
Abstract
Parkinson’s disease (PD) is a neurodegenerative motor disorder which is largely sporadic; however, some familial forms have been identified. Genetic PD can be inherited by autosomal, dominant or recessive mutations. While the dominant mutations mirror the prototype of PD with adult-onset and L-dopa-responsive cases, autosomal recessive PD (ARPD) exhibit atypical phenotypes with additional clinical manifestations. Young-onset PD is also very common with mutations in recessive gene loci. The main genes associated with ARPD are Parkin, PINK1, DJ-1, ATP13A2, FBXO7 and PLA2G6. Calcium dyshomeostasis is a mainstay in all types of PD, be it genetic or sporadic. Intriguingly, calcium imbalances manifesting as altered Store-Operated Calcium Entry (SOCE) is suggested in PLA2G6-linked PARK 14 PD. The common pathways underlying ARPD pathology, including mitochondrial abnormalities and autophagic dysfunction, can be investigated ex vivo using induced pluripotent stem cell (iPSC) technology and are discussed here. PD pathophysiology is not faithfully replicated by animal models, and, therefore, nigral dopaminergic neurons generated from iPSC serve as improved human cellular models. With no cure to date and treatments aiming at symptomatic relief, these in vitro models derived through midbrain floor-plate induction provide a platform to understand the molecular and biochemical pathways underlying PD etiology in a patient-specific manner.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Autophagic–lysosomal pathway
- Calcium
- Cellular reprogramming
- Dopaminergic neurons
- Lewy bodies
- Mitophagy
- PARK-14
- Phospholipase A2
- SOCE
1 Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder characterized by motor symptoms such as resting tremor, bradykinesia, rigidity, postural instability, stooped posture and freezing, as well as non-motor symptoms including cognitive and behavioural symptoms, sleep disorders, autonomic dysfunction, sensory symptoms and fatigue (Tolosa et al. 2006; de Lau et al. 2006; Jankovic 2008; O’Sullivan et al. 2008; Kalinderi et al. 2016). The pathological hallmark of PD is the progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and the subsequent loss of dopamine inputs to forebrain striatal structures along with the appearance of protein inclusions called Lewy bodies (LB) composed of α-synuclein fibrils (de Lau et al. 2006; Jankovic 2008). PD is classified into two genetic subtypes including monogenic familial forms with Mendelian inheritance and sporadic forms with no underlying genetic factors (Karimi-Moghadam et al. 2018). The sporadic forms of PD are highly prevalent whereas familial PD accounts for only 5–10% of the reported cases. Monogenic familial forms of PD are rare, caused by highly penetrant disease-causing mutations. Parkinsonism caused by dominant mutations including alpha-synuclein (SNCA), leucine-rich repeat kinase 2 (LRRK2), vacuolar protein sorting 35 (VPS35) and the like are largely similar to the common, late-onset sporadic PD (Bonifati 2012, 2014). Autosomal recessive PD (ARPD) results from mutations in different loci which have clinical signs typical of PD or can exhibit a wide range of other complex symptoms. The common pathways underlying ARPD are mitochondrial quality control, protein degradation processes and oxidative stress responses, among others (Scott et al. 2017; van der Merwe et al. 2015).
The current knowledge of PD is mostly from postmortem studies and animal models. While the former represent only the end-stage of the disease, the latter fail to reflect human disease pathology due to interspecies differences. In this context, human pluripotent stem cells (both embryonic stem cells, ESC; and induced pluripotent stem cells, iPSC) are an excellent source of cells for differentiation to DA neurons in vitro. ‘Disease modelling in a dish’ by recapitulating the disease phenotypes in defined cell populations would make it possible to understand the cellular and molecular mechanisms of PD, along with providing a high-throughput drug screening platform (Marchetto et al. 2011; Badger et al. 2014; Martínez-Morales and Liste 2012).
2 Autosomal Recessive Parkinsonism
The hereditary forms of parkinsonism which are transmitted in an autosomal recessive fashion are given in Table 1. Mutations have been identified most commonly in three genes in several ethnic groups spanning different geographical locations: parkin (PRKN, PARK2), PTEN- induced putative kinase 1 (PINK1, PARK6), and Parkinson protein 7 (DJ-1, PARK7). Point mutations, large genomic rearrangements, leading to deletions or multiplications presenting as homozygous or compound heterozygous variations are reported, particularly for parkin (Lucking et al. 2000). Recessive mutations in several genes, including ATPase type 13A2 (ATP13A2, PARK9), phospholipase A2, group VI (PLA2G6, PARK14), F-box only protein 7 (FBXO7, PARK15), spatacsin (SPG11), and DNA polymerase gamma (POLG), cause young- or juvenile-onset PD. These present with other clinical manifestations like dystonia, dementia and other disturbances (Bonifati 2012). DNAJ subfamily C member 6 (DNAJC6, PARK19), synaptojanin-1 (SYNJ1, PARK 20) and vacuolar protein sorting 13C (VPS13C, PARK23) are also reported to have mutations causing autosomal recessive PD (Karimi-Moghadam et al. 2018).
2.1 PLA2G6 (PARK14)
Phospholipase A2 group 6 (PLA2G6, iPLA2β) gene encodes a calcium-independent group 6 phospholipase A2 enzyme, which hydrolyzes the sn-2 ester bond of the membrane glycerophospholipids to produce free fatty acids and lysophospholipids (Pérez et al. 2004). Various mutations in this gene have been discovered in patients with neurodegenerative disorders such as infantile neuroaxonal dystrophy (INAD), atypical neuroaxonal dystrophy (ANAD), adult-onset dystonia-parkinsonism (DP) and autosomal recessive early-onset parkinsonism (AREP), together known as PLA2G6-asscociated neurodegeneration, PLAN (Gregory et al. 1993). PLAN can be classified as neurodegeneration with brain iron accumulation II (NBIA II); however, a wide range of clinical variability is exhibited in these phenotypes with most PD cases devoid of brain iron deposition or cortical atrophy (Guo et al. 2018). iPLA2β protein contains an N-terminal domain, Ankyrin repeats and catalytic domains (Fig. 1). iPLA2β is predominantly localized in the cytosol but can translocate to the Golgi, ER, mitochondria and nucleus under stimulation (Turk and Ramanadham 2004; Kinghorn et al. 2015; Balsinde and Balboa 2005; Ramanadham et al. 2015). Two distinct 85 kDa (VIA-1) and 88 kDa (VIA-2) human iPLA2β isoforms have been discovered along with many N-terminal truncated forms due to proteolytic cleavage and alternate splicing (Ramanadham et al. 2015). It is highly expressed in the human brain including SNpc (http://www.proteinatlas.org). The PLA2G6 gene mutations was associated with parkinsonism almost a decade ago, with R741Q and R747W being the first to be reported in adult-onset levodopa-responsive dystonia-parkinsonism (Paisan-Ruiz et al. 2009, 2010; Sina et al. 2009). p.R741Q has also been indicated in early-onset PD without dystonia (Bohlega et al. 2016). Though PD-associated mutations in this gene are mostly homozygous, some of them are rare and specific to geographic areas (Gui et al. 2013; Shen et al. 2018; Kapoor et al. 2016; Lu et al. 2012) while others are compound heterozygous (Shen et al. 2019; Chu et al. 2020; Ferese et al. 2018). The mutations that are pathogenic and causal for PD in PLA2G6 are detailed in Table 2.

Structure of PLA2G6 (iPLA2β) protein. The full-length protein is shown with seven ankyrin repeats (pink circles), a proline-rich motif (blue box), a glycine-rich nucleotide-binding motif (magenta), a lipase motif (orange with the active site highlighted), and a proposed C-terminal calcium-dependent calmodulin binding domain (purple). Numbers indicate amino acids
Widespread LB pathology is seen with PLA2G6-linked PD (Paisán-Ruiz et al. 2012; Miki et al. 2017). The loss of PLA2G6 in Drosophila results in impaired retromer function, ceramide accumulation and lysosomal dysfunction, leading to age-dependent loss of neuronal activity (Lin et al. 2018). Dysfunction of mitochondria and increased lipid peroxidation have also been reported in PLA2G6-deficient Drosophila mimicking the human fibroblasts with R747W mutation (Kinghorn et al. 2015). Increased sensitivity to oxidative stress, progressive neurodegeneration and a severely reduced lifespan and impaired motor co-ordination is seen in PLA2G6-knockout flies (Iliadi et al. 2018). In yet another fly model, the loss of PLA2G6 leads to shortening of phospholipid acyl chains, resulting in ER stress and impaired neuronal activity as well as formation of α-synuclein fibrils, demonstrating that phospholipid remodeling by PLA2G6 is essential for DA neuron survival and function (Mori et al. 2019). Similarly, a rodent knockin model of D331Y PLA2G6 mutation exhibited early degeneration of SNpc DA possibly via mitochondrial and ER stress, impaired autophagic mechanisms and gene expression changes (Chiu et al. 2019). Elevated levels of both α-synuclein and phosphorylated α-synuclein are seen in PLA2G6 knockout mice models, facilitating the formation of LB and eventually death of affected DA neurons (Sumi-Akamaru et al. 2016). In an in vitro study examining the catalytic activity of PLA2G6 proteins, recombinant proteins containing the three mutations associated with dystonia-parkinsonism (R632W, R741Q and R747W) did not show any altered catalytic activity, whereas the mutations associated with INAD led to a significant loss of enzyme activity (Engel et al. 2010). In addition, PLA2G6-PD mutants of SHSY5Y, a neuroblastoma cell line, failed to prevent rotenone-induced death of dopaminergic cells (Chiu et al. 2017). PLA2G6-PD does not present with a typical clinical scenario and continues to evolve with a wide phenotypic spectrum. It is safe to infer that the PD-relevant mutations do not significantly alter the catalytic activity of the enzyme but induce damage through parallel mechanisms like oxidative stress, mitochondrial dysfunction or even compromised lipid remodeling.
2.1.1 PLA2G6 and Store-Operated Calcium Entry (SOCE)
Store-operated calcium entry (SOCE) is an arm of calcium signaling activated by depletion of ER stores that triggers influx of calcium across the plasma membrane (PM) brought about by the calcium sensor STIM and the PM pore channel Orai (Feske et al. 2005, 2006; Vig et al. 2006). Interestingly, in a genetic screening of Drosophila, not only STIM1 and Orai1, but also a fly orthologue of PLA2G6 encoded by the CG6718 gene, were identified as SOCE activators (Vig et al. 2006). Many groups have from this time identified PLA2G6 as an endogenous activator of SOCE (Smani et al. 2016; Schäfer et al. 2012; Singaravelu 2006; Bolotina and Orai 2008). The physiological relevance of neuronal SOCE is disputed (Lu and Fivaz 2016) and its role in DA neurons, particularly, is not known. In PD, abnormal calcium homeostasis triggers a cascade of downstream events that eventually leads to cell death (Michel et al. 2016). Interestingly, primary skin fibroblasts from idiopathic and PLA2G6-PD (R747W) patients revealed a significant deficit in endogenous SOCE and similarly, MEFs from the exon2-knockout mice exhibited deficient store-operated PLA2G6-dependent calcium signaling (Zhou et al. 2016a). This was also mirrored in the iPSC-derived DA neurons along with low ER calcium levels and deficient autophagic flux. The knockout mice also showed age-dependent loss of DA neurons. This study for the first time arrived at a causal relationship between PLA2G6-dependent SOCE, depleted stores, dysfunctional autophagy in DA neurons and a PD-like phenotype (Zhou et al. 2016a). Recently, in a patient-derived (D331Y) DA neuron model, imbalance of calcium homeostasis, markedly deficient SOCE, increase of UPR proteins, mitochondrial dysfunction, increase of ROS, and apoptosis was reported. Interestingly, the UPR modulator, azoramide rescued the phenotype of the mutant DA neurons, possibly via CREB signaling (Ke et al. 2020). These recent developments have opened new exciting areas to study the significant contributions of PLA2G6 and SOCE in PD, and may involve new undiscovered molecules providing a yet unexplored arena for PD-drug discovery.
2.2 Parkin, PINK1 and DJ-1
The E3 ubiquitin ligase parkin (PARK2) and the serine/threonine kinase PINK1 (phosphatase and tensin homolog-induced putative kinase1, PARK6), act together in a mitochondrial quality control pathway and promote the selective autophagy of depolarized mitochondria (mitophagy) (Narendra et al. 2012). PINK1 levels are low in healthy cells as it is continually cleaved inside the mitochondria in a sequential manner by proteases (Yamano and Youle 2013). The import of PINK1 into mitochondria is stopped when the organelle loses its inner membrane electrochemical gradient (depolarization), which leads to the stabilization of the protein on the mitochondrial outer membrane (Lin and Kang 2008). This accumulation of PINK1 kinase on the mitochondria triggers parkin recruitment and activation resulting in ubiquitination of various outer mitochondrial membrane proteins (Taanman and Protasoni 2017; Matsuda et al. 2010). The damaged mitochondria are eventually eliminated by autophagy. Pathogenic PD-associated mutations in either Parkin or PINK1 causes accumulation of impaired mitochondria, increased ROS and neuronal cell death (Seirafi et al. 2015). More than 100 different Parkin mutations have been reported from PD patients, including deletions, insertions, multiplications, missense and truncating mutations, and over 40 point mutations and, rarely, large deletions, have been detected in PINK1 (Lesage and Brice 2009). Clinically, both cause young-onset PD and show responsiveness to levodopa. The phenotype associated with the oncogene DJ-1 mutations has been studied in a smaller number of patients but it is overall indistinguishable from that of the patients with PINK1 or Parkin mutations (Bonifati et al. 2003). DJ-1 is thought to be involved in the regulation of the integrity and calcium crosstalk between endoplasmic reticulum (ER) and mitochondria, and pathogenic mutations lead to impaired ER-mitochondria association in PD (Liu et al. 2019).
2.3 ATP13A2, FBXO7, SPG11 and POLG
Mutations in ATP13A2, FBXO7, spatacsin and POLG cause juvenile-onset ARPD along with PLA2G6 (Bonifati 2012). Mutations in ATP13A2 or PARK9, were first identified in 2006 in a Chilean family and are associated with a juvenile-onset, levodopa-responsive type of parkinsonism called Kufor–Rakeb syndrome (KRS). KRS involves pyramidal degeneration, supranuclear palsy, and cognitive impairment (Ramirez et al. 2006). The ATP13A2 gene encodes a large lysosomal protein, belonging to the P5-type ATPase family of transmembrane active transporters. Its substrate specificity remains unknown. It is suggested that ATP13A2 recruits HDAC6 to lysosomes to promote autophagosome–lysosome fusion and maintain normal autophagic flux (Wang et al. 2019). This, in turn, is required for preventing α-synuclein aggregation in neurons. FBXO7, in turn is an adaptor protein in SCFFBXO7 ubiquitin E3 ligase complex that mediates degradative or non-degradative ubiquitination of substrates. FBXO7 mutations aggravate protein aggregation in mitochondria and inhibit mitophagy (Zhou et al. 2018). Parkin- and FBXO7-linked PD have overlapping pathophysiologic mechanisms and clinical features. Wild-type FBXO7, but not PD-linked FBXO7 mutants, has been shown to rescue DA neuron degeneration in Parkin null Drosophila (Zhou et al. 2016b). Loss of activity of FBXO7 in seen in patients with PARK 15-PD and is therefore crucial for the maintenance of neurons (Zhao et al. 2011). SPG11 or spatacsin mutations present with bilateral symmetric parkinsonism at an early age with rapid deterioration and development of spastic paraplegia and thinning of the corpus callosum on MRI, typical of spastic paraplegia 11 (Guidubaldi et al. 2011; Cao et al. 2013). An involvement of POLG, the mitochondrial DNA polymerase that is responsible for replication of the mitochondrial genome is considered in early-onset PD especially in the presence of additional symptoms, such as ophthalmoparesis, non-vascular white matter lesions and psychiatric comorbidity (Anett et al. 2020). A role of mitochondrial DNA defects in the pathogenesis of neurodegenerative parkinsonism with POLG mutations is speculated (Miguel et al. 2014).
3 Common Pathways in ARPD
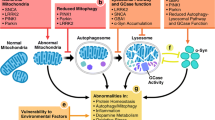
There are many converging features seen at the molecular and clinical levels in ARPD that are discussed in the following section. Understanding these causal molecular mechanisms is crucial to identify common targets and devise therapeutic approaches. However, some fundamental underlying processes still remain unclear. The contributions of the intracellular organelle ER in ARPD pathology via the calcium signaling pathway, SOCE is poorly understood. The mitochondrion, which is the star player in PD pathogenesis, regulates SOCE activity possibly via sub-plasmalemmal calcium buffering, the generation of mediators, local ATP modulation and regulation of STIM1 (Malli and Graier 2017). In turn, SOCE-derived calcium significantly affects mitochondrial metabolism. Hence, the communication between SOCE and mitochondria is hypothesized to be interdependent and complex leading to fine-tuning of both SOCE and mitochondrial function (Spät and Szanda 2017). Though PD-relevant mitochondrial processes are studied extensively, SOCE and its role in PD are largely unexplored. The microbiota–gut–brain axis and its imbalance by alterations in the human microbiome also represent a risk factor for PD (Sampson et al. 2016). Such pathways that are not well-proven are omitted from this section, for ease of understanding.
3.1 Mitochondrial Pathways
The most compelling evidence for loss of mitochondrial fidelity comes from the genes PINK1 and Parkin. As mentioned earlier, PINK1 accumulates on the outer membrane of damaged mitochondria and activates Parkin’s E3 ubiquitin ligase activity. Parkin recruited to the damaged mitochondrion ubiquitinates the outer mitochondrial membrane proteins to trigger selective autophagy. In the late 1970s when accidental exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was found to cause PD and neurodegeneration, the first causal mechanism speculated was mitochondrial dysfunction (William Langston et al. 1983). A specific defect of Complex I activity is also seen in the substantia nigra of patients with PD (Schapira et al. 1990). We now understand that the pathways included in mitochondrial quality control system are fission/fusion, mitochondrial transport, mitophagy and mitochondrial biogenesis (Scott et al. 2017). The precise mechanisms by which Parkin and PINK1 regulate fission and fusion is debated, but studies from Drosophila and mammalian culture systems, though contradictory, indicate unbalanced mitochondrial fission and fusion in PINK1 mutants (Scott et al. 2017; Chen and Chan 2009; Pryde et al. 2016; Scarffe et al. 2014; Yu et al. 2011). DJ-1 (Irrcher et al. 2010) and ATP13A2 (Park et al. 2014) mutants also show fragmented mitochondria. The combined effects of Parkin and PGC-1α in the maintenance of mitochondrial homeostasis in dopaminergic neurons is demonstrated (Zheng et al. 2017). PINK1 is also involved in mitochondrial motility along axons and dendrites of neurons. PINK1 interacts with Miro, a component of the motor/adaptor complex binding mitochondria to microtubules and allowing their movement to and from cellular processes (Brunelli et al. 2020). Miro is phosphorylated by PINK1 and ubiquitinated by parkin, leading to its degradation and halting mitochondrial transport promoting clearance of damaged mitochondria (Liu et al. 2012). Parkin/PINK1 is hence involved in mitochondrial trafficking (Scott et al. 2017; Weihofen et al. 2009).
The clearance of damaged mitochondria or mitophagy is a pathway common to mostly all ARPD-related genes. The role of Pink1-Parkin in mitophagy is well-established (Deas et al. 2011; Yamano and Youle 2020). Fbxo7 is also shown to induce mitophagy in response to mitochondrial depolarization in a common pathway with Parkin and PINK1, and PD-associated mutations interfere in this mechanism (Burchell et al. 2013). Parkin (Kuroda et al. 2006) and PINK1 (Pirooznia et al. 2020) is also linked to mitochondrial biogenesis, therefore probably being a part of mitochondrial transcription/replication.
3.2 Autophagy–Lysosomal Pathways
In addition to impaired mitophagy, protein degradation pathways, especially the autophagy–lysosomal pathway, are affected in PD. ATP13A2 is suggested as a regulator of the autophagy–lysosome pathway. ATP13A2 acts in concert with another PD-protein SYT11 and its loss of function results in dysfunctional autophagy–lysosomal pathway as seen in PD (Bento et al. 2016). α-synuclein-independent neurotoxicity due to endolysosomal dysfunction has also been demonstrated in ATP13A2 null mice (Kett et al. 2015). Parkin knockout neurons too show perturbed lysosomal morphology and mitochondrial stress (Okarmus et al. 2020). DJ-1 is associated to chaperone-mediated autophagy (CMA) and its deficiency aggravates α-synuclein aggregation by inhibiting CMA activation (Xu et al. 2017). Loss of DJ-1 could also lead to impaired autophagy and accumulation of dysfunctional mitochondria (Krebiehl et al. 2010). Autophagic defects are a mainstay in PLA2G6-PD. Genetic or molecular impairment of PLA2G6-dependent calcium signaling is a trigger for autophagic dysfunction, progressive loss of DA neurons and age-dependent L-DOPA-sensitive motor dysfunction in a mouse knockout model (Zhou et al. 2016a).
3.3 Cell Death and Oxidative Stress
Oxidative stress and apoptosis are frequently involved in ARPD pathogenesis. ROS accumulation plays a key role in the initiation and acceleration of cell death, compromising neuronal function and structural integrity (Schieber and Chandel 2014). The protein products of Parkin, PINK1 and DJ-1 are associated with disrupted oxidoreductive homeostasis in DA neurons. Impaired cell survival in part due to defective oxidative stress response is implicated in PARK2 knockout neurons (Bogetofte et al. 2019). Further, transgenic overexpression of the parkin substrate, aminoacyl-tRNA synthetase complex interacting multifunctional protein-2 (AIMP2) leads to a selective, age-dependent progressive loss of dopaminergic neurons via activation of poly(ADP-ribose) polymerase-1 (PARP1) (Lee et al. 2013). Similarly, PINK1 is also shown to exert a neuroprotective effect by inhibiting ROS formation and maintaining normal mitochondrial membrane potential and morphology in cultured SN dopaminergic neurons (Wang et al. 2011). The profiles of oxidative damage in the whole brain and neurochemical metabolites in the striatum of PINK1 knockout rats at different ages and genders were studied and oxidative damage revealed as a crucial factor for PD (Ren et al. 2019). Loss of PINK1 inhibits the mitochondrial Na(+)/Ca(2+) exchanger (NCLX), resulting in impaired mitochondrial calcium extrusion, which was, however, fully rescued by activation of the protein kinase A (PKA) pathway (Kostic et al. 2015). DJ-1 also has a role in cell death and combating oxidative stress. It suppresses PTEN activity, thereby promoting cell growth and promoting cellular defence against ROS through PI3K/Akt signaling (Chan and Chan 2015). Reduced anti-oxidative stress mechanisms have been reported in PD patients with mutant DJ-1 protein (Takahashi-Niki et al. 2004). It is also described that Daxx, the death-associated protein, translocated to the cytosol selectively in SNpc neurons due to MPTP-mediated down-regulation of DJ-1 after treatment with the neurotoxin in mouse models (Karunakaran et al. 2007). DJ-1 is also hypothesized to regulate the expression of UCP4 by oxidation and partially via NF-κB pathway in its protective response to oxidative stress (Xu et al. 2018). DJ-1, particularly in its oxidized form, is documented as a biomarker for many diseases including PD. DJ-1 may also work by increasing microRNA-221expression through the MAPK/ERK pathway, subsequently leading to repression of apoptotic molecules (Oh et al. 2018). Additionally, cell-permeable Tat-DJ-1 protein exerts neuroprotective effects in cell lines and mouse models of PD (Jeong et al. 2012). Data from the field indicate that DJ-1 may become activated in the presence of ROS or oxidative stress, but also as part of physiological receptor-mediated signal transduction and acts as a transcriptional regulator of antioxidative gene batteries (Kahle et al. 2009). ATP13A2, on the contrary, is thought to protect against hypoxia-induced oxidative stress (Xu et al. 2012). A recent study revealed a conserved neuroprotective mechanism that counters mitochondrial oxidative stress via ATP13A2-mediated lysosomal spermine export (Vrijsen et al. 2020). PLA2G6 protein is also indicated in oxidative stress-related pathways (Kinghorn et al. 2015; Ke et al. 2020).
4 Induced Pluripotent Stem Cells (iPSC) in Parkinson’s Disease Research
Yamanaka’s discovery in 2007 where key transcriptional factors (Oct4, Sox2, Klf4 and c-Myc) were used to reprogramme adult cells to a de-differentiated, poised cell type called Induced pluripotent stem cells (iPSCs) revolutionized the field of human disease modeling (Takahashi et al. 2007). Reprogrammed iPSCs are similar to embryonic stem cells or ESCs, are pluripotent and can differentiate to multiple lineages. iPSCs when derived from a PD patient has the patient’s complete genetic background and provides a valuable platform to study the impact of genetic mutations. Within a year of the discovery of iPSCs, PD-patient-derived iPSCs (Park et al. 2008) and DA neuron differentiation from iPSCs were reported (Soldner et al. 2009). iPSC models have successively been established from various sporadic and familial PD patients. To date, iPSCs are the most robust cellular system to understand PD and generate disease-relevant cell types for PD (Playne and Connor 2017).
iPSC studies typically involve few participants and random selection of cases and controls, which results in heterogeneous models in vitro. Consequently, a large sample size is required to increase statistical power and sample sizes of 10–30 individuals per iPSC study may be required to achieve a statistical power of 80% (Hoekstra et al. 2017; Tran et al. 2020). These are, in turn, labour-intensive and expensive; hence, it is unlikely that these requirements are met. A smaller number may be used if clinically and genotypically homogeneous subjects are used to reduce the variance in the cellular phenotypes. Hence, a preponderance of familial PD is seen in these studies. A recent report elegantly summarizes a meta-analysis of 385 iPSC-derived neuronal lines modeling mutations/deletions/triplications in LRRK2, PRKN, PINK1, GBA and SNCA (Tran et al. 2020). The authors discuss the importance of using the right controls in such studies. When healthy subjects are used as controls, differences in genetic background may give rise to variance in neuronal phenotypes studies that are not caused by disease mutations. Gene-editing techniques (TALEN, ZFN, CRISPR/Cas9) aid in the generation of isogenic lines that differ in only one single mutant gene, and this circumvents the issue of variance due to genetic background. CRISPR/Cas9 system, an RNA-based endonuclease, is the most common and effective tool used in the iPSC model for introducing the genetic changes seen in PD, including but not limited to knockout, knockin and gene correction (Arias-Fuenzalida et al. 2017; Qing et al. 2017; Soldner et al. 2016; Vermilyea et al. 2020). Disease-causing mutations, therefore, can be inserted in healthy ESC or iPSC lines or gene-correction of a single mutation can be performed in PD-lines to include comparative isogenic control lines (Tran et al. 2020).
Differentiation protocols for DA neurons mimic embryologic development in utero. Unlike cortical neurons, midbrain DA neurons are derived from the ventral floor plate of the neural tube (Ono et al. 2007). The molecular mechanisms that regulate the development of midbrain DA neurons in vivo, and how taking cues from this, one can generate in vitro human midbrain DA neurons from iPSCs was systematically reviewed previously (Arenas et al. 2015). Dual-SMAD inhibition along with modulation of sonic hedgehog (SHH) and WNT signaling by CHIR99021 (GSK3β inhibitor), and addition of FGF8 is routinely used to generate midbrain floor-plate precursors (Kirkeby et al. 2012; Kriks et al. 2011; Reinhardt et al. 2013). BDNF, GDNF, TGFβ3, dbcAMP, ascorbic acid, DAPT and ActivinA are used to enhance the purity and maturity of DA neurons which express the key marker TH (tyrosine hydroxylase) (Monzel et al. 2017a; Smits and Schwamborn 2020; Smits et al. 2019). A schematic and generalized diagram outlining the midbrain DA differentiation protocol from iPSCs is shown in Fig. 2 (the starting population can also be ESCs). It is important to note that, irrespective of the protocols used a heterogeneous cell population is attained, with neurons, glia and NSCs. To attain a high percentage of TH+ DA neuron population, several strategies have been employed. Sorting of DA progenitors which are CD184high/CD44−(Suzuki et al. 2017) or CORIN+(Kikuchi et al. 2017) is shown to increase the TH+ DA neuron yield. CRISPR/ Cas9-based knockin of a fluorescent reporter to visually identify and purify TH+ DA neurons has also been attempted (Calatayud et al. 2019). A monolayer-based neural differentiation protocol was described recently that reproducibly generates ~70–80% midbrain DA neurons (Stathakos et al. 2020; Stathakos et al. 2019). A higher concentration of 300 ng/ml SHH (100–200 ng/ml is used normally) in combination with a lower concentration of 0.6 μM CHIR99021 (0.8 μM–3 μM is used normally) and passaging and replating in the early differentiation and patterning stages maximized the yield of midbrain DA neurons as early as day 30. This monolayer platform is amenable to imaging and functional assessments of autophagy/mitophagy (Stathakos et al. 2020). In an interesting study, autophagic dysfunction and premature aging was shown by PD-patient-derived NSCs (Zhu et al. 2019). One of these patients had early-onset PD with a novel mutation in PLA2G6 gene. The authors hypothesize that developmental defects, and the subsequent depletion of NSC pool size could lead to lower DA neuron number and this impacts the onset and severity of the disease progression (Zhu et al. 2019). Hence, not only iPSC-derived DA neurons, but also the developmentally upstream NSCs could be a disease-relevant phenotype for prediction analyses and design of intervention therapies.

A generalized schematic protocol for differentiation of human induced pluripotent stem cells (iPSCs) into midbrain dopaminergic (DA) neurons. Human iPSCs are treated with small molecules, such as GSK3β inhibitors (GSK3i) and SMAD inhibitors, along with Shh/FGF8b, to induce midbrain floor-plate formation and subsequent midbrain DA specification. This is done either by means of direct differentiation from iPSCs or through embryoid bodies (EBs). The DA progenitors can be sorted with a midbrain cell surface marker like CORIN to achieve higher purity of DA neurons via elimination of unwanted contaminant cells. Mature midbrain DA neurons are generated from these progenitors by addition of mentioned factors at the end of 40–70 DIV (days in vitro) in total. * indicates factors that may be used in the final differentiation step, but not compulsory. ActA, activin A; AA, ascorbic acid; BDNF, brain-derived neurotrophic factor; DAPT, γ-secretase inhibitor; dbcAMP, dibutyryl cyclic adenosine monophosphate; FGF8, fibroblast growth factor 8; GDNF, glial-cell-derived neurotrophic factor; Shh, sonic hedgehog; TGFβ3, transforming growth factor beta-3
4.1 iPSC-Derived Two-Dimensional (2D) and Three-Dimensional (3D) Culture Models of ARPD
Mutations in the PARK2 gene, encoding the protein parkin, have been identified as the most common cause of ARPD. Unsurprisingly, the limited in vitro iPSC-derived ARPD models primarily examine the cellular pathologies of this gene. Human iPSC-derived neurons with PARK2 knockout is known to demonstrate severe mitochondrial dysfunction even in the absence of external stressors. PARK2 patient iPSC-derived neurons showed increased oxidative stress, higher α-synuclein accumulation and enhanced activity of the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway (Imaizumi et al. 2012). Interestingly, iPSC-derived neurons, but not fibroblasts or iPSCs, exhibited abnormal mitochondrial morphology and impaired mitochondrial homeostasis in their study. In a similar study, the loss of parkin significantly increased the spontaneous DA release independent of extracellular calcium and showed decreased dopamine uptake by reducing the total amount of correctly folded DAT along with DA-dependent oxidative stress. All these phenotypes could be rescued by overexpression of parkin, but not its PD-linked T240R mutant or GFP (Jiang et al. 2012). Mitochondrial dysfunction, elevated α-synuclein, synaptic dysfunction, DA accumulation, and increased oxidative stress and ROS have been reported in PARK2- and PINK1-patient-derived neurons in a floor-plate-based but not a neural-rosette-based directed differentiation strategy (Chung et al. 2016). Impairment of mitophagy via formation of S-nitrosylated PINK1 (SNO-PINK1) has also been shown in iPSC-derived parkin-mutant neurons (Oh et al. 2017). In a recent study, PARK2 knockout neurons from isogenic lines exhibited lysosomal impairments and autophagic perturbations, suggesting an impairment of the autophagy–lysosomal pathway in parkin-deficient cells (Okarmus et al. 2020). The same group had earlier shown disturbances in oxidative stress defence, mitochondrial respiration and morphology, cell cycle control and cell viability of parkin-deficient neurons (Bogetofte et al. 2019).
Midbrain-specific 3D cultures are at present a powerful tool for modeling PD in vitro. The use of microwells by Tieng et al. was the very first attempt in this direction to generate embryoid bodies, which were then placed on an orbital shaker before being seeded and grown at air–liquid interface (Tieng et al. 2014). DA progenitor cells expressed FOXA2 and LMX1A as well as TH within a short span of 3 weeks. Subsequently, a number of reports have been published for midbrain organoids with neuromelanin expression seen in long-term cultures (Smits and Schwamborn 2020; Kim et al. 2019; Jo et al. 2016; Monzel et al. 2017b; Qian et al. 2016). However, PD modeling with midbrain organoids is largely focused on dominant mutations like LRRK2 (Smits et al. 2019; Kim et al. 2019), SNCA (Jan et al. 2018) and also an only report on sporadic PD (Chlebanowska et al. 2020). A very recent study used CRISPR-Cas9 genome editing to develop isogenic loss-of-function 3D models of early-onset autosomal recessive PD (PARKIN−/−, DJ-1−/−, and ATP13A2−/−) to identify common pathways (Ahfeldt et al. 2020). The DA neuronal population was markedly reduced in PRKN−/− organoids but no significant differences were observed in the other two cell lines. The death of newly differentiated TH+ neurons and higher expression of VTA marker CALB1 in the PRKN−/− organoids were indicative of A9-like neurons being more severely affected than others. A dysregulation of the autophagy–lysosomal pathway and upregulated ROS in all cell lines and an upregulation of pathways associated with oxidative phosphorylation, mitochondrial dysfunction, and Sirtuin signaling, as well as a significant depletion of mitochondrial proteins were seen in the PRKN−/− DA neurons (Ahfeldt et al. 2020). Astrocytic pathologies in human PRKN-mutated iPSC-derived midbrain organoids were revealed for the first time, suggesting a non-autonomous cell death mechanism for dopaminergic neurons in brains of PRKN-mutated patients (Kano et al. 2020). Mutations in PINK1 have also been reported to generate reduced TH+ counts in midbrain organoids (Jarazo et al. 2019). Human midbrain organoid/spheroid cultures are a scalable and reproducible system to obtain DA neurons expressing markers of terminal differentiation along with neuromelanin production in a 3D environment that replicates the neuronal and glial cytoarchitecture of the human midbrain (Galet et al. 2020). They can hence provide a crucial platform to explore the molecular basis of ARPD, and also to delineate the associated cellular pathologies.
Cell Replacement Therapy with iPSC-Based DA Derivatives
The various challenges pertaining to the safety and efficacy of stem-cell-based cell transplantations in PD have been elegantly reviewed and described (Fan et al. 2020). The right neural cell type for transplantation is of utmost importance. FGF8b inclusion in the differentiation protocols helps in acquisition of a caudal midbrain fate and promotes high dopaminergic graft volume, density and yield as evidenced by deep sequencing of more than 30 human ESC-derived midbrain tissues (Kirkeby et al. 2017). Dopaminergic precursors beyond the floor-plate progenitor stage but before formation of TH+ dopaminergic neurons are found to be most efficient for graft survival, integration and function in animal models (Kikuchi et al. 2017; Nolbrant et al. 2017). Grafting of these precursors into the putamen area, where SNpc dopaminergic neurons innervate, is an approach most likely to succeed (Fan et al. 2020). The number of cells to be transplanted is still debated. Takahashi’s group reported a minimum of 16,000 TH+ cells in a primate model to see improvements in PD score and motor function (Kikuchi et al. 2017). The generation and implantation of iPSC-derived autologous dopaminergic progenitor cells in a patient with idiopathic PD is reported with clinical and imaging results suggesting possible benefit over a period of 24 months (Schweitzer et al. 2020). A global consortium, GForce-PD (http://www.gforce-pd.com), was set up in 2014, with major academic networks in Europe, the United States and Japan working on developing stem-cell-derived neural cell therapies for PD (Barker et al. 2015). The clinical trials using human ESCs are ongoing in Australia (NCT02452723) and China (NCT03119636), with their pre-clinical data published (Garitaonandia et al. 2016; Wang et al. 2018). A clinical trial (JMA-IIA00384, UMIN000033564) in Japan to treat PD patients by using iPSC-derived DA progenitors (DAPs) was started in 2018 by Takahashi and colleagues (Barker et al. 2017; Doi et al. 2020). The results of these trials are eagerly anticipated.
5 Limitations to iPSC-Based Disease Modeling of PD
Although iPSCs and their derivatives are currently in the forefront as PD models, there are many challenges which remain unaddressed. The most significant drawback of in vitro models is the absence of LB formation in PD iPSC-derived DA neurons. Increased levels of phosphorylated pS129 α-synuclein, however, have been observed (Lin et al. 2016). Additionally, the efficiency of generating DA neurons varies significantly between different methods and approximately 20–30% of the final cells are identified as DA neurons even with the most robust method such as the floor-plate induction protocol (Cui et al. 2016). Sorting of DA progenitors with markers such as CORIN seems to aid in a better yield of mature and functional DA neurons (Doi et al. 2020; Paik et al. 2018). Knocking in a reporter gene in the endogenous TH locus has been attempted to quantify the final yield of DA neurons (Cui et al. 2016) to understand the efficiency of different published protocols. However, no significant progress has been made to analyze if the DA neurons derived in vitro are similar to the SNpc neurons impacted in a PD patient. A TH+ DA neuron is necessarily not a representation of the A9 or SNpc nuclei of the brain, though GIRK2/TH positivity and low Calbindin is considered as an A9 signature (Hartfield et al. 2014; Sánchez-Danés et al. 2012; Woodard et al. 2014). A reliable strategy would be multiplexing markers for reliable subtype identification (Kim et al. 2020). Another difficulty in modeling PD with iPSCs is the induction of ‘aging’ in a culture dish. Pharmacological inhibition of telomerase by the inhibitor BIBR1532 demonstrates moderate disease-relevant phenotypes in PINK1 and PARKIN DA neurons (Vera et al. 2016). Progerin (the protein associated with premature aging) overexpression as a strategy to induce aging is also reported (Miller et al. 2013) but interpretation is complex as disease-relevant phenotypes and progerin-phenotypes are indistinguishable (Sison et al. 2018). Moreover, contrary to what is seen in PD pathology, an exogenous stressor is always necessary to observe disease phenotypes in an iPSC–DA system. In a PD-patient-derived iPSC model, DA neurons exhibit apoptosis only after exposure to stressors including hydrogen peroxidase, MG132 and 6-OHDA (Cooper et al. 2012). Lastly, 2D culture systems that are normally used to differentiate DA neurons do not mimic the complex in vivo environment. 3D organoids fill this gap by representing a more physiologically relevant model system. However, the tremendous progress seen in the field is largely limited to cortical or cerebral organoids. A few midbrain spheroid or organoid culture systems are nonetheless reported (Smits et al. 2019; Kim et al. 2019; Jo et al. 2016; Monzel et al. 2017b). Results from these studies indicate that 3D midbrain cultures are definitely an improvement over 2D cultures to model PD. A recent study describes the robust generation of midbrain organoids with homogeneous distribution of midbrain DA neurons along with other neuronal subtypes as well as functional glial cells, including astrocytes and oligodendrocytes (Kwak et al. 2020). Nevertheless, an overall low efficiency of generation and heterogeneity within the midbrain organoids along with its ethical considerations raises contentious questions towards a bench-to-bedside approach.
6 Conclusions
Human pluripotent stem cells, iPSCs in particular, are an invaluable tool to help us better understand PD pathology by generating functional DA neurons with A9-like identity and also reproducing the midbrain cell composition. Improvement in differentiation protocols and 3D culturing techniques combined with genome-editing technologies aids in better PD modeling studies. Additionally, these cultures exhibit key features of PD, such as α-syn accumulation, autophagic defects, oxidative stress and impairment of mitochondrial function. However, it may be advantageous to include other cell types like microglia in PD studies rather than focusing on midbrain-specific organoids to understand the disease pathology in a relevant way. The blood-brain barrier (BBB) and its dysfunction in PD should also be emphasized. Further, the future direction in investigating PD should make use of the organ-on-chip or organoids-on-chip model with a multi-organ configuration to study different cell types and involvement of various organs in PD progression and pathology. Lastly, ARPD genes other than PRKN and PINK1, though rare, may provide insights into the common molecular pathways of the monogenic disease forms and should be included in such detailed studies.
Abbreviations
- ARPD:
-
autosomal recessive Parkinson’s disease
- DA:
-
dopaminergic
- ER:
-
endoplasmic reticulum
- ESC:
-
embryonic stem cell
- iPSC:
-
induced pluripotent stem cell
- LB:
-
Lewy bodies
- NSC:
-
neural stem cell
- PD:
-
Parkinson’s disease
- PM:
-
plasma membrane
- ROS:
-
reactive oxygen species
- SNpc:
-
substantia nigra pars compacta
- SOCE:
-
store-operated calcium entry
- TH:
-
tyrosine hydroxylase
Bibliography
Ahfeldt T et al (2020) Pathogenic pathways in early-onset autosomal recessive Parkinson’s disease discovered using isogenic human dopaminergic neurons. Stem Cell Rep. https://doi.org/10.1016/j.stemcr.2019.12.005
Anett I et al (2020) Hereditary Parkinson’s disease as a new clinical manifestation of the damaged POLG gene. Orv Hetil. https://doi.org/10.1556/650.2020.31724
Arenas E, Denham M, Villaescusa JC (2015) How to make a midbrain dopaminergic neuron. Dev. https://doi.org/10.1242/dev.097394
Arias-Fuenzalida J et al (2017) FACS-assisted CRISPR-Cas9 genome editing facilitates Parkinson’s disease modeling. Stem Cell Reports 9:1423–1431
Badger JL, Cordero-Llana O, Hartfield EM, Wade-Martins R (2014) Parkinson’s disease in a dish – using stem cells as a molecular tool. Neuropharmacology 76:88–96
Balsinde J, Balboa MA (2005) Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell Signal. https://doi.org/10.1016/j.cellsig.2005.03.002
Barker RA, Studer L, Cattaneo E, Takahashi J (2015) G-Force PD: a global initiative in coordinating stem cell-based dopamine treatments for Parkinson’s disease. NPJ Park Dis. https://doi.org/10.1038/npjparkd.2015.17
Barker RA, Parmar M, Studer L, Takahashi J (2017) Human trials of stem cell-derived dopamine neurons for Parkinson’s disease: dawn of a new era. Cell Stem Cell 21:569–573
Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC (2016) The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun. https://doi.org/10.1038/ncomms11803
Bogetofte H et al (2019) PARK2 mutation causes metabolic disturbances and impaired survival of human iPSC-derived neurons. Front Cell Neurosci. https://doi.org/10.3389/fncel.2019.00297
Bohlega SA et al (2016) Clinical heterogeneity of PLA2G6-related Parkinsonism: analysis of two Saudi families. BMC Res Notes. https://doi.org/10.1186/s13104-016-2102-7
Bolotina V, Orai M (2008) STIM1 and iPLA2 β: a view from a different perspective. J Physiol. https://doi.org/10.1113/jphysiol.2008.154997
Bonifati V (2012) Autosomal recessive parkinsonism. Park Relat Disord. https://doi.org/10.1016/s1353-8020(11)70004-9
Bonifati V (2014) Genetics of Parkinson’s disease - state of the art, 2013. Park Relat Disord. https://doi.org/10.1016/S1353-8020(13)70009-9
Bonifati V et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. https://doi.org/10.1126/science.1077209
Brunelli F, Valente EM, Arena G (2020) Mechanisms of neurodegeneration in Parkinson’s disease: keep neurons in the PINK1. Mech Ageing Dev. https://doi.org/10.1016/j.mad.2020.111277
Burchell VS et al (2013) The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci. https://doi.org/10.1038/nn.3489
Calatayud C et al (2019) CRISPR/Cas9-mediated generation of a tyrosine hydroxylase reporter iPSC line for live imaging and isolation of dopaminergic neurons. Sci Rep. https://doi.org/10.1038/s41598-019-43080-2
Cao L et al (2013) Novel SPG11 mutations in Chinese families with hereditary spastic paraplegia with thin corpus callosum. Park Relat Disord. https://doi.org/10.1016/j.parkreldis.2012.10.007
Chan JYH, Chan SHH (2015) Activation of endogenous antioxidants as a common therapeutic strategy against cancer, neurodegeneration and cardiovascular diseases: a lesson learnt from DJ-1. Pharmacol Ther. https://doi.org/10.1016/j.pharmthera.2015.09.005
Chen H, Chan DC (2009) Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum Mol Genet. https://doi.org/10.1093/hmg/ddp326
Chiu CC et al (2017) PARK14 PLA2G6 mutants are defective in preventing rotenoneinduced mitochondrial dysfunction, ROS generation and activation of mitochondrial apoptotic pathway. Oncotarget. https://doi.org/10.18632/oncotarget.20893
Chiu CC et al (2019) PARK14 (D331Y) PLA2G6 causes early-onset degeneration of substantia nigra dopaminergic neurons by inducing mitochondrial dysfunction, ER stress, mitophagy impairment and transcriptional dysregulation in a knockin mouse model. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1118-5
Chlebanowska P, Tejchman A, Sułkowski M, Skrzypek K, Majka M (2020) Use of 3D organoids as a model to study idiopathic form of parkinson’s disease. Int J Mol Sci. https://doi.org/10.3390/ijms21030694
Chu YT, Lin HY, Chen PL, Lin CH (2020) Genotype-phenotype correlations of adult-onset PLA2G6-associated neurodegeneration: case series and literature review. BMC Neurol. https://doi.org/10.1186/s12883-020-01684-6
Chung SY et al (2016) Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and α-synuclein accumulation. Stem Cell Rep. https://doi.org/10.1016/j.stemcr.2016.08.012
Cooper O et al (2012) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. https://doi.org/10.1126/scitranslmed.3003985
Cui J et al (2016) Quantification of dopaminergic neuron differentiation and neurotoxicity via a genetic reporter. Sci Rep. https://doi.org/10.1038/srep25181
de Lau L, Breteler MMMB, de Lau LML, Breteler MMMB (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5:525–535
Deas E, Wood NW, Plun-Favreau H (2011) Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim Biophys Acta, Mol Cell Res. https://doi.org/10.1016/j.bbamcr.2010.08.007
Doi D et al (2020) Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat Commun. https://doi.org/10.1038/s41467-020-17165-w
Engel LA, Jing Z, O’Brien DE, Sun M, Kotzbauer PT (2010) Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One 5
Fan Y, Winanto, Ng SY (2020) Replacing what’s lost: A new era of stem cell therapy for Parkinson’s disease. Transl Neurodegener. https://doi.org/10.1186/s40035-019-0180-x
Ferese R et al (2018) Heterozygous PLA2G6 mutation leads to iron accumulation within basal ganglia and Parkinson’s disease. Front Neurol. https://doi.org/10.3389/fneur.2018.00536
Feske S, Prakriya M, Rao A, Lewis RS (2005) A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med 202:651–662
Feske S et al (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441:179–185
Galet B, Cheval H, Ravassard P (2020) Patient-derived midbrain organoids to explore the molecular basis of Parkinson’s disease. Front Neurol. https://doi.org/10.3389/fneur.2020.01005
Garitaonandia I et al (2016) Neural stem cell tumorigenicity and biodistribution assessment for phase I clinical trial in Parkinson’s disease. Sci Rep. https://doi.org/10.1038/srep34478
Gregory A, Kurian MA, Maher ER, Hogarth P, Hayflick SJ (1993) PLA2G6-associated neurodegeneration. GeneReviews(®). NBK1675 [bookaccession]
Gui YX et al (2013) Four novel rare mutations of PLA2G6 in Chinese population with Parkinson’s disease. Park Relat Disord. https://doi.org/10.1016/j.parkreldis.2012.07.016
Guidubaldi A et al (2011) Novel mutations in SPG11 cause hereditary spastic paraplegia associated with early-onset levodopa-responsive Parkinsonism. Mov Disord. https://doi.org/10.1002/mds.23552
Guo Y, Tang B, Guo J (2018) PLA2G6-associated neurodegeneration (PLAN): review of clinical phenotypes and genotypes. Front Neurol. https://doi.org/10.3389/fneur.2018.01100
Hartfield EM et al (2014) Physiological characterisation of human iPS-derived dopaminergic neurons. PLoS One 9
Hoekstra SD, Stringer S, Heine VM, Posthuma D (2017) Genetically-informed patient selection for iPSC studies of complex diseases may aid in reducing cellular heterogeneity. Front Cell Neurosci. https://doi.org/10.3389/fncel.2017.00164
Iliadi KG, Gluscencova OB, Iliadi N, Boulianne GL (2018) Mutations in the Drosophila homolog of human PLA2G6 give rise to age-dependent loss of psychomotor activity and neurodegeneration. Sci Rep. https://doi.org/10.1038/s41598-018-21343-8
Imaizumi Y et al (2012) Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain. https://doi.org/10.1186/1756-6606-5-35
Irrcher I et al (2010) Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. https://doi.org/10.1093/hmg/ddq288
Jan A et al (2018) Activity of translation regulator eukaryotic elongation factor-2 kinase is increased in Parkinson disease brain and its inhibition reduces alpha synuclein toxicity. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-018-0554-9
Jankovic J (2008) Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp.2007.131045
Jarazo J et al (2019) Parkinson’s disease phenotypes in patient specific brain organoids are improved by HP-β-CD treatment. bioRxiv. https://doi.org/10.1101/813089
Jeong HJ et al (2012) Transduced tat-DJ-1 protein protects against oxidative stress-induced SH-SY5Y cell death and Parkinson disease in a mouse model. Mol Cells. https://doi.org/10.1007/s10059-012-2255-8
Jiang H et al (2012) Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun. https://doi.org/10.1038/ncomms1669
Jo J et al (2016) Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell. https://doi.org/10.1016/j.stem.2016.07.005
Kahle PJ, Waak J, Gasser T (2009) DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med. https://doi.org/10.1016/j.freeradbiomed.2009.08.003
Kalinderi K, Bostantjopoulou S, Fidani L (2016) The genetic background of Parkinson’s disease: current progress and future prospects. Acta Neurol Scand. https://doi.org/10.1111/ane.12563
Kano M et al (2020) Reduced astrocytic reactivity in human brains and midbrain organoids with PRKN mutations. NPJ Park Dis. https://doi.org/10.1038/s41531-020-00137-8
Kapoor S et al (2016) Genetic analysis of PLA2G6 in 22 Indian families with infantile neuroaxonal dystrophy, atypical late-onset neuroaxonal dystrophy and dystonia parkinsonism complex. PLoS One 11
Karimi-Moghadam A, Charsouei S, Bell B, Jabalameli MR (2018) Parkinson disease from Mendelian forms to genetic susceptibility: new molecular insights into the neurodegeneration process. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-018-0587-4
Karunakaran S et al (2007) Activation of apoptosis signal regulating kinase 1 (ASK1) and translocation of death-associated protein, Daxx, in substantia nigra pars compacta in a mouse model of Parkinson’s disease: protection by α-lipoic acid. FASEB J. https://doi.org/10.1096/fj.06-7580com
Ke M et al (2020) Azoramide protects iPSC-derived dopaminergic neurons with PLA2G6 D331Y mutation through restoring ER function and CREB signaling. Cell Death Dis. https://doi.org/10.1038/s41419-020-2312-8
Kett LR et al (2015) α-synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal parkinsonism protein Atp13a2. J Neurosci. https://doi.org/10.1523/JNEUROSCI.0632-14.2015
Kikuchi T et al (2017) Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature. https://doi.org/10.1038/nature23664
Kim H et al (2019) Modeling G2019S-LRRK2 sporadic Parkinson’s disease in 3D midbrain organoids. Stem Cell Rep. https://doi.org/10.1016/j.stemcr.2019.01.020
Kim TW, Koo SY, Studer L (2020) Pluripotent stem cell therapies for Parkinson disease: present challenges and future opportunities. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2020.00729
Kinghorn KJ et al (2015) Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain. https://doi.org/10.1093/brain/awv132
Kirkeby A et al (2012) Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep 1:703–714
Kirkeby A et al (2017) Predictive markers guide differentiation to improve graft outcome in clinical translation of hESC-based therapy for Parkinson’s disease. Cell Stem Cell. https://doi.org/10.1016/j.stem.2016.09.004
Kostic M et al (2015) PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. https://doi.org/10.1016/j.celrep.2015.08.079
Krebiehl G et al (2010) Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One. https://doi.org/10.1371/journal.pone.0009367
Kriks S et al (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480:547–551
Kuroda Y et al (2006) Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. https://doi.org/10.1093/hmg/ddl006
Kwak TH et al (2020) Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson’s disease modeling. Stem Cells. https://doi.org/10.1002/stem.3163
Lee Y et al (2013) Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci. https://doi.org/10.1038/nn.3500
Lesage S, Brice A (2009) Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. https://doi.org/10.1093/hmg/ddp012
Lin W, Kang UJ (2008) Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem. https://doi.org/10.1111/j.1471-4159.2008.05398.x
Lin L et al (2016) Molecular features underlying neurodegeneration identified through in vitro modeling of genetically diverse Parkinson’s disease patients. Cell Rep. https://doi.org/10.1016/j.celrep.2016.05.022
Lin G et al (2018) Phospholipase PLA2G6, a parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide levels, similar to α-synuclein gain. Cell Metab. https://doi.org/10.1016/j.cmet.2018.05.019
Liu S et al (2012) Parkinson’s disease-associated kinase PINK1 regulates miro protein level and axonal transport of mitochondria. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002537
Liu Y et al (2019) DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1906565116
Lu B, Fivaz M (2016) Neuronal SOCE: myth or reality? Trends Cell Biol. https://doi.org/10.1016/j.tcb.2016.09.008
Lu CS et al (2012) PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson’s disease. Am J Med Genet Part B Neuropsychiatr Genet. https://doi.org/10.1002/ajmg.b.32012
Lucking CB et al (2000) Association between early-onset Parkinson’s disease and mutations in the parkin gene. French Parkinson’s Disease Genetics Study Group. N Engl J Med
Malli R, Graier WF (2017) The role of mitochondria in the activation/maintenance of SOCE: the contribution of mitochondrial Ca2+ uptake, mitochondrial motility, and location to store-operated Ca2+ entry. In: Advances in experimental medicine and biology. https://doi.org/10.1007/978-3-319-57732-6_16
Marchetto MC, Brennand KJ, Boyer LF, Gage FH (2011) Induced pluripotent stem cells (iPSCs) and neurological disease modeling: Progress and promises. Hum Mol Genet. https://doi.org/10.1093/hmg/ddr336
Martínez-Morales PL, Liste I (2012) Stem cells as in vitro model of Parkinson’s disease. Stem Cells Int. https://doi.org/10.1155/2012/980941
Matsuda N et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. https://doi.org/10.1083/jcb.200910140
Michel PP, Hirsch EC, Hunot S (2016) Understanding dopaminergic cell death pathways in Parkinson disease. Neuron. https://doi.org/10.1016/j.neuron.2016.03.038
Miguel R et al (2014) POLG1-related levodopa-responsive parkinsonism. Clin Neurol Neurosurg. https://doi.org/10.1016/j.clineuro.2014.08.020
Miki Y et al (2017) PLA2G6 accumulates in Lewy bodies in PARK14 and idiopathic Parkinson’s disease. Neurosci Lett 645:40–45
Miller JD et al (2013) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. https://doi.org/10.1016/j.stem.2013.11.006
Monzel AS et al (2017a) Derivation of human midbrain-specific organoids from neuroepithelial stem cells. Stem Cell Rep 8:1144–1154
Monzel AS et al (2017b) Derivation of human midbrain-specific organoids from neuroepithelial stem cells. Stem Cell Rep. https://doi.org/10.1016/j.stemcr.2017.03.010
Mori A et al (2019) Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1902958116
Narendra D, Walker JE, Youle R (2012) Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a011338
Nolbrant S, Heuer A, Parmar M, Kirkeby A (2017) Generation of high-purity human ventral midbrain dopaminergic progenitors for in vitro maturation and intracerebral transplantation. Nat Protoc. https://doi.org/10.1038/nprot.2017.078
O’Sullivan SS et al (2008) Nonmotor symptoms as presenting complaints in Parkinson’s disease: a clinicopathological study. Mov Disord. https://doi.org/10.1002/mds.21813
Oh CK et al (2017) S-nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiPSC-based Parkinson’s disease models. Cell Rep. https://doi.org/10.1016/j.celrep.2017.10.068
Oh SE et al (2018) The Parkinson’s disease gene product DJ-1 modulates miR-221 to promote neuronal survival against oxidative stress. Redox Biol. https://doi.org/10.1016/j.redox.2018.07.021
Okarmus J et al (2020) Lysosomal perturbations in human dopaminergic neurons derived from induced pluripotent stem cells with PARK2 mutation. Sci Rep. https://doi.org/10.1038/s41598-020-67091-6
Ono Y et al (2007) Differences in neurogenic potential in floor plate cells along an anteroposterior location: midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development. https://doi.org/10.1242/dev.02879
Paik EJ, O’Neil AL, Ng SY, Sun C, Rubin LL (2018) Using intracellular markers to identify a novel set of surface markers for live cell purification from a heterogeneous hIPSC culture. Sci Rep. https://doi.org/10.1038/s41598-018-19291-4
Paisan-Ruiz C et al (2009) Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 65:19–23
Paisán-Ruiz C et al (2010) Early-onset L-dopa-responsive Parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and Spatacsin mutations. Mov Disord. https://doi.org/10.1002/mds.23221
Paisán-Ruiz C et al (2012) Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. https://doi.org/10.1016/j.neurobiolaging.2010.05.009
Park IH et al (2008) Disease-specific induced pluripotent stem cells. Cell. https://doi.org/10.1016/j.cell.2008.07.041
Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM (2014) Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet. https://doi.org/10.1093/hmg/ddt623
Pérez R, Melero R, Balboa MA, Balsinde J (2004) Role of group VIA calcium-independent phospholipase A2 in arachidonic acid release, phospholipid fatty acid incorporation, and apoptosis in U937 cells responding to hydrogen peroxide. J Biol Chem 279:40385–40391
Pirooznia SK et al (2020) PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol Neurodegener. https://doi.org/10.1186/s13024-020-00363-x
Playne R, Connor B (2017) Understanding Parkinson’s disease through the use of cell reprogramming. Stem Cell Rev Rep. https://doi.org/10.1007/s12015-017-9717-5
Pryde KR, Smith HL, Chau KY, Schapira AHV (2016) PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol. https://doi.org/10.1083/jcb.201509003
Qian X et al (2016) Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. https://doi.org/10.1016/j.cell.2016.04.032
Qing X et al (2017) CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and α-synuclein modulation in dopaminergic neurons. Stem Cell Res. https://doi.org/10.1016/j.scr.2017.08.013
Ramanadham S et al (2015) Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J Lipid Res. https://doi.org/10.1194/jlr.R058701
Ramirez A et al (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. https://doi.org/10.1038/ng1884
Reinhardt P et al (2013) Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One 8
Ren X, Hinchie A, Swomley A, Powell DK, Butterfield DA (2019) Profiles of brain oxidative damage, ventricular alterations, and neurochemical metabolites in the striatum of PINK1 knockout rats as functions of age and gender: relevance to Parkinson disease. Free Radic Biol Med. https://doi.org/10.1016/j.freeradbiomed.2019.08.008
Sampson TR et al (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. https://doi.org/10.1016/j.cell.2016.11.018
Sánchez-Danés A et al (2012) Efficient generation of A9 midbrain dopaminergic neurons by lentiviral delivery of LMX1A in human embryonic stem cells and induced pluripotent stem cells. Hum Gene Ther. https://doi.org/10.1089/hum.2011.054
Scarffe LA, Stevens DA, Dawson VL, Dawson TM (2014) Parkin and PINK1: much more than mitophagy. Trends Neurosci. https://doi.org/10.1016/j.tins.2014.03.004
Schäfer C, Rymarczyk G, Ding L, Kirber MT, Bolotina VM (2012) Role of molecular determinants of store-operated Ca(2+) entry (Orai1, phospholipase A2 group 6, and STIM1) in focal adhesion formation and cell migration. J Biol Chem 287:40745–40757
Schapira AHV et al (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. https://doi.org/10.1111/j.1471-4159.1990.tb02325.x
Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol. https://doi.org/10.1016/j.cub.2014.03.034
Schweitzer JS et al (2020) Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N Engl J Med. https://doi.org/10.1056/nejmoa1915872
Scott L, Dawson VL, Dawson TM (2017) Trumping neurodegeneration: targeting common pathways regulated by autosomal recessive Parkinson’s disease genes. Exp Neurol. https://doi.org/10.1016/j.expneurol.2017.04.008
Seirafi M, Kozlov G, Gehring K (2015) Parkin structure and function. FEBS J. https://doi.org/10.1111/febs.13249
Shen T et al (2018) Genetic analysis of ATP13A2, PLA2G6 and FBXO7 in a cohort of Chinese patients with early-onset Parkinson’s disease. Sci Rep. https://doi.org/10.1038/s41598-018-32217-4
Shen T et al (2019) Early-onset parkinson’s disease caused by pla2g6 compound heterozygous mutation, a case report and literature review. Front Neurol. https://doi.org/10.3389/fneur.2019.00915
Sina F, Shojaee S, Elahi E, Paisán-Ruiz C (2009) R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol. https://doi.org/10.1111/j.1468-1331.2008.02356.x
Singaravelu K (2006) Regulation of store-operated calcium entry by calcium-independent phospholipase A2 in rat cerebellar astrocytes. J Neurosci 26:9579–9592
Sison SL, Vermilyea SC, Emborg ME, Ebert AD (2018) Using patient-derived induced pluripotent stem cells to identify Parkinson’s disease-relevant phenotypes. Curr Neurol Neurosci Rep. https://doi.org/10.1007/s11910-018-0893-8
Smani T, Dominguez-Rodriguez A, Callejo-Garcia P, Rosado JA, Avila-Medina J (2016) Phospholipase A2 as a molecular determinant of store-operated calcium entry. Adv Exp Med Biol 898:111–131
Smits LM, Schwamborn JC (2020) Midbrain organoids: a new tool to investigate Parkinson’s disease. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2020.00359
Smits LM et al (2019) Modeling Parkinson’s disease in midbrain-like organoids. NPJ Park Dis. https://doi.org/10.1038/s41531-019-0078-4
Soldner F et al (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. https://doi.org/10.1016/j.cell.2009.02.013
Soldner F et al (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature. https://doi.org/10.1038/nature17939
Spät A, Szanda G (2017) The role of mitochondria in the activation/maintenance of SOCE: store-operated Ca2+ entry and mitochondria. In: Advances in experimental medicine and biology. https://doi.org/10.1007/978-3-319-57732-6_14
Stathakos P et al (2019) Imaging autophagy in hiPSC-derived midbrain dopaminergic neuronal cultures for parkinson’s disease research. Methods Mol Biol. https://doi.org/10.1007/978-1-4939-8873-0_17
Stathakos P et al (2020) A monolayer hiPSC culture system for autophagy/mitophagy studies in human dopaminergic neurons. Autophagy. https://doi.org/10.1080/15548627.2020.1739441
Sumi-Akamaru H et al (2016) High expression of α-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-016-0298-3
Suzuki S et al (2017) Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2016.12.188
Taanman JW, Protasoni M (2017) Loss of PINK1 or parkin function results in a progressive loss of mitochondrial function. In: Autophagy: cancer, other pathologies, inflammation, immunity, infection, and aging, vol 12. Academic, London. https://doi.org/10.1016/B978-0-12-812146-7.00007-X
Takahashi K et al (2007) Induction of {pluripotent} {stem} {cells} from {adult} {human} {fibroblasts} by {defined} {factors}. Cell 131:861–872
Takahashi-Niki K, Niki T, Taira T, Iguchi-Ariga SMM, Ariga H (2004) Reduced anti-oxidative stress activities of DJ-1 mutants found in Parkinson’s disease patients. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2004.05.187
Tieng V et al (2014) Engineering of midbrain organoids containing long-lived dopaminergic neurons. Stem Cells Dev. https://doi.org/10.1089/scd.2013.0442
Tolosa E, Wenning G, Poewe W (2006) The diagnosis of Parkinson’s disease. Lancet Neurol. https://doi.org/10.1016/S1474-4422(05)70285-4
Tran J, Anastacio H, Bardy C (2020) Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. NPJ Park Dis. https://doi.org/10.1038/s41531-020-0110-8
Turk J, Ramanadham S (2004) The expression and function of a group VIA calcium-independent phospholipase A2 (iPLA2β) in β-cells. Can J Physiol Pharmacol. https://doi.org/10.1139/y04-064
van der Merwe C, Jalali Sefid Dashti Z, Christoffels A, Loos B, Bardien S (2015) Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur J Neurosci. https://doi.org/10.1111/ejn.12872
Vera E, Bosco N, Studer L (2016) Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation. Cell Rep. https://doi.org/10.1016/j.celrep.2016.09.062
Vermilyea SC et al (2020) In vitro CRISPR/Cas9-directed gene editing to model LRRK2 G2019S Parkinson’s disease in common marmosets. Sci Rep. https://doi.org/10.1038/s41598-020-60273-2
Vig M et al (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312:1220–1223
Vrijsen S et al (2020) ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1922342117
Wang HL et al (2011) PARK6 PINK1 mutants are defective in maintaining mitochondrial membrane potential and inhibiting ROS formation of substantia nigra dopaminergic neurons. Biochim Biophys Acta Mol Basis Dis. https://doi.org/10.1016/j.bbadis.2011.03.007
Wang YK et al (2018) Human clinical-grade parthenogenetic ESC-derived dopaminergic neurons recover locomotive defects of nonhuman primate models of Parkinson’s disease. Stem Cell Rep. https://doi.org/10.1016/j.stemcr.2018.05.010
Wang R et al (2019) ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome–lysosome fusion. J Cell Biol. https://doi.org/10.1083/jcb.201804165
Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ (2009) Pink1 forms a multiprotein complex with miro and milton, linking Pink1 function to mitochondrial trafficking. Biochemistry. https://doi.org/10.1021/bi8019178
William Langston J, Ballard P, Tetrud JW, Irwin I (1983) Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. https://doi.org/10.1126/science.6823561
Woodard CM et al (2014) IPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for parkinson’s disease. Cell Rep. https://doi.org/10.1016/j.celrep.2014.10.023
Xu Q et al (2012) Hypoxia regulation of ATP13A2 (PARK9) gene transcription. J Neurochem. https://doi.org/10.1111/j.1471-4159.2012.07676.x
Xu CY et al (2017) DJ-1 inhibits α-synuclein aggregation by regulating chaperone-mediated autophagy. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2017.00308
Xu S, Yang X, Qian Y, Xiao Q (2018) Parkinson’s disease-related DJ-1 modulates the expression of uncoupling protein 4 against oxidative stress. J Neurochem. https://doi.org/10.1111/jnc.14297
Yamano K, Youle RJ (2013) PINK1 is degraded through the N-end rule pathway. Autophagy. https://doi.org/10.4161/auto.24633
Yamano K, Youle RJ (2020) Two different axes CALCOCO2-RB1CC1 and OPTN-ATG9A initiate PRKN-mediated mitophagy. Autophagy. https://doi.org/10.1080/15548627.2020.1815457
Yu W, Sun Y, Guo S, Lu B (2011) The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet. https://doi.org/10.1093/hmg/ddr235
Zhao T et al (2011) Loss of nuclear activity of the FBXO7 protein in patients with parkinsonian-pyramidal syndrome (PARK15). PLoS One. https://doi.org/10.1371/journal.pone.0016983
Zheng L et al (2017) Parkin functionally interacts with PGC-1α to preserve mitochondria and protect dopaminergic neurons. Hum Mol Genet. https://doi.org/10.1093/hmg/ddw418
Zhou Q et al (2016a) Impairment of PARK14-dependent Ca2+signalling is a novel determinant of Parkinson’s disease. Nat Commun 7
Zhou ZD, Sathiyamoorthy S, Angeles DC, Tan EK (2016b) Linking F-box protein 7 and parkin to neuronal degeneration in Parkinson’s disease (PD). Mol Brain. https://doi.org/10.1186/s13041-016-0218-2
Zhou ZD, Lee JCT, Tan EK (2018) Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat Res Rev Mutat Res. https://doi.org/10.1016/j.mrrev.2018.10.001
Zhu L et al (2019) Stress-induced precocious aging in PD-patient iPSC-derived NSCs may underlie the pathophysiology of Parkinson’s disease. Cell Death Dis. https://doi.org/10.1038/s41419-019-1313-y
Acknowledgements
This work was supported by the DBT/Wellcome Trust India Alliance Early Career Fellowship [IA/E/18/1/504319] awarded to the author. Figures were created with BioRender. Prof. Gaiti Hasan, National Centre for Biological Sciences (NCBS), TIFR provided critical inputs for the manuscript.
Conflict of Interests
The author declares no competing interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gopurappilly, R. (2021). Pluripotent Stem Cell Derived Neurons as In Vitro Models for Studying Autosomal Recessive Parkinson’s Disease (ARPD): PLA2G6 and Other Gene Loci. In: Turksen, K. (eds) Cell Biology and Translational Medicine, Volume 14. Advances in Experimental Medicine and Biology(), vol 1347. Springer, Cham. https://doi.org/10.1007/5584_2021_643
Download citation
DOI: https://doi.org/10.1007/5584_2021_643
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-80491-6
Online ISBN: 978-3-030-80492-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)