
Abstract
Axon regeneration in the mature mammalian central nervous system (CNS) is extremely limited after injury. Consequently, functional deficits persist after spinal cord injury (SCI), traumatic brain injury, stroke, and related conditions that involve axonal disconnection. This situation differs from that in the mammalian peripheral nervous system (PNS), where long-distance axon regeneration and substantial functional recovery can occur in the adult. Both extracellular molecules and the intrinsic growth capacity of the neuron influence regenerative success. This chapter discusses determinants of axon regeneration in the PNS and CNS.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Spinal Cord Injury
- Dorsal Root Ganglion Neuron
- Axon Regeneration
- Central Nervous System Injury
- Central Nervous System Neuron
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
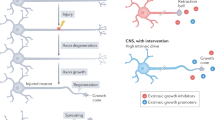
Central nervous system (CNS) axons do not spontaneously regenerate after injury in adult mammals. In contrast, peripheral nervous system (PNS) axons readily regenerate, allowing recovery of function after peripheral nerve damage. Aguayo and colleagues demonstrated that at least some mature CNS neurons retain the capacity to regenerate when provided with a permissive peripheral nerve graft (Richardson et al. 1980, 1984; David and Aguayo, 1981; Benfey and Aguayo, 1982). This work suggested that the PNS environment is stimulatory and/or that the CNS environment is inhibitory for axon growth. Subsequent studies identified both growth-promoting factors in the PNS and growth-inhibiting factors in the CNS. Inhibitors of regeneration include specific proteins in CNS myelin and molecules associated with the astroglial scar. In addition, slower debris clearance in the CNS relative to the PNS may impede axonal re-growth. The failure of axotomized CNS neurons to induce growth-promoting genes also limits brain and spinal cord repair. An understanding of factors which influence axon growth is critical for the development of therapeutics to promote CNS regeneration.
2 Axon Regeneration in the Peripheral Nervous System
2.1 Overview of Peripheral Nervous System Regeneration
After peripheral nerve injury, axons readily regenerate. The distal portion of the axon, which is disconnected from the cell body, undergoes Wallerian degeneration. This active process results in fragmentation and disintegration of the axon. Debris is removed by glial cells, predominantly macrophages. Proximal axons can then regenerate and re-innervate their targets, allowing recovery of function.
2.2 Regeneration-Associated Genes
Following axotomy, PNS neurons upregulate numerous regeneration-associated genes (RAGs). Some of these genes have a direct role in axon regeneration, while others do not. A number of RAGs have been shown to be important for neurite outgrowth and/or regeneration. These include c-Jun (Raivich et al. 2004), activating transcription factor-3 (ATF-3) (Seijffers et al. 2006), SRY-box containing gene 11 (Sox11) (Jankowski et al. 2009), small proline-repeat protein 1A (SPRR1A) (Bonilla et al. 2002), growth-associated protein-43 (GAP-43) and CAP-23 (Bomze et al. 2001).
One strategy to identify RAGs involves injuring a peripheral nerve, and then observing gene expression changes in the corresponding cell bodies (Bonilla et al. 2002; Tanabe et al. 2003; Costigan et al. 2002). A number of such studies have used gene profiling technology to examine gene expression changes in sensory neurons following axotomy. For example, Bonilla et al. (2002) demonstrated that SPRR1A is highly induced in dorsal root ganglion (DRG) neurons one week after sciatic nerve transection (protein increased more than 60-fold from whole DRGs). Immunohistochemistry demonstrated expression of SPRR1A in DRG neuronal cell bodies and regenerating peripheral axons. SPRR1A expression is also increased after sciatic nerve injury in the ventral horn motor neuron cell bodies and sensory fibers within the spinal cord (Fig. 1). Herpes simplex virus-mediated overexpression of SPRR1A in embryonic chick DRG neurons promotes neurite outgrowth. The association of SPRR1A expression with regeneration and its ability to promote neurite outgrowth suggest that it may have a role in axon regeneration.

SPRR1A upregulation in the central process of primary afferent sensory neurons and in motoneurons after sciatic nerve injury. The sciatic nerve was crushed at the mid-thigh on one side of an adult mouse. Seven days later, the animal was sacrificed, and L5 spinal cord transverse sections were processed for anti-SPRR1A immunohistology (red) and for Nissl Stain (blue). Note the intense SPRR1A protein upregulation in afferent terminals in the dorsal horn (arrowheads) and in ventral horn motoneurons (arrow). Upregulation is confined to the injured side (left) with very low levels of SPRR1A on the intact side (right). Dorsal is up. Methods as in Bonilla et al. (2002). Image courtesy of Dr. William B. Cafferty
ATF-3 is a transcriptional factor induced in sensory neurons after injury (Tanabe et al. 2003; Boeshore et al. 2004). Over expression of ATF-3 promotes neurite outgrowth (Seijffers et al. 2006). Sox11 and c-Jun are injury-induced transcription factors and are required for efficient nerve regeneration (Jankowski et al. 2009; Raivich et al. 2004). Transcriptional factors associated with regeneration, such as c-Jun, appear to induce the expression of other RAGs and thereby may promote a growth state (Raivich et al. 2004).
3 Axon Regeneration in the Central Nervous System
3.1 Overview of Central Nervous System Regeneration
Pioneering work by Aguayo and colleagues demonstrated that adult mammalian CNS neurons, which normally do not regenerate, are able to grow for long distances into the permissive environment of a peripheral nerve graft (Richardson et al. 1980, 1984; David and Aguayo 1981; Benfey and Aguayo 1982). These studies demonstrated that the environment is a critical determinant of axon regeneration. Subsequently, numerous molecules were identified within the CNS that limit regeneration.
The two major classes of CNS regeneration inhibitors are the myelin-associated inhibitors (MAIs) and the chondroitin sulfate proteoglycans (CSPGs). These molecules limit axon regeneration, and, by interfering with their function, some degree of growth in the adult CNS is achieved.
Cell-autonomous factors are also important determinants of CNS regeneration failure. CNS neurons do not upregulate growth-associated genes to the same extent as do PNS neurons. Consequently, their ability to regenerate is limited even in the absence of inhibitors. Increasing the intrinsic growth capacity of neurons allows modest axon regeneration within the CNS (Bomze et al. 2001; Neumann and Woolf 1999).
Axon regeneration is one of many factors influencing recovery after CNS damage. Sprouting of uninjured axons can also contribute dramatically to functional improvements. Additionally, plasticity at the synaptic level may underlie a certain degree of recovery seen even in the absence of treatments (i.e., learning to use spared neuronal circuitry in new ways). CNS regeneration studies do not always distinguish between these different mechanisms, and, for the purpose of this discussion, they will be considered together. Replacement of lost neuronal cell bodies, a prominent component of many CNS disorders, is beyond the scope of this chapter.
3.2 Myelin-Associated Inhibitors
MAIs are proteins expressed by oligodendrocytes as components of CNS myelin. MAIs impair neurite outgrowth in vitro and are thought to limit axon growth in vivo after CNS damage. MAIs include Nogo-A (Chen et al. 2000; GrandPre et al., 2000), myelin-associated glycoprotein (MAG) (McKerracher et al., 1994), oligodendrocyte myelin glycoprotein (OMgp) (Kottis et al. 2002), ephrin-B3 (Benson et al. 2005) and Semaphorin 4D (Sema4D) (Moreau-Fauvarque et al. 2003). Three of these (Nogo-A, MAG and OMgp) interact with a neuronal Nogo-66 receptor 1 (NgR1) to limit axon growth. These three structurally unrelated ligands also show affinity for a second axon growth-inhibiting receptor, paired immunoglobulin-like receptor B (PirB) (Atwal et al. 2008). For the most part, MAIs are not found in PNS myelin, which is produced by Schwann cells rather than oligodendrocytes. An exception is MAG, which is present in PNS myelin but is cleared much more rapidly by glial cells in the periphery than in the brain and spinal cord.
One of the most well- characterized MAIs is Nogo-A. Genetic deletion of Nogo-A promotes corticospinal (Fig. 2) and raphespinal tract growth and enhances functional recovery after SCI, although this phenotype is modulated by strain background, age and axonal tract (Kim et al. 2003b; Simonen et al. 2003; Zheng et al. 2003; Dimou et al., 2006). Even after controlling for these factors, certain targeted mutations create a greater axon growth response than others (Cafferty et al. 2007b). However, even the least growth-promoting mutation of the Nogo gene has an enhanced growth phenotype after pyramidotomy (Cafferty and Strittmatter 2006). In addition, antibodies that target Nogo-A promote axonal growth and functional recovery after CNS injury (Z’Graggen et al., 2000; Wiessner et al. 2003; Seymour et al. 2005). An anti-Nogo-A antibody has advanced to clinical trials for SCI.

Corticospinal tract (CST) axonal tracing in mice lacking Nogo-A/B after mid-thoracic spinal cord dorsal hemisection. A parasagittal section of thoracic spinal cord from a mouse with a mutation in the Nogo gene that prevents Nogo-A and Nogo-B expression. The CST is traced from a cortical biotin dextran amine (BDA) injection after dorsal hemisection. Rostral is left; dorsal is up. The lesion is indicated by the asterisk. Note the evidence of CST fiber growth caudal to the lesion site (arrows). Significantly less BDA tracing is present caudal to the lesion in control animals (not shown). This photographic montage is a different image from the same mice described in Kim et al. (2003b)
Two inhibitory portions of Nogo-A have been identified. The first, termed Nogo-66, is a 66 amino acid fragment which interacts with NgR1 on the neuronal membrane (Fournier et al. 2001). An adjacent 24 amino acid sequence, although not inhibitory in itself, facilitates picomolar-affinity binding of the Nogo-66 to NgR1 (Hu et al. 2005). The Nogo-66 domain can also interact directly with a secondary receptor, PirB, on the surface of neurons (Atwal et al. 2008). The other inhibitory portion of Nogo-A Amino-Nogo, acts via an independent mechanism to disrupt neuronal integrin function (Hu and Strittmatter 2008).
Two other Nogo isoforms exist (Nogo-B and Nogo-C), which contain the inhibitory Nogo-66 loop found in Nogo-A but lack the Amino-Nogo sequence. These isoforms are not found naturally in myelin, but transgenic overexpression of Nogo-C in Schwann cells, which normally do not express any Nogo isoform, delays peripheral nerve regeneration. This demonstrates the ability of Nogo-66 to limit axon regeneration in vivo (Kim et al. 2003a).
MAG is another inhibitory protein present in CNS myelin (McKerracher et al., 1994). MAG interacts with several neuronal receptors to limit neurite outgrowth in vitro, including NgR1 (Liu et al. 2002; Domeniconi et al. 2002), gangliosides (Vyas et al. 2002; Mehta et al. 2007), Nogo receptor 2 (NgR2) (Venkatesh et al. 2005) and PirB (Atwal et al. 2008). The relative importance of each receptor varies with neuronal type. For example, in postnatal DRG neurons, NgR1 mediates the majority of inhibition by MAG, whereas in postnatal cerebellar granule neurons, GT1b appears to be more important (Mehta et al. 2007). Although MAG inhibits neurite outgrowth in vitro, no evidence of enhanced corticospinal tract (CST) regeneration or sprouting after SCI or optic nerve injury is observed in MAG knockout mice (Bartsch et al. 1995). Thus, MAG appears to have a less important role in limiting CNS axon growth than other inhibitors such as Nogo-A.
OMgp, another MAI that interacts with NgR1 and PirB, also limits neurite outgrowth in vitro (Kottis et al. 2002). CNS regeneration studies with OMgp knockout mice have not been described.
NgR1 is a glycosylphosphatidylinositol (GPI)-linked membrane receptor, which lacks a transmembrane or cytoplasmic domain. It interacts with coreceptors LINGO-1 (Mi et al. 2004) and p75 (Wang et al. 2002) or TAJ/TROY (Park et al. 2005; Shao et al. 2005), depending on neuronal type, to limit axon growth. Enhanced rubrospinal and raphespinal, but not corticospinal, axon regeneration is observed in NgR1 knockout mice after SCI (Kim et al. 2004). The enhanced axonal growth is correlated with improved functional recovery. Although CST regeneration is not observed after SCI in mice lacking NgR1, corticofugal axons do show enhanced growth in these mice after a stroke (Li et al. 2004) or pyramidotomy (Cafferty and Strittmatter 2006). Thus, NgR1 limits axonal growth and functional recovery after CNS damage.
NgR1 function can be blocked by a soluble form of extracellular NgR1 fused to human Fc (NgR(310)ecto-Fc). NgR(310)ecto-Fc promotes corticospinal and raphespinal growth and functional recovery after SCI in rats (Li et al. 2004). In addition, transgenic mice which secrete NgR(310)ecto under control of the GFAP promoter (causing reactive astrocytes to secrete high levels of the protein after CNS injury) show enhanced functional recovery after SCI (Li et al. 2005).
A competitive NgR1 antagonist, Nogo-extracellular peptide, residues 1–40 (NEP1-40), binds to, but does not activate, NgR1. NEP1-40 attenuates inhibition of neurite outgrowth by Nogo-A and CNS myelin. After SCI, NEP1-40 promotes corticospinal and raphespinal regeneration and functional recovery, even when the initiation of treatment is delayed for one week (GrandPre et al. 2002; Li and Strittmatter, 2003). Because NEP1-40 blocks Nogo-66, but not other NgR1 ligands, it is less effective than NgR(310)ecto-Fc.
The studies described above confirm the importance of NgR1 and its ligands in limiting CNS regeneration. In vivo functional studies of other MAI receptors have not yet been reported.
3.3 Chondroitin Sulfate Proteoglycans
The astroglial scar, which forms after CNS injury, is a physical barrier to regeneration, and also contains inhibitory molecules that impede axon growth. CSPGs are the main inhibitory molecules found in the glial scar (reviewed in Morgenstern et al. 2002). CSPGs are upregulated by reactive astrocytes after CNS damage and are both membrane bound and secreted into the extracellular space. Members of this class of inhibitors include neurocan (Asher et al. 2000), versican (Schmalfeldt et al. 2000), brevican (Yamada et al. 1997), phosphacan (Inatani et al. 2001), aggrecan (Chan et al. 2008) and NG2 (Dou and Levine, 1994). A receptor for CSPGs has not been identified.
Interfering with CSPG function promotes axon regeneration in the CNS. CPSGs contain core proteins with attached glycosaminoglycan (GAG) side chains, which can be cleaved by the bacterial enzyme Chondroitinase ABC. This enzyme reduces the inhibitory activity of CSPGs in vitro (McKeon et al. 1995). Moreover, when Chondroitinase ABC is administered after spinal contusion in rats, regeneration of both descending CST and ascending sensory fibers can be detected (Bradbury et al. 2002). This axonal growth is accompanied by enhanced recovery of associated locomotor and proprioceptive functions. Several other studies have confirmed that Chondrotinase ABC promotes axonal growth after CNS injury (Barritt et al. 2006; Massey et al. 2006; Cafferty et al. 2007a).
3.4 Other Axon Regeneration Inhibitors
Axon regeneration inhibitors (ARIs) found in the CNS that are not present in myelin or the glial scar include repulsive guidance molecule (RGM) and semaphorin 3A (Sema3A). Evidence that these molecules limit CNS regeneration include studies demonstrating that an anti-RGMa antibody (Hata et al. 2006) or a small molecule inhibitor of Sema3A (Kaneko et al. 2007) promote functional recovery after SCI in rats.
3.5 Inhibitory Signaling Pathways
Multiple ARIs have been shown to activate the small GTPase ras homolog gene family, member A (RhoA) (Niederost et al. 2002; Fournier et al. 2003; Shao et al. 2005). Activated RhoA, in turn, activates Rho-associated coiled-coil containing protein kinase 2 (ROCK2), a kinase that regulates actin cytoskeletal dynamics (reviewed in Schmandke et al. 2007). Activation of ROCK2 results in cessation of neurite growth. Interfering with RhoA or ROCK2 activity promotes CNS axon regeneration and functional recovery.
Ibuprofen, which inhibits RhoA, promotes corticospinal and raphespinal sprouting after spinal contusion (Fu et al. 2007; Wang et al. 2009), as well as long- distance raphespinal axon regeneration after a complete spinal cord transection (Wang et al. 2009). Tissue sparing at the lesion site is also enhanced by ibuprofen and thus may contribute to functional recovery (Wang et al. 2009).
The ROCK2 inhibitor Y27632 promotes CST sprouting and locomotor recovery after dorsal hemi section spinal injury in rats (Fournier et al. 2003). In addition, ROCK2 knockout mice show enhanced functional recovery after SCI (Duffy PJ, Schmandke A, and Strittmatter SM, unpublished observations). Thus, ROCK2 is an important mediator of CNS regeneration failure.
Some evidence suggests that epidermal growth factor receptor (EGFR) contributes to CNS regeneration failure. One study demonstrated enhanced optic nerve regeneration after treatment with the irreversible EGFR inhibitor PD168393 (Koprivica et al. 2005). This study provides evidence that trans-activation of EGFR mediates inhibition of neurite outgrowth by MAIs and CSPGs. Another study observed that PD168393 enhances sparing, and/or regeneration of 5-hydroxytryptophan-immunoreactive (serotonergic) fibers caudal to a spinal cord lesion (Erschbamer et al. 2007). Thus, EGFR activation appears to limit recovery after CNS trauma.
Other molecules that have been implicated in ARI- signaling include protein kinase C, (Sivasankaran et al. 2004), LIM kinase, Slingshot phosphatase and cofilin (Hsieh et al. 2006).
3.6 Intrinsic Growth State of the Neuron
In contrast to the PNS, the upregulation of peripheral RAGs (see Sect. 2.2) is relatively modest in the CNS after injury (Fernandes et al. 1999; Marklund et al. 2006). This paucity of RAG expression appears to be partially responsible for the limited ability of CNS neurons to regenerate. Increasing RAG expression in CNS neurons improves their regenerative ability. For example, Bomze et al. (2001) demonstrated that overexpressing GAP-43 and CAP-23 together promotes sensory axon regeneration after SCI.
DRG neurons have a peripheral process and a central process. Injury to the peripheral process results in robust upregulation of RAGs, as described above. However, injury to the central process by dorsal rhizotomy or spinal cord dorsal hemi section does not induce nearly as robust of a regenerative response, and central processes fail to regenerate in the CNS. Injury of peripheral axons one week prior to central injury (termed a conditioning lesion) allows some degree of sensory fiber regeneration within the spinal cord (Neumann and Woolf 1999). The conditioning lesion appears to enhance the growth state of the neuron such that its central process is able to regenerate in the CNS environment.
Cyclic adenosine monophosphate (cAMP) is a second messenger molecule which influences the growth state of the neuron. cAMP levels are increased by a peripheral conditioning lesion (Qiu et al. 2002). Elevation of cAMP levels by intraganglionic injection of a membrane-permeable cAMP analog, dibutyryl cAMP (db-cAMP), mimics the growth-promoting effects of a conditioning lesion, promoting regeneration of sensory axons within the spinal cord (Neumann et al. 2002; Qiu et al. 2002). In vivo injection of db-cAMP prior to DRG removal also improves the ability of dissociated DRG cultures to grow on MAG or CNS myelin in vitro, indicating that cAMP elevation can promote growth in the presence of MAIs (Qiu et al. 2002).
Rolipram, a phosphodiesterase 4 inhibitor, increases cAMP by interfering with its hydrolysis. When delivered 2 weeks after spinal cord hemisection, rolipram increases serotonergic axon regeneration into embryonic spinal tissue grafts implanted at the lesion site at the time of injury (Nikulina et al. 2004). Reactive gliosis is reduced by rolipram, and this might contribute to the functional recovery observed. Additionally, enhanced axonal sparing and myelination are induced by cAMP elevation in combination with Schwann cell grafts after SCI, compared to Schwann cell grafts alone (Pearse et al. 2004), suggesting additional mechanisms by which cAMP elevation could lead to functional improvements. Nonetheless, the demonstration of serotonergic axon growth into grafts at the lesion site in both of these studies indicates that cAMP elevation can induce CNS axon regeneration.
cAMP elevation activates protein kinase A (PKA) and induces CREB-mediated transcription of various growth-associated genes, including IL-6 and arginase I. Subsequent synthesis of polyamines by arginase I has been proposed as a possible mechanism by which cAMP increases neurite growth (Cai et al. 2002).
4 Conclusion
Regeneration of the injured mammalian CNS was once thought to be an unachievable goal. Recent advances in our understanding of factors which limit CNS regeneration and those which facilitate PNS regeneration have lead to therapies which allow some degree of recovery from brain and SCI in animal models. These findings open the possibility of promoting regeneration of the damaged human CNS.
References
Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW (2000) Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci 20:2427–2438
Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322:967–970
Barritt AW, Davies M, Marchand F, Hartley R, Grist J, Yip P, McMahon SB, Bradbury EJ (2006) Chondroitinase ABC promotes sprouting of intact and injured spinal systems after spinal cord injury. J Neurosci 26:10856–10867
Bartsch U, Bandtlow CE, Schnell L, Bartsch S, Spillmann AA, Rubin BP, Hillenbrand R, Montag D, Schwab ME, Schachner M (1995) Lack of evidence that myelin-associated glycoprotein is a major inhibitor of axonal regeneration in the CNS. Neuron 15:1375–1381
Benfey M, Aguayo AJ (1982) Extensive elongation of axons from rat brain into peripheral nerve grafts. Nature 296:150–152
Benson MD, Romero MI, Lush ME, Lu QR, Henkemeyer M, Parada LF (2005) Ephrin-B3 is a myelin-based inhibitor of neurite outgrowth. Proc Nat Acad Sci USA 102:10694–10699
Boeshore KL, Schreiber RC, Vaccariello SA, Sachs HH, Salazar R, Lee J, Ratan RR, Leahy P, Zigmond RE (2004) Novel changes in gene expression following axotomy of a sympathetic ganglion: a microarray analysis. J Neurobiol 59:216–235
Bomze HM, Bulsara KR, Iskandar BJ, Caroni P, Pate Skene JH (2001) Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci 4:38–43
Bonilla IE, Tanabe K, Strittmatter SM (2002) Small proline-rich repeat protein 1A Is expressed by axotomized neurons and promotes axonal outgrowth. J Neurosci 22:1303–1315
Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB (2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416:636–640
Cafferty WB, Strittmatter SM (2006) The Nogo-Nogo receptor pathway limits a spectrum of adult cns axonal growth. J Neurosci 26:12242–12250
Cafferty WB, Yang S-H, Duffy PJ, Li S, Strittmatter SM (2007a) Functional axonal regeneration through astrocytic scar genetically modified to digest chondroitin sulfate proteoglycans. J Neurosci 27:2176–2185
Cafferty WB, Kim J-E, Lee J-K, Strittmatter SM (2007b) Response to correspondence: Kim et al. “Axon regeneration in young adult mice lacking Nogo-A/B.” Neuron 38, 187–199. Neuron 54:195–199
Cai D, Deng K, Mellado W, Lee J, Ratan RR, Filbin MT (2002) Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 35:711–719
Chan CC, Roberts CR, Steeves JD, Tetzlaff W (2008) Aggrecan components differentially modulate nerve growth factor-responsive and neurotrophin-3-responsive dorsal root ganglion neurite growth. J Neurosci Res 86:581–592
Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME (2000) Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403:434–439
Costigan M, Befort K, Karchewski L, Griffin R, D’Urso D, Allchorne A, Sitarski J, Mannion J, Pratt R, Woolf C (2002) Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci 3:16
David S, Aguayo AJ (1981) Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science 214:931–933
Dimou L, Schnell L, Montani L, Duncan C, Simonen M, Schneider R, Liebscher T, Gullo M, Schwab ME (2006) Nogo-A-deficient mice reveal strain-dependent differences in axonal regeneration. J Neurosci 26:5591–5603
Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang KC, Nikulina E, Kimura N, Cai H, Deng K, Gao Y, He Z, Filbin MT (2002) Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron 35:283–290
Dou CL, Levine JM (1994) Inhibition of neurite growth by the NG2 chondroitin sulfate proteoglycan. J Neurosci 14:7616–7628
Erschbamer M, Pernold K, Olson L (2007) Inhibiting epidermal growth factor receptor improves structural, locomotor, sensory, and bladder recovery from experimental spinal cord injury. J Neurosci 27:6428–6435
Fernandes KJ, Fan DP, Tsui BJ, Cassar SL, Tetzlaff W (1999) Influence of the axotomy to cell body distance in rat rubrospinal and spinal motoneurons: differential regulation of GAP-43, tubulins, and neurofilament-M. J Comp Neurol 414:495–510
Fournier AE, GrandPre T, Strittmatter SM (2001) Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409:341–346
Fournier AE, Takizawa BT, Strittmatter SM (2003) Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci 23:1416–1423
Fu Q, Hue J, Li S (2007) Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J Neurosci 27:4154–4164
GrandPre T, Nakamura F, Vartanian T, Strittmatter SM (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403:439–444
GrandPre T, Li S, Strittmatter SM (2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417:547–551
Hata K, Fujitani M, Yasuda Y, Doya H, Saito T, Yamagishi S, Mueller BK, Yamashita T (2006) RGMa inhibition promotes axonal growth and recovery after spinal cord injury. J Cell Biol 173:47–58
Hsieh SH, Ferraro GB, Fournier AE (2006) Myelin-associated inhibitors regulate cofilin phosphorylation and neuronal inhibition through LIM kinase and slingshot phosphatase. J Neurosci 26:1006–1015
Hu F, Strittmatter SM (2008) The N-terminal domain of Nogo-A inhibits cell adhesion and axonal outgrowth by an integrin-specific mechanism. J Neurosci 28:1262–1269
Hu F, Liu BP, Budel S, Liao J, Chin J, Fournier A, Strittmatter SM (2005) Nogo-A interacts with the Nogo-66 receptor through multiple sites to create an isoform-selective subnanomolar agonist. J Neurosci 25:5298–5304
Inatani M, Honjo M, Otori Y, Oohira A, Kido N, Tano Y, Honda Y, Tanihara H (2001) Inhibitory effects of neurocan and phosphacan on neurite outgrowth from retinal ganglion cells in culture. Invest Ophthalmol Vis Sci 42:1930–1938
Jankowski MP, McIlwrath SL, Jing X, Cornuet PK, Salerno KM, Koerber HR, Albers KM (2009) Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Research 1256:43–54
Kaneko S, Iwanami A, Nakamura M, Kishino A, Kikuchi K, Shibata S, Okano HJ, Ikegami T, Moriya A, Konishi O, Nakayama C, Kumagai K, Kimura T, Sato Y, Goshima Y, Taniguchi M, Ito M, He Z, Toyama Y, Okano H (2007) A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med 12:1380–1389
Kim J-E, Bonilla IE, Qiu D, Strittmatter SM (2003a) Nogo-C is sufficient to delay nerve regeneration. Mol Cell Neurosci 23:451–459
Kim J-E, Li S, GrandPré T, Qiu D, Strittmatter SM (2003b) Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38:187–199
Kim J-E, Liu BP, Park JH, Strittmatter SM (2004) Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron 44:439–451
Koprivica V, Cho K-S, Park JB, Yiu G, Atwal J, Gore B, Kim JA, Lin E, Tessier-Lavigne M, Chen DF, He Z (2005) EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science 310:106–110
Kottis V, Thibault P, Mikol D, Xiao ZC, Zhang R, Dergham P, Braun PE (2002) Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J Neurochem 82:1566–1569
Li S, Strittmatter SM (2003) Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. J Neurosci 23:4219–4227
Li S, Liu BP, Budel S, Li M, Ji B, Walus L, Li W, Jirik A, Rabacchi S, Choi E, Worley D, Sah DWY, Pepinsky B, Lee D, Relton J, Strittmatter SM (2004) Blockade of Nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble Nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci 24:10511–10520
Li S, Kim J-E, Budel S, Hampton TG, Strittmatter SM (2005) Transgenic inhibition of Nogo-66 receptor function allows axonal sprouting and improved locomotion after spinal injury. Mol Cell Neurosci 29:26–39
Liu BP, Fournier A, GrandPre T, Strittmatter SM (2002) Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 297:1190–1193
Marklund N, Fulp CT, Shimizu S, Puri R, McMillan A, Strittmatter SM, McIntosh TK (2006) Selective temporal and regional alterations of Nogo-A and small proline-rich repeat protein 1A (SPRR1A) but not Nogo-66 receptor (NgR) occur following traumatic brain injury in the rat. Exp Neurol 197:70–83
Massey JM, Hubscher CH, Wagoner MR, Decker JA, Amps J, Silver J, Onifer SM (2006) Chondroitinase ABC digestion of the perineuronal net promotes functional collateral sprouting in the Cuneate nucleus after cervical spinal cord injury. J Neurosci 26:4406–4414
McKeon RJ, Höke A, Silver J (1995) Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Experimental Neurology 136:32–43
McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE (1994) Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13:805–811
Mehta NR, Lopez PH, Vyas AA, Schnaar RL (2007) Gangliosides and Nogo Receptors Independently Mediate Myelin-associated Glycoprotein Inhibition of Neurite Outgrowth in Different Nerve Cells. J Biol Chem 282:27875–27886
Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB (2004) LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 7:221–228
Moreau-Fauvarque C, Kumanogoh A, Camand E, Jaillard C, Barbin G, Boquet I, Love C, Jones EY, Kikutani H, Lubetzki C, Dusart I, Chedotal A (2003) The Transmembrane Semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J Neurosci 23:9229–9239
Morgenstern DA, Asher RA, Fawcett JW (2002) Chondroitin sulphate proteoglycans in the CNS injury response. Prog Brain Res 137:313–332
Neumann S, Woolf CJ (1999) Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23:83–91
Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34:885–893
Niederost B, Oertle T, Fritsche J, McKinney RA, Bandtlow CE (2002) Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J Neurosci 22:10368–10376
Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT (2004) The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci USA 101:8786–8790
Park JB, Yiu G, Kaneko S, Wang J, Chang J, He Z (2005) A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 45:345–351
Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB (2004) cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med 10:610–616
Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34:895–903
Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Ls R-S, Wolfer DP, Lipp H-P, Aguzzi A, Wagner EF, Behrens A (2004) The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43:57–67
Richardson PM, McGuinness UM, Aguayo AJ (1980) Axons from CNS neurons regenerate into PNS grafts. Nature 284:264–265
Richardson PM, Issa VM, Aguayo AJ (1984) Regeneration of long spinal axons in the rat. J Neurocytol 13:165–182
Schmalfeldt M, Bandtlow CE, Dours-Zimmermann MT, Winterhalter KH, Zimmermann DR (2000) Brain derived versican V2 is a potent inhibitor of axonal growth. J Cell Sci 113:807–816
Schmandke A, Schmandke A, Strittmatter SM (2007) ROCK and Rho: biochemistry and neuronal functions of Rho-associated protein kinases. Neuroscientist 13:454–469
Seijffers R, Allchorne AJ, Woolf CJ (2006) The transcription factor ATF-3 promotes neurite outgrowth. Mol Cell Neurosci 32:143–154
Seymour AB, Andrews EM, Tsai S-Y, Markus TM, Bollnow MR, Brenneman MM, O’Brien TE, Castro AJ, Schwab ME, Kartje GL (2005) Delayed treatment with monoclonal antibody IN-1 1 week after stroke results in recovery of function and corticorubral plasticity in adult rats. J Cereb Blood Flow Metab 25:1366–1375
Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, Murray B, Jung V, Pepinsky RB, Mi S (2005) TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron 45:353–359
Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME (2003) Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron 38:201–211
Sivasankaran R, Pei J, Wang KC, Zhang YP, Shields CB, Xu X-M, He Z (2004) PKC mediates inhibitory effects of myelin and chondroitin sulfate proteoglycans on axonal regeneration. Nat Neurosci 7:261–268
Tanabe K, Bonilla I, Winkles JA, Strittmatter SM (2003) Fibroblast growth factor-inducible-14 is induced in axotomized neurons and promotes neurite outgrowth. J Neurosci 23:9675–9686
Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ (2005) The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci 25:808–822
Vyas AA, Patel HV, Fromholt SE, Heffer-Lauc M, Vyas KA, Dang J, Schachner M, Schnaar RL (2002) Gangliosides are functional nerve cell ligands for myelin-associated glycoprotein (MAG), an inhibitor of nerve regeneration. Proc Natl Acad Sci USA 99:8412–8417
Wang KC, Kim JA, Sivasankaran R, Segal R, He Z (2002) p75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420:74–78
Wang X, Budel S, Baughman K, Gould G, Song KH, Strittmatter SM (2009) Ibuprofen enhances recovery from spinal cord injury by limiting tissue loss and stimulating axonal growth. J Neurotrauma 26:81–95
Wiessner C, Bareyre FM, Allegrini PR, Mir AK, Frentzel S, Zurini M, Schnell L, Oertle T, Schwab ME (2003) Anti-Nogo-A antibody infusion 24 hours after experimental stroke improved behavioral outcome and corticospinal plasticity in normotensive and spontaneously hypertensive rats. J Cereb Blood Flow Metab 23:154–165
Yamada H, Fredette B, Shitara K, Hagihara K, Miura R, Ranscht B, Stallcup WB, Yamaguchi Y (1997) The brain chondroitin sulfate proteoglycan brevican associates with astrocytes ensheathing cerebellar glomeruli and inhibits neurite outgrowth from granule neurons. J Neurosci 17:7784–7795
Z’Graggen WJ, Fouad K, Raineteau O, Metz GAS, Schwab ME, Kartje GL (2000) Compensatory sprouting and impulse rerouting after unilateral pyramidal tract lesion in neonatal rats. J Neurosci 20:6561–6569
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Huebner, E.A., Strittmatter, S.M. (2009). Axon Regeneration in the Peripheral and Central Nervous Systems. In: Koenig, E. (eds) Cell Biology of the Axon. Results and Problems in Cell Differentiation, vol 48. Springer, Berlin, Heidelberg. https://doi.org/10.1007/400_2009_19
Download citation
DOI: https://doi.org/10.1007/400_2009_19
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-03018-5
Online ISBN: 978-3-642-03019-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)