca2+ sources often act in conjunction resulting in a cumulative elevation in [Ca2+]i and emergent response properties [45–48].
The NMDAR-dependent pathways of cytosolic Ca2+ regulation are complex as both excessive activation and blockade of NMDARs promote neuronal death [5, 49–51], while moderate activation of NMDARs is absolutely required for normal neuronal development and function. As a result, a key challenge in development of NMDAR-based therapies is introduced by a possibility that the same agent (e.g., NMDAR antagonist) or process (e.g., NMDAR activation) can be both neuroprotective and neurotoxic depending on the neuronal status and the phase, intensity and duration of ongoing neuronal damage. Therefore, the therapeutic index (i.e., the ratio of the lethal dose to the therapeutic dose) of many NMDAR agents would be expected to be variable, case-dependent and ≤1 on average.
A pool of functional NMDARs can be subdivided into synaptic and extrasynaptic based on their location relative to the synaptic cleft. Recent studies have started to explore an intriguing possibility that activity of synaptic and extrasynaptic NMDARs defines neuronal fate [50, 51]: activation of synaptic NMDARs leads to neuroprotection, while activation of extrasynaptic NMDARs is neurotoxic. Therefore, the overall intensity of NMDAR activation may not be as defining for the fate of neurons as the fraction of synaptic vs. extrasynaptic NMDAR activation. According to this hypothesis, Ca2+ ions entering neurons through extrasynaptic NMDARs are the most harmful. The basis for differences between the effects of synaptic and extrasynaptic NMDARs is not well-understood, but may include at least three factors, as discussed by [50]: (1) differences in the intracellular signaling pathways; (2) differences in the NMDAR subunit composition; and (3) differences in the activation profiles (e.g., synaptic NMDARs are typically activated by high transient concentrations of synaptic glutamate (∼1 mM); while extrasynaptic NMDARs are activated by persistent, but relatively low concentrations (≤1 μM) of ambient glutamate). However, the division of NMDARs into synaptic and extrasynaptic may be rather provisional because NMDARs can move laterally between synaptic and extrasynaptic sites [52]. This behavior is not unique to NMDARs and has also been observed in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) [53] and α3-/α7-containing nAChRs [54].
Moreover, direct measurements of extracellular glutamate levels [55] as well as experimental and computer modeling of glutamatergic synaptic transmission and spillover [56–58] suggest that even after relocation to extrasynaptic sites (i.e., up to several micrometers away from presynaptic release site), NMDARs do not become independent of synaptic stimulation as they can still be activated by synchronous glutamate spillovers originating from multiple active glutamatergic synapses [59, 60]. The effectiveness of glutamate spillover in activation of extrasynaptic NMDARs and cross-talk between adjacent synapses directly results from morphological and release properties of central punctate glutamatergic synapses [56, 61] and kinetic properties of NMDARs: i.e., high potency (EC50 ∼ 3 μM, [62]) and incomplete desensitization [63, 64]. Consistent with these views, the otherwise low levels of extracellular glutamate in hippocampal slices (e.g., ∼25 nM; [65]) can be substantially enhanced in the vicinity of active glutamatergic synapses [55] or during the reversal of neuronal/glial glutamate transporters that may take place under ischemia and other pathological conditions [66, 67]. However, what happens to intracellular pathways linked to an individual receptor as it switches teams (i.e., from synaptic to extrasynaptic) remains unknown (see more discussions on this topic in [50, 68, 69]).
This apparent ambiguity in the role of NMDARs in neuronal death and survival should not derail the ongoing search for a therapeutic optimum in the level of NMDAR activation and Ca2+ entry while the fact that, to date, clinical trials have been mostly unsuccessful in identifying effective NMDAR-based therapies against ischemia and other neurodegenerative conditions invites discoveries of new approaches and nontrivial solutions like never before. One of these promising emergent approaches termed “pathologically activated therapeutics” [70] makes use of low-potency open-channel NMDAR blockers, such as memantine [71]. These compounds may have neuroprotective properties as their inhibitory effects do not preclude the physiologically beneficial low-intensity activation of NMDARs, but substantially reduce the excessive activation of NMDARs which is neurotoxic. However, memantine has been also shown to inhibit α7 nAChRs with a similar or even greater potency (IC50 ∼ 0.3–5 μM) than NMDARs (IC50 ∼ 1–10 μM) [72–74]. In some cases, inhibition of α7 nAChRs by memantine may be counterproductive because moderate activation of α7 nAChRs is usually neuroprotective and cognitively beneficial (see below). Despite numerous reports of positive effects of memantine on patients with AD, non-AD dementias and other neurodegenerative disorders [75–81], the effectiveness, consistency and safety of memantine-based therapies have been questioned on multiple occasions [72, 82–85] and neurotoxic effects of therapeutic doses of memantine (∼20 mg/kg) have been reported, for example, due to a drug interaction between memantine and common acetylcholine esterase inhibitors, such as donepezil [82, 86]. Accordingly, targeting intracellular sites downstream of NMDAR activation may present an alternative and possibly, more promising therapeutic approach [87].
α7 nAChRs
Background
Neuronal nicotinic AChRs are cation-selective and Ca2+ permeable ion channel complexes. Twelve genes encoding for neuronal nAChR subunits have been identified to date [88]. Four of these genes encode for α7, α8, α9, and α10 subunits that may form functional homomeric nAChRs when expressed alone. The family of functional heteromeric nAChRs is more diverse: these functional receptors are required to have two principal α subunits (i.e., α2, α3, α4 or α6) and two or three complementary β subunits (i.e., β2 or β4). In addition, one structural subunit (i.e., α5 or β3) may also be present [89]. Among nAChRs, the α7 nAChR exhibits the highest permeability ratio of Ca2+ over Na+ ions (PCa/PNa) [90–97]. The high Ca2+ permeability of α7 nAChRs suggests important roles for this receptor in modulation of neurotransmitter release, gene expression, neuroprotection and neurotoxicity [98–101]. The existing evidence indicates that α7 nAChRs maintain a high degree of functional homology, including Ca2+ permeability, across species and preparations [102, 103]. Therefore, the properties of α7 nAChRs expressed in heterologous systems are expected to be comparable to native α7 nAChRs expressed in various brain regions. However, although α7 nAChRs can form functional homomeric nAChRs, there is a growing pool of evidence for the existence of functional heteromeric α7-containing nAChRs resulting from co-expression of α7 and non-α7 subunits (e.g., α5, β2 and β3 subunits). These native α7-containing heteromeric receptor ion channel complexes exhibit pharmacological, kinetic and desensitization properties somewhat different from those of homomeric α7 nAChRs expressed in heterologous systems [104–112].
The early studies of Ca2+ permeability of α7 nAChRs used primarily heterologous systems expressing homomeric α7 nAChRs and reported the permeability ratios for Ca2+ over Na+ ions substantially greater than those for NMDARs: PCa/PNa (α7R) ∼ 15–20 vs. PCa/PNa (NMDAR) ∼ 8–10 [93–95, 113]. However, more recent studies used hippocampal cultured neurons and acutely dissociated hippocampal and hypothalamic neurons to report more modest values: PCa/PNa(α7R) ∼ 6 vs. PCa/PNa (NMDAR) ∼ 8–10 [90, 97]. Moreover, in these experiments the Ca2+ permeability of NMDARs was found to be significantly greater than that of α7 nAChRs [97]. The observed discrepancies between the early and more recent studies may have resulted from differences in agonist application techniques, data analysis and estimates of ionic activities and liquid junction potentials. Alternatively, it is possible that native, possibly heteromeric, α7-containing nAChRs exhibit a lower Ca2+ permeability than homomeric α7 nAChRs. However, a direct comparison of Ca2+ permeabilities of native and heterologous α7 nAChRs using identical experimental techniques and data analysis has not been conducted.
Because of their high permeability to Ca2+ ions, NMDARs and α7 nAChRs form excellent examples of ligand-gated Ca2+ ion channels. As discussed, moderate activation of these receptors and thus, moderate elevation in [Ca2+]i have been found to be neuroprotective in a number of in vitro and ex vivo toxicity models as well as in vivo settings [18, 21, 23, 25–27, 41–44, 114–116]. Moreover, both types of receptors appear to employ Ca2+-PI3K-Akt-dependent pathways for mediation of neuroprotective effects [41–43, 49, 101, 117]. However, despite these important similarities, NMDARs and α7 nAChRs belong to different families of ligand-gated receptors [62, 118] and their kinetic and pharmacological properties are quite different. For instance, the mean open time of α7 nAChR-mediated channels (∼100–400 μs, [119–121]) is at least tenfold shorter than that of NMDAR channels [63, 122]. In addition, in the continuous presence of agonist, α7 nAChR-mediated currents (but not NMDAR-mediated currents) can be completely inhibited by desensitization and/or agonist-mediated open channel block [123, 124]. The short open time and rapid desensitization act as mechanisms that protect α7 nAChR-expressing cells from excessive and thus, damaging Ca2+ influx. The open channel Mg2+ block plays an analogous role for NMDAR-mediated ion channels. By contrast, Mg2+ ions do not significantly alter the function of α7 nAChRs at negative membrane potentials, although they induce rectification at depolarized membrane potentials [125].
Ca2+ Permeability of α 7 nAChRs and NMDARs
The sensitivity of α7 nAChR- and NMDAR-mediated whole-cell responses to external Ca2+ ions (i.e., [Ca2+]o) have also been found to be different (Fig. 27.1, [97]). The whole-cell conductance of α7 nAChR-mediated responses in tuberomammmillary (TM) neurons was significantly greater at low [Ca2+]o (i.e., 2 mM) than at high [Ca2+]o (i.e., 20 mM) [97]. This difference was not due to a current rundown because experiments in low [Ca2+]o that gave larger currents were conducted after experiments in high [Ca2+]o that gave smaller currents [97]. By contrast, a tenfold increase in [Ca2+]o from 2 to 20 mM did not significantly reduce the whole-cell conductance of NMDAR-mediated responses near their reversal potential in acutely dissociated hippocampal CA1 neurons [97]. Similar observations have been made in single-channel [126] and whole-cell [90, 127] experiments in cultured hippocampal neurons. However, a 67-fold increase in [Ca2+]o from 0.3 to 20 mM has been reported to reduce the whole-cell conductance of NMDAR-mediated currents by 32% in cultured spinal cord and hippocampal neurons [128]. These differences in Ca2+ sensitivity of α7 nAChR- and NMDAR-mediated ion channels may reflect different affinities with which Ca2+ ions block monovalent permeation [129], and/or a potential Ca2+-dependent modulation of α7 nAChR-channel kinetics and/or binding. All of these effects would be expected to make excessive activation of α7 nAChRs somewhat less damaging than equivalent activation of NMDARs. These views are consistent with recent experimental results [41, 43]: in these experiments, pre-conditioning of retinal ganglion cells in very high concentrations of nicotine (i.e., <500 μM), but not glutamate, was neuroprotective against glutamate toxicity.

The whole-cell conductances of α7 nAChR- and NMDAR-mediated responses near the reversal potential. The mean and standard deviation of the slope conductance near Vrev built for TM α7 nAChR- (a) and hippocampal CA1 pyramidal NMDAR-mediated responses (b). A significant [Ca2+]o–dependent decrease in the whole-cell conductance of TM α7 nAChR-, but not CA1 NMDAR-mediated responses was observed [97]. This decrease was not due to a current rundown because it persisted in experiments where high (i.e., 20 mM) [Ca2+]o was used before low (i.e., 2 mM) [Ca2+]o [97]. Examples of TM α7 nAChR-mediated currents obtained by applications of choline at various positive and negative membrane voltages in voltage-clamp in 2 mM [Ca2+]o (c) and 20 mM [Ca2+]o (d). The whole-cell conductance of TM α7 nAChR channels in high [Ca2+]o was always lower than that in low [Ca2+]o, presumably due to a Ca2+-dependent block of monovalent ion permeation. (e) The current–voltage relationship for responses illustrated in (c) and (d). No considerable current rectification was observed owing to Mg2+-free external and internal solutions and the presence of F− ions in the internal solution. The I-V curves were fitted with second-order polynomial equations. Panels (c–e) illustrate data obtained from the same acutely dissociated TM neuron. Examples of CA1 NMDAR-mediated currents obtained by applications of NMDA plus glycine at various positive and negative membrane voltages in voltage-clamp in 2 mM [Ca2+]o (f) and 20 mM [Ca2+]o (g). (h) The current–voltage relationship for responses illustrated in (f) and (g). The whole-cell conductance of NMDAR channels in 20 mM [Ca2+]o was similar to that in 2 mM [Ca2+]o, indicating a lack of significant Ca2+-dependent block of monovalent ion permeation. The I-V curves were fitted with second-order polynomial equations. Panels (f–h) illustrate data obtained from the same acutely dissociated hippocampal CA1 neuron. Note that although the application pipettes were filled with 40 mM choline or 200 μM NMDA + 20 μM glycine, the effective concentrations of choline or NMDA+glycine near the recorded neurons were unknown and considerably lower than the concentrations of agonists in application pipettes. However, in each given experiment these concentrations were very stable evidenced by stable responses [97] (Reprinted from Uteshev [97] with permission from Blackwell Publishing in the format Journal via Copyright Clearance Center)
In addition to Ca2+ permeability, the impact of activation of ligand-gated Ca2+ channels on cellular behavior and survival is affected by the channel distribution within the cell and the cell surface [50, 54, 130–132]. As mentioned, synaptic NMDARs promote neuroprotection, while extrasynaptic NMDARs may be neurotoxic [133]. By contrast, functional neuronal α7 nAChRs are predominantly pre- or extrasynaptic with only a handful of known exceptions [134–137] and yet, moderate activation of α7 nAChRs is usually neuroprotective. The reason for this important difference between NMDARs and α7 nAChRs is unknown and it is likely that other receptor properties (e.g., kinetic and desensitization properties) in addition to receptor location and ion channel Ca2+ permeability contribute to determining the receptor role in neuronal survival.
Desensitization vs. Open-Channel Block of α 7 nAChRs
In the continuous presence of nicotinic agonists, activation of α7 nAChRs is reduced naturally by two independent processes: desensitization and open channel block by agonist molecules. It is important to distinguish between these processes, especially if high concentrations of agonists are used (e.g., >2 mM ACh). At negative membrane voltages, positively charged agonists (e.g., ACh, choline) elicit both desensitization and open channel block of α7 nAChR ion channels [123]. The desensitization component of α7 nAChR-mediated responses elicited by ACh or choline can be isolated by conducting electrophysiological experiments at positive membrane voltages [123]. At negative membrane voltages, when high agonists concentrations are used (e.g., >2 mM ACh), open channel block is nearly complete although fully reversible. To minimize open channel block at negative membrane voltages, lower agonist concentrations should be used (e.g., <200 μM ACh) because the block is low-potency. By contrast, if weakly charged agonists are used (e.g., [3-(2,4-dimethoxybenzylidene)-anabaseine, i.e., DMXBA, the code name GTS-21], pKa ∼ 7.4, [138]), the separation of desensitization from open channel block is more challenging as open channel block is less dependent on the membrane voltage. In these cases, low agonist concentrations (e.g., <30 μM DMXBA) need to be used to reduce the contribution of open channel block to current decay [123].
Effects of Activation and Inactivation of α 7 nAChRs
While in some models of neurotoxicity high concentrations of α7 nAChR agonists caused cellular death [25]; in other models, even very high concentrations of nicotine (e.g., 500 μM) promoted neuronal survival [41]. These discrepancies in results may be linked to differences in the agonist concentration and time course of agonist application, as well as inactivation, desensitization and other kinetic properties of α7 nAChRs, e.g., open channel block by nicotinic agonists [123, 124, 139, 140]. Notably, low concentrations of nicotinic agonists such as those observed in the cerebrospinal fluid (CSF) in vivo (e.g., <1 μM nicotine or <100 μM choline) are more likely to cause desensitization than activation of α7 nAChRs [124, 140]. Accordingly, it has been hypothesized that it is desensitization or inhibition and not activation of α7 nAChRs that may trigger intracellular events responsible for neuroprotection and cognitive benefits [141–143]. This hypothesis, however, cannot explain a number of recent experimental findings. For instance, systemic administration of PNU-120596, a nicotinic agent that considerably reduces α7 nAChR desensitization (see below), produced positive behavioral effects restoring auditory gating deficit in a mouse model of schizophrenia [32]. Moreover, a direct testing of this hypothesis using structurally similar high-efficacy (i.e., full) and low-efficacy (i.e., partial) α7 nAChR agonists clearly demonstrated that activation of α7 nAChRs is essential for cognitive enhancement in a rat model of inhibitory avoidance [144]. Similarly, the eye-blink conditioning response is improved by α7 nAChR agonists, but impaired by antagonists [145–147] and in α7 knock-out animals [148]. Finally, cell death induced by excessive, but not moderate activity of α7 nAChRs in the NGF/serum-withdrawal toxicity model in pheochromocytoma-12 (PC-12) cells expressing functional α7 nAChRs supports the need for activation rather than desensitization of α7 nAChRs for survival of PC-12 cells [25].
By contrast, the role of α7 nAChRs in the pathophysiology of AD is less defined, primarily because of the limited understanding of how α7 nAChRs interact with Aβ1-42. For example, both activation and blockade of α7 nAChRs inhibits Aβ1-42-induced phosphorylation of tau proteins in PC-12 cells [143]. One hypothesis is that although activation of α7 nAChRs is neuroprotective and cognitively beneficial in some experimental models [23, 149–153], in mouse models of late stages of AD, which correlate with an excessive accumulation of Aβ1-42, the role of α7 nAChRs reverses. The mechanism of this role reversal may include continuing high-affinity binding of Aβ1-42 to α7 nAChRs and formation of α7-Aβ1-42 complexes which inhibit and even reverse the physiological function of α7 nAChRs and thus, the neuroprotective binding of nicotinic agonists to α7 nAChRs becomes impaired [150, 154–161]. This hypothesis received additional support from a number of recent studies that demonstrated that blocking or eliminating α7 nAChRs could alleviate some symptoms of AD. Specifically, (1) deletion of the α7 nAChR gene ameliorates certain behavioral deficits in a transgenic mouse model of AD [162]; (2) intracellular accumulation of Aβ1–42 that occurs predominantly in α7 nAChR-expressing neurons is blocked by α-bungarotoxin, a selective α7 nAChRs antagonist and by phenylarsine, an inhibitor of endocytosis [163]; and (3) α7 nAChRs mediate Aβ1-42-induced phosphorylation of tau proteins [154, 155]. These experiments supported the idea of high-affinity binding of Aβ1–42 to α7 nAChRs on neuronal cell surfaces [164], subsequent endocytosis of the resulting α7-Aβ1-42 complex and its accumulation within the lysosomal compartment provoking intracellular toxicity [163, 165].
α 7 nAChRs as a Therapeutic Tool
There is a substantial body of supportive evidence linking age-, disease- and trauma-related alterations in the expression and function of α7 nAChRs to neurodegenerative, sensorimotor and psychiatric disorders associated with cognitive decline and attention deficits [101, 166–180]. By contrast, activation of α7 nAChRs by nicotine and selective α7 nAChR agents has been shown to produce neuroprotection in vivo [26, 150, 181], ex vivo and in vitro [18, 21, 23, 25–27, 182–189] and enhance cognitive performance in patients and animal models of neurodegenerative disorders including AD, schizophrenia, brain trauma and aging [32, 101, 148, 181, 183, 189–209].
Deficits in hippocampal α7 nAChR activation are a key accompanying factor in certain cognitive disorders and enhancing this activation by nicotinic agonists has been shown to produce neuroprotection and cognitive benefits. Currently available therapeutic approaches aimed at rescuing the brain α7 nAChR activation include (Fig. 27.2): (1) ACh esterase inhibitors (AChE; e.g., donepezil) – the left most pathway; (2) α7 nAChR agonists – the right most pathway; and (3) positive allosteric modulators (PAMs) of α7 nAChRs – the middle pathway. The rationale for therapeutic use of α7 nAChR agonists and modulators arrives from observations that in neurological disorders such as dementia and schizophrenia as well as after brain trauma, functional α7 nAChRs expressed in central neurons do not vanish but their number may decline in a region-specific manner [167, 168, 171, 173, 177, 178, 180, 210]. Therefore, a moderately enhanced activation of α7 nAChRs can be achieved by pharmacological tools and this enhancement may benefit patients with neurodegeneration and cognitive decline (see Sects. 3.1, 3.2, 3.3, 3.4, 3.5).

Therapeutic approaches aimed at rescuing the brain α7 nAChR activation. The left most pathway: ACh esterase inhibitors (e.g., donepezil) increase the CSF level of ACh and promote activation of both nAChRs and mAChRs. Despite cognitive benefits (dashed line), the lack of selectivity may cause considerable side effects (e.g., autonomic). The right most pathway: α7 nAChR agonists. A moderate activation of α7 nAChRs by selective agonists (e.g., DMXBA) protects neurons, benefits cognition and appears to be clinically safe. The middle pathway: positive allosteric modulators (PAMs) of α7 nAChRs. Choline is a low-potency endogenous selective agonist of α7 nAChRs, but its potency can be considerably increased by Type-II α7-PAMs, such as PNU-120596. α7-PAMs do not activate α7 nAChRs in the absence of nicotinic agonists. Instead, α7-PAMs lower the energy barrier, allowing lower concentrations of nicotinic agonists to activate the receptor. In the presence of Type-II α7-PAMs, endogenous choline may become effective in producing moderate persistent activation of native α7 nAChRs. This type of activation of α7 nAChRs may promote neuroprotection and benefit cognition
Positive cognitive effects of inhibitors of AChE result from inhibition of the hydrolysis of ACh and thus, enhanced activation of both muscarinic AChRs (i.e., mAChRs) and nAChRs, including α7 subtype (Fig. 27.2, the left most pathway). Similar to α7 nAChRs, activation of mAChRs and non-α7 nAChRs has been reported to be cognitively beneficial (horizontal dashed path, Fig. 27.2) [211–217]. However, the lack of specificity may cause autonomic adverse effects. For example, donepezil and other AChE inhibitors have been reported to cause centrally-mediated nausea, vomiting and diarrhea [218, 219].
As discussed earlier, a moderate activation of α7 nAChRs by selective agonists (e.g., DMXBA, the right most pathway, Fig. 27.2) protects neurons, benefits cognition and appears to be clinically safe. For example, no major central side effects have been linked to oral administration of large doses of DMXBA (e.g., <450 mg/day, [138, 192]). In hippocampal slices, activation of α7 nAChRs by therapeutic nicotinic agonists, such as DMXBA, can be potentiated by PAMs [220]. PAMs would also be expected to enhance activation of α7 nAChRs by physiological levels of endogenous nicotinic agonists (i.e., ACh and choline) [34, 35] released naturally as needed.
Effects of PAMs on α7 nAChR Activation and Ca2+ Influx
PAM Hypothesis
Choline is an endogenous selective agonist of α7 nAChRs [221, 222]. The cerebrospinal fluid (CSF) contains choline at concentrations much lower (∼5–10 μM, [169, 223–227]) than its EC50 (∼0.5–1.5 mM; [222, 228]). Moreover, choline exhibits a much greater potency for desensitization (IC50 ∼ 40 μM, [124]) than activation of α7 nAChRs. Therefore, the endogenous concentration of choline in the CSF appears to be too low to activate α7 nAChRs [34, 35, 124] and in the past, endogenous choline has not been seriously considered as a therapeutic candidate [186]. However, the ambient levels of choline can be elevated 3–4-fold under conditions associated with ischemia, stroke, and substantial plasma membrane damage [223, 224, 226, 227, 229]. Cell death also creates a large source of choline causing a breakdown of phosphatidylcholine, the principle plasma membrane phospholipid, into choline and diacylglycerol. Given the low ambient concentrations of choline in the CSF under physiological conditions [169, 225], it is unlikely that in the absence of cholinergic synaptic inputs or exogenous nicotinic agents, native α7 nAChRs are persistently activated by endogenous choline [124]. However, the effects of endogenous choline may be notably different in the presence of Type-II α7-PAMs, such as PNU-120596, which significantly enhances the responsiveness of α7 nAChRs to nicotinic agents (see Sects. 3.2, 3.3, 3.4). PNU-120596 is a positive allosteric modulator of α7 nAChRs that reduces desensitization of α7 nAChRs and thus, increases the potency of nicotinic agonists enhancing the responsiveness of functional α7 nAChRs [32, 34, 220, 230, 231] and producing behavioral improvements in animal models [32]. PNU-120596 has been shown to increase the mean open time of α7 nAChR channels without producing significant changes in ion channel selectivity, single channel conductance and Ca2+ permeability [32]. PNU-120596 does not activate α7 nAChRs in the absence of nicotinic agonists. Instead, it lowers the energy barrier, allowing lower concentrations of nicotinic agonists to activate the receptor [232]. Intravenous administration of 1 mg/kg PNU-120596 elevates the concentration of PNU-120596 in the brains of rats to ∼1.5 μM [32]. This value falls near the EC50 for potentiating effects of PNU-120596 (EC50 ∼ 1.5 μM) [233, 234]. Concentrations slightly lower than the EC50 (i.e., 1 μM PNU-120596) have been shown to enhance the effects of sub-threshold concentrations of choline allowing physiological levels of choline to become effective in activation of native α7 nAChRs in the absence of exogenous nicotinic agents [34, 35]. Therefore, in the presence of PNU-120596, endogenous choline may become effective in producing moderate persistent activation of α7 nAChRs and the corresponding elevation in the Ca2+ influx and neuronal excitability (see Sects. 3.3 and 3.4) supporting neuroprotection and cognition (see Sect. 2.5).
There are two types of PAMs [235]: Type I – these compounds enhance the amplitude of α7 nAChR-mediated currents without affecting the current duration; and Type II – these compounds dramatically reduce desensitization and thus, prolong the duration of activation of α7 nAChRs in the constant presence of agonists (Fig. 27.3). The Type-II PAMs (e.g., PNU-120596) are most interesting because these compounds not only reduce desensitization of α7 nAChRs but also allow nicotinic agonists to activate already desensitized α7 nAChRs [32]. Therefore, in the presence of Type-II α7-PAMs, desensitization does not contribute to α7 nAChR activation deficits and previously desensitized α7 nAChRs can be successfully recruited for activation. Recent studies have also demonstrated that PNU-120596 is able to increase the activation potency of choline, allowing low sub-threshold (for activation) physiological concentrations of choline (∼10 μM) to become effective in activation of α7 nAChRs [34, 35]. This finding suggests an intriguing possibility of using endogenous choline (in the presence of Type-II α7-PAMs) as a therapeutic agent for enhancing activation of α7 nAChRs and thus, Ca2+ influx in neuronal systems characterized by cholinergic deficiency.

Examples and illustrative effects of Type-I and Type-II α7-PAMs. (a) NS-1738, 5-HI, Invermectin and Genistein represent the family of Type-I α7-PAMs. Schematic current traces illustrate the effects of Type-I α7-PAMs on α7 nAChRs: Type-I α7-PAMs increase the peak of α7 nAChR-mediated responses but do not alter the rate of desensitization of α7 nAChRs. (b) PNU-120596, TQS, A867744, JNJ-1930942 represent the family of Type-II α7-PAMs. Schematic current traces illustrate the effects of Type-II α7-PAMs on α7 nAChRs: Type-II α7-PAMs increase the peak of α7 nAChR-mediated responses and considerably reduce the desensitization of α7 nAChRs
A reduced version of this hypothesis has been tested in ex vivo electrophysiological experiments using hypothalamic and hippocampal brain slices [34, 35]. Under this scenario, endogenous levels of choline were modeled by the addition of physiological concentrations of choline (5–10 μM) to artificial cerebrospinal solution (ACSF) and whole-cell voltage- and current-clamp recordings were conducted in the presence and absence of 1–5 μM PNU-120596 to determine the effects of enhanced activation of native α7 nAChRs by choline on the electrical activity of hypothalamic and hippocampal neurons in brain slices (Figs. 27.4 and 27.5).

Step-like current and voltage deviations in the presence of 10 μM choline and 1 μM PNU-120596 in ACSF. (a–c) Current deviations were completely and reversibly blocked by 20 nM MLA, confirming the involvement α7 nAChRs. All current traces in (a–c) were obtained from the same TM neuron. (d, e) Step-like responses were observed in both voltage- (d) and current-clamp (e) recordings. Traces in (d) and (e) were obtained from the same TM neuron 1 min apart. In these experiments, the frequency of step-like current events appeared to be sensitive and rapidly responsive to changes in the ACSF concentrations of choline and PNU-120596 [34, 35]. Activation of α7 nAChRs in current-clamp elicited transient repetitive step-like depolarizations: ∼4 mV for individual events and ∼25 mV for simultaneous multiple events (e). The bottom trace in (d) and the top trace in (e) share the same time scale shown between these traces. The vertical scale bar indicates either 20 pA (for traces in d) or 20 mV (for traces in e). In experiments shown in (d, e), 0.3 μM TTX was continuously present in ACSF and the internal pipette solution contained CsMeSO3. In voltage-clamp experiments, the membrane voltage was held at −60 mV. (f) To visualize individual step-like depolarizations, a small continuous hyperpolarizing current (−5 pA) was injected into the recorded neuron resulting in cessation of spontaneous firing. Under these silent conditions, transient step-like depolarizations triggered short trains of action potentials (open arrows). However, occasionally, depolarizations did not trigger action potentials or triggered only a single action potential per depolarization (filled arrows). Step-like voltage and current deviations were resistant to 20 μM gabazine, 15 μM DNQX, 50 μM AP-5, 40 μM picrotoxin, and 0.3 μM TTX applied to ACSF (Reprinted from Gusev and Uteshev [34] with permission from ASPET)

Activation of TM α7 nAChRs by 10 μM choline plus 1 μM PNU-120596 enhances spontaneous firing of TM neurons in current-clamp. The spontaneous firing of TM neurons was native as current injections were not applied (i.e., 0 pA). Horizontal bars indicate −65 mV. In current-clamp, in the absence of PNU-120596 and choline, TM neurons exhibited regular patterns of spontaneous firing (a). In these control experiments, when the membrane voltage was hyperpolarized to −65 mV by injections of a small current, step-like depolarizations were not observed (b). Recordings in (a) and (b) were obtained from the same TM neuron 1 min apart. After the sustained repetitive activation of TM nAChRs was observed in voltage-clamp upon administration of 10 μM choline plus 1 μM PNU-120596 (c), current-clamp recordings were conducted using the same TM neuron (d). In current clamp, activation of TM α7 nAChRs resulted in transient repetitive increases in the frequency of spontaneous firing of TM neurons (d, filled arrows). Traces shown in (c) and (d) were obtained from the same TM neuron 1 min apart. The framed insert in (d) illustrates at a higher time resolution a portion of recording containing one transient excitation. (e) The effects of individual step-like depolarizations in current clamp. When a hyperpolarizing current (∼ −40 pA) was injected in the recorded TM neuron (the injection time is marked by *) during a prolonged interval of increased frequency (the interval between open and filled triangles), it resulted in cessation of spontaneous firing, allowing detection of the final portion of an underlying step-like depolarization. Therefore, a prolonged depolarization was observed as both an increase in spontaneous firing in the beginning of depolarization (open triangle) and a depolarizing step at the end of depolarization (filled triangle). Subsequent step-like depolarizations are also seen between the two dashed lines in insert. The insert illustrates this transition process at a higher resolution. In these experiments, ACSF contained 20 μM, gabazine, 15 μM DNQX, 50 μM AP-5 and 40 μM picrotoxin. The internal solution was K-gluconate-based (Reprinted from Gusev and Uteshev [34]. With permission from ASPET)
Synergistic Action of Physiological Choline and PNU-120596
Intriguingly, current and voltage deviations recorded in voltage- and current-clamp, respectively, resulting from a synergistic action of 10 μM choline plus 1–2 μM PNU-120596 were step-like and thus, reminiscent of and postulated to be single α7 nAChR ion channel openings detectable in whole-cell patch-clamp configuration (Fig. 27.4a–e). These experiments revealed that in the presence of PNU-120596 and 5–10 μM choline, even very low densities of α7 nAChRs such as the expression found in hippocampal CA1 pyramidal neurons (only ∼5% of that found in hippocampal CA1 interneurons [35]) generate persistent step-like currents which cause transient step-like depolarizations and occasionally, trigger bursts of action potentials. This persistent current would be expected to generate a persistent Ca2+ influx (see Sects. 3.4 and 3.5). A similar activity was detected under slightly hyperpolarized conditions in hypothalamic TM neurons (Fig. 27.4f). Moreover, activation of TM α7 nAChRs by 10 μM choline plus 1 μM PNU-120596 enhances spontaneous firing of TM neurons (Fig. 27.5a–d). In current-clamp, when a hyperpolarizing current (∼ −40 pA) was injected in the recorded TM neuron (the injection time is marked by * (Fig. 27.5e)) during a prolonged interval of increased frequency (the interval between open and filled triangles), it resulted in cessation of spontaneous firing, allowing detection of the final portion of an underlying step-like depolarization. Therefore, a prolonged step-like depolarization was observed as an increase in spontaneous firing in the beginning of depolarization (Fig. 27.5e, open triangle) and a depolarizing step at the end of depolarization (Fig. 27.5e, filled triangle).
In these experiments, the frequency of step-like current events appeared to be sensitive and rapidly responsive to changes in the ACSF concentrations of choline and PNU-120596 [34, 35]. Therefore, the synergistic action of endogenous choline and Type-II α7-PAMs may cause a sustained activation of α7 nAChRs and the corresponding persistent Ca2+ influx (see Sects. 3.4 and 3.5). These observations suggest that the net depolarization, excitation and Ca2+ influx could be modulated and optimized by tuning the administration doses of dietary choline [189] and Type-II α7-PAMs [34, 35].
Detection of Activity of Individual α 7 nAChRs in Whole-Cell
It is this capability of as few as only one individual functional α7 nAChR to depolarize and excite the entire neuron that makes it possible for a low density expression of functional α7 nAChRs to be effective in enhancing the excitability of hippocampal CA1 pyramidal neurons in the presence of PNU-120596 [35]. Therefore, high levels of expression of α7 nAChRs and synchronization of their activity may not be required for significant depolarizing and excitatory effects of physiological concentrations of choline in the presence of PNU-120596. The excitability of hippocampal CA1 pyramidal neurons positively correlate with cognitive performance and has been shown to decline with age likely due to an age-dependent enhancement of inhibitory effects of the Ca2+-dependent potassium conductance [236, 237]. Therefore, therapeutic approaches that provide neuroprotection and restore excitability of hippocampal CA1 pyramidal neurons may benefit patients with various forms of dementia and brain trauma.
Detecting activity of individual α7 nAChR ion channels in whole-cell patch-clamp experiments appears to be possible if the probability of ion channel openings is sufficiently low and the channels remain open for a prolonged period of time during which the ionic gradient across the membrane and thus, the ionic current, remain relatively constant. These requirements appear to be fulfilled for α7 nAChRs activated by physiological concentrations of choline in the presence of 1–5 μM PNU-120596 in hippocampal CA1 pyramidal neurons [35], hippocampal CA1 interneurons (Kalappa and Uteshev, unpublished observations) and hypothalamic TM α7 nAChRs [34].
In current-clamp patch-clamp experiments using hippocampal CA1 pyramidal neurons that express a very low density of functional α7 nAChRs [35], individual step-like voltage deviations triggered action potentials in 7 out of 13 cells tested (Fig. 27.4b, c). When these deviations failed to cause action potentials, they generated small step-like depolarizations whose amplitudes (∼3–5 mV) could be predicted from the neuronal input resistance (∼500 MΩ), the amplitude of step-like currents (∼8 pA) and the Ohm’s law (500 MΩ × 8 pA ∼4 mV). These estimates support the hypothesis that the observed single channel openings were most likely generated by α7 nAChRs expressed in both proximal and distal regions of the neuronal membrane and not generated only by α7 nAChRs located in the immediate vicinity of the recording patch electrode. An additional support to this hypothesis comes from the observation that in current-clamp experiments with hippocampal CA1 pyramidal neurons, recorded action potentials were triggered by α7 nAChR-mediated step-like depolarizations, while action potentials in between step-like depolarizations were not detected [35]. Therefore, it is unlikely that step-like depolarizations generated by distal α7 nAChRs (e.g., located far away from the recording pipette) have been routinely undetected (due to, for example, electrotonic filtering) because action potentials generated by distal α7 nAChRs would have occurred randomly including in between detected step-like depolarizations and this has not been observed.
These findings support the hypothesis that in the presence of PNU-120596, whole-cell patch-clamp recordings are able to detect α7 nAChR-mediated single ion channel openings from the entire cell surface. This conclusion justifies use of this approach for estimation of the total whole-cell influx of Ca2+ ions (see Sect. 3.4).
Current Net Charge and Ca2+ Influx
The mean net charge per min generated by hippocampal CA1 pyramidal α7 nAChR ion channels in response to 10 μM choline plus 2 μM PNU-120596 was estimated to be ∼9.3 pC/min = 0.16 pA [35]. This value is nearly tenfold smaller than the mean net charge of TM α7 nAChR-mediated responses elicited by 10 μM choline plus 1 μM PNU-120596 which was estimated to be ∼84 pC/min = 1.4 pA [34]. Therefore, given the 10% fractional Ca2+ current, Ca2+ ions would be expected to enter hippocampal and TM neurons at a rate of ∼0.93 pC/min and ∼8.4 pC/min, respectively, which translates into a sustained Ca2+ current ∼0.016 pA and ∼0.14 pA, respectively. These Ca2+ currents were elicited by physiological concentrations of choline and concentrations of PNU-120596 that restored the auditory gating deficit in mice [32]. Therefore, it is reasonable to expect that in in vivo settings, similar rates of Ca2+ entry in neurons expressing very low (such as hippocampal CA1 pyramidal neurons) and very high (such as hypothalamic TM neurons) densities of functional α7 nAChRs would contribute to behavioral improvements. However, a prolonged exposure of neurons to nicotinic agonists in the presence of Type-II α7-PAMs may be cytotoxic because of excessive accumulation of Ca2+ in the cytosol and possible activation of Ca2+-dependent apoptotic pathways (see Sects. 1.1 and 1.2).
The mean number of α7 nAChR ion channels opened in hippocampal CA1 pyramidal and hypothalamic TM neurons at any given time were estimated to be NpyrPopen ∼ 0.029 (i.e., 0.16 pA/5.5 pA) and NTMPopen ∼ 0.27 (i.e., 1.4 pA/5.1 pA), respectively, where Npyr and NTM are the total number of detectable functional α7 nAChRs in a pyramidal and TM neuron, respectively. Note that in experiments with TM neurons, 10 μM choline plus 1 μM PNU-120596 were used [34], whereas in the hippocampal study, the concentration of PNU-120596 was increased to 2 μM because of the substantially lower levels of expression of functional α7 nAChRs in hippocampal CA1 pyramidal neurons compared to TM neurons [35].
Direct Measurements of a 7 nAChR-Mediated Ca2+ Influx in the Presence of PNU-120596
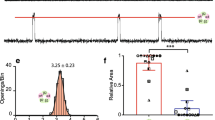
Openings of individual α7 nAChR-mediated ion channels recorded in whole-cell configuration would be expected to produce transient focal entries of Ca2+ ions. These near-membrane Ca2+ blinks have indeed been observed in fluorescent Ca2+ imaging experiments conducted in filopodia of human neuroblastoma SH-SY5Y cells and in chick retinal ganglion cells expressing α7-nAChR [238]. In the presence of PNU-120596, activation of individual and/or clusters of α7 nAChRs by nicotine resulted in transient and very focal elevations of [Ca2+]i (Fig. 27.6). These Ca2+ blinks lasted for a few seconds and were clearly observed in the presence and absence of PNU-120596, but in the presence of PNU-120596, the frequency and the duration of Ca2+ blinks were considerably increased [238]. The Ca2+ blinks were resistant to hyperpolarization induced by valinomycin (a K+ ionophore), but vanished upon removal of external Ca2+ [238]. Ryanodine (1 μM) failed to inhibit the Ca2+ blinks indicating that Ca2+ ions do not enter cells from ryanodine-sensitive cytosolic Ca2+ stores [238]. Figure 27.6 illustrates that, although the location and amplitudes of the Ca2+ blinks were variable in the presence of PNU-120596, spatiotemporally discrete Ca2+ blinks could be clearly resolved in the same filopodia during nicotine application. While certain distinct regions (#2 and #4) produced repetitive Ca2+ blinks, neighboring regions (#1, #3, and #5) did not display any Ca2+ events (Fig. 27.6a, b). The regions of brief Ca2+ elevations were localized to a sub-micron dimension (Fig. 27.6c). These observations further support the novel concept (see Sects. 3.1–3.4) that in the presence of Type-II α7-PAMs, individual functional α7 nAChRs generate distinct current events that may affect the behavior of the entire neuron [34, 35, 238].

The spatiotemporal profile of the unitary Ca 2+ events (“blinks”). (a) Sequential images from a time series showing two Ca2+ blinks separated by 1.1 μm in a single filopodia. Top left image shows the regions used for measurements overlaid on the fluorescence image, subsequent F/F0 images were captured every second during application of nicotine + PNU-120596. (b) Time-course of the F/F0 in two regions (#2 and #4) that exhibit repetitive Ca2+ elevations lasting ∼3 s and in contiguous regions (#1, #3, and #5) that did not display considerable Ca2+ activity. (c) Intensity profile of the F/F0 signal at t = 1 s in regions #2 and #4, showing the spatial spread of the Ca2+ elevations. The cross-section at >20% of the peak fluorescence averaged 0.67 μm and 0.64 μm for regions #1 and #2, respectively. Cell calcium by CHURCHILL LIVINGSTONE (Reproduced from (Gilbert et al., 2009) [238] with permission of CHURCHILL LIVINGSTONE in the format Journal via Copyright Clearance Center)
Non-neuronal NMDARs and α7 nAChRs
In addition to being broadly expressed in the central and peripheral nervous systems of mammals, functional NMDARs and α7 nAChRs are expressed in the immune system [186, 239–250], cancer cells [251–257] and other non-neuronal cells that promote angiogenesis and proliferation of cancer. Activation of α7 nAChRs in non-neuronal systems inhibits inflammation and promotes development of cancer. Although the exact role of NMDARs and α7 nAChRs in immune and cancer cells is not well understood, the high permeability of these receptor ion channels to Ca2+ ions suggest important implications for cellular function, survival and proliferation. Therefore, activation, inhibition and modulation of NMDARs and α7 nAChRs in immune and cancer cells can be used for therapeutic purposes to regulate immune defense mechanisms, reduce inflammation, inhibit proliferation or induce apoptosis of cancer cells.
Conclusions and Future Directions
In central neurons, there appear to be multiple ways of achieving optimal levels of Ca2+ entrance and [Ca2+]i to support neuronal function and survival. Among these are inhibition of excessive Ca2+ influx through NMDAR channels by low-potency use-dependent blockers, such as memantine, and enhancement of deficient Ca2+ influx through α7 nAChR channels by partial agonists of α7 nAChRs, such as DMXBA. Moderate activation of highly Ca2+-permeable NMDAR- and α7 nAChR-mediated ion channels has been shown to support neuronal function and is crucial for neuronal survival. Recently, positive allosteric modulators (PAMs) of α7 nAChRs have been identified as a promising pharmacological tool that can be used to enhance deficient activation of α7 nAChRs associated with certain neurodegenerative disorders. α7-PAMs do not activate α7 nAChRs and thus, α7 nAChRs are activated by endogenous cholinergic agonists released naturally as needed. Activation of functional α7 nAChRs is neuroprotective and thus, beneficial to neurons that express these receptors. Although some neurons that experience age- or trauma-related deficits in excitability (e.g., hippocampal CA1 pyramidal neurons [236, 237, 258]) express only very low densities of functional α7 nAChRs [35], in the presence of Type-II α7-PAMs, these neurons may also become eligible for benefits from expression and activation of functional α7 nAChRs [35].
Recent experimental results indicated that Type-II α7-PAMs may convert endogenous choline and ACh into efficacious therapeutic agents by enhancing their potency for activation of α7 nAChRs. Therefore, in the presence of Type-II PAMs, such as 1 mg/kg PNU-120596, endogenous choline may produce moderate persistent activation of α7 nAChRs and thus, moderately enhance Ca2+ influx and neuronal excitability in the absence of exogenous nicotinic agonists – effects that in in vivo settings may produce neuroprotection and cognitive benefits. Treatments involving endogenous choline may be safer than those involving synthetic α7 nAChR agonists. Hypothetically, activation of α7 nAChRs by endogenous nicotinic agonists can be moderately enhanced by optimal doses of α7-PAMs and a balanced choline diet [189]. Ideally, α7-PAM-based therapeutic interventions should be able to deliver neuroprotective and cognitive benefits by optimizing activation of α7 nAChRs and α7 nAChR-mediated Ca2+ influx in neuronal systems characterized by deficient activation of α7 nAChRs. In addition, an intriguing possibility exists for α7-PAMs to join a cohort of projected drug candidates for enhancement of cognition in healthy individuals [259].
Interestingly, only ∼10% of hippocampal α7 proteins are surface-expressed [132] and therefore, the CA1 hippocampal region may contain a large pool of unused α7 proteins. It is intriguing to speculate that under certain physiological conditions, this pool of dormant α7 proteins could be recruited to become functional and cell surface-expressed. It is also reasonable to expect that certain endogenous compounds could enhance α7 nAChR activity in a manner similar to α7-PAMs. Finding these conditions and mechanisms of regulation of α7 nAChR surface expression and function may have a very positive impact on the future of cholinergic therapies aimed at restoring and boosting cognition in dementia patients and healthy individuals.
References
Franklin JL, Johnson EM Jr (1992) Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci 15:501–508
Freir DB, Herron CE (2003) Inhibition of L-type voltage dependent calcium channels causes impairment of long-term potentiation in the hippocampal CA1 region in vivo. Brain Res 967:27–36
Fu H, Li W, Lao Y, Luo J, Lee NT, Kan KK, Tsang HW, Tsim KW, Pang Y, Li Z, Chang DC, Li M, Han Y (2006) Bis(7)-tacrine attenuates beta amyloid-induced neuronal apoptosis by regulating L-type calcium channels. J Neurochem 98:1400–1410
Harkany T, Abraham I, Timmerman W, Laskay G, Toth B, Sasvari M, Konya C, Sebens JB, Korf J, Nyakas C, Zarandi M, Soos K, Penke B, Luiten PG (2000) Beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci 12:2735–2745
Ikonomidou C, Stefovska V, Turski L (2000) Neuronal death enhanced by N-methyl-D-aspartate antagonists. Proc Natl Acad Sci USA 97:12885–12890
Lopez JR, Lyckman A, Oddo S, Laferla FM, Querfurth HW, Shtifman A (2008) Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J Neurochem 105:262–271
Mattson MP (1990) Antigenic changes similar to those seen in neurofibrillary tangles are elicited by glutamate and Ca 2+ influx in cultured hippocampal neurons. Neuron 4:105–117
Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C (2008) Amyloid beta oligomers (A beta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci 28:788–797
Papadia S, Hardingham GE (2007) The dichotomy of NMDA receptor signaling. Neuroscientist 13:572–579
Pierrot N, Ghisdal P, Caumont AS, Octave JN (2004) Intraneuronal amyloid-beta1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem 88:1140–1150
Scragg JL, Fearon IM, Boyle JP, Ball SG, Varadi G, Peers C (2005) Alzheimer’s amyloid peptides mediate hypoxic up-regulation of L-type Ca2+ channels. FASEB J 19:150–152
Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31:454–463
Davare MA, Hell JW (2003) Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1.2 during aging. Proc Natl Acad Sci USA 100:16018–16023
Thibault O, Hadley R, Landfield PW (2001) Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci 21:9744–9756
Thibault O, Landfield PW (1996) Increase in single L-type calcium channels in hippocampal neurons during aging. Science 272:1017–1020
Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K (1997) Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem 68:265–271
Weiss JH, Pike CJ, Cotman CW (1994) Ca2+ channel blockers attenuate beta-amyloid peptide toxicity to cortical neurons in culture. J Neurochem 62:372–375
Akaike A, Tamura Y, Yokota T, Shimohama S, Kimura J (1994) Nicotine-induced protection of cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res 644:181–187
Bok J, Wang Q, Huang J, Green SH (2007) CaMKII and CaMKIV mediate distinct prosurvival signaling pathways in response to depolarization in neurons. Mol Cell Neurosci 36:13–26
Collins F, Schmidt MF, Guthrie PB, Kater SB (1991) Sustained increase in intracellular calcium promotes neuronal survival. J Neurosci 11:2582–2587
Egea J, Rosa AO, Sobrado M, Gandia L, Lopez MG, Garcia AG (2007) Neuroprotection afforded by nicotine against oxygen and glucose deprivation in hippocampal slices is lost in alpha7 nicotinic receptor knockout mice. Neuroscience 145:866–872
Franklin JL, Johnson EM (1998) Control of neuronal size homeostasis by trophic factor-mediated coupling of protein degradation to protein synthesis. J Cell Biol 142:1313–1324
Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, Maeda T, Akaike A (1997) Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann Neurol 42:159–163
Koike T, Martin DP, Johnson EM Jr (1989) Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic-factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci USA 86:6421–6425
Li Y, Papke RL, He YJ, Millard WJ, Meyer EM (1999) Characterization of the neuroprotective and toxic effects of alpha7 nicotinic receptor activation in PC12 cells. Brain Res 830:218–225
Shimohama S, Greenwald DL, Shafron DH, Akaike A, Maeda T, Kaneko S, Kimura J, Simpkins CE, Day AL, Meyer EM (1998) Nicotinic à7 receptors protect against glutamate neurotoxicity and neuronal ischemic damage. Brain Res 779:359–363
Shimohama S, Kihara T (2001) Nicotinic receptor-mediated protection against beta-amyloid neurotoxicity. Biol Psychiatry 49:233–239
Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD (1999) Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol 146:955–966
Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH, Simpkins JW (2008) Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci USA 105:15148–15153
Blair LA, Bence-Hanulec KK, Mehta S, Franke T, Kaplan D, Marshall J (1999) Akt-dependent potentiation of L channels by insulin-like growth factor-1 is required for neuronal survival. J Neurosci 19:1940–1951
Smith CC, McMahon LL (2006) Estradiol-induced increase in the magnitude of long-term potentiation is prevented by blocking NR2B-containing receptors. J Neurosci 26:8517–8522
Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, Rutherford-Root KL, Berkenpas MB, Hoffmann WE, Piotrowski DW, Groppi VE, Allaman G, Ogier R, Bertrand S, Bertrand D, Arneric SP (2005) A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci 25:4396–4405
Timmermann DB, Gronlien JH, Kohlhaas KL, Nielsen EO, Dam E, Jorgensen TD, Ahring PK, Peters D, Holst D, Christensen JK, Malysz J, Briggs CA, Gopalakrishnan M, Olsen GM (2007) An allosteric modulator of the alpha7 nicotinic acetylcholine receptor possessing cognition-enhancing properties in vivo. J Pharmacol Exp Ther 323:294–307
Gusev AG, Uteshev VV (2010) Physiological concentrations of choline activate native alpha7-containing nicotinic acetylcholine receptors in the presence of PNU-120596 [1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)-urea]. J Pharmacol Exp Ther 332:588–598
Kalappa BI, Gusev AG, Uteshev VV (2010) Activation of functional alpha7-containing nAChRs in hippocampal CA1 pyramidal neurons by physiological levels of choline in the presence of PNU-120596. PLoS One 5:e13964
Malysz J, Gronlien JH, Anderson DJ, Hakerud M, Thorin-Hagene K, Ween H, Wetterstrand C, Briggs CA, Faghih R, Bunnelle WH, Gopalakrishnan M (2009) In vitro pharmacological characterization of a novel allosteric modulator of alpha 7 neuronal acetylcholine receptor, 4-(5-(4-chlorophenyl)-2-methyl-3-propionyl-1H-pyrrol-1-yl)benzenesulfonami de (A-867744), exhibiting unique pharmacological profile. J Pharmacol Exp Ther 330:257–267
Dinklo T, Shaban H, Thuring JW, Lavreysen H, Stevens KE, Zheng L, Mackie C, Grantham C, Vandenberk I, Meulders G, Peeters L, Verachtert H, De Prins E, Lesage AS (2011) Characterization of 2-[[4-fluoro-3-(trifluoromethyl)phenyl]amino]-4-(4-pyridinyl)-5-thiazoleme thanol (JNJ-1930942), a novel positive allosteric modulator of the {alpha}7 nicotinic acetylcholine receptor. J Pharmacol Exp Ther 336:560–574
Choi DW (1988) Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci 11:465–469
Mennerick S, Zorumski CF (2000) Neural activity and survival in the developing nervous system. Mol Neurobiol 22:41–54
Tymianski M, Charlton MP, Carlen PL, Tator CH (1993) Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci 13:2085–2104
Brandt SK, Weatherly ME, Ware L, Linn DM, Linn CL (2011) Calcium preconditioning triggers neuroprotection in retinal ganglion cells. Neuroscience 172:387–397
Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE (2006) Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci 26:4509–4518
Asomugha CO, Linn DM, Linn CL (2010) ACh receptors link two signaling pathways to neuroprotection against glutamate-induced excitotoxicity in isolated RGCs. J Neurochem 112:214–226
Ogita K, Okuda H, Yamamoto Y, Nishiyama N, Yoneda Y (2003) In vivo neuroprotective role of NMDA receptors against kainate-induced excitotoxicity in murine hippocampal pyramidal neurons. J Neurochem 85:1336–1346
Nakazawa H, Murphy TH (1999) Activation of nuclear calcium dynamics by synaptic stimulation in cultured cortical neurons. J Neurochem 73:1075–1083
Hardingham GE, Arnold FJ, Bading H (2001) A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci 4:565–566
Pokorska A, Vanhoutte P, Arnold FJ, Silvagno F, Hardingham GE, Bading H (2003) Synaptic activity induces signalling to CREB without increasing global levels of cAMP in hippocampal neurons. J Neurochem 84:447–452
Uteshev VV, Knot HJ (2005) Somatic Ca(2+) dynamics in response to choline-mediated excitation in histaminergic tuberomammillary neurons. Neuroscience 134:133–143
Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 11:476–487
Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696
Hardingham GE, Bading H (2003) The Yin and Yang of NMDA receptor signalling. Trends Neurosci 26:81–89
Tovar KR, Westbrook GL (2002) Mobile NMDA receptors at hippocampal synapses. Neuron 34:255–264
Borgdorff AJ, Choquet D (2002) Regulation of AMPA receptor lateral movements. Nature 417:649–653
Fernandes CC, Berg DK, Gomez-Varela D (2010) Lateral mobility of nicotinic acetylcholine receptors on neurons is determined by receptor composition, local domain, and cell type. J Neurosci 30:8841–8851
Okubo Y, Sekiya H, Namiki S, Sakamoto H, Iinuma S, Yamasaki M, Watanabe M, Hirose K, Iino M (2010) Imaging extrasynaptic glutamate dynamics in the brain. Proc Natl Acad Sci USA 107:6526–6531
Uteshev VV, Pennefather PS (1997) Analytical description of the activation of multi-state receptors by continuous neurotransmitter signals at brain synapses. Biophys J 72:1127–1134
Rusakov DA, Wuerz A, Kullmann DM (2004) Heterogeneity and specificity of presynaptic Ca2+ current modulation by mGluRs at individual hippocampal synapses. Cereb Cortex 14:748–758
Zheng K, Scimemi A, Rusakov DA (2008) Receptor actions of synaptically released glutamate: the role of transporters on the scale from nanometers to microns. Biophys J 95:4584–4596
Asztely F, Erdemli G, Kullmann DM (1997) Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron 18:281–293
Kullmann DM (2000) Spillover and synaptic cross talk mediated by glutamate and GABA in the mammalian brain. Prog Brain Res 125:339–351
Uteshev VV, Pennefather PS (1996) A mathematical description of miniature postsynaptic current generation at central nervous system synapses. Biophys J 71:1256–1266
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496
Lester RA, Jahr CE (1992) NMDA channel behavior depends on agonist affinity. J Neurosci 12:635–643
Clements AM, Westbrook GL (1991) Activation kinetics reveal the number of glutamate and glycine binding sites on the N -methyl-D-aspartate receptor. Neuron 7:605–613
Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27:9736–9741
Rossi DJ, Oshima T, Attwell D (2000) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403:316–321
Camacho A, Massieu L (2006) Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res 37:11–18
Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D (2006) NMDA receptor surface mobility depends on NR2A-2B subunits. Proc Natl Acad Sci USA 103:18769–18774
Groc L, Bard L, Choquet D (2009) Surface trafficking of N-methyl-D-aspartate receptors: physiological and pathological perspectives. Neuroscience 158:4–18
Lipton SA (2007) Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci 8:803–808
Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA (1992) Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci 12:4427–4436
Aracava Y, Pereira EF, Maelicke A, Albuquerque EX (2005) Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther 312:1195–1205
Maskell PD, Speder P, Newberry NR, Bermudez I (2003) Inhibition of human alpha 7 nicotinic acetylcholine receptors by open channel blockers of N-methyl-D-aspartate receptors. Br J Pharmacol 140:1313–1319
Kotermanski SE, Johnson JW (2009) Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J Neurosci 29:2774–2779
Aarsland D, Ballard C, Walker Z, Bostrom F, Alves G, Kossakowski K, Leroi I, Pozo-Rodriguez F, Minthon L, Londos E (2009) Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol 8:613–618
Atri A, Shaughnessy LW, Locascio JJ, Growdon JH (2008) Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Assoc Disord 22:209–221
Leroi I, Overshott R, Byrne EJ, Daniel E, Burns A (2009) Randomized controlled trial of memantine in dementia associated with Parkinson’s disease. Mov Disord 24:1217–1221
Levin OS, Batukaeva LA, Smolentseva IG, Amosova NA (2009) Efficacy and safety of memantine in Lewy body dementia. Neurosci Behav Physiol 39:597–604
Parsons CG, Danysz W, Quack G (1999) Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist–a review of preclinical data. Neuropharmacology 38:735–767
Plosker GL, Lyseng-Williamson KA (2005) Memantine: a pharmacoeconomic review of its use in moderate-to-severe Alzheimer’s disease. Pharmacoeconomics 23:193–206
Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ (2003) Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348:1333–1341
Creeley CE, Wozniak DF, Nardi A, Farber NB, Olney JW (2008) Donepezil markedly potentiates memantine neurotoxicity in the adult rat brain. Neurobiol Aging 29:153–167
Schugens MM, Egerter R, Daum I, Schepelmann K, Klockgether T, Loschmann PA (1997) The NMDA antagonist memantine impairs classical eyeblink conditioning in humans. Neurosci Lett 224:57–60
Swerdlow NR, van Bergeijk DP, Bergsma F, Weber E, Talledo J (2009) The effects of memantine on prepulse inhibition. Neuropsychopharmacology 34:1854–1864
Vercelletto M, Boutoleau-Bretonniere C, Volteau C, Puel M, Auriacombe S, Sarazin M, Michel BF, Couratier P, Thomas-Anterion C, Verpillat P, Gabelle A, Golfier V, Cerato E, Lacomblez L (2011) Memantine in behavioral variant frontotemporal dementia: negative results. J Alzheimers Dis 23:749–759
Schneider LS, Insel PS, Weiner MW (2011) Treatment with cholinesterase inhibitors and memantine of patients in the Alzheimer’s Disease neuroimaging initiative. Arch Neurol 68:58–66
Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M (2002) Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 298:846–850
Hogg RC, Raggenbass M, Bertrand D (2003) Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol 147:1–46
Corringer PJ, Le Novere N, Changeux JP (2000) Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol 40:431–458
Castro NG, Albuquerque EX (1995) à-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys J 68:516–524
Fucile S (2004) Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 35:1–8
Seguela P, Wadiche J, Dinely-Miller K, Dani JA, Patrick JW (1993) Molecular cloning, functional properties and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 13(2):596–604
Sands SB, Costa ACS, Patrick JW (1993) Barium permeability of neuronal nicotinic acetylcholine receptor alpha 7 expressed in Xenopus oocytes. Biophys J 65:2614–2621
Vernino S, Amador M, Luetje CW, Patrick J, Dani JA (1992) Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron 8:127–134
Bertrand D, Galzi JL, Devillers-Thiery A, Bertrand S, Changeux JP (1993) Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci USA 90:6971–6975
Nutter TJ, Adams DJ (1995) Monovalent and divalent cation permeability and block of neuronal nicotinic receptor channels in rat parasympathetic ganglia. J Gen Physiol 105:701–723
Uteshev VV (2010) Evaluation of Ca2+ permeability of nicotinic acetylcholine receptors in hypothalamic histaminergic neurons. Acta Biochim Biophys Sin (Shanghai) 42:8–20
Meyer EM, Tay ET, Zoltewicz JA, Papke RL, Meyers C, King M, Fiebre CMd (1998) Neuroprotective and memory-related actions of novel à7 nicotinic agents with different mixed agonist/antagonist properties. J Pharmacol Exp Ther 284:1026–1032
Role LW, Berg DK (1996) Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16:1077–1085
Dajas-Bailador F, Wonnacott S (2004) Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci 25:317–324
Thomsen MS, Hansen HH, Timmerman DB, Mikkelsen JD (2010) Cognitive improvement by activation of alpha7 nicotinic acetylcholine receptors: from animal models to human pathophysiology. Curr Pharm Des 16:323–343
Albuquerque EX, Pereira EF, Mike A, Eisenberg HM, Maelicke A, Alkondon M (2000) Neuronal nicotinic receptors in synaptic functions in humans and rats: physiological and clinical relevance. Behav Brain Res 113:131–141
Fucile S, Renzi M, Lax P, Eusebi F (2003) Fractional Ca(2+) current through human neuronal alpha7 nicotinic acetylcholine receptors. Cell Calcium 34:205–209
El-Hajj RA, McKay SB, McKay DB (2007) Pharmacological and immunological identification of native alpha7 nicotinic receptors: evidence for homomeric and heteromeric alpha7 receptors. Life Sci 81:1317–1322
Khiroug SS, Harkness PC, Lamb PW, Sudweeks SN, Khiroug L, Millar NS, Yakel JL (2002) Rat nicotinic ACh receptor alpha7 and beta2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol 540:425–434
Listerud M, Brussaard AB, Devay P, Colman DR, Role LW (1991) Functional contribution of neuronal AChR subunits revealed by antisense oligonucleotides. Science 254:1518–1521
Palma E, Maggi L, Barabino B, Eusebi F, Ballivet M (1999) Nicotinic acetylcholine receptors assembled from the alpha7 and beta3 subunits. J Biol Chem 274:18335–18340
Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L (1996) Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature 380:347–351
Sudweeks SN, Yakel JL (2000) Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J Physiol 527(Pt 3):515–528
Virginio C, Giacometti A, Aldegheri L, Rimland JM, Terstappen GC (2002) Pharmacological properties of rat alpha 7 nicotinic receptors expressed in native and recombinant cell systems. Eur J Pharmacol 445:153–161
Yu CR, Role LW (1998) Functional contribution of the alpha5 subunit to neuronal nicotinic channels expressed by chick sympathetic ganglion neurones. J Physiol 509(Pt 3):667–681
Yu CR, Role LW (1998) Functional contribution of the alpha7 subunit to multiple subtypes of nicotinic receptors in embryonic chick sympathetic neurones. J Physiol 509(Pt 3):651–665
Patrick J, Sequela P, Vernino S, Amador M, Luetje C, Dani JA (1993) Functional diversity of neuronal nicotinic acetylcholine receptors. Prog Brain Res 98:113–120
Levin ED (2002) Nicotinic receptor subtypes and cognitive function. J Neurobiol 53:633–640
Marini AM, Rabin SJ, Lipsky RH, Mocchetti I (1998) Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-D-aspartate. J Biol Chem 273:29394–29399
Valera E, Sanchez-Martin FJ, Ferrer-Montiel AV, Messeguer A, Merino JM (2008) NMDA-induced neuroprotection in hippocampal neurons is mediated through the protein kinase A and CREB (cAMP-response element-binding protein) pathway. Neurochem Int 53:148–154
Akaike A, Takada-Takatori Y, Kume T, Izumi Y (2010) Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci 40:211–216
Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA (2004) Cys-loop receptors: new twists and turns. Trends Neurosci 27:329–336
Fucile S, Palma E, Martinez-Torres A, Miledi R, Eusebi F (2002) The single-channel properties of human acetylcholine alpha 7 receptors are altered by fusing alpha 7 to the green fluorescent protein. Proc Natl Acad Sci USA 99:3956–3961
Mike A, Castro NG, Albuquerque EX (2000) Choline and acetylcholine have similar kinetic properties of activation and desensitization on the alpha7 nicotinic receptors in rat hippocampal neurons. Brain Res 882:155–168
Shao Z, Yakel JL (2000) Single channel properties of neuronal nicotinic ACh receptors in stratum radiatum interneurons of rat hippocampal slices. J Physiol 527(Pt 3):507–513
Ascher P, Bregestovski P, Nowak L (1988) N -methyl-D-aspartate-activated channels of mouse central neurones in magnesium-free solutions. J Physiol 399:207–226
Uteshev VV, Meyer EM, Papke RL (2002) Activation and inhibition of native neuronal alpha-bungarotoxin-sensitive nicotinic ACh receptors. Brain Res 948:33–46
Uteshev VV, Meyer EM, Papke RL (2003) Regulation of neuronal function by choline and 4OH-GTS-21 through alpha 7 nicotinic receptors. J Neurophysiol 89:1797–1806
Alkondon M, Reinhardt S, Lobron C, Hermsen B, Maelicke A, Albuquerque EX (1994) Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. II. The rundown and inward rectification of agonist-elicited whole cell currents and identification of receptor subunits by in situ hybridization. J Pharmacol Exp Ther 271:494–506
Jahr CE, Stevens CF (1993) Calcium permeability of the N-methyl-D-aspartate receptor channel in hippocampal neurons in culture. Proc Natl Acad Sci USA 90:11573–11577
Iino M, Ozawa S, Tsuzuki K (1990) Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurons. J Physiol 424:151–165
Mayer ML, Westbrook GL (1987) Permeation and block of N -methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurons. J Physiol 394:501
Lyford LK, Lee JW, Rosenberg RL (2002) Low-affinity Ca(2+) and Ba(2+) binding sites in the pore of alpha7 nicotinic acetylcholine receptors. Biochim Biophys Acta 1559:69–78
Conroy WG, Liu Z, Nai Q, Coggan JS, Berg DK (2003) PDZ-containing proteins provide a functional postsynaptic scaffold for nicotinic receptors in neurons. Neuron 38:759–771
Ehlers MD, Heine M, Groc L, Lee MC, Choquet D (2007) Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron 54:447–460
Mielke JG, Mealing GA (2009) Cellular distribution of the nicotinic acetylcholine receptor alpha7 subunit in rat hippocampus. Neurosci Res 65:296–306
Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414
Zhang Z, Coggan JS, Berg DK (1996) Synaptic currents generated by neuronal acetylcholine receptors sensitive to alpha-bungarotoxin. Neuron 17:1231–1240
Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV (1998) Synaptic potentials mediated via alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci 18:8228–8235
Hefft S, Hulo S, Bertrand D, Muller D (1999) Synaptic transmission at nicotinic acetylcholine receptors in rat hippocampal organotypic cultures and slices. J Physiol 515(Pt 3):769–776
Hatton GI, Yang QZ (2002) Synaptic potentials mediated by alpha 7 nicotinic acetylcholine receptors in supraoptic nucleus. J Neurosci 22:29–37
Kem WR (2000) The brain alpha7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer’s disease: studies with DMXBA (GTS-21). Behav Brain Res 113:169–181
Uteshev VV, Stevens DR, Haas HL (1996) Alpha-Bungarotoxin-sensitive nicotinic responses in rat tuberomammillary neurons. Pflugers Arch 432:607–613
Alkondon M, Pereira EF, Almeida LE, Randall WR, Albuquerque EX (2000) Nicotine at concentrations found in cigarette smokers activates and desensitizes nicotinic acetylcholine receptors in CA1 interneurons of rat hippocampus. Neuropharmacology 39:2726–2739
Fujii S, Ji Z, Sumikawa K (2000) Inactivation of alpha7 ACh receptors and activation of non-alpha7 ACh receptors both contribute to long term potentiation induction in the hippocampal CA1 region. Neurosci Lett 286:134–138
Ferchmin PA, Perez D, Eterovic VA, de Vellis J (2003) Nicotinic receptors differentially regulate N-methyl-D-aspartate damage in acute hippocampal slices. J Pharmacol Exp Ther 305:1071–1078
Hu M, Gopalakrishnan M, Li J (2009) Positive allosteric modulation of alpha7 neuronal nicotinic acetylcholine receptors: lack of cytotoxicity in PC12 cells and rat primary cortical neurons. Br J Pharmacol 158:1857–1864
Briggs CA, Gronlien JH, Curzon P, Timmermann DB, Ween H, Thorin-Hagene K, Kerr P, Anderson DJ, Malysz J, Dyhring T, Olsen GM, Peters D, Bunnelle WH, Gopalakrishnan M (2009) Role of channel activation in cognitive enhancement mediated by alpha7 nicotinic acetylcholine receptors. Br J Pharmacol 158:1486–1494
Leon SF, Suwazono S, Takenaga S, Arimura K, Osame M (1997) The effects of tobacco smoking on the short, middle, and long latency responses of the blink reflex in humans. J Clin Neurophysiol 14:144–149
Woodruff-Pak DS, Green JT, Coleman-Valencia C, Pak JT (2000) A nicotinic cholinergic agonist (GTS-21) and eyeblink classical conditioning: acquisition, retention, and relearning in older rabbits. Exp Aging Res 26:323–336
Woodruff-Pak DS (2003) Mecamylamine reversal by nicotine and by a partial alpha7 nicotinic acetylcholine receptor agonist (GTS-21) in rabbits tested with delay eyeblink classical conditioning. Behav Brain Res 143:159–167
Brown KL, Comalli DM, Biasi MD, Woodruff-Pak DS (2010) Trace eyeblink conditioning is impaired in alpha7 but not in beta2 nicotinic acetylcholine receptor knockout mice. Front Behav Neurosci 4:166
Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT (2010) Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J Neurosci 30:2442–2453
Ren K, King MA, Liu J, Siemann J, Altman M, Meyers C, Hughes JA, Meyer EM (2007) The alpha7 nicotinic receptor agonist 4OH-GTS-21 protects axotomized septohippocampal cholinergic neurons in wild type but not amyloid-overexpressing transgenic mice. Neuroscience 148:230–237
Jonnala RR, Buccafusco JJ (2001) Relationship between the increased cell surface alpha7 nicotinic receptor expression and neuroprotection induced by several nicotinic receptor agonists. J Neurosci Res 66:565–572
Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li J (2007) High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Res 1151:227–235
Qi XL, Nordberg A, Xiu J, Guan ZZ (2007) The consequences of reducing expression of the alpha7 nicotinic receptor by RNA interference and of stimulating its activity with an alpha7 agonist in SH-SY5Y cells indicate that this receptor plays a neuroprotective role in connection with the pathogenesis of Alzheimer’s disease. Neurochem Int 51:377–383
Wang HY, Li W, Benedetti NJ, Lee DH (2003) Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J Biol Chem 278:31547–31553
Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD (2001) Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci 21:4125–4133
Dineley KT, Bell KA, Bui D, Sweatt JD (2002) Beta -Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem 277:25056–25061
Dineley KT (2007) Beta-amyloid peptide–nicotinic acetylcholine receptor interaction: the two faces of health and disease. Front Biosci 12:5030–5038
Clifford PM, Siu G, Kosciuk M, Levin EC, Venkataraman V, D’Andrea MR, Nagele RG (2008) Alpha7 nicotinic acetylcholine receptor expression by vascular smooth muscle cells facilitates the deposition of Abeta peptides and promotes cerebrovascular amyloid angiopathy. Brain Res 1234:158–171
Soderman A, Thomsen MS, Hansen HH, Nielsen EO, Jensen MS, West MJ, Mikkelsen JD (2008) The nicotinic alpha7 acetylcholine receptor agonist ssr180711 is unable to activate limbic neurons in mice overexpressing human amyloid-beta1-42. Brain Res 1227:240–247
Wang HY, Stucky A, Liu J, Shen C, Trocme-Thibierge C, Morain P (2009) Dissociating beta-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer’s disease brain. J Neurosci 29:10961–10973
Martin SE, de Fiebre NE, de Fiebre CM (2004) The alpha7 nicotinic acetylcholine receptor-selective antagonist, methyllycaconitine, partially protects against beta-amyloid1-42 toxicity in primary neuron-enriched cultures. Brain Res 1022:254–256
Dziewczapolski G, Glogowski CM, Masliah E, Heinemann SF (2009) Deletion of the alpha 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. J Neurosci 29:8805–8815
Nagele RG, D’Andrea MR, Anderson WJ, Wang HY (2002) Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110:199–211
Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB (2000) Beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem 275:5626–5632
D’Andrea MR, Nagele RG (2006) Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer’s disease pyramidal neurons. Curr Pharm Des 12:677–684
Jenden DJ, Scremin OU, Roch M, Li G (1996) The influence of aging on whole body choline release and clearance. Life Sci 58:2003–2009
Guan ZZ, Zhang X, Ravid R, Nordberg A (2000) Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J Neurochem 74:237–243
Nordberg A (2001) Nicotinic receptor abnormalities of Alzheimer’s disease: therapeutic implications. Biol Psychiatry 49:200–210
Sarter M, Parikh V (2005) Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci 6:48–56
Martin LF, Freedman R (2007) Schizophrenia and the alpha7 nicotinic acetylcholine receptor. Int Rev Neurobiol 78:225–246
Leonard S, Breese C, Adams C, Benhammou K, Gault J, Stevens K, Lee M, Adler L, Olincy A, Ross R, Freedman R (2000) Smoking and schizophrenia: abnormal nicotinic receptor expression. Eur J Pharmacol 393:237–242
Freedman R, Adams CE, Leonard S (2000) The alpha7-nicotinic acetylcholine receptor and the pathology of hippocampal interneurons in schizophrenia. J Chem Neuroanat 20:299–306
Stevens KE, Freedman R, Collins AC, Hall M, Leonard S, Marks MJ, Rose GM (1996) Genetic correlation of inhibitory gating of hippocampal auditory evoked response and alpha-bungarotoxin-binding nicotinic cholinergic receptors in inbred mouse strains. Neuropsychopharmacology 15:152–162
Felix R, Levin ED (1997) Nicotinic antagonist administration into the ventral hippocampus and spatial working memory in rats. Neuroscience 81:1009–1017
Wevers A, Witter B, Moser N, Burghaus L, Banerjee C, Steinlein OK, Schutz U, de Vos RA, Steur EN, Lindstrom J, Schroder H (2000) Classical Alzheimer features and cholinergic dysfunction: towards a unifying hypothesis? Acta Neurol Scand Suppl 176:42–48
Freedman R, Hall M, Adler LE, Leonard S (1995) Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry 38:22–33
Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH (1995) Alteration in nicotine binding sites in Parkinson’s disease. Lewy body dementia and Alzheimer’s disease: possible index of early neuropathology. Neuroscience 64:385–395
Nordberg A, Winblad B (1986) Reduced number of [3H]nicotine and [3H]acetylcholine binding sites in the frontal cortex of Alzheimer brains. Neurosci Lett 72:115–119
Shimohama S, Taniguchi T, Fujiwara M, Kameyama M (1986) Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J Neurochem 46:288–293
London ED, Ball MJ, Waller SB (1989) Nicotinic binding sites in cerebral cortex and hippocampus in Alzheimer’s dementia. Neurochem Res 14:745–750
Takeuchi H, Yanagida T, Inden M, Takata K, Kitamura Y, Yamakawa K, Sawada H, Izumi Y, Yamamoto N, Kihara T, Uemura K, Inoue H, Taniguchi T, Akaike A, Takahashi R, Shimohama S (2009) Nicotinic receptor stimulation protects nigral dopaminergic neurons in rotenone-induced Parkinson’s disease models. J Neurosci Res 87:576–585
Kaneko S, Maeda T, Kume T, Kochiyama H, Akaike A, Shimohama S, Kimura J (1997) Nicotine protects cultured cortical neurons against glutamate-induced cytotoxicity via alpha7-neuronal receptors and neuronal CNS receptors. Brain Res 765:135–140
Meyer EM, King MA, Meyers C (1998) Neuroprotective effects of 2,4-dimethoxybenzylidene anabaseine (DMXB) and tetrahydroaminoacridine (THA) in neocortices of nucleus basalis lesioned rats. Brain Res 786:252–254
Li Y, Meyer EM, Walker DW, Millard WJ, He YJ, King MA (2002) Alpha7 nicotinic receptor activation inhibits ethanol-induced mitochondrial dysfunction, cytochrome c release and neurotoxicity in primary rat hippocampal neuronal cultures. J Neurochem 81:853–858
Verbois SL, Scheff SW, Pauly JR (2003) Chronic nicotine treatment attenuates alpha 7 nicotinic receptor deficits following traumatic brain injury. Neuropharmacology 44:224–233
Buccafusco JJ, Beach JW, Terry AV Jr, Doad GS, Sood A, Arias E, Misawa H, Masai M, Fujii T, Kawashima K (2004) Novel analogs of choline as potential neuroprotective agents. J Alzheimers Dis 6:S85–S92
Fucile S, Renzi M, Lauro C, Limatola C, Ciotti T, Eusebi F (2004) Nicotinic cholinergic stimulation promotes survival and reduces motility of cultured rat cerebellar granule cells. Neuroscience 127:53–61
Rosa AO, Egea J, Gandia L, Lopez MG, Garcia AG (2006) Neuroprotection by nicotine in hippocampal slices subjected to oxygen-glucose deprivation: involvement of the alpha7 nAChR subtype. J Mol Neurosci 30:61–62
Guseva MV, Hopkins DM, Scheff SW, Pauly JR (2008) Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma 25:975–983
Buccafusco JJ, Letchworth SR, Bencherif M, Lippiello PM (2005) Long-lasting cognitive improvement with nicotinic receptor agonists: mechanisms of pharmacokinetic-pharmacodynamic discordance. Trends Pharmacol Sci 26:352–360
Buccafusco JJ, Terry AV Jr, Decker MW, Gopalakrishnan M (2007) Profile of nicotinic acetylcholine receptor agonists ABT-594 and A-582941, with differential subtype selectivity, on delayed matching accuracy by young monkeys. Biochem Pharmacol 74:1202–1211
Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, Burnett AL (2003) Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology 28:542–551
Leiser SC, Bowlby MR, Comery TA, Dunlop J (2009) A cog in cognition: how the alpha 7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol Ther 122:302–311
Olincy A, Stevens KE (2007) Treating schizophrenia symptoms with an alpha7 nicotinic agonist, from mice to men. Biochem Pharmacol 74:1192–1201
Arendash GW, Sengstock GJ, Sanberg PR, Kem WR (1995) Improved learning and memory in aged rats with chronic administration of the nicotinic receptor agonist GTS-21. Brain Res 674:252–259
Meyer EM, Tay ET, Papke RL, Meyers C, Huang G, de Fiebre CM (1997) Effects of 3-[2,4-dimethoxybenzylidene]anabaseine (DMXB) on rat nicotinic receptors and memory-related behaviors. Brain Res 768:49–56
Ross RG, Stevens KE, Proctor WR, Leonard S, Kisley MA, Hunter SK, Freedman R, Adams CE (2010) Research review: cholinergic mechanisms, early brain development, and risk for schizophrenia. J Child Psychol Psychiatry 51:535–549
Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, Ellis J, Zerbe GO, Leonard S, Stevens KE, Stevens JO, Martin L, Adler LE, Soti F, Kem WR, Freedman R (2006) Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry 63:630–638
Martin EJ, Panikar KS, King MA, Deyrup M, Hunter B, Wang G, Meyer E (1994) Cytoprotective actions of 2,4-dimethoxybenzylidene anabaseine in differentiated PC12 cells and septal cholinergic cells. Drug Dev Res 31:134–141
Briggs CA, Anderson DJ, Brioni JD, Buccafusco JJ, Buckley MJ, Campbell JE, Decker MW, Donnelly-Roberts D, Elliot RL, Gopalakrishnan M, Holladay MW, Hui Y, Jackson W, Kim DJB, Marsh KC, O’Neill AO, Pendergast MA, Ryther KB, Sullivan JP, Arneric SP (1997) Functional characterization of the novel nicotinic receptor ligand GTS-21 in vitro and in vivo. Pharmacol Biochem Behav 57:231–241
Van Kampen M, Selbach K, Schneider R, Schiegel E, Boess F, Schreiber R (2004) AR-R 17779 improves social recognition in rats by activation of nicotinic alpha7 receptors. Psychopharmacology 172:375–383
Woodruff-Pak DS, Li Y, Kem WR (1994) A nicotinic agonist (GTS-21), eyeblink classical conditioning, and nicotinic receptor binding in rabbit brain. Brain Res 645:309–317
Wishka DG, Walker DP, Yates KM, Reitz SC, Jia S, Myers JK, Olson KL, Jacobsen EJ, Wolfe ML, Groppi VE, Hanchar AJ, Thornburgh BA, Cortes-Burgos LA, Wong EH, Staton BA, Raub TJ, Higdon NR, Wall TM, Hurst RS, Walters RR, Hoffmann WE, Hajos M, Franklin S, Carey G, Gold LH, Cook KK, Sands SB, Zhao SX, Soglia JR, Kalgutkar AS, Arneric SP, Rogers BN (2006) Discovery of N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide, an agonist of the alpha7 nicotinic acetylcholine receptor, for the potential treatment of cognitive deficits in schizophrenia: synthesis and structure-activity relationship. J Med Chem 49:4425–4436
Bitner RS, Bunnelle WH, Decker MW, Drescher KU, Kohlhaas KL, Markosyan S, Marsh KC, Nikkel AL, Browman K, Radek R, Anderson DJ, Buccafusco J, Gopalakrishnan M (2010) In vivo pharmacological characterization of a novel selective alpha7 neuronal nicotinic acetylcholine receptor agonist ABT-107: preclinical considerations in Alzheimer’s disease. J Pharmacol Exp Ther 334:875–886
Bitner RS, Bunnelle WH, Anderson DJ, Briggs CA, Buccafusco J, Curzon P, Decker MW, Frost JM, Gronlien JH, Gubbins E, Li J, Malysz J, Markosyan S, Marsh K, Meyer MD, Nikkel AL, Radek RJ, Robb HM, Timmermann D, Sullivan JP, Gopalakrishnan M (2007) Broad-spectrum efficacy across cognitive domains by alpha7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J Neurosci 27:10578–10587
Boess FG, De Vry J, Erb C, Flessner T, Hendrix M, Luithle J, Methfessel C, Riedl B, Schnizler K, van der Staay FJ, van Kampen M, Wiese WB, Koenig G (2007) The novel alpha7 nicotinic acetylcholine receptor agonist N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-7-[2-(methoxy)phenyl]-1-benzofuran-2- carboxamide improves working and recognition memory in rodents. J Pharmacol Exp Ther 321:716–725
Pichat P, Bergis OE, Terranova JP, Urani A, Duarte C, Santucci V, Gueudet C, Voltz C, Steinberg R, Stemmelin J, Oury-Donat F, Avenet P, Griebel G, Scatton B (2007) SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (II) efficacy in experimental models predictive of activity against cognitive symptoms of schizophrenia. Neuropsychopharmacology 32:17–34
Tatsumi R, Fujio M, Takanashi S, Numata A, Katayama J, Satoh H, Shiigi Y, Maeda J, Kuriyama M, Horikawa T, Murozono T, Hashimoto K, Tanaka H (2006) (R)-3’-(3-methylbenzo[b]thiophen-5-yl)spiro[1-azabicyclo[2,2,2]octane-3,5′ -oxazolidin]-2′-one, a novel and potent alpha7 nicotinic acetylcholine receptor partial agonist displays cognitive enhancing properties. J Med Chem 49:4374–4383
Ren K, Thinschmidt J, Liu J, Ai L, Papke RL, King MA, Hughes JA, Meyer EM (2007) Alpha7 Nicotinic receptor gene delivery into mouse hippocampal neurons leads to functional receptor expression, improved spatial memory-related performance, and tau hyperphosphorylation. Neuroscience 145:314–322
Banerjee C, Nyengaard JR, Wevers A, de Vos RA, Jansen Steur EN, Lindstrom J, Pilz K, Nowacki S, Bloch W, Schroder H (2000) Cellular expression of alpha7 nicotinic acetylcholine receptor protein in the temporal cortex in Alzheimer’s and Parkinson’s disease-a stereological approac. Neurobiol Dis 7:666–672
Loughead J, Ray R, Wileyto EP, Ruparel K, Sanborn P, Siegel S, Gur RC, Lerman C (2010) Effects of the alpha4beta2 partial agonist varenicline on brain activity and working memory in abstinent smokers. Biol Psychiatry 67:715–721
Furey ML, Pietrini P, Haxby JV, Alexander GE, Lee HC, VanMeter J, Grady CL, Shetty U, Rapoport SI, Schapiro MB, Freo U (1997) Cholinergic stimulation alters performance and task-specific regional cerebral blood flow during working memory. Proc Natl Acad Sci USA 94:6512–6516
Kirrane RM, Mitropoulou V, Nunn M, Silverman J, Siever LJ (2001) Physostigmine and cognition in schizotypal personality disorder. Schizophr Res 48:1–5
Koller G, Satzger W, Adam M, Wagner M, Kathmann N, Soyka M, Engel R (2003) Effects of scopolamine on matching to sample paradigm and related tests in human subjects. Neuropsychobiology 48:87–94
Green A, Ellis KA, Ellis J, Bartholomeusz CF, Ilic S, Croft RJ, Phan KL, Nathan PJ (2005) Muscarinic and nicotinic receptor modulation of object and spatial n-back working memory in humans. Pharmacol Biochem Behav 81:575–584
Ellis JR, Ellis KA, Bartholomeusz CF, Harrison BJ, Wesnes KA, Erskine FF, Vitetta L, Nathan PJ (2006) Muscarinic and nicotinic receptors synergistically modulate working memory and attention in humans. Int J Neuropsychopharmacol 9:175–189
Dunbar G, Kuchibhatla R, Lee G (2011) A randomized double-blind study comparing 25 and 50 mg TC-1734 (AZD3480) with placebo, in older subjects with age-associated memory impairment. J Psychopharmacol 25:1020–1029
Farlow MR, Salloway S, Tariot PN, Yardley J, Moline ML, Wang Q, Brand-Schieber E, Zou H, Hsu T, Satlin A (2010) Effectiveness and tolerability of high-dose (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: a 24-week, randomized, double-blind study. Clin Ther 32:1234–1251
Alva G, Cummings JL (2008) Relative tolerability of Alzheimer’s disease treatments. Psychiatry (Edgmont) 5:27–36
Lopez-Hernandez GY, Thinschmidt JS, Morain P, Trocme-Thibierge C, Kem WR, Soti F, Papke RL (2009) Positive modulation of alpha7 nAChR responses in rat hippocampal interneurons to full agonists and the alpha7-selective partial agonists, 4OH-GTS-21 and S 24795. Neuropharmacology 56:821–830
Papke RL, Bencherif M, Lippiello P (1996) An evaluation of neuronal nicotinic acetylcholine receptor activation by quaternary nitrogen compounds indicates that choline is selective for the alpha 7 subtype. Neurosci Lett 213:201–204
Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX (1997) Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci 9:2734–2742
Bertrand N, Ishii H, Spatz M (1996) Cerebral ischemia in young and adult gerbils: effects on cholinergic metabolism. Neurochem Int 28:293–297
Jope RS, Gu X (1991) Seizures increase acetylcholine and choline concentrations in rat brain regions. Neurochem Res 16:1219–1226
Parikh V, Sarter M (2006) Cortical choline transporter function measured in vivo using choline-sensitive microelectrodes: clearance of endogenous and exogenous choline and effects of removal of cholinergic terminals. J Neurochem 97:488–503
Rao AM, Hatcher JF, Dempsey RJ (2000) Lipid alterations in transient forebrain ischemia: possible new mechanisms of CDP-choline neuroprotection. J Neurochem 75:2528–2535
Scremin OU, Jenden DJ (1991) Time-dependent changes in cerebral choline and acetylcholine induced by transient global ischemia in rats. Stroke 22:643–647
Papke RL, Papke JKP (2002) Comparative pharmacology of rat and human alpha7 nAChR conducted with net charge analysis. Br J Pharmacol 137:49–61
Klein J, Koppen A, Loffelholz K (1998) Regulation of free choline in rat brain: dietary and pharmacological manipulations. Neurochem Int 32:479–485
Faghih R, Gfesser GA, Gopalakrishnan M (2007) Advances in the discovery of novel positive allosteric modulators of the alpha7 nicotinic acetylcholine receptor. Recent Patents CNS Drug Discov 2:99–106
Roncarati R, Seredenina T, Jow B, Jow F, Papini S, Kramer A, Bothmann H, Dunlop J, Terstappen GC (2008) Functional properties of alpha7 nicotinic acetylcholine receptors co-expressed with RIC-3 in a stable recombinant CHO-K1 cell line. Assay Drug Dev Technol 6:181–193
Barron SC, McLaughlin JT, See JA, Richards VL, Rosenberg RL (2009) The allosteric modulator of {alpha}7 nicotinic receptors, PNU-120596, causes conformational changes in the extracellular ligand binding domain similar to acetylcholine. Mol Pharmacol 76:253–263
Young GT, Zwart R, Walker AS, Sher E, Millar NS (2008) Potentiation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci USA 105:14686–14691
Gronlien JH, Hakerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, Malysz J (2007) Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol 72:715–724
Bertrand D, Bertrand S, Cassar S, Gubbins E, Li J, Gopalakrishnan M (2008) Positive allosteric modulation of the alpha7 nicotinic acetylcholine receptor: ligand interactions with distinct binding sites and evidence for a prominent role of the M2-M3 segment. Mol Pharmacol 74:1407–1416
Disterhoft JF, Oh MM (2007) Alterations in intrinsic neuronal excitability during normal aging. Aging Cell 6:327–336
Kaczorowski CC, Disterhoft JF (2009) Memory deficits are associated with impaired ability to modulate neuronal excitability in middle-aged mice. Learn Mem 16:362–366
Gilbert D, Lecchi M, Arnaudeau S, Bertrand D, Demaurex N (2009) Local and global calcium signals associated with the opening of neuronal alpha7 nicotinic acetylcholine receptors. Cell Calcium 45:198–207
De Rosa MJ, Dionisio L, Agriello E, Bouzat C, Esandi Mdel C (2009) Alpha 7 nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci 85:444–449
Hao J, Simard AR, Turner GH, Wu J, Whiteaker P, Lukas RJ, Shi FD (2010) Attenuation of CNS inflammatory responses by nicotine involves alpha7 and non-alpha7 nicotinic receptors. Exp Neurol 227:110–119
Kawashima K, Fujii T (2003) The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci 74:675–696
Koval L, Lykhmus O, Zhmak M, Khruschov A, Tsetlin V, Magrini E, Viola A, Chernyavsky A, Qian J, Grando S, Komisarenko S, Skok M (2011) Differential involvement of alpha4beta2, alpha7 and alpha9alpha10 nicotinic acetylcholine receptors in B lymphocyte activation in vitro. Int J Biochem Cell Biol 43:516–524
Mashkina AP, Cizkova D, Vanicky I, Boldyrev AA (2010) NMDA receptors are expressed in lymphocytes activated both in vitro and in vivo. Cell Mol Neurobiol 30:901–907
Mashkina AP, Tyulina OV, Solovyova TI, Kovalenko EI, Kanevski LM, Johnson P, Boldyrev AA (2007) The excitotoxic effect of NMDA on human lymphocyte immune function. Neurochem Int 51:356–360
Nizri E, Hamra-Amitay Y, Sicsic C, Lavon I, Brenner T (2006) Anti-inflammatory properties of cholinergic up-regulation: a new role for acetylcholinesterase inhibitors. Neuropharmacology 50:540–547
Nizri E, Irony-Tur-Sinai M, Lory O, Orr-Urtreger A, Lavi E, Brenner T (2009) Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol 183:6681–6688
Sharma G, Vijayaraghavan S (2002) Nicotinic receptor signaling in nonexcitable cells. J Neurobiol 53:524–534
Skok MV, Grailhe R, Agenes F, Changeux JP (2007) The role of nicotinic receptors in B-lymphocyte development and activation. Life Sci 80:2334–2336
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421:384–388
Yawata I, Takeuchi H, Doi Y, Liang J, Mizuno T, Suzumura A (2008) Macrophage-induced neurotoxicity is mediated by glutamate and attenuated by glutaminase inhibitors and gap junction inhibitors. Life Sci 82:1111–1116
Catassi A, Paleari L, Servent D, Sessa F, Dominioni L, Ognio E, Cilli M, Vacca P, Mingari M, Gaudino G, Bertino P, Paolucci M, Calcaterra A, Cesario A, Granone P, Costa R, Ciarlo M, Alama A, Russo P (2008) Targeting alpha7-nicotinic receptor for the treatment of pleural mesothelioma. Eur J Cancer 44:2296–2311
Catassi A, Servent D, Paleari L, Cesario A, Russo P (2008) Multiple roles of nicotine on cell proliferation and inhibition of apoptosis: implications on lung carcinogenesis. Mutat Res 659:221–231
Davis R, Rizwani W, Banerjee S, Kovacs M, Haura E, Coppola D, Chellappan S (2009) Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS One 4:e7524
Egleton RD, Brown KC, Dasgupta P (2008) Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol Sci 29:151–158
North WG, Gao G, Memoli VA, Pang RH, Lynch L (2010) Breast cancer expresses functional NMDA receptors. Breast Cancer Res Treat 122:307–314
Paleari L, Catassi A, Ciarlo M, Cavalieri Z, Bruzzo C, Servent D, Cesario A, Chessa L, Cilli M, Piccardi F, Granone P, Russo P (2008) Role of alpha7-nicotinic acetylcholine receptor in human non-small cell lung cancer proliferation. Cell Prolif 41:936–959
Tachibana N, Shirakawa T, Ishii K, Takahashi Y, Tanaka K, Arima K, Yoshida T, Ikeda S (2010) Expression of various glutamate receptors including N-methyl-D-aspartate receptor (NMDAR) in an ovarian teratoma removed from a young woman with anti-NMDAR encephalitis. Intern Med 49:2167–2173
Oh MM, Wu WW, Power JM, Disterhoft JF (2006) Galantamine increases excitability of CA1 hippocampal pyramidal neurons. Neuroscience 137:113–123
Lynch G, Palmer LC, Gall CM (2011) The likelihood of cognitive enhancement. Pharmacol Biochem Behav 99:116–129
Acknowledgements
I thank Dr. William Kem and Dr. Hong Xing for providing images of chemical structures of PNU-120596 and 5-HI. This work was supported by the NIH grant R01 DK082625 to VU.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Uteshev, V.V. (2012). α7 Nicotinic ACh Receptors as a Ligand-Gated Source of Ca2+ Ions: The Search for a Ca2+ Optimum. In: Islam, M. (eds) Calcium Signaling. Advances in Experimental Medicine and Biology, vol 740. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2888-2_27
Download citation
DOI: https://doi.org/10.1007/978-94-007-2888-2_27
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-2887-5
Online ISBN: 978-94-007-2888-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)