
Abstract
Emerging evidence suggests the growing importance of “nongenetic factors” in the pathogenesis of atherosclerotic vascular disease. Indeed, the inherited genome determines only part of the risk profile as genomic approaches do not take into account additional layers of biological regulation by “epi”-genetic changes. Epigenetic modifications are defined as plastic chemical changes of DNA/histone complexes which critically affect gene activity without altering the DNA sequence. These modifications include DNA methylation, histone posttranslational modifications, and non-coding RNAs and have the ability to modulate gene expression at both transcriptional and posttranscriptional level. Notably, epigenetic signals are mainly induced by environmental factors (i.e., pollution, smoking, noise) and, once acquired, may be transmitted to the offspring. The inheritance of adverse epigenetic changes may lead to premature deregulation of pathways involved in vascular damage and endothelial dysfunction. Here, we describe the emerging role of epigenetic modifications as fine-tuners of gene transcription in atherosclerosis. Specifically, the following aspects are described in detail: (1) discovery and impact of the epigenome in cardiovascular disease, (2) the epigenetic landscape in atherosclerosis; (3) inheritance of epigenetic signals and premature vascular disease; (4) epigenetic control of lipid metabolism, vascular oxidative stress, inflammation, autophagy, and apoptosis; (5) epigenetic biomarkers in patients with atherosclerosis; (6) novel therapeutic strategies to modulate epigenetic marks. Understanding the individual epigenetic profile may pave the way for new approaches to determine cardiovascular risk and to develop personalized therapies to treat atherosclerosis and its complications.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Discovery and Impact of Epigenetics
Although inborn genetic variation can influence disease susceptibility, accumulating evidence supports the notion that cardiovascular diseases (CVD) are heavily affected by non-genetic factors. Indeed, the inherited genome determines only part of the risk profile as genomic approaches do not take into account additional layers of biological regulation by “epi”-genetic changes – defined as acquired modifications to the genome subject to influence by the environment (Handy et al. 2011). The discovery of epigenetic modifications dates back to 1956, when the British developmental biologist Conrad Waddington demonstrated for the first time the inheritance of a characteristic acquired in a population in response to an environmental stimulus (Slack 2002). Waddington’s experimental work showed that embryo fruit flies carrying an identical genetic background displayed different thorax and wing structures when exposed to different temperature or chemical stimuli (Noble 2015). While the term “epigenetics” initially embraced the process by which a fertilized zygote develops into a complex organism, the concept was refined over the next years by the observations that cells sharing the same genetic information can exhibit clear differences in gene expression. Waddington’s work has contributed to understand that heritable traits can associate not only with changes in nucleotide sequence, but also with chemical modifications of DNA or proteins interacting with DNA. Later experiments in 1975 showed that epigenetic changes could be transmitted to daughter cells, thus regulating gene expression across multiple generations (Gonzalez-Recio et al. 2015). There are several examples outlining the importance of epigenetic processing in health and disease. First, epigenetic regulation of gene expression is a major determinant of cell fate. Epigenetic signals occurring at the levels of DNA and histones have the ability to license regions of the genome while shutting down others, thus favoring specific transcriptional programs which enable the differentiation of genetically identical pluripotent stem cells toward different cell types (i.e. myocytes, endothelial cells, adipocytes) (Brunet and Berger 2014). Along the same line, monozygotic twins who share an identical genetic make-up may display different physical and behavioral traits when raised under different environmental conditions (Fraga et al. 2005). Chronic exposure to different stimuli (i.e., pollution, noise) will enable the induction of specific epigenetic programs eventually leading to different gene expression profiles over the lifetime (Baccarelli and Ghosh 2012). Of note, epigenetic-induced deregulation of longevity genes was shown to accelerate cellular senescence and vascular aging (Sen et al. 2016). On the other hand, specific interventions such as caloric restriction have shown to delay age-dependent onset of diseases mostly via an epigenetic reprogramming. Taken together, epigenetic regulation of gene transcription represents a pivotal mechanism of adaptation to different environmental conditions and is among the most important mechanisms underlying cardiovascular diseases. A deep understanding of the epigenetic machinery may enable the characterization of cell-specific transcriptional programs in patients with atherosclerosis and could offer the tools for personalized therapeutic approaches to prevent cardiovascular diseases. The availability of new technologies for the study of our genome has indeed allowed a deep characterization of chromatin structure and function thus unveiling an array of epigenetic changes at the level of DNA and histones. This important information has enabled to understand inter-individual diversity and has prompted us to strive for an ever greater level of personalization in cardiovascular medicine.
2 Classification of Epigenetic Modifications
Epigenetic modifications fall into three main categories: (1) chemical modifications of DNA (i.e., methylation); (2) posttranslational modifications of histone tails; (3) regulation of gene expression by non-coding RNAs [i.e., microRNAs, PIWI-interacting RNAs, endogenous short interfering RNAs, long non-coding RNAs (lncRNAs)] (Fig. 1). The present chapter will focus on the role of chromatin modifications in atherosclerosis. The function of ncRNAs in this setting is described extensively in a different chapter.

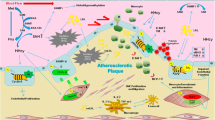
Role of epigenetic inheritance in vascular disease. Environmental factors induce epigenetic changes which are transmitted to the offspring, thus leading to maladaptive transcriptional programs responsible for early vascular damage
2.1 DNA Methylation
Nuclear DNA is tightly wrapped around a core of eight histone proteins (two copies each of H2A, H2B, H3, and H4), generating repetitive units known as nucleosomes. These structures enable a very efficient packaging of DNA within the cell nucleus, through the formation of chromatin. Despite its complex three-dimensional structure, chromatin is a very dynamic entity where DNA and histones can be chemically modified. This alters the accessibility of transcriptional factors to genes and, thus, modulates transcription (Goldberg et al. 2007).
DNA methylation plays an important role in the regulation of chromatin structure and gene expression, and therefore, participates in a variety of biological processes (Hamidi et al. 2015), including tissue-specific regulation of gene expression, genomic imprinting, X chromosome inactivation, silencing of transposable elements, and defense against viral sequences (Prasher et al. 2019). This epigenetic modification is relatively stable, it is tissue-specific and can be transmitted to the offspring (Izquierdo and Crujeiras 2019). Methylation of DNA mainly occurs through attachment of methyl group (CH3) from S-adenosyl methionine (SAM) to the C5 position in the cytosine-paired-with-guanine (CpG) dinucleotide sequences thus forming 5-methylcytosine (5mC) (Davis and Gallagher 2019). Furthermore, non-CpG methylation (i.e., CHG or CHH region) may also occur and this signature is usually observed in embryonic stem cells (Bernstein et al. 2007).
CpG sequences are generally located into promoter regions of genes, however, they can also be located within gene bodies (Costantino et al. 2015). Promoter methylation is generally associated with transcriptional repression, while gene body methylation is associated with enhanced transcription (Maunakea et al. 2010). Although both transcriptional repression and activation have been reported, DNA methylation leads – in >95% of cases – to gene silencing (Xiao et al. 2019). Specifically, methylated cytosines are recognized by DNA methyl-binding proteins (MBPs) that prevent the binding of transcription factors to DNA (Prasher et al. 2019). Alternatively, DNA methylation may recruit specific proteins, such as the methyl-CpG binding protein 2 (MeCP2), that specifically bind to methylated regions to foster transcriptional repression (Jones et al. 1998). Besides methylation, other chemical modifications of cytosine have been identified, such as hydroxymethylation, formylation, and carboxylation, but their interrelation with methylation is not completely understood yet (Elia and Condorelli 2019).
DNA methylation is catalyzed by the DNA methyltransferase 1 (DNMT1), 3a (DNMT3a), and 3b (DNMT3b) and is reversed by Tet methylcytosine dioxygenases (TET1, 2, and 3) (Xu et al. 2018). Among these enzymes, Dnmt1 is responsible for the maintenance of methylation patterns in the genome by replicating the hemi-methylated CpG sites (Vilkaitis et al. 2005), whereas Dnmt3a/b are considered de novo methyltransferases (Okano et al. 1999). The activity of DNMT1 has been demonstrated to be dependent on a co-factor that recognizes hemi-methylated DNA. The Ubiquitin-like with PHD and Ring Finger Domains 1 (UHRF1), were shown to be essential for maintenance of the methylation status of DNA by directing the recruitment of DNMT1 to the replication forks (Bostick et al. 2007). DNMT3 can also methylate non-CpG sites (at CpA and CHG) and the presence of methylated non-CpG sites in the human genome indicates that there is ongoing de novo DNA methylation at each round of genome replication (Aavik et al. 2019).
DNA demethylation can be achieved by either passive or active mechanisms (Costantino et al. 2015). Passive demethylation can be the result of Dnmt1 inhibition during cell replication (Wolffe et al. 1999) while active demethylation is modulated by DNA demethylases. DNA demethylation may follow two main pathways: the first is dependent on cytosine deamination (AID, APOBEC3G, FTO) while the second is dependent on the oxidation of methylated cytosines (Aavik et al. 2019). The latter reaction is catalyzed by members of the Ten-eleven translocation (TET) proteins family (TET1-3) which convert 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC) (Wu and Zhang 2010). TET1 is mostly found in embryonic stem cells, whereas TET2 and TET3 are ubiquitously expressed (Costantino et al. 2015). TET1 is low in normal tissues, whereas TET2 is an abundant protein in most tissues/cell types and TET3 expression is overlapping with TET2 in many tissues (Kohli and Zhang 2013). The observed abundance of TET2 and TET3 enzymes in various tissues indicates that DNA demethylation must have an important role in cellular homeostasis (Aavik et al. 2019). The TET family enzymes represent promising therapeutic targets given their ability to oxidize 5mC thus leading to the reversal of gene repression by DNA methylation (Greco et al. 2016).
2.2 Histone Posttranslational Modifications
DNA is packaged into repeating units called nucleosomes by wrapping around multimeric histone proteins. When nucleosomes are organized into tightly packed bundles (heterochromatin), the transcriptional machinery is hampered by a reduction of chromatin accessibility. Conversely, when chromatin is relaxed (euchromatin), DNA is more accessible to transcription factors and gene transcription may occur. Each histone (H) protein is an octamer comprised of 2 sets of H2A, H2B, H3, and H4 proteins with a single histone H1 linker protein between nucleosomes. Each histone subunit has an N-terminal tail containing a lysine (K) residue that protrudes away from the surface of the histone octamer creating an exposed surface (Davis and Gallagher 2019). Histones are amenable to many posttranslational modifications (PTMs), which include methylation, acetylation, ubiquitination, phosphorylation, SUMOylation, GlcNAcylation, carbonylation, and ADP-ribosylation (Zhang et al. 2018). Histone modifications are induced by specific enzymes that act predominantly at histone N-terminal tails primarily involving the amino acids lysine (K) and arginine (R). These modifications can be present on multiple (but specific) sites on histones (Kouzarides 2007).
In addition to DNA modifications, PTMs of histone tails may serve as a repressive or activating signal depending on the specific histone tail, the number of groups added, and the region of chromatin where the modification occurs (Bauer and Martin 2017). Although the biological significance of many PTMs remains to be elucidated, considerable advances have been made in the understanding of lysine acetylation and methylation (Bernstein et al. 2007). The first PTM reported is histone acetylation, an epigenetic signature characterized by the addition of positively charged acetyl groups to amino acid residues on the histones, which neutralizes the negative charges of DNA. This modification reduces the affinity of the histone for DNA and consequently results in the formation of a more relaxed chromatin structure, eventually increasing chromatin accessibility (Nicorescu et al. 2019). Acetylation occurs mainly on lysine residues on histones H3 and H4; this mark mainly associates with activation of transcription by enhancing chromatin accessibility (Gillette and Hill 2015). In this context, bromodomain and extra-terminal proteins recognize histone acetylation marks and initiate the assembly of the transcriptional machinery (Filippakopoulos and Knapp 2014). Acetylation is modulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs) which are involved in the addition or removal of an acetyl group, respectively (Baccarelli and Ghosh 2012). This modification is driven by recognition and binding of transcription factors able to recruit one of a growing family of HATs, namely CBP/p300, MYST, and GNAT (Kouzarides 2007). Under physiological conditions, lysine residues on histone tails are positively charged and can bind DNA which is a negatively charged molecule due to its phosphate components. The interaction between histone and DNA favors a compact chromatin structure, with scarce accessibility to transcription factors. Conversely, when lysine residues are acetylated they lose their positive charge, thus favoring an open chromatin and, hence, enhanced gene expression (Costantino et al. 2015). HATs catalyze the addition of two-carbon acetyl groups to lysine residues from acetyl-CoA (Carrozza et al. 2003). On the other hand, removal of acetyl groups from histone residues by HDACs represses gene transcription (Stratton et al. 2019). Specifically, HDACs catalyze removal of acetyl groups from lysine residues and are distributed among four classes (Class I, IIa, IIb, III, and IV) (Haberland et al. 2009).
In contrast to lysine acetylation, a process known to enhance gene expression, histone methylation is a more complex modification which may result in different chromatin states according to the methylated residue and the number of added methyl groups (Cooper and El-Osta 2010). For instance, the lysine residues at positions 4, 9, and 27 on histone H3 can be methylated to different levels (mono-, bi-, and tri-methylation), resulting in different states of chromatin accessibility (Elia and Condorelli 2019). Histone methylation is defined as the transfer of methyl group from S-adenosyl-L-methionine to lysine or arginine residues of histone proteins by histone methyltransferases (HMTs). While lysines may be mono-, di-, or tri-methylated, arginines may be monomethylated or demethylated (symmetrically or asymmetrically) (Bannister and Kouzarides 2011). Histone methyltransferases (HMTs) include the EZH, SETD, PRDM, PRMT, METTL, and MLL enzyme families (Stratton et al. 2019).
Over the last few years, a well-established theory was that histone turnover was the only mechanism regulating histone methylation. More recently, histone demethylases (HDMs) have been reported as a novel mechanism capable of demethylating lysine residues on histone tails (Handy et al. 2011). Indeed, histone methylation is a very dynamic process regulated by histone demethylases, which remove the methyl groups from lysine residues with a high gene specificity (Tsukada et al. 2006). HDMs include members of UTX/Y, JARID1, JMJD, LSD, PHF, and FBXL enzyme families (Stratton et al. 2019).
Two important studies have identified the first HDM, lysine demethylase 1 (LSD1) which is able to remove methyl group from H3K4 and H3K9, suggesting that lysine demethylation is subjected to dynamic modulation (Metzger et al. 2005). Several lysine demethylases specific for diverse histone lysine residues have been identified (Costantino et al. 2015). A better comprehension of how such demethylases are regulated during disease states is of paramount importance.
Interestingly, studies have shown that DNA methylase (DNMTs), histone methyltransferase (HMTs), and histone acetyltransferase (HATs) are closely interconnected to regulate chromatin remodelling under specific stimuli (Li et al. 2007). For example, H3K9 methylation induces DNA methylation while CpG methylation favors H3K9m (Cedar and Bergman 2009). Therefore, chromatin modifications may influence each other and can propagate.
3 Epigenetic Inheritance and Vascular Disease
An important aspect to consider when dealing with epigenetics is that epigenetic modifications are heritable. Epigenetic signals acquired during the lifetime are transmitted to the offspring, where they participate to determine substantial changes in phenotype (Fig. 2) (Heard and Martienssen 2014). In the Överkalix study, conducted in northern Sweden to investigate the physiological effects of various environmental factors on transgenerational epigenetic inheritance, three cohorts born in 1890, 1905, and 1920 were studied until death. The study showed that overeating during a child’s slow growth period (SGP), before their prepubertal peak in growth velocity, influences descendants’ risk of death from cardiovascular disease and diabetes (Pembrey et al. 2006). If food was not readily available during the father’s slow growth period, then cardiovascular disease mortality of the proband was low. By contrast, diabetes mortality increased if the paternal grandfather was exposed to a surfeit of food during his slow growth period (Odds Ratio 4.1, 95% confidence interval 1.33–12.93, P = 0.01) (Pembrey et al. 2006). This study was one of the first to demonstrate that a nutrition-linked mechanism through the male line seems to have influenced the risk of developing cardiovascular and metabolic diseases. Periconceptional diet was shown to influence DNA methylation levels with phenotypic consequences. A genome-scale analysis of differential DNA methylation in whole blood after periconceptional exposure to the Dutch Hunger Winter famine revealed changes of differentially methylated regions (Tobi et al. 2014). These changes were preferentially observed at regulatory regions of genes relevant for metabolic homeostasis, inflammation and longevity, key processes underlying the pathogenesis of atherosclerosis. Specifically, individuals conceived during the Dutch famine showed, six decades later, reduced methylation levels at the promoter of insulin-like growth factor type 2 (IGF-2), a gene regulating of glucose homeostasis, cardiovascular function, and lipid metabolism (Heijmans et al. 2008). Overall, these data suggest that epigenetic modulation of pathways by prenatal malnutrition may promote adverse cardiometabolic phenotypes during childhood. Inheritance of specific epigenetic patterns may contribute to explain the alarming increase in childhood obesity from 32 million in 1990 to around 42 million in 2016 (Brown et al. 2015). Notably, early metabolic alterations in obese children are associated with low-grade inflammation, high oxidative stress levels, impaired nitric oxide bioavailability, endothelial dysfunction, and arterial stiffness (Suglia et al. 2018). Macrophage polarization and adipose tissue inflammation are also important features commonly found in obese children (Singer and Lumeng 2017). Epigenetic modulation of inflammation is supported by the notion that promoter methylation of TNF-α, pyruvate dehydrogenase kinase 4 (PDK4), and leptin (LEP) are all reduced in obese as compared to lean children, while methylation of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and proopiomelanocortin (POMC) genes are increased (Garcia-Cardona et al. 2014). As a consequence of these alterations, risk of diabetes, dyslipidemia, and silent heart disease are significantly higher in obese as compared to non-obese children (Juonala et al. 2011). Despite emerging evidence indicates the existence of a transgenerational epigenetic inheritance in humans, data still remain inconclusive due to relevant confounders represented by genetic, ecological, and cultural inheritance. Parents and offspring may share the same epigenomic features, but it is challenging to understand whether these features are being transmitted through the germline or are newly established in each generation by the action of shared genes and shared environments (Horsthemke 2018). The most difficult aspect in humans is the demonstration that a specific epigenetic factor in the germ cells is responsible for the phenotypic effect in the next generation. While this phenomenon was clearly described in plants, nematodes, and fruit flies, its occurrence in humans remains controversial. This important aspect of epigenetic regulation is the object of intense investigation and future studies will contribute to clarify the contribution of inheritable epigenetic traits to global vascular risk in humans.

Epigenetic changes and environmental factors. Exposure to different environmental factors induces several epigenetic modifications. These entail changes of DNA methylation, posttranslational histone modifications, and altered expression of non-coding RNAs. Reproduced with permission from Costantino et al. Eur Heart J. 2018;39:4150–4158
4 Epigenetic Processing in Atherosclerotic Vascular Disease
4.1 DNA Methylation
Reduced DNA methylation is an important signature of atherosclerotic lesions both in humans and in experimental models, such as apolipoprotein E knockout mice (Apoe−/−) and neointima of balloon-denuded New Zealand White (NZW) rabbit aortas (Napoli et al. 2012). Several studies have shown profound changes of atherosclerosis-specific methylated CpGs in human atherosclerotic plaques (hypomethylation of 3,997 promoter sites = 84%) with a progressive increase in gene methylation as the lesions matured (Aavik et al. 2015). Global hypomethylation of DNA in human aortic lesions was the result of a near-complete demethylation of the subset of CpG islands that were hypermethylated in control aortas (Aavik et al. 2019). Beside global variations in methylation status, alterations in the methylation pattern of specific genes have been causally implicated in the pathogenesis of atherosclerosis. Methylation of genes regulating cellular proliferation, namely genes encoding for estrogen receptor-α (ERα) was found in atherosclerotic plaques from human subjects (Fig. 3). Moreover, ERα gene was also shown to be methylated in vascular smooth muscle cells (SMCs) in vitro during their phenotypic switch (Aavik et al. 2019). Methylation-dependent increase in proliferation of SMCs may represent an important epigenetic mechanism underpinning atherogenesis. Indeed – under physiological conditions – arterial SMCs are terminally differentiated and replication does not occur. This associates with a low activity of the epigenetic machinery. By contrast, during lesion development, changes in DNA methylation affect transcriptional programs fostering SMCs proliferation. The effects of DNA methylation may be the direct result of gene hypomethylation or can be due to methylation-induced alterations of DNA integrity and function. DNA hypomethylation of 15-lipoxygenase and extracellular superoxide dismutase (eSOD) genes were found during SMCs replications (Napoli et al. 2012). Epigenetic remodelling of growth regulatory genes such as platelet-derived growth factor and c-myc might also contribute to SMCs proliferation (Hiltunen and Yla-Herttuala 2003). De novo methylation of genes is the most plausible mechanism to explain the dynamic effects of epigenetic processing in atherosclerosis. Intervention studies in cellular models have clearly shown that treatment with the methylation inhibitor 5-azadeoxycytidine heavily affects gene expression in vitro and in vivo (Yang et al. 2010). The importance of de novo methylation is supported by the notion that overexpression of Dnmt, an enzyme which lacks de novo methylation activity, does not reverse genomic hypomethylation in cancer (Zhang and Xu 2017).

Alterations of the chromatin landscape in atherosclerosis. HAT histone methyltransferase, HDAC histone deacetylase, HMT histone methyltransferase, HDM histone demethylase, H3 histone 3, K lysine residue
The accumulation of reactive oxygen species (ROS) is a key promoter of epigenetic signatures. It has been shown that ROS may foster changes in DNA methylation, and in turn, methylation chances may induce the upregulation of pro-oxidant genes. Reduced methylation of CpG islands at the promoter of the shc1 gene, encoding for the mitochondrial adaptor p66Shc, has been associated with high oxidative stress levels, mitochondrial insufficiency, apoptosis, and endothelial dysfunction (Paneni et al. 2012). Both diabetic mice and humans display reduced DNA methylation of shc1 promoter in the vasculature and the heart, and this epigenetic signature persists despite intensive glycemic control (Paneni et al. 2012; Costantino et al. 2017). Interestingly, epigenetic reprogramming by selective targeting of dnmt3b restores promoter methylation thus repressing p66Shc transcription (Costantino et al. 2018a). Other genes implicated in oxidative stress and vascular disease, namely the AP-1 transcription factor JunD and the prolyil-isomerase-1 (Pin1) were also shown to be modulated by specific changes of CpG methylation (Paneni et al. 2013, 2015a). DNA methylation was also reported to affect the expression of inflammatory genes including TNF-α, COX-2, and IL-1β (Stylianou 2019). Genetic deletion of Tet methylcytosine dioxygenase 2 (TET2) in macrophages leads to increased expression of inflammatory cytokines and chemokines in response to native LDL (Peng et al. 2016). A similar increase in plasma interleukin-8 was observed in people with TET2 mutations but not in those without the mutations suggesting that control of DNA methylation by TET2 regulates inflammation in atherosclerosis (Fuster et al. 2017).
Atherosclerosis and ageing are highly interconnected, and age-dependent epigenetic modifications may significantly contribute to vascular disease (Paneni et al. 2017). Aging is generally associated with a progressive decline in global DNA methylation (Hannum et al. 2013). Indeed, DNA methylation content assessed by whole-genome bisulfite is heavily reduced in centenarians as compared with newborns (Heyn et al. 2012). Reduced DNA methylation at the promoter of genes associated with self-renewal was also found in stem cells isolated from aged versus young subjects and associated with functional defects and reduced ability to differentiate (Sun et al. 2014). DNMT3A, TET2, and ASXL1 are key regulators of DNA methylation status and their deregulation is associated with clonal expansion in hematopoiesis and functional defects, a phenomenon known as clonal hematopoiesis of indeterminate potential (CHIP). CHIP was found to be associated with an increased risk of CVD, stroke, coronary calcification, malignancies, and all-cause mortality (Jaiswal et al. 2017). Dnmt3a gene was the most recurrently mutated in individuals with CHIP. Hematopoietic tissue-specific conditional Dnmt3a deletion in mouse models led to a progressive expansion of long-term HSC with impaired differentiation by incomplete epigenetic repression of HSC-specific genes (Challen et al. 2011). Hematopoietic tissue-specific Tet2 loss leads to decreased levels of 5hmC, increased stem cell self-renewal, delayed HSC differentiation, and skewed development toward the monocyte/macrophage lineage (Moran-Crusio et al. 2011). Thus, hematopoietic stressors can induce stable epigenetic reprogramming of bone marrow HSCs contributing to clonal hematopoiesis and myeloid skewing. Albeit DNMT3A and TET2 exert opposite functions in terms of DNA methylation, a recent study showed that these chromatin modifiers both repress the erythroid regulator Klf1 suggesting a model of cooperative inhibition resulting in HSCs transformation (Zhang et al. 2016). Methylation changes at the promoter of eNOS, GATA-2, and GATA-3 during aging were also associated with functional defects and reduced differentiation potential of endothelial progenitor cells (EPCs), whereas reprogramming approaches aimed at restoring promoter methylation of these genes contributed to cell rejuvenation and longevity (Chan et al. 2004). Beside modulating EPCs functionality, DNA methylation directly regulates endothelial cell function mainly via TET2 signaling. TET2 promotes endothelial autophagy and downregulates inflammatory factors such as VCAM1, ICAM1, MCP1, and IL-1β in lesions of high fat fed ApoE−/− mice (Peng et al. 2016). Moreover, TET2 overexpression decreased the methylation levels of Beclin-1 promoter, thus leading to increased endothelial cell autophagy and decreased inflammatory factors in endothelial cells exposed to oxidized LDL (Li et al. 2015). Epigenetic changes of p66Shc and eNOS have also shown to affect endothelial function in vivo as well as in patients with obesity and type 2 diabetes (Costantino et al. 2017, 2019).
4.2 Histone Posttranslational Modifications
Growing evidence indicates that posttranslational modifications of histones, mainly at lysine and arginine residues, significantly affect chromatin accessibility thus enabling cell-specific transcriptional programs implicated in the pathophysiology of atherosclerotic vascular disease (Jiang et al. 2018) (Fig. 3). Chromatin modifications play a pivotal role in regulating vascular inflammation by epigenetic modulation of nuclear factor kappa-B (NF-kB) signaling. Several cardiovascular risk factors have shown to modulate histone marks and gene expression. In primary human aortic endothelial cells, hyperglycemia induces a specific mono-methylation of histone 3 at lysine 4 (H3K4m1) thus enabling chromatin accessibility at the level of NF-kB p65 promoter (El-Osta et al. 2008). Hyperglycemia-related H3K4m1 is induced by the mammalian methyltransferase SETD7, which is emerging as a key player in the regulation of vascular inflammation and oxidative stress (Okabe et al. 2012). Several stimuli, including reactive oxygen species are able to increase SETD7 activity. Indeed, overexpression of scavenger enzymes (e.g., uncoupling protein-1) blunted SETD7-dependent H3K4m1 and subsequent activation of NF-kB p65 and downstream inflammatory genes, namely MCP-1, VCAM-1, and ICAM-1 (El-Osta et al. 2008). Of note, SETD7 expression is increased in peripheral blood monocytes from patients with type 2 diabetes, and correlated with endothelial function and oxidative stress levels, as assessed by flow-mediated dilation (FMD) of the brachial artery and urinary levels of isoPGF2α, respectively (Paneni et al. 2015b). Moreover, SETD7 induction positively correlated with the expression of proatherosclerotic genes including COX-2, iNOS, and VCAM-1 (Paneni et al. 2015b). Chromatin immunoprecipitation experiments revealed that SETD7 specifically binds a specific region of the human NF-kB p65 promoter thus fostering the expression of inflammatory genes. Beside SETD7, other chromatin modifiers such as the methyltransferase Suv39h1 and LSD1 have shown their ability to modulate NF-kB-dependent inflammatory genes (IL-6 and MCP-1) by reducing the levels of dimethylation (H3K9m2) and trimethylation of H3 at lysine 9 (H3K9m3) in SMCs (Brasacchio et al. 2009). Suv39h1 gain- and loss-of-function approaches in experimental models of type 2 diabetes showed that H3K9m2 and H3K9m3 can be dynamically modulated thus reverting the SMCs phenotype. Histone acetyltransferases, such as CBP/p300 and p/CAF, were found to be critically involved in the NF-kB signaling and upregulation of TNF-α and COX-2, master regulators of the atherosclerotic process (Deng et al. 2004). Mechanistically, p300 interacts with the p65 (RELA) subunit of NF-kB in the nucleus to displace repressive p50– HDAC1 interactions (Ashburner et al. 2001). This results in increased acetylation of both p65 itself and histones in proximity of NF-kB promoter. The p65–p300 interaction has been found to be critical for the inflammatory activation of both macrophages and endothelial cells (Jiang et al. 2018). The relevance of this epigenetic route is supported by the notion that inhibition of p300 by curcumin enhances macrophage cholesterol efflux in human and mouse macrophages in vitro, and exerts an anti-inflammatory effect by suppressing NF-kB activity (Khyzha et al. 2017). Deregulation of histone deacetylases, namely sirtuins, also represents a major underpinning of vascular disease and atherosclerosis via modulation of oxidative stress, endothelial function, inflammation, mitochondrial bioenergetics, and cellular metabolism (Winnik et al. 2015). SIRT1 activity is reduced in patients with obesity and significantly contributes to endothelial dysfunction (Masi et al. 2020). By contrast, pharmacological activation of SIRT1 by resveratrol restores endothelial function and NO availability and attenuates dyslipidemia and obesity-induced metabolic alterations in human subjects (Carrizzo et al. 2013). Overexpression of SIRT1 in endothelial cells increases their sprouting and migration, whereas SIRT1 depletion impairs angiogenic response, mainly via FLT1 and CXCR4 (Potente et al. 2007). SIRT1 was also found to regulate the transcription of the mitochondrial adaptor p66Shc, thus leading to oxidative stress and endothelial dysfunction (Zhou et al. 2011; Mohammed et al. 2020). Furthermore, pharmacological activation of SIRT1 by visfatin has shown to attenuate oxLDL-induced senescence of EPCs through the PI3K/Akt/ERK pathway (Ming et al. 2016). SIRT3 and SIRT6 have also recently emerged as an essential regulator of endothelial cell senescence and angiogenesis, key alterations commonly observed in atherosclerosis (Costantino et al. 2018b). Deregulation of chromatin modifying enzymes SUV39H1, JMJD2C, and SRC-1 in visceral fat arteries from obese patients was associated with increased di-(H3K9me2) and trimethylation (H3K9me3) as well as acetylation (H3K9ac) of histone 3 lysine 9 (H3K9) (Costantino et al. 2019). This epigenetic pattern was found to promote transcriptional signatures involved in endothelial ROS generation and vascular dysfunction. Interestingly, in vivo editing of SUV39H1, JMJD2C, and SRC-1 blunted obesity-related endothelial dysfunction in obese mice (Costantino et al. 2019). Other histone deacetylases have shown to affect the atherosclerotic phenotype, although often with conflicting results. HDAC3, HDAC5, and HDAC7 were shown to repress KLF2 expression in human endothelial cells, while their overexpression fostered a proatherogenic phenotype (Kwon et al. 2014). In mice, HDAC3 and HDAC9 deletion in macrophages favors the upregulation of pro-inflammatory signatures eventually leading to a proatherosclerotic phenotype (Stratton et al. 2019). Consistently, Apoe−/− mice with endothelial specific deletion of HDAC3 displayed increased neointimal formation, suggesting an atheroprotective role of endothelial HDAC3 (Zampetaki et al. 2010). Along the same line, Ldlr−/− mice treated with the HDAC inhibitor Trichostatin A exhibited increased plaque size and enhanced macrophage infiltration (Choi et al. 2005). In rats, HDACs are upregulated in aortic SMCs under mitogenic stimulation and their inhibition alters the expression of cell cycle genes, resulting in diminished SMC proliferation, which might be expected to be atheroprotective (Findeisen et al. 2011).
Specific chromatin modifications occurring in macrophages, namely increased activity of the demethylase JMJD3, were also found to act as pivotal regulator of pro-inflammatory cytokines (i.e., TNFα), by erasing the H3K27me3 mark (Yan et al. 2014). Further work is needed to test whether chemical inhibition of JMJD3 has a protective effect in atherosclerotic mouse models. Technologies to probe H3K27me3 and other histone PTMs genome-wide in small samples will be vital to fully understand the cause and consequence of epigenetic changes that occur during atherogenesis.
5 Chromatin Signatures as Epigenetic Biomarkers in Atherosclerosis
Epigenetic signals acquired during the life course can be employed as potential biomarkers of cardiovascular disease. Indeed, modifications of the epigenetic landscape may reflect dynamic changes in expression of genes relevant to vascular homeostasis. A global reduction of DNA methylation was reported in patients with coronary atherosclerosis. The evaluation of the methylation status of LINE-1 due to its high representation in the human genome (55%) and its high methylation levels can be used to evaluate global methylation changes (Aavik et al. 2019). Hypomethylation of the IL-6 promoter, leading to its upregulation and systemic inflammation, was also associated with a higher risk of coronary heart disease (Zuo et al. 2016). The promoter of two atheroprotective genes, namely the estrogen receptor α (ESR1) and estrogen receptor β (ESR2), was hypermethylated in atherosclerotic vascular tissues as well as in cultured vascular SMCs (Napoli et al. 2012). Age-related mutations of key epigenetic regulators including DNMT3A, TET2, and ASXL1 in hematopoietic stem cells (HSCs) is emerging as a major mechanism underlying clonal expansion of HSCs, a phenomenon known as clonal hematopoiesis of indeterminate potential (CHIP) (Li et al. 2018). Notably, the presence of CHIP was associated with an increased risk of CVD and all-cause mortality (Jaiswal et al. 2017). In the northern Sweden population health study, individuals with a history of MI showed differential DNA methylation at 211 CpG-sites representing genes related to CVD, cardiac function, cardiogenesis and recovery after ischemic injury (Rask-Andersen et al. 2016). Hence, epigenetic information may explain the alterations in cardiovascular gene expression trajectories and offer biomarkers for the follow-up of these patients. Overall, available evidence suggests that the epigenome represents a fundamental biological layer in the regulation of gene expression, and epigenetic information may contribute to advance CV risk stratification. The “epigenetic landscape” may provide a real post-genomic snapshot offering the tools to build individual maps of CV risk (Costantino et al. 2018c). Hence, epigenetic changes accumulated during the lifetime may be employed to customize diagnostic and therapeutic approaches in primary and secondary prevention of CVD. Individual epigenetic maps will be invaluable to define new risk scores predicting beyond traditional calculators. This innovative approach will be invaluable for the development of personalized therapies in patients with atherosclerosis (Fig. 4).

Approaches to detect epigenetic biomarkers in patients with atherosclerosis. PBMCs peripheral blood mononuclear cells, WGBS whole-genome bisulfite sequencing, MSREs methylation sensitive restriction enzymes, HPLC high-performance liquid chromatography, ChIP chromatin immunoprecipitation, miRNAs microRNAs, RNA-seq RNA sequencing. Modified from Costantino et al. Eur Heart J. 2018;39:4150-4158
6 Epigenetic Drugs
Targeting epigenetic modifications is a highly promising approach to restore gene expression and to rescue or prevent alterations of vascular phenotype (Masi et al. 2020; Landmesser et al. 2020). There are several examples of how specific interventions can be employed to modify the landscape of DNA/histone modifications (Table 1). Emerging evidence indicates that circulating angiogenic cells (CACs), endothelial progenitor cells (EPCs) and, more in general, bone marrow-derived stem cells are critically involved in vascular repair and maintenance of vascular homeostasis via different mechanisms which include local engraftment and paracrine actions. Epigenetic mechanisms substantially contribute to the functional decline of these cells during life (Costantino et al. 2018b). Indeed, DNA methylation is decreased at the promoter of genes associated with self-renewal, whereas promoters of genes regulating differentiation are generally hypermethylated (Sun et al. 2014). Pharmacological blockade of the DNA methyltransferase by RG108 has shown to rejuvenate BM-derived stem cells by resetting the expression of senescence-related genes (Oh et al. 2015). These findings may open perspectives for the ex vivo epigenetic reprogramming of BM-derived cells for the treatment of atherosclerosis and ischemic heart failure. However, pharmacological editing of global DNA methylation lacks specificity and may be associated with undesirable effects, such as autoimmune diseases (lupus erythematosus was reported in 25–30% of patients) (Chen et al. 2017). The relevance of epigenetic drugs is outlined by the notion that several of these compounds are being tested in clinical trials for the treatment of cancer, neurological and cardiovascular diseases (Costantino et al. 2015). More than 20 years ago, Decitabine (5-aza-2′-deoxycytidine) was found to reactivate ESR1 and ESR2 expression in cancer cells and used to treat myelodysplastic syndromes. However almost 20 years later this drug was reported to blunt inflammation in endothelial cells in a mouse model of atherosclerosis (Dunn et al. 2014). The FDA-approved drug Vorinostat (suberoylanilide hydroxamic acid), a histone deacetylase inhibitor, has shown to prevent eNOS uncoupling, NF-kB signaling, and oxidative stress in experimental diabetes (Advani et al. 2011). Vorinostat also promotes the autophagic flux, a process which is defective in atherosclerosis (Xie et al. 2014). In experimental models of atherosclerosis, the HDAC inhibitor sodium butyrate was shown to blunt NF-kB signaling and inflammatory molecules, namely TNF-α, IL-6, VCAM-1, and ICAM-1 (Hu et al. 2014). Chronic treatment with the SIRT1 activator resveratrol improves endothelial function and insulin sensitivity in patients with obesity and T2D (Pollack and Crandall 2013). Metformin and glucagon-like peptide 1 (GLP-1), widely-used anti-diabetic medications, have also shown to modulate SIRT1 activity, thus affecting histone acetylation and transcription of genes implicated in beta cell function and preservation of insulin signaling (Costantino et al. 2015). The histone acetyltransferase p300 inhibitor curcumin has shown to reduce lipopolysaccharide-induced expression of key proatherogenic cytokines such as MCP-1, IL-1β, and TNF-α in primary human monocytes as well as to reduce macrophage polarization (Moss and Ramji 2016). Chronic treatment (4 months) with curcumin reduced atherosclerotic lesion size in ApoE and LDLr double knockout mice (Hasan et al. 2014). Moreover, a randomized double-blind trial involving 240 individuals with T2D reported a decrease in CVD risk with 6 months of curcumin dietary supplementation, exemplified through a lower pulse wave velocity and improved metabolic profile (Chuengsamarn et al. 2014). In T2D patients, chronic supplementation with curcumin (1–2 months) ameliorated proteinuria, reduced pro-fibrotic cytokines (i.e., TGF-β and IL-8), and improved microangiopathy (Khajehdehi et al. 2011). Current limitations in the use of curcumin include its poor bioavailability due to a very rapid excretion from the body (Lopresti 2018). The use of liposomal curcumin, nanoparticles, and a curcumin phospholipid complex represents current strategies to boost the clinical applicability of this compound. Several other compounds including folates, apicidin, PPARγ agonists, and valproic acid have shown the ability to revert chromatin modifications in cardiometabolic states (Napoli et al. 2012). Recent evidence indicates that apabetalone (RVX-208) – an epigenetic regulator targeting bromodomain and extra-terminal (BET) proteins – stimulates reverse cholesterol transport by inducing ApoAI expression and increasing HDL levels (Ghosh et al. 2017). This drug has also shown to decrease systemic inflammation in humans, as assessed by C-reactive protein as well as to act as an anti-thrombotic agent (Schooling and Zhao 2019). Pooled analysis of short-term non-randomized studies showed fewer cardiovascular events among patients treated with apabetalone as compared to placebo (Nicholls et al. 2018). The recent phase III BETonMACE trial, designed to investigate the impact of apabetalone on cardiovascular outcomes in 2425 patients with diabetes after an ACS, failed to meet the primary endpoint (cardiovascular death, non-fatal myocardial infarction, or stroke) (Ray et al. 2020). However, the drug showed to significantly impact on secondary endpoints. Undoubtedly, larger clinical trials are needed to better explore the safety and efficacy of apabetalone for the treatment of cardiovascular disease.
7 Conclusions
Evidence discussed here suggests that epigenetic processing plays a central role in the pathogenesis of atherosclerosis. Specific epigenetic patterns, characterized by modifications of DNA/histone complexes as well as by ncRNAs, have shown to promote transcriptional programs involved in inflammation, oxidative stress, and endothelial dysfunction. Plastic changes may also contribute to the phenotypic switch of SMCs, a key factor involved in the pathophysiology of atherosclerosis. A careful analysis of the individual epigenetic landscape may furnish novel biomarkers of atherosclerotic vascular disease. An important challenge is the demonstration that epigenetic signals are causally linked to vascular disease phenotypes. The causality of presumed epigenetic events and atherosclerotic phenotypes can be assessed by using different strategies. One possibility would be using longitudinal studies whereby the epigenome can be profiled before the onset of the disease and during follow-up. Alternatively, specific tools, such as the framework of Mendelian randomization which explores the causal link between exposure, epigenetic marks and outcome, can be employed to discriminate between epigenetic phenomena and epi-phenomena. Ongoing epigenomic studies will help to unveil the complex relationship among genetics, epigenetics, and CVD, and will contribute to characterize the incremental value of epigenetic information over established clinical and genetic scores. Epigenetic information can also be used to develop new mechanism-based therapies. The growing understanding of chromatin architecture and metabolism has led to the design of specific molecules able to modulate chromatin accessibility by enhancing or repressing epigenetic marks on DNA/histone complexes. Noteworthy, some of these drugs have been already approved for the treatment of several conditions including cancer, neurological and cardiovascular disease. Taken together, epigenetic information could advance individualized risk assessment and personalized therapeutic approaches in patients with cardiometabolic disturbances.
References
Aavik E, Lumivuori H, Leppanen O, Wirth T, Hakkinen SK, Brasen JH, Beschorner U, Zeller T, Braspenning M, van Criekinge W, Makinen K, Yla-Herttuala S (2015) Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster. Eur Heart J 36(16):993–1000
Aavik E, Babu M, Yla-Herttuala S (2019) DNA methylation processes in atheosclerotic plaque. Atherosclerosis 281:168–179
Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, Yuen DA, Connelly KA, Marsden PA, Gilbert RE (2011) Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol 178(5):2205–2214
Ashburner BP, Westerheide SD, Baldwin AS Jr (2001) The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol 21(20):7065–7077
Baccarelli A, Ghosh S (2012) Environmental exposures, epigenetics and cardiovascular disease. Curr Opin Clin Nutr Metab Care 15(4):323–329
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395
Bauer AJ, Martin KA (2017) Coordinating regulation of gene expression in cardiovascular disease: interactions between chromatin modifiers and transcription factors. Front Cardiovasc Med 4:19
Bernstein BE, Meissner A, Lander ES (2007) The mammalian epigenome. Cell 128(4):669–681
Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317(5845):1760–1764
Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, Calkin AC, Brownlee M, Cooper ME, El-Osta A (2009) Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58(5):1229–1236
Brown CL, Halvorson EE, Cohen GM, Lazorick S, Skelton JA (2015) Addressing childhood obesity: opportunities for prevention. Pediatr Clin North Am 62(5):1241–1261
Brunet A, Berger SL (2014) Epigenetics of aging and aging-related disease. J Gerontol A Biol Sci Med Sci 69(Suppl 1):S17–S20
Carrizzo A, Puca A, Damato A, Marino M, Franco E, Pompeo F, Traficante A, Civitillo F, Santini L, Trimarco V, Vecchione C (2013) Resveratrol improves vascular function in patients with hypertension and dyslipidemia by modulating NO metabolism. Hypertension 62(2):359–366
Carrozza MJ, Utley RT, Workman JL, Cote J (2003) The diverse functions of histone acetyltransferase complexes. Trends Genet 19(6):321–329
Cedar H, Bergman Y (2009) Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10(5):295–304
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, Liang S, Lu Y, Darlington GJ, Meissner A, Issa JP, Godley LA, Li W, Goodell MA (2011) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44(1):23–31
Chan Y, Fish JE, D’Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM, Marsden PA (2004) The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem 279(33):35087–35100
Chen SH, Lv QL, Hu L, Peng MJ, Wang GH, Sun B (2017) DNA methylation alterations in the pathogenesis of lupus. Clin Exp Immunol 187(2):185–192
Choi JH, Nam KH, Kim J, Baek MW, Park JE, Park HY, Kwon HJ, Kwon OS, Kim DY, Oh GT (2005) Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 25(11):2404–2409
Chuengsamarn S, Rattanamongkolgul S, Phonrat B, Tungtrongchitr R, Jirawatnotai S (2014) Reduction of atherogenic risk in patients with type 2 diabetes by curcuminoid extract: a randomized controlled trial. J Nutr Biochem 25(2):144–150
Cooper ME, El-Osta A (2010) Epigenetics: mechanisms and implications for diabetic complications. Circ Res 107(12):1403–1413
Costantino S, Paneni F, Cosentino F (2015) Targeting chromatin remodeling to prevent cardiovascular disease in diabetes. Curr Pharm Biotechnol 16(6):531–543
Costantino S, Paneni F, Battista R, Castello L, Capretti G, Chiandotto S, Tanese L, Russo G, Pitocco D, Lanza GA, Volpe M, Luscher TF, Cosentino F (2017) Impact of glycemic variability on chromatin remodeling, oxidative stress, and endothelial dysfunction in patients with type 2 diabetes and with target HbA1c levels. Diabetes 66(9):2472–2482
Costantino S, Paneni F, Mitchell K, Mohammed SA, Hussain S, Gkolfos C, Berrino L, Volpe M, Schwarzwald C, Luscher TF, Cosentino F (2018a) Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66(Shc). Int J Cardiol 268:179–186
Costantino S, Camici GG, Mohammed SA, Volpe M, Luscher TF, Paneni F (2018b) Epigenetics and cardiovascular regenerative medicine in the elderly. Int J Cardiol 250:207–214
Costantino S, Libby P, Kishore R, Tardif JC, El-Osta A, Paneni F (2018c) Epigenetics and precision medicine in cardiovascular patients: from basic concepts to the clinical arena. Eur Heart J 39(47):4150–4158
Costantino S, Paneni F, Virdis A, Hussain S, Mohammed SA, Capretti G, Akhmedov A, Dalgaard K, Chiandotto S, Pospisilik JA, Jenuwein T, Giorgio M, Volpe M, Taddei S, Luscher TF, Cosentino F (2019) Interplay among H3K9-editing enzymes SUV39H1, JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative stress in obesity. Eur Heart J 40(4):383–391
Davis FM, Gallagher KA (2019) Epigenetic mechanisms in monocytes/macrophages regulate inflammation in cardiometabolic and vascular disease. Arterioscler Thromb Vasc Biol 39(4):623–634
Deng WG, Zhu Y, Wu KK (2004) Role of p300 and PCAF in regulating cyclooxygenase-2 promoter activation by inflammatory mediators. Blood 103(6):2135–2142
Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, Jang I, Son DJ, Kim D, Pan C, Fan Y, Jordan IK, Jo H (2014) Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest 124(7):3187–3199
Elia L, Condorelli G (2019) The involvement of epigenetics in vascular disease development. Int J Biochem Cell Biol 107:27–31
El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M (2008) Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 205(10):2409–2417
Filippakopoulos P, Knapp S (2014) Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov 13(5):337–356
Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D, Bruemmer D (2011) Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol 31(4):851–860
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102(30):10604–10609
Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA, Walsh K (2017) Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355(6327):842–847
Garcia-Cardona MC, Huang F, Garcia-Vivas JM, Lopez-Camarillo C, Del Rio Navarro BE, Navarro Olivos E, Hong-Chong E, Bolanos-Jimenez F, Marchat LA (2014) DNA methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int J Obes 38(11):1457–1465
Ghosh GC, Bhadra R, Ghosh RK, Banerjee K, Gupta A (2017) RVX 208: a novel BET protein inhibitor, role as an inducer of apo A-I/HDL and beyond. Cardiovasc Ther 35(4):e12265
Gillette TG, Hill JA (2015) Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ Res 116(7):1245–1253
Goldberg AD, Allis CD, Bernstein E (2007) Epigenetics: a landscape takes shape. Cell 128(4):635–638
Gonzalez-Recio O, Toro MA, Bach A (2015) Past, present, and future of epigenetics applied to livestock breeding. Front Genet 6:305
Greco CM, Kunderfranco P, Rubino M, Larcher V, Carullo P, Anselmo A, Kurz K, Carell T, Angius A, Latronico MV, Papait R, Condorelli G (2016) DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy. Nat Commun 7:12418
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10(1):32–42
Hamidi T, Singh AK, Chen T (2015) Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 7(2):247–265
Handy DE, Castro R, Loscalzo J (2011) Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation 123(19):2145–2156
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K (2013) Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 49(2):359–367
Hasan ST, Zingg JM, Kwan P, Noble T, Smith D, Meydani M (2014) Curcumin modulation of high fat diet-induced atherosclerosis and steatohepatosis in LDL receptor deficient mice. Atherosclerosis 232(1):40–51
Heard E, Martienssen RA (2014) Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157(1):95–109
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 105(44):17046–17049
Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, Puca AA, Sayols S, Pujana MA, Serra-Musach J, Iglesias-Platas I, Formiga F, Fernandez AF, Fraga MF, Heath SC, Valencia A, Gut IG, Wang J, Esteller M (2012) Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A 109(26):10522–10527
Hiltunen MO, Yla-Herttuala S (2003) DNA methylation, smooth muscle cells, and atherogenesis. Arterioscler Thromb Vasc Biol 23(10):1750–1753
Horsthemke B (2018) A critical view on transgenerational epigenetic inheritance in humans. Nat Commun 9(1):2973
Hu X, Zhang K, Xu C, Chen Z, Jiang H (2014) Anti-inflammatory effect of sodium butyrate preconditioning during myocardial ischemia/reperfusion. Exp Ther Med 8(1):229–232
Izquierdo AG, Crujeiras AB (2019) Role of epigenomic mechanisms in the onset and management of insulin resistance. Rev Endocr Metab Disord 20(1):89–102
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL (2017) Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 377(2):111–121
Jiang W, Agrawal DK, Boosani CS (2018) Cellspecific histone modifications in atherosclerosis (review). Mol Med Rep 18(2):1215–1224
Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19(2):187–191
Juonala M, Magnussen CG, Berenson GS, Venn A, Burns TL, Sabin MA, Srinivasan SR, Daniels SR, Davis PH, Chen W, Sun C, Cheung M, Viikari JS, Dwyer T, Raitakari OT (2011) Childhood adiposity, adult adiposity, and cardiovascular risk factors. N Engl J Med 365(20):1876–1885
Khajehdehi P, Pakfetrat M, Javidnia K, Azad F, Malekmakan L, Nasab MH, Dehghanzadeh G (2011) Oral supplementation of turmeric attenuates proteinuria, transforming growth factor-beta and interleukin-8 levels in patients with overt type 2 diabetic nephropathy: a randomized, double-blind and placebo-controlled study. Scand J Urol Nephrol 45(5):365–370
Khyzha N, Alizada A, Wilson MD, Fish JE (2017) Epigenetics of atherosclerosis: emerging mechanisms and methods. Trends Mol Med 23(4):332–347
Kohli RM, Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502(7472):472–479
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705
Kwon IS, Wang W, Xu S, Jin ZG (2014) Histone deacetylase 5 interacts with Kruppel-like factor 2 and inhibits its transcriptional activity in endothelium. Cardiovasc Res 104(1):127–137
Landmesser U, Poller W, Tsimikas S, Most P, Paneni F, Luscher TF (2020) From traditional pharmacological towards nucleic acid-based therapies for cardiovascular diseases. Eur Heart J. https://doi.org/10.1093/eurheartj/ehaa229
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128(4):707–719
Li G, Peng J, Liu Y, Li X, Yang Q, Li Y, Tang Z, Wang Z, Jiang Z, Wei D (2015) Oxidized low-density lipoprotein inhibits THP-1-derived macrophage autophagy via TET2 down-regulation. Lipids 50(2):177–183
Li F, Wu X, Zhou Q, Zhu DW (2018) Clonal hematopoiesis of indeterminate potential (CHIP): a potential contributor to atherlosclerotic cardio/cerebro-vascular diseases? Genes Dis 5(2):75–76
Lopresti AL (2018) The problem of curcumin and its bioavailability: could its gastrointestinal influence contribute to its overall health-enhancing effects? Adv Nutr 9(1):41–50
Masi S, Ambrosini S, Mohammed SA, Sciarretta S, Luscher TF, Paneni F, Costantino S (2020) Epigenetic remodeling in obesity-related vascular disease. Antioxid Redox Signal. https://doi.org/10.1089/ars.2020.8040
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, Turecki G, Delaney A, Varhol R, Thiessen N, Shchors K, Heine VM, Rowitch DH, Xing X, Fiore C, Schillebeeckx M, Jones SJ, Haussler D, Marra MA, Hirst M, Wang T, Costello JF (2010) Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466(7303):253–257
Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437(7057):436–439
Ming GF, Tang YJ, Hu K, Chen Y, Huang WH, Xiao J (2016) Visfatin attenuates the ox-LDL-induced senescence of endothelial progenitor cells by upregulating SIRT1 expression through the PI3K/Akt/ERK pathway. Int J Mol Med 38(2):643–649
Mohammed SA, Ambrosini S, Luscher T, Paneni F, Costantino S (2020) Epigenetic control of mitochondrial function in the vasculature. Front Cardiovasc Med 7:28
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, Perna F, Pandey S, Madzo J, Song C, Dai Q, He C, Ibrahim S, Beran M, Zavadil J, Nimer SD, Melnick A, Godley LA, Aifantis I, Levine RL (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20(1):11–24
Moss JW, Ramji DP (2016) Nutraceutical therapies for atherosclerosis. Nat Rev Cardiol 13(9):513–532
Napoli C, Crudele V, Soricelli A, Al-Omran M, Vitale N, Infante T, Mancini FP (2012) Primary prevention of atherosclerosis: a clinical challenge for the reversal of epigenetic mechanisms? Circulation 125(19):2363–2373
Nicholls SJ, Ray KK, Johansson JO, Gordon A, Sweeney M, Halliday C, Kulikowski E, Wong N, Kim SW, Schwartz GG (2018) Selective BET protein inhibition with apabetalone and cardiovascular events: a pooled analysis of trials in patients with coronary artery disease. Am J Cardiovasc Drugs 18(2):109–115
Nicorescu I, Dallinga GM, de Winther MPJ, Stroes ESG, Bahjat M (2019) Potential epigenetic therapeutics for atherosclerosis treatment. Atherosclerosis 281:189–197
Noble D (2015) Conrad Waddington and the origin of epigenetics. J Exp Biol 218(Pt 6):816–818
Oh YS, Jeong SG, Cho GW (2015) Anti-senescence effects of DNA methyltransferase inhibitor RG108 in human bone marrow mesenchymal stromal cells. Biotechnol Appl Biochem 62(5):583–590
Okabe J, Orlowski C, Balcerczyk A, Tikellis C, Thomas MC, Cooper ME, El-Osta A (2012) Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ Res 110(8):1067–1076
Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99(3):247–257
Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, Luscher TF, Cosentino F (2012) Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res 111(3):278–289
Paneni F, Osto E, Costantino S, Mateescu B, Briand S, Coppolino G, Perna E, Mocharla P, Akhmedov A, Kubant R, Rohrer L, Malinski T, Camici GG, Matter CM, Mechta-Grigoriou F, Volpe M, Luscher TF, Cosentino F (2013) Deletion of the activated protein-1 transcription factor JunD induces oxidative stress and accelerates age-related endothelial dysfunction. Circulation 127(11):1229–1240. e1221-1221
Paneni F, Costantino S, Castello L, Battista R, Capretti G, Chiandotto S, D’Amario D, Scavone G, Villano A, Rustighi A, Crea F, Pitocco D, Lanza G, Volpe M, Del Sal G, Luscher TF, Cosentino F (2015a) Targeting prolyl-isomerase Pin1 prevents mitochondrial oxidative stress and vascular dysfunction: insights in patients with diabetes. Eur Heart J 36(13):817–828
Paneni F, Costantino S, Battista R, Castello L, Capretti G, Chiandotto S, Scavone G, Villano A, Pitocco D, Lanza G, Volpe M, Luscher TF, Cosentino F (2015b) Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Genet 8(1):150–158
Paneni F, Diaz Canestro C, Libby P, Luscher TF, Camici GG (2017) The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol 69(15):1952–1967
Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjostrom M, Golding J, Team AS (2006) Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet 14(2):159–166
Peng J, Yang Q, Li AF, Li RQ, Wang Z, Liu LS, Ren Z, Zheng XL, Tang XQ, Li GH, Tang ZH, Jiang ZS, Wei DH (2016) Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE−/− mice. Oncotarget 7(47):76423–76436
Pollack RM, Crandall JP (2013) Resveratrol: therapeutic potential for improving cardiometabolic health. Am J Hypertens 26(11):1260–1268
Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, Zeiher AM, Dimmeler S (2007) SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev 21(20):2644–2658
Prasher D, Greenway SC, Singh RB (2019) The impact of epigenetics on cardiovascular disease. Biochem Cell Biol 98(1):12–22. https://doi.org/10.1139/bcb-2019-0045
Rask-Andersen M, Martinsson D, Ahsan M, Enroth S, Ek WE, Gyllensten U, Johansson A (2016) Epigenome-wide association study reveals differential DNA methylation in individuals with a history of myocardial infarction. Hum Mol Genet 25(21):4739–4748
Ray KK, Nicholls SJ, Buhr KA, Ginsberg HN, Johansson JO, Kalantar-Zadeh K, Kulikowski E, Toth PP, Wong N, Sweeney M, Schwartz GG, Investigators BE Committees (2020) Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: a randomized clinical trial. JAMA 323(16):1565–1573. https://doi.org/10.1001/jama.2020.3308
Schooling CM, Zhao JV (2019) How might bromodomain and extra-terminal (BET) inhibitors operate in cardiovascular disease? Am J Cardiovasc Drugs 19(2):107–111
Sen P, Shah PP, Nativio R, Berger SL (2016) Epigenetic mechanisms of longevity and aging. Cell 166(4):822–839
Singer K, Lumeng CN (2017) The initiation of metabolic inflammation in childhood obesity. J Clin Invest 127(1):65–73
Slack JM (2002) Conrad Hal Waddington: the last renaissance biologist? Nat Rev Genet 3(11):889–895
Stratton MS, Farina FM, Elia L (2019) Epigenetics and vascular diseases. J Mol Cell Cardiol 133:148–163
Stylianou E (2019) Epigenetics of chronic inflammatory diseases. J Inflamm Res 12:1–14
Suglia SF, Koenen KC, Boynton-Jarrett R, Chan PS, Clark CJ, Danese A, Faith MS, Goldstein BI, Hayman LL, Isasi CR, Pratt CA, Slopen N, Sumner JA, Turer A, Turer CB, Zachariah JP, American Heart Association Council on E, Prevention, Council on Cardiovascular Disease in the Y, Council on Functional G, Translational B, Council on C, Stroke N, Council on Quality of C, Outcomes R (2018) Childhood and adolescent adversity and cardiometabolic outcomes: a scientific statement from the American Heart Association. Circulation 137(5):e15–e28
Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, Gu H, Bock C, Meissner A, Gottgens B, Darlington GJ, Li W, Goodell MA (2014) Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14(5):673–688
Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, Slieker RC, Stok AP, Thijssen PE, Muller F, van Zwet EW, Bock C, Meissner A, Lumey LH, Eline Slagboom P, Heijmans BT (2014) DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun 5:5592
Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439(7078):811–816
Vilkaitis G, Suetake I, Klimasauskas S, Tajima S (2005) Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J Biol Chem 280(1):64–72
Winnik S, Auwerx J, Sinclair DA, Matter CM (2015) Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J 36(48):3404–3412
Wolffe AP, Jones PL, Wade PA (1999) DNA demethylation. Proc Natl Acad Sci U S A 96(11):5894–5896
Wu SC, Zhang Y (2010) Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 11(9):607–620
Xiao FH, Wang HT, Kong QP (2019) Dynamic DNA methylation during aging: a “prophet” of age-related outcomes. Front Genet 10:107
Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, Jiang N, Jessen ME, Warner JJ, Lavandero S, Gillette TG, Turer AT, Hill JA (2014) Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129(10):1139–1151
Xu S, Pelisek J, Jin ZG (2018) Atherosclerosis is an epigenetic disease. Trends Endocrinol Metab 29(11):739–742
Yan Q, Sun L, Zhu Z, Wang L, Li S, Ye RD (2014) Jmjd3-mediated epigenetic regulation of inflammatory cytokine gene expression in serum amyloid A-stimulated macrophages. Cell Signal 26(9):1783–1791
Yang X, Lay F, Han H, Jones PA (2010) Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci 31(11):536–546
Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, Pepe AE, Wang G, Habi O, deFalco E, Cockerill G, Mason JC, Hu Y, Xu Q (2010) Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation 121(1):132–142
Zhang W, Xu J (2017) DNA methyltransferases and their roles in tumorigenesis. Biomark Res 5:1
Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, Guzman A, Lei Y, Huang YH, Rao A, Li W, Goodell MA (2016) DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet 48(9):1014–1023
Zhang W, Song M, Qu J, Liu GH (2018) Epigenetic modifications in cardiovascular aging and diseases. Circ Res 123(7):773–786
Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, Huang S, Liu JJ, Lu YB, Zhang ZQ, Yang RF, Zhang R, Cai H, Liu DP, Liang CC (2011) Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res 109(6):639–648
Zuo HP, Guo YY, Che L, Wu XZ (2016) Hypomethylation of interleukin-6 promoter is associated with the risk of coronary heart disease. Arq Bras Cardiol 107(2):131–136
Acknowledgments
F.P. is the recipient of a H.H. Sheikh Khalifa bin Hamad Al Thani Foundation Assistant Professorship at the Faculty of Medicine, University of Zurich. The present work is supported by the Zürich Heart House, the Swiss Heart Foundation, Swiss Life Foundation, Kurt und Senta-Hermann Stiftung, the EMDO Stiftung, the Olga Mayenfisch Stiftung and the Schweizerische Diabetes-Stiftung to F.P; the Holcim Foundation and the Swiss Heart Foundation to S.C
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this chapter

Cite this chapter
Costantino, S., Paneni, F. (2020). The Epigenome in Atherosclerosis. In: von Eckardstein, A., Binder, C.J. (eds) Prevention and Treatment of Atherosclerosis . Handbook of Experimental Pharmacology, vol 270. Springer, Cham. https://doi.org/10.1007/164_2020_422
Download citation
DOI: https://doi.org/10.1007/164_2020_422
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-86075-2
Online ISBN: 978-3-030-86076-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)