
Abstract
The mechanisms responsible for vascular disease development have been investigated for many decades, but we are far from a complete identification of all involved molecular processes. This still remains a major unmet need and despite significant improvements in diagnosis, prevention, and early intervention, cardiovascular pathologies are still the leading cause of death and disability worldwide. Epigenetics guides gene expression through the regulation of transcription independently of the genetic code. Those regulatory mechanisms are essential to numerous processes, such as cell growth, development, and differentiation, and they might depend on environmental adaptation, aging, and disease states. The current knowledge on the epigenetic mechanisms regulating vascular physiopathology has uncovered new potential targets for intervention. Herein, we provide an overview of the epigenetic landscape and its role in vascular diseases, highlighting the impact of DNA methylation and histone modification as well as non-coding RNA mechanisms.
Ignacio Fernando Hall, Montserrat Climent, Floriana Maria Farina have been equally contribute to this work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- DNA methylation
- Histone modification
- Non-coding RNAs
- Atherosclerosis
- Vascular diseases
- Epigenetics
- Transcription
- Gene expression
1 Introduction: The Constant Burden of Cardiovascular Disease
All vasculature-associated diseases, including atherosclerosis and pulmonary hypertension, belong to the vast category of cardiovascular diseases (CVDs). CVDs are the principal cause of death and disability worldwide [1], and are indeed responsible for 31% of global deaths (https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)). Over the last decade, progresses in diagnosis, prevention, and treatment have considerably improved overall survival. However, the number of people diagnosed with CVDs remains very constant worldwide [2]. This fact already triggering some innovative ideas many years ago, suggesting that not all cases of CVD morbidity could be explained by common risk factors, such as blood lipids, hypertension, and diabetes [3]. Nonetheless, the probability of developing vasculature-associated diseases might depend, besides the environmental risk factors cited above, also on genetic variations, which should not to be confused with genetic mutations, such as those observed in pathologies like familial hypercholesterolemia [4]. Among those variations are the single nucleotide polymorphisms (SNPs), whose association with vascular pathologies is a complex challenge that must be integrated with specific sets of risk factors. However, all this information is not sufficient to explain the great penetrance of vasculature-associated diseases in the global population; additional elements need to be included in the global picture. The first theorization of such innovative events was proposed in the middle of the twentieth century by the British developmental biologist Conrad Waddington. Indeed, with no idea of the mechanisms, he coined a new term: epigenetic landscape [5]. Waddington proposed the existence of biological phenomena on top of genetic variability, as essential modulators of cell fate determination [6].
In eukaryotic organisms, organ development is tightly regulated by coordinated steps of gene expression activation and repression that follow precise time and space events in cells sharing the same DNA sequence [7]. This phenomenon modulates the extreme level of folding of chromatin needed to fit chromosomes into the nucleus and also the changes necessary so that genes can be either accessible to regulator elements and be transcribed (areas called euchromatin) or tightly packed and inactive (heterochromatin) [8]. Transformation of euchromatin to heterochromatin and vice versa contributes to gene regulation and defines today’s meaning of the term epigenetics, which is more complex compared to Waddington’s definition: epigenetics now refers to those heritable changes that, rather than depending on changes of the DNA sequence, influence how chromatin structure affects gene expression. Those are principally based on chemical modification of DNA—in particular, on cytosine—and histone proteins; more recently, processes regulated by non-coding RNAs have also been added to those classified as epigenetic mechanisms [9, 10].
Although the understanding of the epigenetic mechanisms associated with vasculature-associated diseases is still in its infancy, it is advancing very fast due to great improvements in DNA sequencing capacity. Thus, today we foresee that the knowledge generated by studying the role of epigenetics in vasculature-associated diseases may be soon translated into new therapeutic approaches [11, 12]. The concept of epigenetics is quite intriguing for the field of human epidemiology. As a plethora of studies has assessed the central role of epigenetic modifications in human disease etiology, it is becoming evident how epigenetics might link environmental factors and lifestyle to pathology onset and progression [13]. In light of the crucial role of epigenetics, the study of its epidemiology permits a better identification of the interindividual variables impacting differential gene expression and disease susceptibility, augmenting the armamentarium of physicians and researchers working in several scientific fields, including the cardiovascular one [14].
In this chapter, we give an overview of the role of epigenetics as the main non-genetic component of vascular disease risk, focusing on the chemical alteration of DNA and histones and the activity of non-coding RNAs in the development of these pathologies.
1.1 Etiology and Pathobiology of Vascular Diseases
Vasculature-associated diseases comprise a wide variety of conditions affecting primarily blood vessels, including atherosclerosis, restenosis, aneurysm, and different hypertensive syndromes, such as pulmonary artery hypertension (PAH), among others [15].
While the specific pathophysiologic processes underpinning injury development might differ from one pathology to another, they still share common genetic and non-genetic molecular determinants [16]. Disease initiation and progression are defined by alterations in the transcriptional program of the cells residing within the vessel wall—namely endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and fibroblasts—and the impaired communication with inflammatory cells recruited to the site of injury. Indeed, sustained exposure to stressors fosters vascular remodeling through the establishment of disease-prone cellular phenotypes, by disrupting intra- and inter-cellular responses [11].
EC de-regulation and activation leads to endothelial dysfunction and deterioration [17]. Similarly, VSMCs undergo profound phenotypic changes, transitioning towards a synthetic, non-contractile state while gaining migratory properties along with excessive extracellular matrix (ECM) deposition and alteration of the apoptotic program [18]. Furthermore, immune cell (mainly monocytes/macrophages) differentiation, activation, and polarization within vascular lesions nurture disease progression [19].
Of note, several lines of research have elucidated that the impact of VSMC plasticity on vascular disease might vary in a context-dependent fashion. As such, VSMC proliferation and migration can be harmful or beneficial, whereas apoptosis, senescence, and switching to a more macrophage-like phenotype can promote inflammation and disease progression [20].
To further complicate matters, these pathologies can, in turn, lead to secondary injures, such as heart failure, myocardial infarction, and ventricular hypertrophy. Additionally, their progression might be hastened by complications such as diabetes and metabolic disorders [21].
Here, we will discuss the role of epigenetics in two specific pathologies: atherosclerosis and PAH.
1.2 Atherosclerosis
Atherosclerosis is the main clinical manifestation of CVDs and underlies the majority of cardiovascular complications, remaining the leading cause of morbidity and mortality worldwide [22, 23]. It is a chronic inflammatory disease that preferentially develops in specific points of vessels, known as predisposition sites, such as the branching points of large arterial trees and the inner curvature of the aortic arch [24].
Atherogenesis onset is characterized by the infiltration in the arterial wall of low-density lipoproteins (LDLs), which are then retained in the endothelium and oxidized. These oxidized LDLs (oxLDLs) trigger endothelial dysfunction at atheroprone sites, impairing laminar flow, promoting inflammatory cell infiltration and adhesion, and rearranging VSMC phenotype, eventually leading to arterial wall thickening, narrowing of the vascular lumen, accumulation of lipids, and the formation of plaques [23, 25]. Lesion progression can trigger plaque rupture or erosion, causing ischemic events, such as ischemic stroke and myocardial infarction (MI). Indeed, as the atheromatous plaque evolves, it is stabilized by a fibrous cap, but at later stages becomes thinner and susceptible to rupture, resulting in the aforementioned acute events [11, 15].
Additionally, pathologies such as coronary artery disease (CAD) and restenosis display similar or related features, being therefore classified as atherosclerosis-related conditions. More in detail, CAD is a localized form of atherosclerosis affecting coronary vessels. The main treatment option is surgical angioplasty associated with drug-eluting stent placement [26]. However, the efficacy of this solution can be temporary, favoring the formation of in-stent obstructions, called stenosis/restenosis, in the first 6 months post-surgery, as reported in 25–50% of cases [27]. At the cellular level, restenotic obstructions mainly rely on EC activation and the re-activation/de-differentiation of VSMCs that migrate to the site of injury, causing neointimal hyperplasia [28].
1.3 Pulmonary Artery Hypertension
Hypertension is a disorder that depends on complex genetic and environmental factors. PAH is characterized by progressive pulmonary vascular remodeling that gradually leads to narrowing of the vessel, increased pulmonary artery resistance, and imbalance between vasoconstriction and vasodilatation, events that eventually result in right ventricular maladaptive hypertrophy, heart failure, and premature death [29,30,31].
PAH can be classified into different subcategories, including idiopathic PAH, heritable PAH, and PAH associated with other diseases [32]. However, as mentioned above, all types of PAH share common aspects, including PAEC proliferation; PASMC proliferation, migration, and contraction; inflammation; and fibroblast proliferation, activation, and migration [32].
During PAH pathogenesis, vascular remodeling is guided by several molecular processes, with dysregulation evident across the different layers of the vessel wall; such mechanisms include, but are not limited to, a hypoxic microenvironment, growth factor changes, signaling pathway modulation, metabolic imbalance, and epigenetic modifications [33]. At the cellular level, it is characterized by alterations of PAEC biology, leading to a dysfunctional endothelium and endothelial-to-mesenchymal transition (EndMT). Further alterations pertain to apoptosis resistance, phenotype conversion, proliferation, and migration of PASMCs, alongside fibroblast accumulation [34,35,36].
Of note, PASMCs and PAECs are the principal cell types contributing to the pathogenesis of PAH, and their dysregulation is significantly involved in the vascular remodeling occurring during systemic hypertension and PAH, correlating with the detection of specific sub-cellular populations [15].
2 DNA Methylation and Vascular Diseases
Through the chemical modification of nuclear DNA and its interactors, epigenetics translates the influence of the environment into gene expression regulation. Appropriate packaging of genomic DNA within the nucleus is of pivotal importance for proper gene transcription. DNA is tightly organized in wrapped structures, known as chromatin, containing an equal mass of proteins (histones) [37]. DNA rolls up with histone octamers, consisting of two H2A-H2B dimers surrounding H3-H4 dimers, to form nucleosomes [38]. The solid interactions among nuclear DNA, histones, and linker RNA lead to the formation of a nucleoprotein complex: this chromatin can be defined as euchromatin or heterochromatin, depending on its level of compaction [39].
DNA methylation is the best-characterized chemical modification. It is a ubiquitous epigenetic mechanism that occurs when a methyl group derived from S-adenosyl-methionine is bound to position 5 of the cytosine ring, forming 5-methyl-cytosine (5mC) [40]. This heritable modification can occur at cytosine-guanine dinucleotide sites, called CpG islands, and it is commonly associated with repression and gene silencing [41], especially if it is happening at transcription start sites (TSSs), by preventing transcription factors (TFs) from approaching DNA [42]. This modification is commonly catalyzed by the activity of a family of enzymes named DNA methyltransferases (DNMTs) [43]. This family comprises three active members, identified in mammalian cells as DNMT1, DNMT3A, and DNMT3B [43, 44]. Among them, DNMT1 is broadly expressed in mammalian cells and has a role in the maintenance of the mitotic inheritance of methylated DNA [45]. The Ubiquitin-like with PHD and Ring Finger Domains 1 (UHRF1) has a crucial role in this process, favoring DNMT1 in the recognition of hemimethylated DNA [46]. On the other hand, DNMT3A and DNMT3B are the main factors responsible for de novo methylation [47], which commonly occurs in early embryos [48] and at different stages of development [49].
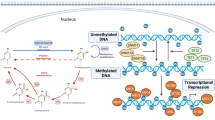
Conversely, the removal of 5mC, also known as DNA demethylation, leads to DNA transcription and can be achieved through passive or active mechanisms. As easily perceivable, passive demethylation occurs due to a lack of activity of DNMT1 during cell division [50]. On the other hand, active demethylation occurs through direct enzymatic removal of the methyl group from 5mC. First described in 2009, it was reported that ten-eleven translocation (TET) methylcytosine dioxygenases family plays a crucial role in DNA demethylation via the enzymatic oxidation of 5mC to generate 5-hydroxymethylcytosine (5hmC) in a Fe(II)- and α-ketoglutarate (α-KG)-dependent manner [51]. 5hmC is a crucial intermediate that is gradually replaced by unmethylated cytosines through several mechanisms. In particular, the oxidation reaction can proceed to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) [52, 53]. Furthermore, the pathway is continued by thymine-DNA glycosylase (TDG), which recognizes and excises 5caC (and also 5fC) from DNA. The latter reaction depends on base excision repair (BER), engaged for completing the DNA demethylation cycle [54] (Fig. 20.1). Although 5hmC has broadly been characterized as an intermediate state of the demethylation process, it emerged to represent a crucial epigenetic mark; increasing data are suggesting that 5hmC may represent a stable epigenetic mark [55]. 5hmC distribution was reported to have a pivotal role in the acquisition of cellular imprinting during cellular differentiation, but of outmost importance, it is associated with the recruitment of TFs, and therefore is entitled to actively regulate gene transcription processes [56, 57].

Illustration of DNA methylation and histone modification effects on gene expression. Top: Mechanisms of DNA methylation and demethylation. During differentiation processes, DNA undergoes different chemical modifications. De novo methylation is mediated primarily by DNMT3A and DNMT3B. Upon DNA replication, newly synthesized strands lack methylation marks, but DNMT1 rapidly restores the correct methylation on a newly synthesized DNA. Adult patterns of methylation are erased by epigenetic mechanism involving TET enzymes. Bottom: The nucleosome is composed of double-stranded DNA wrapped around a core of histone proteins. Modifications such as acetylation and methylation of lysine residues on core histones are mutually exclusive. Acetylation on lysines is always associated with increased gene expression. Lysine methylation is associated with gene activation when found at H3K4, and H4K40, but is associated with gene silencing at H3K27, H3K9, and H4K20
Besides its involvement in physiological processes, the regulation of DNA methylation is one of the crucial epigenetic mechanisms in the development and progression of cancer, where its patterns are globally disrupted [58]. In recent decades, many other pathologies have broadly been related to DNA methylation impairment; among them, cardiovascular pathologies are prominent.
2.1 Atherosclerosis and DNA Methylation
Due to their strong dependence on environmental stimuli, epigenetic modifications have been progressively associated with atherosclerosis development. Thus, they are of pivotal interest for therapeutic and biomarker outcomes.
Decades of studies described that DNA hypomethylation seems to be preponderated during atherosclerosis progression [59, 60]. Nonetheless, focal DNA hypermethylation is now considered of crucial importance in atherosclerosis development [61, 62]. In particular, several reports describe how the regulation of DNA methylation can broadly affect atherosclerosis development and progression by directly targeting all the layers within the vessel, affecting ECs, immune cells, and VSMCs.
EC damage can be considered the first pathological trigger for atherosclerosis development, where oxidative stress, lipid deposition, and transcription of inflammatory mediators play a crucial role [63]. In the context of oxidative stress, the aberrant transcription of Src homology 2 domain-containing protein (p66Shc) contributes to mitochondrial dysfunction, increased apoptosis, and endothelial functional alteration [64]. This pathological effect was prevented by the restoration of DNMT3B activity that resulted in p66Shc repressed transcription [65].
Treatment of ECs in vitro with oxLDL was reported to directly induce DNMT1-mediated methylation of Kruppel-like factor 2 (KLF2), abrogating endothelium-dependent vascular homeostasis, and so inducing a pro-atherogenic phenotype [66]. In addition, it induced increased methylation of the promoter regions of Src homology 2-containing protein tyrosine phosphatase 1 (SHP-1) [67]. Atherogenic stimuli are reported to induce a strong increase in expression of antioxidative enzymes (SOD2, catalase, and GPx) in infiltrating immune cells and migrating VSMCs, via altering 5mC status [68, 69].
Nonetheless, VSMCs were described to display a pro-atherogenic phenotype after SOD2 inhibition mediated by induction of DNA methylation; this effect was completely prevented with 5-azacytidine (5-aza), a very well-known DNMT1 inhibitor [70]. In line with this finding, UHRF1 was found to regulate the pro-atherogenic phenotype of VSMCs, directly cooperating with DNMT1, as a downstream target of platelet growth factor-BB (PDGF-BB) treatment [71].
2.2 Pulmonary Artery Hypertension and DNA Methylation
Epigenetics is involved in the development of PAH [72], so it may be a potential therapeutic target to ameliorate the clinical outcome of this severe disease [73]. Global alteration of DNA methylation status is reported to be clearly linked with PAH development and progression. In particular, the promoter region of the superoxide dismutase (SOD)2 gene was found to be hypermethylated through the activity of DNMT1 and DNMT3B in PASMCs isolated from spontaneously developing PAH (fawn-hooded) rats [74] and plexiform regions of PAH patients [75], overall contributing to the activation of hypoxia-inducible factor (HIF)1-α, and thus inducing a pro-proliferative and apoptosis-resistant state. This pathological phenotype can be abolished with the administration of 5-aza, which blunted DNA methylation on the SOD2 gene at different loci [75].
Furthermore, in PAECs, strong hypermethylation was found at the promoter of several genes and microRNAs involved in lipid metabolism, including ABCA1, ABCB4, ADIPOQ, miR-26A, and BCL2L11. ABCA1 was found to have reduced expression at the mRNA and protein levels, leading to novel therapeutic possibilities [76].
Global DNA methylation level reduction was also observed in fetal lambs exposed to long-term high-altitude hypoxia: this was associated with loss of the cyclin dependent kinase inhibitor (CDKN1A, p21), which caused aberrant PASMC proliferation in these fetuses, leading to PAH in the newborn [77].
Moreover, the presence of differential epigenetic marks was demonstrated between pulmonary venous-occlusive disease (PVOD) patients and PAH patients. This was due to hypermethylation of the granulysin (GNLY) gene, a cytosolic antimicrobial peptide found in the gDNA of explanted lungs and peripheral blood mononuclear cells [78]. This finding is noteworthy due to the lack of knowledge on this pathology, allowing early diagnosis and rapid discrimination of the two groups of patients.
3 The Histone Code and Vascular Diseases
As already described in previous paragraphs, genetic information is finely regulated by environmental stimuli and the accessibility of gene promoters to TFs.
Histones are the key component of chromatin and are rearranged in a spiral secondary structure known as a solenoid [79]. There are four types of histone protein: H2A, H2B, H3, and H4. They share a common structure, composed of a globular domain surrounded by flexible, protruding domains (also known as amino(N)- and carbon(C)-terminal tails). An octamer of dimers of each type of histone forms the main essence of chromatin: the nucleosome [37]. Within the three-dimensional structure of the solenoid, additional H1 histone proteins cooperate in the maintenance of chromosomal stability [80] As first discovered by the epigenetic pioneer Emil Heitz [81], there are two different chromatin states characterized by different accessibility capacities: euchromatin is related to active transcription because of its less compact and more accessible structure; on the other hand, heterochromatin is a tightly compact structure where transcription is prevented.
The protrusion of the N-terminal tails of all histones and of the C-terminal tails of H2A histones permits reversible covalent modifications, known as post-translational modifications (PTM), to control DNA accessibility and to regulate gene transcription [82]. Histone PTMs (hPTMs) include a wide variety of chemical modifications and constitute the so-called “histone code” [83] (Fig. 20.1). More in depth, lysine (K) was the first discovered to be acetylated or methylated [84], but further acylation, biotinylation, crotonylation, formylation, malonylation, sumoylation, and ubiquitination were discovered to collectively regulate conversions on this amino acid [85]. Arginine (R) can be either deiminated (converted to citrulline) or (mono-, di-, and tri-) methylated [86]. Serine (S), threonine (T), and tyrosine (Y) can be phosphorylated [87] and S and T can also be glycosylated [88]. In this complex scenario, hPTMs are the product of the dynamic activity of particular enzymes that deposit chemical modifications (writers), decipher the mark (readers), or proficiently remove the labels (erasers).
Mass spectrometry (MS) was the first approach able to decipher how epigenetic modifications could affect chromatin accessibility and, thus, regulate gene transcription. Taking advantage of the vastly technical benefits of high-throughput technologies, a more precise overview of histone marks has been possible in recent decades. Here, we will discuss the three main hPMTs: acetylation, methylation, and phosphorylation.
Histone Acetylation
Acetylation occurs predominantly on K residues of H3 and H4 histones. Histones exhibit a positive ionic charge that strongly interacts with negative acetyl groups, leading to a weak interaction with DNA, and enhancing chromatin accessibility. Acetylated histones are favorable binding sites for bromodomain-containing proteins (BRDPs), which orchestrate assembly of the transcriptional machinery [89]. Acetylation is predominantly enriched in peculiar regions inside the promoter and at the 5′ side of the coding sequences. Of note, H3 acetylation on K9 and K27 (H3K9ac and H3K27ac) is generally associated with enhancers and promoters of active genes.
Histone deacetylation is mediated by histone deacetylases (HDACs), generating a more compact structure of the nucleosome and, thus, leading to transcription impairment [90]. This mark can be reverted through the action of histone acetyl transferases (HATs; CBP/p300, MYST, and GNAT), which induce chromatin relaxation [90]. Of note, unbalance in the activity of these two families of “switchers” leads to aberrant acetylation patterns and the development of CVDs [91]. Indeed, abnormal histone acetylation is implicated in many diseases, including hypertension [92], PAH [93] and ventricular hypertrophy [94]. Therefore, approaches aimed at manipulating the acetylation/deacetylation balance, preventing an aberrant PTM state, might be an innovative therapeutic strategy for these pathologies. Overall, HDAC inhibitors have demonstrated to be protective agents in the development of several CVDs [95].
Histone Methylation
In contrast to acetylation, which is a clear mark of transcriptional activation, methylation of K and R residues can have different impacts on gene transcription. The most extensively studied histone lysine methylation sites are H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20, although many other methylated lysine residues are present on H1, H2A, H2B, and at other H3 and H4 loci [96].
Histone methylation is mediated by lysine- and arginine-methyltransferases (KMTs, and PRMTs, respectively), taking advantage of S-adenosyl-L-methionine (SAM) as the methyl-donor. These enzymes can add up to three methyl groups, resulting in mono-, di-, or tri-methylation of lysines, and mono- or di-methylation of arginines. Generally, monomethylated lysine improves transcription, whereas the addition of further methyl groups can have a different effect depending on the residue (e.g., H3K4me2–3, H3K36me3, and H3K79me2–3). In addition, high plasticity in the recruitment of reader enzymes induces either gene transcription or repression based on the methylation level at the residue (e.g., H3K4me2 and H3K4me3) [97].
Histone demethylases (HDMs) are the principal effectors of methyl group removal from arginine and lysine residues [98]. This process takes place through the activity of FAD-dependent amine oxidases [99] or FeII and α-KG-dependent dioxygenases [100].
Histone Phosphorylation
Despite being the first modification to be characterized, histone phosphorylation is somewhat less well understood than acetylation and methylation. Like with acetylation, phosphorylation produces a negative charge, reducing the positive charge carried by histones and leading to increased accessibility of chromatin. Histone phosphorylation is transient and dynamically coordinated by kinases and is mainly associated with gene activation mediated by specific TFs [101]. Histone phosphorylation has broadly been related to apoptosis, DNA repair, chromatin rearrangements during cellular division, and gene transcription [102]. Of note, one of the most studied phosphorylation sites is H3S10, which once phosphorylated by Aurora B, recruits enzymes that block H3K9 methylation; HATs then acetylate the lysine residue, further increasing transcriptional activity, in a mechanism known as phosphor-acetylation [103].
3.1 Atherosclerosis and Histone Modifications
Epigenetic de-regulation is a clear and well-known hallmark of atherosclerosis development. Global reduction in H3K27 tri-methylation was observed at a late stage in atherosclerotic plaques. Nevertheless, this was not related to alterations in global levels of the corresponding histone methyltransferase EZH2, the catalytic subunit of the polycomb repressive complex 2 (PRC2). Similarly, neither alterations in BMI1, a PRC1 complex component, which binds to H3K27me3, nor the expression of the histone demethylase JMJD3, which removes the methyl marks on H3K27, were reported [104]. In addition, H3K27me3 reduction was also reported to affect NF-κB activation in human coronary artery ECs, resulting in an increase in inflammatory status due to the aberrant transcription of adhesion molecules and cytokines [105].
G9a, another well-known epigenetic enzyme, is able to operate H3K9 mono- and di-methylation. In hyperhomocysteinemic ApoE−/− mice treated with methionine, G9a was reduced together with H3K9me2, resulting in the promotion of apoptosis in macrophages and increased plaque stability [106].
In VSMCs and macrophages of human carotid arteries, histone hyperacetylation—with a consequent reduction in methylation levels at H3K9 and H3K27—was reported to be a clear mark of atherosclerotic severity [107]. Furthermore, increased HAT-mediated histone acetylation was a potential regulator of VSMC differentiation due to the recruitment of serum response factor (SRF) and its cofactor, myocardin [108], that further mediate the activation of CArG box elements of VSMC-specific marker genes [109]. Moreover, HDAC3 deficiency in myeloid cells was reported to improve plaque stability by increasing collagen deposition, and has been proposed as a macrophage polarization inducer, favoring the alternatively activated macrophage phenotype. These cells secrete TGFβ1, which can be considered a trigger promoting collagen deposition by VSMCs [110].
HDAC inhibitors have been proposed as intriguing suppressors of atherosclerosis development due to transcriptional reduction of a subset of inflammatory genes (e.g., TLR-induced IL-12p40 secretion in dendritic cells). They have been also described to induce other pro-atherogenic genes (e.g., COX-2 expression), suggesting that further efforts are needed to better describe their role in atherosclerosis [111].
Nonetheless, epigenetic mechanisms dynamically arrange gene expression, with the mutual relation between hPTMs and DNA modifications being of outmost importance. In particular, synergistic effects have been reported between DNMT3A/3B and H3K9 or H3K36 methylases (SETDB1, SUV39H1/2, EHMT1/2, and SETD2); in contrast, DNA methylation is completely abrogated by H3K4 mono-, di-, and tri-methylation [112].
In addition, H3K27 methylation (operated by EZH2) stimulated foam cell formation in ApoE−/− mice, improving atherosclerosis development. In particular, EZH2 enrolls DNMT1 and methyl CpG-binding protein-2 (MeCP2). The DNMT1-MeCP2 complex then promotes the methylation of the ATP-binding cassette transporter A1 (ABCA1) promoter, whose inhibition improves atherosclerosis [113].
3.2 Pulmonary Artery Hypertension and Histone Modifications
Over the past decades, increasing evidence has suggested that hPTMs, and especially aberrant histone acetylation, influences PAH development. Of note, epigenetics-based therapies directly targeting histone acetylation could be innovative approaches for the treatment of PAH.
Firstly, EZH2, which operates H3K27-specific methylation, was found upregulated in human PASMCs, regulating proliferation, migration, and anti-apoptotic processes [114].
Transcriptional regulator myocardin-related transcription factor A (MRTF-A) interacts with NF-κB, halting H3K4 methyltransferase on cell adhesion molecule (CAM) promoters in response to hypoxia. This complex then promotes ICAM-1, VCAM-1, and E-selectin transcription, improving leukocyte migration to the pulmonary vascular wall [115].
Moreover, in the monocrotaline (MCT)-induced PAH rat model, the histone methyltransferase nuclear receptor binding SET domain 2 (NSD2) was recently found to be involved in the development of the disease. In particular, NSD2 knockdown reduced H3K36me2, with an effect on pulmonary arterial remodeling through autophagy inhibition, pulmonary artery pressure normalization, and right ventricular hypertrophy reversion [116].
Histone acetylation also plays a critical role in PAH. Idiopathic PAH patients and chronically hypoxic rats had upregulated HDAC1 and HDAC5 in several tissues, including heart, lung, and pulmonary arteries [117]. Increased histone acetylation is further induced by hypoxia and modulates hypoxia-inducible factor-1α (HIF-1α) binding levels in the endothelin-1 (ET-1) gene core promoter region in spontaneously hypertensive rats (SHR) [118]. In line with this, HDAC inhibitors were reported to attenuate hypertensive responses in SHRs [119]. Furthermore, valproic acid (VPA) prevented hypertension development via reduction of mineralocorticoid receptor (MR) transcription by increasing its acetylation [120]. In addition, trichostatin A (TSA), another HDAC inhibitor, effectively reduced blood pressure and vascular inflammation in SHRs [121].
An increased accumulation of HDAC4 and HDAC5 in the PAEC nuclei of PAH patients was reported to impair myocyte enhancer factor 2 (MEF2) activity, inducing connexin 37 (Cx37), connexin 40 (Cx40), KLF2, and KLF4, and resulting in an improvement in cell proliferation [122]. Moreover, PASMCs had increased levels of HDAC3 and HDAC6, regulating SOD3 expression and inhibiting BAX-induced cell death programs, and thus enhancing cell proliferation [123]. Furthermore, a global increase in acetylation was observed in Angiotensin (AII)-treated VSMCs. AII is a strong vasoconstrictor directly responsible for triggering hypertension. AII-treated VSMCs had elevated HDAC5 phosphorylation in a time- and dose-dependent manner. This led to HDAC5 export out of the nucleus, causing a reduction in MEF2 transcription [124].
4 ncRNAs and Vascular Diseases
Once acknowledged that only approximately 2% of the mammalian genome possesses a coding potential, the past decades have seen increased interest in non-coding transcripts. Initially considered to be artifacts, these molecules are now recognized as active players in the gene regulation machinery [125]. With the advent of new-generation sequencing, the catalog of non-coding RNAs (ncRNAs) and the understanding of their functions in cardiovascular health and disease has grown exponentially [126,127,128].
ncRNAs can be classified into two major groups based on their size: small (<200 nucleotides) and long (>200 nucleotides) ncRNAs [129, 130]. Small ncRNAs comprise regulatory elements, such as microRNAs (miRNAs, miRs) and Piwi RNAs (piRNAs), and the more stably expressed housekeeping ncRNAs, such as small nucleolar RNAs (snoRNAs) and transfer RNAs (tRNAs). The classification of long ncRNAs (lncRNAs) is more complex and can be based on genomic localization, specific function, and mechanism of biogenesis. Of note, a particular class of lncRNA defined as circular RNAs (circRNAs) has gained increasing interest in experimental biology because of its peculiar features [131,132,133,134]. Alternatively, ncRNAs can be classified on the basis of their role into infrastructural (i.e., small nuclear and nucleolar RNAs, ribosomal RNAs) and regulatory RNAs (i.e., miRNAs, lncRNAs, piRNAs, and small interfering RNAs) [135]. Notably, it has been reported that regulatory ncRNAs may actively participate in gene regulation by modulating chromatin structure, adding another layer of complexity to the epigenetic landscape of vessel biology [136, 137].
Increasing evidence has demonstrated the importance of ncRNAs during the development of different pathologies, including those of the vascular system [15]. While the current knowledge on miRNAs is extensive, further investigation is required to elucidate the contribution of lncRNAs to vascular pathophysiology [125, 138].
miRNAs are small ncRNAs with a size of ~20 nucleotides (nt) that regulate gene expression at the post-transcriptional level. miRNAs play an essential role in controlling several pathways and biological processes, such as development, differentiation, apoptosis, and survival [139]. Because they have critical roles in the regulation of gene expression during normal physiology, being involved in many different biological processes, their malfunction has been directly associated with many diseases, including those of the vascular system [140, 141].
In the nucleus, miRNAs are first transcribed into a typical hairpin primary miRNA (pri-miRNA) structure by RNA polymerase enzymes, namely Pol II. Then, the pri-miRNA is specifically recognized and cleaved by the microprocessor complex DGCR8/Drosha, producing precursor miRNAs (pre-miRNAs). Pre-miRNAs are exported to the cytoplasm by the Exportin 5 (XPO5)/RanGTP complex, where they are further processed by Dicer, which removes the terminal loop, resulting in an intermediate miRNA duplex. Interestingly, the directionality of the strands determines the name of the mature miRNA form: the 5p strand arises from the 5′ end of the pre-miRNA hairpin, while the 3p originates from the 3′ end. This duplex is further processed by the Argonaute (AGO) protein family. Although both strands can be loaded onto AGO proteins, generally, the strand with lower 5′ stability or 5′ uracil is preferentially utilized and is considered the guide strand. The non-loaded strand, called the passenger strand, is unwound from the guide strand and eventually degraded. The guide strand is loaded onto the RNA inducing silencing complex (RISC), finally enabling mRNA target recognition. miRNA and RISC promote the recruitment of poly(A)-binding proteins, which shorten the poly(A) tail of mRNAs [141] (Fig. 20.2). Once the miRNA is loaded onto the RISC, miRNA:mRNA interaction can occur. The miRNA binds its mRNA target in a sequence-specific manner, suppressing gene expression by either blocking the translational mechanism or activating nucleolytic mRNA degradation mainly through targeting the 3′-untranslated region (3’UTR) of the target gene [129]. The specific sequence in the 3’UTR mRNA target recognized by the miRNA is known as the miRNA Recognition Element (MRE), which includes a region of ~8 nt at the 5′ end called the seed sequence [129, 142, 143] (Fig. 20.2).

MicroRNA biogenesis. A microRNA is transcribed as pri-microRNA, which matured first in a pre-microRNA and finally in the mature microRNA. The latest binds the 3’UTR of the target gene inhibiting gene expression
LncRNAs represent a large and heterogeneous class of ncRNAs, comprising a wide catalog of transcripts that differ in their biogenesis, genomic origin, and function [126, 144]. Most lncRNAs are synthesized via spliceosome-mediated processes on Pol II and Pol III, and present 5′-caps as well as 3′ polyadenylated (polyA) tails [145, 146]. LncRNAs are usually subject to splicing events, being presumably transcribed and processed similarly to mRNAs [145], from which they differ mainly by the absence of translational coding potential. Mature transcripts are low-copy number molecules with specific sub-cellular localization [131, 147]. Of note, this is merely a general definition, as several exceptions have been reported so far. For instance, lncRNAs of intergenic origin differ from mRNA-like lncRNAs due to the lack of a polyA tail. Additionally, a few lncRNAs have been shown to contain functional cryptic open reading frames (ORFs) that may give rise to small peptides [148].
Generally, lncRNA sequences are poorly conserved among species; however, this might be compensated by the maintenance of structural properties warranting conserved functionality [149]. Additionally, their expression is closely modulated at developmental stages, displaying tissue and cell specificity as well, this being suggestive of their involvement in gene regulation [130, 150].
Based on their direction with respect to the coding gene, lncRNAs transcripts are categorized as sense, antisense, exonic, and intronic. Other well-known forms are promoter bidirectional non-coding lncRNA, enhancer lncRNAs (eRNAs), intergenic lncRNA (lincRNAs), and circular RNAs (circRNAs) [151, 152] (Fig. 20.3). Moreover, these transcripts can act via cis- or trans-regulatory circuits [22, 153]. Herein, we present a partial description of their biological functions, which might be only the tip of the iceberg of the roles of lncRNAs in biological systems. LncRNAs can function as: (1) signals, responding to specific stimuli in the intra- and/or extracellular milieu; (2) decoys, sequestering biological active molecules and altering chromatin accessibility; (3) scaffolds, participating in specific functional multi-riboprotein complexes; (4) guides, orchestrating chromatin-modifying complexes; (5) mediators, contributing to 3D nuclear organization; and (6) molecular sponges, sequestering active miRNA molecules to inhibit and fine-tune their function [131, 144] (Fig. 20.3).

Schematic classification of the different types and functions of lncRNAs. Left: LncRNAs are classified according to their genomic position: Exon are represented in green and orange, enhancer in light green, promoter in pink, while intron in gray. Arrows indicate transcription starting sites. Right: LncRNAs modulate gene expression by different mechanisms: in the nucleus they might guide TFs to promoter region (1) or sequester TFs inhibiting their function (2); They can also modulate chromatin structure by acting as scaffold (3) or guide (4)
This class of ncRNA is, therefore, emerging as a fundamental player in the regulation of gene expression at the chromatin, DNA, transcriptional, and post-transcriptional levels in health and disease [15, 154]. As lncRNAs impact incidence and outcome of human diseases, a better understanding of their putative roles in vascular pathobiology merits further investigation [125, 155]. Indeed, key findings in the field warrant the potential use of these molecules in a clinical setting, as further described in this chapter.
Finally, among lncRNA transcripts, it is worth mentioning circRNAs in further detail. Like the vast majority of RNAs, circRNAs are generally transcribed by Pol II as a pre-messenger RNA (pre-mRNA) [156]. However, unlike linear transcripts, circRNAs undergo an alternative splicing event, namely back-splicing, promoting the formation of RNA loops. This tail-to-head folding exploits canonical splicing sites and proceeds from an upstream 5′ splice site (acceptor) to a downstream 3′ one (donor) [133, 134]. Consequently, covalently closed RNAs, displaying neither 5–3′ polarity nor a polyA tail, are generated. Given their structure, circRNAs are more stable than linear RNAs and less sensitive to RNAse R exoribonuclease degradation [157]. Finally, RNA loops can be secreted in exosome vesicles in response to specific stimuli and, subsequently, detectable in body fluids, such as serum and saliva [22]. Hence, circRNAs are promising candidates for human disease prognosis, prevention, and treatment.
In conclusion, compelling evidence has shown that ncRNAs such as miRNAs, lncRNAs, and circRNAs could be exploited for the development of new RNA-based therapeutics. Indeed, despite the limitations of such approaches, they represent a valuable tool for the generation of novel strategies in vascular disease.
4.1 Atherosclerosis and miRNAs
Atherosclerosis is an inflammatory disease in which hypercholesterolemia plays a central role in pathologically activating vascular cells and the immune system [158, 159]. These processes might be further accelerated by conditions such as diabetes mellitus and hypertension [160]. Carotid and coronary artery disease are two major atherosclerotic conditions and are the primary cause of stroke and heart attack, respectively. miRNAs are involved in every stage of the biological processes responsible for atherosclerosis development. Indeed, numerous miRNAs (miR-21-5p, −34a-5p, −146a-5p, −146b-5p, −210-3p) have been found to be upregulated in plaques from aorta, carotid, and femoral artery [161].
A detailed analysis of the single vessel layers shows how miRNAs are specific for the endothelium, infiltrating immune cells, and VSMCs. For instance, several miRNAs have been associated with dysfunction of the endothelium in vascular diseases. miRNAs involved in endothelium senescence include miR-34a, −217, and -146a, whereas others like miR-200c, −126, −10a, and -181b are modulated by oxidative stress and pro-inflammatory factors [160, 162]. Adhesion molecules are dysregulated by the misexpression of miRNAs such as miR-126, −31, and −17–3p [162]. Interestingly, the endothelium has been also found to secrete microparticles containing miRNAs, such as miR-19b, which affects other cellular components during plaque formation [163].
During atherosclerosis development, oxLDLs are loaded into monocytes/macrophages and VSMCs within the intimal layer, leading to the formation of foam cells and activated VSMCs. The role of macrophages during cellular adhesion, lipid uptake, and inflammatory responses has an important impact on plaque development. Several miRNAs, such as miR-99b, −152, −125a-5p, and −155, have been observed to be modulated in monocytes/macrophages within atherosclerotic plaques, influencing important steps, such as the accumulation of foam cells [160, 162]. Also, miR-10a plays a role in the pro-inflammatory contribution to the malfunctional endothelial phenotype during atherosclerosis [164]. In addition, miR-21 has been found to be increased in monocytes, driving atherosclerosis progression [165]. As for ECs, also macrophages are able to transfer miRNAs (i.e., miR-146a, −128) through extracellular vesicles [165]. However, macrophages might also secret miRNAs in conjunction with high-density lipoprotein (HDL); indeed, in human plasma, miR-210 has been found dysregulated in atherosclerosis patients [165].
Atherosclerotic lesions are also characterized by the presence of migratory VSMCs in the intimal layer. Some examples of miRNAs with an important impact on VSMC phenotypic switching during disease development are miR-143/145, −22, −21, −221/222, and − 128 [159, 160, 165]. Furthermore, during the final phase of atherosclerosis, involving plaque destabilization and rupture, those structures are characterized by a rich content of inflammatory macrophages and VSMCs. The levels of miR-322, −100, −127, −133a/b, and −145 are, for instance, significantly higher in patients with destabilized plaques [160, 165]. Moreover, VSMC differentiation—promoted by the progression of fatty streaks into a fibrous cap—is regulated by miRNAs, such as miR-26a, which targets transforming growth factor-β (TGF β), a critical factor for VSMC differentiation and apoptosis [162]. Other short ncRNAs, such as miR-29b, are involved in the regulation of VSMC migration, by targeting metalloproteases (MMPs), and contractility, by targeting critical genes, such as KLF4 in the case of miR-145 or −128 [162, 166].
4.2 Atherosclerosis and lncRNAs
In contrast to miRNAs, lncRNA investigation has not delivered a wealth of information on their role in atherosclerosis. Nonetheless, based on a handful of key studies, it is understood that lncRNAs can alter the onset and progression of atherosclerosis by affecting a wide array of processes governing the transcriptional program of all three principal cell components of atherosclerotic plaques [28, 150].
A paradigmatic example is the antisense ncRNA known as ANRIL, found in the Inhibitors of Cyclin Dependent Kinase 4 (INK4) locus [167]. ANRIL functions as a guide lncRNA binding specific subunits of the Polycomb repression complexes 1 and 2 (PRC1 and 2) and mediates interaction with target promoters [167]. This fosters H3K27 tri-methylation and transcriptional repression of the INK4 locus in cis. Interestingly, ANRIL exerts its atheroprotective function also in trans [167, 168]. Additionally, it is expressed and modulated by all the three principal cell components, so is directly associated with the severity of atherogenesis and lesion progression [169, 170].
Different lncRNAs (i.e., PUNISHER, LEENE, sONE) have been found to participate in EC differentiation, homeostasis, and function [171,172,173]. Additionally, several others have been identified in atherogenesis, for instance, MALAT1, MEG3, LINC00323–003, and MIR503, as well as the circRNAs cZNF292, cAFF1, cDENND4C, and cTHSD1, which are differentially expressed upon hypoxic conditioning, a stimulus known to evoke EC dysfunction [22, 174, 175]. Nonetheless, only a few have been extensively characterized so far. Human metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was one of the first lncRNAs studied in human disease. It is highly expressed in vascular endothelium and is de-regulated in several CVDs upon distress signals. Indeed, its down-regulation produces a shift from a proliferative to a migratory phenotype, causing aberrant vessel growth in vivo [176]. Coherently, profound cell cycle dysregulation was revealed by gene expression profiling [176]. Nonetheless, recent literature suggests that its role on atherosclerositic deterioration is due to increased accumulation of hematopoietic cells [176, 177].
SENCR is a vascular cell-specific lncRNA expressed in human ECs and VSMCs and plays a role in EC differentiation and homeostasis [178, 179]. Interestingly, Baker’s laboratory uncovered that SENCR may be involved also in endothelium dysfunction. Indeed, its expression is significantly reduced in ECs isolated from patients with premature CAD compared to control subjects, suggesting a central role in atherosclerosis [180]. In addition, SENCR protects against vascular damage by participating in adherent junction maintenance, membrane integrity, and permeability of ECs [179].
Aberrant phenotypic transition and apoptosis impairment of VSMCs are central events in atherogenesis initiation and progression. Along with miRNAs, lncRNAs, and circRNAs participate in the fine-tuning of VSMC biology. This can be achieved either independently or through epigenetic networks. For instance, circLRP6/miR-145 axis modulation is essential for VSMC homeostasis and may play a role in atherogenesis as well [181].
Lnc-Ang362 is an AII-induced lncRNA that enhances aberrant cell growth in VSMCs. It is co-transcribed with miR-221 and -222, well known for their detrimental effect on vessel biology [182]. In light of this, the effects of lnc-Ang362 on proliferation may likely be due to its role as the host transcript of the 2 miRNAs. Noteworthy, as these molecules are modulated in the same manner, further studies might cement lnc-Ang362 as a therapeutic target in atherosclerosis [183]. Similarly, the lncRNA SMILR concurs in atherogenesis by promoting aberrant cell proliferation via modulation of CENPF mRNA. While its overexpression results in neointimal hyperplasia, its disruption ex vivo promoted adverse vascular remodeling [184]. The above-mentioned lncRNA SENCR was first described in human coronary artery VSMCs. In this context, SENCR contributes to maintain cell differentiation by acting on cell contraction-related genes such as myocardin (Myocd), while reducing the pro-migratory gene signature (Midkine/Pleiotrophin). Therefore, it can counteract the pathological migration of VSMCs to the neointima during atherosclerotic lesion formation [178].
Inflammation and innate immune responses are also crucial events in atherosclerosis and act mainly through the modulation of monocyte and macrophage function. Myocardial infarction associated transcript (MIAT) is upregulated in vulnerable atherosclerotic plaques and fosters persistent, aberrant immune responses. Of note, MIAT ablation alleviates injury progression by acting on the miR-149-5p/CD47 axis and promoting macrophage clearance capacity (efferocytosis), thus reducing the inflammatory burden [185]. Concordantly, the monocyte- and macrophage-enriched lncRNA PELATON is upregulated in vulnerable plaques and is localized at inflammation sites. Its knockdown ameliorated atherosclerosis progression by promoting macrophage phagocytosis by acting in trans, and unlike MIAT, PELATON did not affect efferocytosis. Further studies on its coding potential may provide deeper insight into its role in macrophages [186].
RAPIA is a lncRNA that is highly expressed in advanced stages of atherosclerosis; its silencing protects against the worsening of vascular conditions by reducing proliferation and triggering the apoptotic program in macrophages via the miR-183-5p/ITGB1 pathway [187].
Finally, the already mentioned lncRNA MALAT1 is a modulator of the innate immunity response and is atheroprotective. Further studies are needed to better define its mechanism of action; nonetheless, it seems to maintain vascular functionality via a network of ncRNA-mediated processes involving mascRNA and NEAT1 [188].
4.3 Pulmonary Artery Hypertension and miRNAs
Different studies have indicated the involvement of miRNAs during PAH development. Normalization of some of these ncRNAs has been reported to blunt experimental pulmonary hypertension.
Considering lung as a bulk tissue, screening analyses were performed to evaluate miRNA modulation in PAH patients. A first study, for instance, measured 337 miRNAs in PAH and control lungs, identifying six statistically significantly upregulated miRNAs (miR-450a, −145, −302b, −27b, −367, and − 138), while only one, miR-204, was found to be down-regulated in pathological specimens [189]. The authors then linked miR-204 to a fundamental pathway able to sustain PASMC proliferation and resistance to apoptosis, which involves the signal transducer and activator of transcription 3 (STAT3), the protein tyrosine phosphatase non-receptor type 11 (SHP2) expression, the proto-oncogene tyrosine-protein kinase (Src), and nuclear factor of activated T cells (NFAT) [189].
Among miRNAs playing a role in PASMCs, we also have the miR-17–92 cluster. It contains six miRNAs (miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a) transcribed as one common pri-miRNA [190]. miR-17–92 expression was found reduced in PASMCs from patients with PAH [191], and was associated with decreased levels of SMC differentiation proteins, such as α-smooth muscle actin (α-SMA), transgelin (SM22α), and calponin, indicating a direct correlation between an SMC differentiation phenotype and miR-17–92 expression [191].
Another polycistronic miRNA cluster involved in PAH development is miR-143/145, a highly specific ncRNA expressed by SMCs of different origins [192]. Its expression is increased in human PAH as well as in the hypoxia-induced mouse model of PAH [193]. miR-143/145 cluster expression in PAH relies on down-regulation of bone morphogenetic protein receptor type II (BMPR2). Indeed, transforming growth factor-β (TGF-β) and bone morphogenic protein 4 (BMP4) activate Myocd expression and nuclear translocation of Myocd-related transcription factors (MRTFs), respectively, resulting in increased expression of miR-143 or miR-145, with concomitant repression of KLF4 expression, leading to the activation of a contractile gene program in SMCs [194]. Finally, negative manipulation of miR-145, but not miR-143, inhibited hypoxia-induced PAH in mice [193].
The miR-17-92 cluster has been shown also to control endothelium dysfunction in PAH. The development of familial PAH might depend on mutations of BMPR2 [195]. The link between this gene and the pathology relies on miR-17–5p and miR-20a, two members of the miR-17–92 cluster that directly target BMPR2 in PAECs. Interleukin 6 (IL-6) upregulates miR-17/92 expression through increased transcriptional activity of STAT3 [196]. Thus, inhibition of BMPR2 upregulates miR-17–92 expression, which then induces PAEC proliferation and reduces their apoptotic rate, resulting in the development of PAH.
Very recently, a study from Zhang and colleagues demonstrated that miR-483 is down-regulated in the serum of idiopathic PAH patients, with a direct inverse correlation with the severity of the pathology. They also demonstrated that miR-483 directly regulates several genes, including TGF-β, TGF-β receptor 2 (TGFBR2), β-catenin, connective tissue growth factor (CTGF), interleukin-1β (IL-1β), and endothelin-1 (ET-1), all of which are involved in PAH pathogenesis. Finally, an in vivo approach demonstrated that EC-specific miR-483 overexpression in a model of PAH was able to blunt the typical clinical outcomes, such as increased pulmonary vascular pressure and right ventricular hypertrophy [197].
4.4 Pulmonary Artery Hypertension and lncRNAs
The modulation of lncRNAs evoked by stimuli such as hypoxia and oxLDL is involved in de-regulation of PAECs, causing endothelium dysfunction and EndMT. Some examples of modulated ncRNAs are GATA6-AS, H19, MIR210HG, MEG9, MALAT1, and MIR22HG [198, 199], although their contribution to pulmonary hypertrophy is still largely unknown.
Additionally, PASMCs contribute to PAH progression via aberrant proliferative and migration and impairment of apoptosis. These phenotypic changes are sustained by a variety of cytokines and growth factors (i.e., PDGF-BB, NOTCH, Ang II, IL-1, FGF-2, and IGF-1) and lead to aberrant vascular remodeling [33].
The hypoxia-induced lncRNA of the Hoxa cluster antisense RNA 3 (lncRNA Hoxaas3) is upregulated in pulmonary hypertension. It promotes pathology progression by boosting PASMC over-proliferation and accelerating the cell cycle; hence, it may be a suitable therapeutic target to weaken damage progression [200].
The lncRNA regulated by PDGF-BB and TGF-β, namely LnRPT, has been identified in rat PASMCs and described as a putative therapeutic tool. Mechanistically, LnRPT controls PASMC proliferation by inhibiting the Notch pathway. Coherently, this regulatory capacity is reduced upon PDGF-BB-mediated LnRPT down-regulation. Additional studies may provide further insight into the use of LnRPT in PAH treatment [201].
In contrast, the lncRNA H19 is upregulated upon PDGF-BB stimulation in a rodent model of hypoxic-induced damage. It participates in PAH progression by promoting cell proliferation and inflammation through the regulation of the miR-let7b/AT1R axis [202].
The lincRNA Cox2 plays an important role in the modulation of innate immunity by activating or repressing specific responsive genes in mouse macrophages, thereby regulating inflammatory processes [203]. Interestingly, it is upregulated in the peripheral blood of PAH patients and is also modulated in PASMCs upon exposure to hypoxia. Its detrimental effect on PAH development is considered to function via miR-let7A/STAT3 axis regulation [204].
5 Conclusion
The body of evidence discussed in this chapter strongly indicates that chromatin modifications are directly involved in the pathogenesis of CVDs. This is particularly important since, over the past years, the incidence of CVDs has increased greatly. Fortunately, the research focusing on CVDs has grown in step, with many studies aimed at understanding the role of epigenetic processes in CVD development. It is, then, easy to understand that the definition of personalized epigenetic patterns can help to diagnose the causes of such pathologies and, thus, to identify tailor-made therapeutic approaches. Furthermore, recent technologic advances both in terms of data generation and analysis have allowed the scientific community to create detailed epigenetic maps of CVDs, providing clinical practice with new tools to link environmental effects with traditional risk factors: this includes also the possibility to eventually predict individual response to drug treatments.
Fully deciphering the epigenetic networks of the vasculature is, however, still far from completion. Thorough comprehension of the links between epigenetic mechanisms and specific tissue transcriptional programs is needed. This is particularly important for the development of therapies aimed at normalizing altered programs in specific cell types of the vessels. Indeed, the final aim would be to target epigenetic mechanisms in specific tissue compartments to avoid undesirable effects provoked by the modulation of gene expression elsewhere. Therefore, further studies are critically needed in order to personalize these new types of therapies. Because they are able to target all the types of epigenetic mechanisms discussed above, current RNA-based therapeutics might be able to fulfill this unmet need: these include silencing RNAs (siRNAs), antisense oligonucleotides (ASOs), lnRNAs, miRNAs, RNA aptamers, single guide RNAs (sgRNAs) for CRISPR/Cas9 systems, etc. These methodologies can target any gene, including those codifying proteins with no enzymatic activity; this is in contrast with classical drugs, which are not able to do so. This sheds a very positive light on the possibility of being able to tackle the majority of diseases of the cardiovascular system in the near future.
Abbreviations
- 5mC:
-
5-methyl-Cytosine
- 5-aza:
-
5-azacytidine
- 3’UTR:
-
3′-Untranslated region
- circRNA:
-
Circular RNA
- CVD:
-
Cardiovascular disease
- DNMT:
-
DNA methyltransferase
- EC:
-
Endothelial cell
- EndMT:
-
Endothelial-to-mesenchymal transition
- HDL:
-
High-density lipoprotein
- LDL:
-
Low-density lipoprotein
- lncRNA:
-
Long ncRNA
- miRNA, miR:
-
microRNA
- MRE:
-
miRNA Recognition Element
- nt:
-
Nucleotides
- ORF:
-
Open reading frame
- PAH:
-
Pulmonary artery hypertension
- pre-miRNA:
-
Precursor miRNA
- pri-miRNA:
-
Primary miRNA
- PVOD:
-
Venous-occlusive disease
- SNP:
-
Single nucleotide polymorphism
- TSS:
-
Transcription start site
- VSMC:
-
Vascular smooth muscle cell
References
Murray CJ, Lopez AD (1997) Mortality by cause for eight regions of the world: global burden of disease study. Lancet 349(9061):1269–1276. https://doi.org/10.1016/S0140-6736(96)07493-4
Nabel EG, Braunwald E (2012) A tale of coronary artery disease and myocardial infarction. N Engl J Med 366(1):54–63. https://doi.org/10.1056/NEJMra1112570
Rose G (1964) Familial patterns in ischaemic heart disease. Br J Prev Soc Med 18:75–80. https://doi.org/10.1136/jech.18.2.75
Sharifi M, Futema M, Nair D, Humphries SE (2017) Genetic architecture of familial hypercholesterolaemia. Curr Cardiol Rep 19(5):44. https://doi.org/10.1007/s11886-017-0848-8
Waddington CH (2012) The epigenotype. 1942. Int J Epidemiol 41(1):10–13. https://doi.org/10.1093/ije/dyr184
H. WC (1956) The genetic assimilation of the bithorax phenotype. Evolution:1–13
Cantone I, Fisher AG (2013) Epigenetic programming and reprogramming during development. Nat Struct Mol Biol 20(3):282–289. https://doi.org/10.1038/nsmb.2489
Lanctôt C, Cheutin T, Cremer M, Cavalli G, Cremer T (2007) Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet 8(2):104–115. https://doi.org/10.1038/nrg2041
Egger G, Liang G, Aparicio A, Jones PA (2004) Epigenetics in human disease and prospects for epigenetic therapy. Nature 429(6990):457–463. https://doi.org/10.1038/nature02625
Quintavalle M, Condorelli G, Elia L (2011) Arterial remodeling and atherosclerosis: miRNAs involvement. Vasc Pharmacol 55(4):106–110. https://doi.org/10.1016/j.vph.2011.08.216S1537-1891(11)00318-1
van der Harst P, de Windt LJ, Chambers JC (2017) Translational perspective on epigenetics in cardiovascular disease. J Am Coll Cardiol 70(5):590–606. https://doi.org/10.1016/j.jacc.2017.05.067
Greco CM, Condorelli G (2015) Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol 12(8):488–497. https://doi.org/10.1038/nrcardio.2015.71
Cavalli G, Heard E (2019) Advances in epigenetics link genetics to the environment and disease. Nature 571(7766):489–499. https://doi.org/10.1038/s41586-019-1411-0
Suades R, Cosentino F (2019) The environment, epigenetic landscape and cardiovascular risk. Cardiovasc Res 115(13):e147–ee50. https://doi.org/10.1093/cvr/cvz150
Stratton MS, Farina FM, Elia L (2019) Epigenetics and vascular diseases. J Mol Cell Cardiol 133:148–163. https://doi.org/10.1016/j.yjmcc.2019.06.010
Zarzour A, Kim HW, Weintraub NL (2019) Epigenetic regulation of vascular diseases. Arterioscler Thromb Vasc Biol 39(6):984–990. https://doi.org/10.1161/ATVBAHA.119.312193
Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S (2009) Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care 32(Suppl 2):S314–S321. https://doi.org/10.2337/dc09-S330
Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84(3):767–801. https://doi.org/10.1152/physrev.00041.2003
Khoury MK, Yang H, Liu B (2020) Macrophage biology in cardiovascular diseases. Arterioscler Thromb Vasc Biol:ATVBAHA120313584. https://doi.org/10.1161/ATVBAHA.120.313584
Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z (2019) Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 16(12):727–744. https://doi.org/10.1038/s41569-019-0227-9
Wilson PW, D’Agostino RB, Parise H, Sullivan L, Meigs JB (2005) Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation 112(20):3066–3072. https://doi.org/10.1161/CIRCULATIONAHA.105.539528
Fasolo F, Di Gregoli K, Maegdefessel L, Johnson JL (2019) Non-coding RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc Res 115(12):1732–1756. https://doi.org/10.1093/cvr/cvz203
Matouk CC, Marsden PA (2008) Epigenetic regulation of vascular endothelial gene expression. Circ Res 102(8):873–887. https://doi.org/10.1161/CIRCRESAHA.107.171025
Chiu JJ, Chien S (2011) Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91(1):327–387. https://doi.org/10.1152/physrev.00047.2009
Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclerosis. Circulation 105(9):1135–1143. https://doi.org/10.1161/hc0902.104353
Cassar A, Holmes DR, Rihal CS, Gersh BJ (2009) Chronic coronary artery disease: diagnosis and management. Mayo Clin Proc 84(12):1130–1146. https://doi.org/10.4065/mcp.2009.0391
Byrne RA, Joner M, Kastrati A (2015) Stent thrombosis and restenosis: what have we learned and where are we going? The Andreas Grüntzig lecture ESC 2014. Eur Heart J 36(47):3320–3331. https://doi.org/10.1093/eurheartj/ehv511
Pierce JB, Feinberg MW (2020) Long noncoding RNAs in atherosclerosis and vascular injury: pathobiology, biomarkers, and targets for therapy. Arterioscler Thromb Vasc Biol 40(9):2002–2017. https://doi.org/10.1161/ATVBAHA.120.314222
Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A et al (2016) 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37(1):67–119. https://doi.org/10.1093/eurheartj/ehv317
Ryan JJ, Archer SL (2014) The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res 115(1):176–188. https://doi.org/10.1161/CIRCRESAHA.113.301129
Lau EMT, Giannoulatou E, Celermajer DS, Humbert M (2017) Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol 14(10):603–614. https://doi.org/10.1038/nrcardio.2017.84
Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A et al (2013) Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62(25 Suppl):D34–D41. https://doi.org/10.1016/j.jacc.2013.10.029
Zahid KR, Raza U, Chen J, Raj UJ, Gou D (2020) Pathobiology of pulmonary artery hypertension: role of long non-coding RNAs. Cardiovasc Res 116(12):1937–1947. https://doi.org/10.1093/cvr/cvaa050
Dejana E, Hirschi KK, Simons M (2017) The molecular basis of endothelial cell plasticity. Nat Commun 8:14361. https://doi.org/10.1038/ncomms14361
Stenmark KR, Frid MG, Graham BB, Tuder RM (2018) Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res 114(4):551–564. https://doi.org/10.1093/cvr/cvy004
Bisserier M, Janostiak R, Lezoualc’h F, Hadri L (2020) Targeting epigenetic mechanisms as an emerging therapeutic strategy in pulmonary hypertension disease. Vasc Biol 2(1):R17–R34. https://doi.org/10.1530/vb-19-0030
Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184(4139):868–871. https://doi.org/10.1126/science.184.4139.868
Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 a resolution. Nature 389(6648):251–260. https://doi.org/10.1038/38444
Allis CD, Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17(8):487–500. https://doi.org/10.1038/nrg.2016.59
Bird A, Taggart M, Frommer M, Miller OJ, Macleod D (1985) A fraction of the mouse genome that is derived from islands of nonmethylated. CpG-rich DNA Cell 40(1):91–99. https://doi.org/10.1016/0092-8674(85)90312-5
Antequera F, Boyes J, Bird A (1990) High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell 62(3):503–514. https://doi.org/10.1016/0092-8674(90)90015-7
Watt F, Molloy PL (1988) Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev 2(9):1136–1143. https://doi.org/10.1101/gad.2.9.1136
Bestor TH (2000) The DNA methyltransferases of mammals. Hum Mol Genet 9(16):2395–2402. https://doi.org/10.1093/hmg/9.16.2395
Yen RW, Vertino PM, Nelkin BD, Yu JJ, el-Deiry W, Cumaraswamy A et al (1992) Isolation and characterization of the cDNA encoding human DNA methyltransferase. Nucleic Acids Res 20(9):2287–2291. https://doi.org/10.1093/nar/20.9.2287
Hermann A, Goyal R, Jeltsch A (2004) The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem 279(46):48350–48359. https://doi.org/10.1074/jbc.M403427200
Bostick M, Kim JK, Estève PO, Clark A, Pradhan S, Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317(5845):1760–1764. https://doi.org/10.1126/science.1147939
Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99(3):247–257. https://doi.org/10.1016/s0092-8674(00)81656-6
Richard Albert J, Au Yeung WK, Toriyama K, Kobayashi H, Hirasawa R, Brind’Amour J et al (2020) Maternal DNMT3A-dependent de novo methylation of the paternal genome inhibits gene expression in the early embryo. Nat Commun 11(1):5417. https://doi.org/10.1038/s41467-020-19279-7
Yagi M, Kabata M, Tanaka A, Ukai T, Ohta S, Nakabayashi K et al (2020) Identification of distinct loci for de novo DNA methylation by DNMT3A and DNMT3B during mammalian development. Nat Commun 11(1):3199. https://doi.org/10.1038/s41467-020-16989-w
Bhutani N, Burns DM, Blau HM (2011) DNA demethylation dynamics. Cell 146(6):866–872. https://doi.org/10.1016/j.cell.2011.08.042
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324(5929):930–935. https://doi.org/10.1126/science.1170116
He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q et al (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333(6047):1303–1307. https://doi.org/10.1126/science.1210944
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA et al (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333(6047):1300–1303. https://doi.org/10.1126/science.1210597
Maiti A, Drohat AC (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem 286(41):35334–35338. https://doi.org/10.1074/jbc.C111.284620
Hahn MA, Szabó PE, Pfeifer GP (2014) 5-Hydroxymethylcytosine: a stable or transient DNA modification? Genomics 104(5):314–323. https://doi.org/10.1016/j.ygeno.2014.08.015
Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY et al (2014) 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell 56(2):286–297. https://doi.org/10.1016/j.molcel.2014.08.026
Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M et al (2011) Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473(7347):394–397. https://doi.org/10.1038/nature10102
Jair KW, Bachman KE, Suzuki H, Ting AH, Rhee I, Yen RW et al (2006) De novo CpG island methylation in human cancer cells. Cancer Res 66(2):682–692. https://doi.org/10.1158/0008-5472.CAN-05-1980
Greißel A, Culmes M, Napieralski R, Wagner E, Gebhard H, Schmitt M et al (2015) Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb Haemost 114(2):390–402. https://doi.org/10.1160/TH14-10-0852
Aavik E, Lumivuori H, Leppänen O, Wirth T, Häkkinen SK, Bräsen JH et al (2015) Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster. Eur Heart J 36(16):993–1000. https://doi.org/10.1093/eurheartj/ehu437
Valencia-Morales MP, Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S et al (2015) The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genet 8:7. https://doi.org/10.1186/s12920-015-0085-1
Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E et al (2014) DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet 7(5):692–700. https://doi.org/10.1161/CIRCGENETICS.113.000441
Singh U, Jialal I (2006) Oxidative stress and atherosclerosis. Pathophysiology 13(3):129–142. https://doi.org/10.1016/j.pathophys.2006.05.002
Kim YR, Kim CS, Naqvi A, Kumar A, Kumar S, Hoffman TA et al (2012) Epigenetic upregulation of p66shc mediates low-density lipoprotein cholesterol-induced endothelial cell dysfunction. Am J Physiol Heart Circ Physiol 303(2):H189–H196. https://doi.org/10.1152/ajpheart.01218.2011
Costantino S, Paneni F, Mitchell K, Mohammed SA, Hussain S, Gkolfos C et al (2018) Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66. Int J Cardiol 268:179–186. https://doi.org/10.1016/j.ijcard.2018.04.082
Kumar A, Kumar S, Vikram A, Hoffman TA, Naqvi A, Lewarchik CM et al (2013) Histone and DNA methylation-mediated epigenetic downregulation of endothelial Kruppel-like factor 2 by low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol 33(8):1936–1942. https://doi.org/10.1161/ATVBAHA.113.301765
Qi W, Li Q, Liew CW, Rask-Madsen C, Lockhart SM, Rasmussen LM et al (2017) SHP-1 activation inhibits vascular smooth muscle cell proliferation and intimal hyperplasia in a rodent model of insulin resistance and diabetes. Diabetologia 60(3):585–596. https://doi.org/10.1007/s00125-016-4159-1
Kobayashi S, Inoue N, Azumi H, Seno T, Hirata K, Kawashima S et al (2002) Expressional changes of the vascular antioxidant system in atherosclerotic coronary arteries. J Atheroscler Thromb 9(4):184–190. https://doi.org/10.5551/jat.9.184
Weitzman SA, Turk PW, Milkowski DH, Kozlowski K (1994) Free radical adducts induce alterations in DNA cytosine methylation. Proc Natl Acad Sci U S A 91(4):1261–1264. https://doi.org/10.1073/pnas.91.4.1261
Baccarelli A, Bollati V (2009) Epigenetics and environmental chemicals. Curr Opin Pediatr 21(2):243–251. https://doi.org/10.1097/mop.0b013e32832925cc
Elia L, Kunderfranco P, Carullo P, Vacchiano M, Farina FM, Hall IF et al (2018) UHRF1 epigenetically orchestrates smooth muscle cell plasticity in arterial disease. J Clin Invest 128(6):2473–2486. https://doi.org/10.1172/JCI96121
Thenappan T, Ormiston ML, Ryan JJ, Archer SL (2018) Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ 360:j5492. https://doi.org/10.1136/bmj.j5492
Chelladurai P, Seeger W, Pullamsetti SS (2016) Epigenetic mechanisms in pulmonary arterial hypertension: the need for global perspectives. Eur Respir Rev 25(140):135–140. https://doi.org/10.1183/16000617.0036-2016
Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Haromy A et al (2006) An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113(22):2630–2641. https://doi.org/10.1161/CIRCULATIONAHA.105.609008
Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC et al (2010) Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121(24):2661–2671. https://doi.org/10.1161/CIRCULATIONAHA.109.916098
Hautefort A, Chesné J, Preussner J, Pullamsetti SS, Tost J, Looso M et al (2017) Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget 8(32):52995–53016. https://doi.org/10.18632/oncotarget.18031
Yang Q, Lu Z, Ramchandran R, Longo LD, Raj JU (2012) Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high-altitude long-term hypoxia: role of histone acetylation. Am J Physiol Lung Cell Mol Physiol 303(11):L1001–L1010. https://doi.org/10.1152/ajplung.00092.2012
Perros F, Cohen-Kaminsky S, Gambaryan N, Girerd B, Raymond N, Klingelschmitt I et al (2013) Cytotoxic cells and granulysin in pulmonary arterial hypertension and pulmonary veno-occlusive disease. Am J Respir Crit Care Med 187(2):189–196. https://doi.org/10.1164/rccm.201208-1364OC
Finch JT, Klug A (1976) Solenoidal model for superstructure in chromatin. Proc Natl Acad Sci U S A 73(6):1897–1901. https://doi.org/10.1073/pnas.73.6.1897
Hergeth SP, Schneider R (2015) The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep 16(11):1439–1453. https://doi.org/10.15252/embr.201540749
Passarge E (1979) Emil Heitz and the concept of heterochromatin: longitudinal chromosome differentiation was recognized fifty years ago. Am J Hum Genet 31(2):106–115
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705. https://doi.org/10.1016/j.cell.2007.02.005
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403(6765):41–45. https://doi.org/10.1038/47412
Martin C, Zhang Y (2005) The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6(11):838–849. https://doi.org/10.1038/nrm1761
Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E et al (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146(6):1016–1028. https://doi.org/10.1016/j.cell.2011.08.008
Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M et al (2004) Histone deimination antagonizes arginine methylation. Cell 118(5):545–553. https://doi.org/10.1016/j.cell.2004.08.020
Eckhart W, Hutchinson MA, Hunter T (1979) An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 18(4):925–933. https://doi.org/10.1016/0092-8674(79)90205-8
Sakabe K, Wang Z, Hart GW (2010) Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U S A 107(46):19915–19920. https://doi.org/10.1073/pnas.1009023107
Candau R, Zhou JX, Allis CD, Berger SL (1997) Histone acetyltransferase activity and interaction with ADA2 are critical for GCN5 function in vivo. EMBO J 16(3):555–565. https://doi.org/10.1093/emboj/16.3.555
Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY et al (1996) Tetrahymena histone acetyltransferase a: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84(6):843–851. https://doi.org/10.1016/s0092-8674(00)81063-6
Rosa-Garrido M, Chapski DJ, Vondriska TM (2018) Epigenomes in cardiovascular disease. Circ Res 122(11):1586–1607. https://doi.org/10.1161/CIRCRESAHA.118.311597
Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F et al (2011) Epigenetic modulation of the renal β-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med 17(5):573–580. https://doi.org/10.1038/nm.2337
Mumby S, Gambaryan N, Meng C, Perros F, Humbert M, Wort SJ et al (2017) Bromodomain and extra-terminal protein mimic JQ1 decreases inflammation in human vascular endothelial cells: implications for pulmonary arterial hypertension. Respirology 22(1):157–164. https://doi.org/10.1111/resp.12872
Cho YK, Eom GH, Kee HJ, Kim HS, Choi WY, Nam KI et al (2010) Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ J 74(4):760–770. https://doi.org/10.1253/circj.cj-09-0580
Yoon S, Eom GH (2016) HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J 52(1):1–11. https://doi.org/10.4068/cmj.2016.52.1.1
Elia L, Condorelli G (2019) The involvement of epigenetics in vascular disease development. Int J Biochem Cell Biol 107:27–31. https://doi.org/10.1016/j.biocel.2018.12.005
Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T et al (2006) ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442(7098):96–99. https://doi.org/10.1038/nature04835
Costantino S, Paneni F, Cosentino F (2015) Targeting chromatin remodeling to prevent cardiovascular disease in diabetes. Curr Pharm Biotechnol 16(6):531–543. https://doi.org/10.2174/138920101606150407113644
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA et al (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119(7):941–953. https://doi.org/10.1016/j.cell.2004.12.012
Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P et al (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439(7078):811–816. https://doi.org/10.1038/nature04433
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293(5532):1074–1080. https://doi.org/10.1126/science.1063127
Rossetto D, Avvakumov N, Côté J (2012) Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 7(10):1098–1108. https://doi.org/10.4161/epi.21975
Sawicka A, Seiser C (2014) Sensing core histone phosphorylation—a matter of perfect timing. Biochim Biophys Acta 1839(8):711–718. https://doi.org/10.1016/j.bbagrm.2014.04.013
Wierda RJ, Rietveld IM, van Eggermond MC, Belien JA, van Zwet EW, Lindeman JH et al (2015) Global histone H3 lysine 27 triple methylation levels are reduced in vessels with advanced atherosclerotic plaques. Life Sci 129:3–9. https://doi.org/10.1016/j.lfs.2014.10.010
Barroso M, Kao D, Blom HJ, Tavares de Almeida I, Castro R, Loscalzo J et al (2016) S-adenosylhomocysteine induces inflammation through NFkB: a possible role for EZH2 in endothelial cell activation. Biochim Biophys Acta 1862(1):82–92. https://doi.org/10.1016/j.bbadis.2015.10.019
Cong G, Yan R, Huang H, Wang K, Yan N, Jin P et al (2017) Involvement of histone methylation in macrophage apoptosis and unstable plaque formation in methionine-induced hyperhomocysteinemic ApoE. Life Sci 173:135–144. https://doi.org/10.1016/j.lfs.2017.02.003
Greißel A, Culmes M, Burgkart R, Zimmermann A, Eckstein HH, Zernecke A et al (2016) Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol 25(2):79–86. https://doi.org/10.1016/j.carpath.2015.11.001
Manabe I, Owens GK (2001) Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ Res 88(11):1127–1134. https://doi.org/10.1161/hh1101.091339
McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK (2006) Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest 116(1):36–48. https://doi.org/10.1172/JCI26505
Hoeksema MA, Gijbels MJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE et al (2014) Targeting macrophage histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med 6(9):1124–1132. https://doi.org/10.15252/emmm.201404170
Bode KA, Schroder K, Hume DA, Ravasi T, Heeg K, Sweet MJ et al (2007) Histone deacetylase inhibitors decrease toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology 122(4):596–606. https://doi.org/10.1111/j.1365-2567.2007.02678.x
Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z et al (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448(7154):714–717. https://doi.org/10.1038/nature05987
Lv YC, Tang YY, Zhang P, Wan W, Yao F, He PP et al (2016) Histone methyltransferase enhancer of Zeste homolog 2-mediated ABCA1 promoter DNA methylation contributes to the progression of atherosclerosis. PLoS One 11(6):e0157265. https://doi.org/10.1371/journal.pone.0157265
Wang Y, Huang XX, Leng D, Li JF, Liang Y, Jiang T (2021) Effect of EZH2 on pulmonary artery smooth muscle cell migration in pulmonary hypertension. Mol Med Rep 23(2):1. https://doi.org/10.3892/mmr.2020.11768
Chen D, Yang Y, Cheng X, Fang F, Xu G, Yuan Z et al (2015) Megakaryocytic leukemia 1 directs a histone H3 lysine 4 methyltransferase complex to regulate hypoxic pulmonary hypertension. Hypertension 65(4):821–833. https://doi.org/10.1161/HYPERTENSIONAHA.114.04585
Zhou XL, Liu ZB, Zhu RR, Huang H, Xu QR, Xu H et al (2019) NSD2 silencing alleviates pulmonary arterial hypertension by inhibiting trehalose metabolism and autophagy. Clin Sci (Lond) 133(9):1085–1096. https://doi.org/10.1042/CS20190142
Chelladurai P, Dabral S, Basineni SR, Chen CN, Schmoranzer M, Bender N et al (2020) Isoform-specific characterization of class I histone deacetylases and their therapeutic modulation in pulmonary hypertension. Sci Rep 10(1):12864. https://doi.org/10.1038/s41598-020-69737-x
Xu XF, Lv Y, Gu WZ, Tang LL, Wei JK, Zhang LY et al (2013) Epigenetics of hypoxic pulmonary arterial hypertension following intrauterine growth retardation rat: epigenetics in PAH following IUGR. Respir Res 14:20. https://doi.org/10.1186/1465-9921-14-20
Cardinale JP, Sriramula S, Pariaut R, Guggilam A, Mariappan N, Elks CM et al (2010) HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension 56(3):437–444. https://doi.org/10.1161/HYPERTENSIONAHA.110.154567
Lee HA, Lee DY, Cho HM, Kim SY, Iwasaki Y, Kim IK (2013) Histone deacetylase inhibition attenuates transcriptional activity of mineralocorticoid receptor through its acetylation and prevents development of hypertension. Circ Res 112(7):1004–1012. https://doi.org/10.1161/CIRCRESAHA.113.301071
Usui T, Okada M, Mizuno W, Oda M, Ide N, Morita T et al (2012) HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am J Physiol Heart Circ Physiol 302(9):H1894–H1904. https://doi.org/10.1152/ajpheart.01039.2011
Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D et al (2015) Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 131(2):190–199. https://doi.org/10.1161/CIRCULATIONAHA.114.013339
Nozik-Grayck E, Woods C, Stearman RS, Venkataraman S, Ferguson BS, Swain K et al (2016) Histone deacetylation contributes to low extracellular superoxide dismutase expression in human idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 311(1):L124–L134. https://doi.org/10.1152/ajplung.00263.2015
Xu X, Ha CH, Wong C, Wang W, Hausser A, Pfizenmaier K et al (2007) Angiotensin II stimulates protein kinase D-dependent histone deacetylase 5 phosphorylation and nuclear export leading to vascular smooth muscle cell hypertrophy. Arterioscler Thromb Vasc Biol 27(11):2355–2362. https://doi.org/10.1161/ATVBAHA.107.151704
Fiedler J, Baker AH, Dimmeler S, Heymans S, Mayr M, Thum T (2018) Non-coding RNAs in vascular disease—from basic science to clinical applications: scientific update from the working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc Res 114(10):1281–1286. https://doi.org/10.1093/cvr/cvy121
Wapinski O, Chang HY (2011) Long noncoding RNAs and human disease. Trends Cell Biol 21(6):354–361. https://doi.org/10.1016/j.tcb.2011.04.001
Poller W, Dimmeler S, Heymans S, Zeller T, Haas J, Karakas M et al (2018) Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J 39(29):2704–2716. https://doi.org/10.1093/eurheartj/ehx165
Pagiatakis C, Hall IF, Condorelli G (2020) Long non-coding RNA H19: a new avenue for RNA therapeutics in cardiac hypertrophy? Eur Heart J 41(36):3475–3476. https://doi.org/10.1093/eurheartj/ehaa663
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136(2):215–233. https://doi.org/10.1016/j.cell.2009.01.002
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10(3):155–159. https://doi.org/10.1038/nrg2521
Quinn JJ, Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17(1):47–62. https://doi.org/10.1038/nrg.2015.10
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J et al (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19(2):141–157. https://doi.org/10.1261/rna.035667.112
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333–338. https://doi.org/10.1038/nature11928
Barrett SP, Salzman J (2016) Circular RNAs: analysis, expression and potential functions. Development 143(11):1838–1847. https://doi.org/10.1242/dev.128074
Elia L, Condorelli G (2015) RNA (epi)genetics in cardiovascular diseases. J Mol Cell Cardiol 89(Pt A):11–16. https://doi.org/10.1016/j.yjmcc.2015.07.012
Beerman I, Rossi DJ (2015) Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell 16(6):613–625. https://doi.org/10.1016/j.stem.2015.05.009
Elia L, Quintavalle M (2017) Epigenetics and vascular diseases: influence of non-coding RNAs and their clinical implications. Front Cardiovasc Med 4:26. https://doi.org/10.3389/fcvm.2017.00026
Uchida S, Dimmeler S (2015) Long noncoding RNAs in cardiovascular diseases. Circ Res 116(4):737–750. https://doi.org/10.1161/CIRCRESAHA.116.302521
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Lopez-Pedrera C, Barbarroja N, Patiño-Trives AM, Luque-Tévar M, Torres-Granados C, Aguirre-Zamorano MA et al (2020) Role of microRNAs in the development of cardiovascular disease in systemic autoimmune disorders. Int J Mol Sci 21(6). https://doi.org/10.3390/ijms21062012
O’Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) 9:402. https://doi.org/10.3389/fendo.2018.00402
Ghini F, Rubolino C, Climent M, Simeone I, Marzi MJ, Nicassio F (2018) Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat Commun 9(1):3119. https://doi.org/10.1038/s41467-018-05182-9
Shukla GC, Singh J, Barik S (2011) MicroRNAs: processing, maturation, target recognition and regulatory functions. Mol Cell Pharmacol 3(3):83–92
Statello L, Guo CJ, Chen LL, Huarte M (2021) Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22(2):96–118. https://doi.org/10.1038/s41580-020-00315-9
Erdmann VA, Szymanski M, Hochberg A, de Groot N, Barciszewski J (1999) Collection of mRNA-like non-coding RNAs. Nucleic Acids Res 27(1):192–195. https://doi.org/10.1093/nar/27.1.192
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT et al (2007) RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316(5830):1484–1488. https://doi.org/10.1126/science.1138341
Salamon I, Saccani Jotti G, Condorelli G (2018) The long noncoding RNA landscape in cardiovascular disease: a brief update. Curr Opin Cardiol 33(3):282–289. https://doi.org/10.1097/HCO.0000000000000507
Yuan Y, Xu L, Geng Z, Liu J, Zhang L, Wu Y et al (2021) The role of non-coding RNA network in atherosclerosis. Life Sci 265:118756. https://doi.org/10.1016/j.lfs.2020.118756
Pang KC, Frith MC, Mattick JS (2006) Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 22(1):1–5. https://doi.org/10.1016/j.tig.2005.10.003
Leeper NJ, Maegdefessel L (2018) Non-coding RNAs: key regulators of smooth muscle cell fate in vascular disease. Cardiovasc Res 114(4):611–621. https://doi.org/10.1093/cvr/cvx249
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A et al (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25(18):1915–1927. https://doi.org/10.1101/gad.17446611
Boon RA, Jaé N, Holdt L, Dimmeler S (2016) Long noncoding RNAs: from clinical genetics to therapeutic targets? J Am Coll Cardiol 67(10):1214–1226. https://doi.org/10.1016/j.jacc.2015.12.051
Guttman M, Rinn JL (2012) Modular regulatory principles of large non-coding RNAs. Nature 482(7385):339–346. https://doi.org/10.1038/nature10887
Pagiatakis C, Musolino E, Gornati R, Bernardini G, Papait R (2019) Epigenetics of aging and disease: a brief overview. Aging Clin Exp Res. https://doi.org/10.1007/s40520-019-01430-0
Soler-Botija C, Gálvez-Montón C, Bayés-Genís A (2019) Epigenetic biomarkers in cardiovascular diseases. Front Genet 10:950. https://doi.org/10.3389/fgene.2019.00950
Li X, Yang L, Chen LL (2018) The biogenesis, functions, and challenges of circular RNAs. Mol Cell 71(3):428–442. https://doi.org/10.1016/j.molcel.2018.06.034
Lim TB, Lavenniah A, Foo RS (2020) Circles in the heart and cardiovascular system. Cardiovasc Res 116(2):269–278. https://doi.org/10.1093/cvr/cvz227
Aranda JF, Madrigal-Matute J, Rotllan N, Fernández-Hernando C (2013) MicroRNA modulation of lipid metabolism and oxidative stress in cardiometabolic diseases. Free Radic Biol Med 64:31–39. https://doi.org/10.1016/j.freeradbiomed.2013.07.014
Romaine SP, Tomaszewski M, Condorelli G, Samani NJ (2015) MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart 101(12):921–928. https://doi.org/10.1136/heartjnl-2013-305402
Nishiguchi T, Imanishi T, Akasaka T (2015) MicroRNAs and cardiovascular diseases. Biomed Res Int 2015:682857. https://doi.org/10.1155/2015/682857
Collura S, Morsiani C, Vacirca A, Fronterrè S, Ciavarella C, Vasuri F et al (2020) The carotid plaque as paradigmatic case of site-specific acceleration of aging process: the microRNAs and the inflammaging contribution. Ageing Res Rev 61:101090. https://doi.org/10.1016/j.arr.2020.101090
Madrigal-Matute J, Rotllan N, Aranda JF, Fernández-Hernando C (2013) MicroRNAs and atherosclerosis. Curr Atheroscler Rep 15(5):322. https://doi.org/10.1007/s11883-013-0322-z
Li C, Li S, Zhang F, Wu M, Liang H, Song J et al (2018) Endothelial microparticles-mediated transfer of microRNA-19b promotes atherosclerosis via activating perivascular adipose tissue inflammation in apoE. Biochem Biophys Res Commun 495(2):1922–1929. https://doi.org/10.1016/j.bbrc.2017.11.195
Jamaluddin MS, Weakley SM, Zhang L, Kougias P, Lin PH, Yao Q et al (2011) miRNAs: roles and clinical applications in vascular disease. Expert Rev Mol Diagn 11(1):79–89. https://doi.org/10.1586/erm.10.103
Lu Y, Thavarajah T, Gu W, Cai J, Xu Q (2018) Impact of miRNA in atherosclerosis. Arterioscler Thromb Vasc Biol 38(9):e159–ee70. https://doi.org/10.1161/ATVBAHA.118.310227
Farina FM, Hall IF, Serio S, Zani S, Climent M, Salvarani N et al (2020) miR-128-3p is a novel regulator of vascular smooth muscle cell phenotypic switch and vascular diseases. Circ Res 126(12):e120–ee35. https://doi.org/10.1161/CIRCRESAHA.120.316489
Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K et al (2013) Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet 9(7):e1003588. https://doi.org/10.1371/journal.pgen.1003588
Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M et al (2011) Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30(16):1956–1962. https://doi.org/10.1038/onc.2010.568
Holdt LM, Beutner F, Scholz M, Gielen S, Gäbel G, Bergert H et al (2010) ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol 30(3):620–627. https://doi.org/10.1161/ATVBAHA.109.196832
Congrains A, Kamide K, Oguro R, Yasuda O, Miyata K, Yamamoto E et al (2012) Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 220(2):449–455. https://doi.org/10.1016/j.atherosclerosis.2011.11.017
Kurian L, Aguirre A, Sancho-Martinez I, Benner C, Hishida T, Nguyen TB et al (2015) Identification of novel long noncoding RNAs underlying vertebrate cardiovascular development. Circulation 131(14):1278–1290. https://doi.org/10.1161/CIRCULATIONAHA.114.013303
Miao Y, Ajami NE, Huang TS, Lin FM, Lou CH, Wang YT et al (2018) Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat Commun 9(1):292. https://doi.org/10.1038/s41467-017-02113-y
Robb GB, Carson AR, Tai SC, Fish JE, Singh S, Yamada T et al (2004) Post-transcriptional regulation of endothelial nitric-oxide synthase by an overlapping antisense mRNA transcript. J Biol Chem 279(36):37982–37996. https://doi.org/10.1074/jbc.M400271200
Fiedler J, Breckwoldt K, Remmele CW, Hartmann D, Dittrich M, Pfanne A et al (2015) Development of Long noncoding RNA-based strategies to modulate tissue vascularization. J Am Coll Cardiol 66(18):2005–2015. https://doi.org/10.1016/j.jacc.2015.07.081
Boeckel JN, Jaé N, Heumüller AW, Chen W, Boon RA, Stellos K et al (2015) Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res 117(10):884–890. https://doi.org/10.1161/CIRCRESAHA.115.306319
Michalik KM, You X, Manavski Y, Doddaballapur A, Zörnig M, Braun T et al (2014) Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res 114(9):1389–1397. https://doi.org/10.1161/CIRCRESAHA.114.303265
Cremer S, Michalik KM, Fischer A, Pfisterer L, Jaé N, Winter C et al (2019) Hematopoietic deficiency of the Long noncoding RNA MALAT1 promotes atherosclerosis and plaque inflammation. Circulation 139(10):1320–1334. https://doi.org/10.1161/CIRCULATIONAHA.117.029015
Bell RD, Long X, Lin M, Bergmann JH, Nanda V, Cowan SL et al (2014) Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol 34(6):1249–1259. https://doi.org/10.1161/ATVBAHA.114.303240
Lyu Q, Xu S, Lyu Y, Choi M, Christie CK, Slivano OJ et al (2019) Stabilizes vascular endothelial cell adherens junctions through interaction with CKAP4. Proc Natl Acad Sci U S A 116(2):546–555. https://doi.org/10.1073/pnas.1810729116
Boulberdaa M, Scott E, Ballantyne M, Garcia R, Descamps B, Angelini GD et al (2016) A role for the Long noncoding RNA SENCR in commitment and function of endothelial cells. Mol Ther 24(5):978–990. https://doi.org/10.1038/mt.2016.41
Hall IF, Climent M, Quintavalle M, Farina FM, Schorn T, Zani S et al (2019) Circ_Lrp6, a circular RNA enriched in vascular smooth muscle cells, acts as a sponge regulating miRNA-145 function. Circ Res 124(4):498–510. https://doi.org/10.1161/CIRCRESAHA.118.314240
Liu X, Cheng Y, Yang J, Xu L, Zhang C (2012) Cell-specific effects of miR-221/222 in vessels: molecular mechanism and therapeutic application. J Mol Cell Cardiol 52(1):245–255. https://doi.org/10.1016/j.yjmcc.2011.11.008
Leung A, Trac C, Jin W, Lanting L, Akbany A, Sætrom P et al (2013) Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ Res 113(3):266–278. https://doi.org/10.1161/CIRCRESAHA.112.300849
Mahmoud AD, Ballantyne MD, Miscianinov V, Pinel K, Hung J, Scanlon JP et al (2019) The human-specific and smooth muscle cell-enriched LncRNA SMILR promotes proliferation by regulating mitotic CENPF mRNA and drives cell-cycle progression which can be targeted to limit vascular Remodeling. Circ Res 125(5):535–551. https://doi.org/10.1161/CIRCRESAHA.119.314876
Ye ZM, Yang S, Xia YP, Hu RT, Chen S, Li BW et al (2019) LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis 10(2):138. https://doi.org/10.1038/s41419-019-1409-4
Hung J, Scanlon JP, Mahmoud AD, Rodor J, Ballantyne M, Fontaine MAC et al (2020) Novel plaque enriched Long noncoding RNA in atherosclerotic macrophage regulation (PELATON). Arterioscler Thromb Vasc Biol 40(3):697–713. https://doi.org/10.1161/ATVBAHA.119.313430
Sun C, Fu Y, Gu X, Xi X, Peng X, Wang C et al (2020) Macrophage-enriched lncRNA RAPIA: a novel therapeutic target for atherosclerosis. Arterioscler Thromb Vasc Biol 40(6):1464–1478. https://doi.org/10.1161/ATVBAHA.119.313749
Gast M, Rauch BH, Nakagawa S, Haghikia A, Jasina A, Haas J et al (2019) Immune system-mediated atherosclerosis caused by deficiency of long non-coding RNA MALAT1 in ApoE−/−mice. Cardiovasc Res 115(2):302–314. https://doi.org/10.1093/cvr/cvy202
Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J et al (2011) Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 208(3):535–548. https://doi.org/10.1084/jem.20101812
Tanzer A, Stadler PF (2004) Molecular evolution of a microRNA cluster. J Mol Biol 339(2):327–335. https://doi.org/10.1016/j.jmb.2004.03.065
Chen T, Zhou G, Zhou Q, Tang H, Ibe JC, Cheng H et al (2015) Loss of microRNA-17∼92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am J Respir Crit Care Med 191(6):678–692. https://doi.org/10.1164/rccm.201405-0941OC
Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV et al (2009) The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ 16(12):1590–1598. https://doi.org/10.1038/cdd.2009.153
Caruso P, Dempsie Y, Stevens HC, McDonald RA, Long L, Lu R et al (2012) A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ Res 111(3):290–300. https://doi.org/10.1161/CIRCRESAHA.112.267591
Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G et al (2011) Down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J Biol Chem 286(32):28097–28110. https://doi.org/10.1074/jbc.M111.236950
Li W, Dunmore BJ, Morrell NW (2010) Bone morphogenetic protein type II receptor mutations causing protein misfolding in heritable pulmonary arterial hypertension. Proc Am Thorac Soc 7(6):395–398. https://doi.org/10.1513/pats.201002-024AW
Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M et al (2009) Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 104(10):1184–1191. https://doi.org/10.1161/CIRCRESAHA.109.197491
Zhang J, He Y, Yan X, Chen S, He M, Lei Y et al (2020) MicroRNA-483 amelioration of experimental pulmonary hypertension. EMBO Mol Med 12(5):e11303. https://doi.org/10.15252/emmm.201911303
Neumann P, Jaé N, Knau A, Glaser SF, Fouani Y, Rossbach O et al (2018) The lncRNA GATA6-AS epigenetically regulates endothelial gene expression via interaction with LOXL2. Nat Commun 9(1):237. https://doi.org/10.1038/s41467-017-02431-1
Voellenkle C, Garcia-Manteiga JM, Pedrotti S, Perfetti A, De Toma I, Da Silva D et al (2016) Implication of Long noncoding RNAs in the endothelial cell response to hypoxia revealed by RNA-sequencing. Sci Rep 6:24141. https://doi.org/10.1038/srep24141
Zhang H, Liu Y, Yan L, Wang S, Zhang M, Ma C et al (2019) Long noncoding RNA Hoxaas3 contributes to hypoxia-induced pulmonary artery smooth muscle cell proliferation. Cardiovasc Res 115(3):647–657. https://doi.org/10.1093/cvr/cvy250
Chen J, Guo J, Cui X, Dai Y, Tang Z, Qu J et al (2018) The Long noncoding RNA LnRPT is regulated by PDGF-BB and modulates the proliferation of pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 58(2):181–193. https://doi.org/10.1165/rcmb.2017-0111OC
Su H, Xu X, Yan C, Shi Y, Hu Y, Dong L et al (2018) LncRNA H19 promotes the proliferation of pulmonary artery smooth muscle cells through AT. Respir Res 19(1):254. https://doi.org/10.1186/s12931-018-0956-z
Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL et al (2013) A long noncoding RNA mediates both activation and repression of immune response genes. Science 341(6147):789–792. https://doi.org/10.1126/science.1240925
Cheng G, He L, Zhang Y (2020) LincRNA-Cox2 promotes pulmonary arterial hypertension by regulating the let-7a-mediated STAT3 signaling pathway. Mol Cell Biochem 475(1–2):239–247. https://doi.org/10.1007/s11010-020-03877-6
Acknowledgment
This work was supported by grants from the Horizon 2020 Research and Innovation Program under Grant Agreement (No. 828984), the Italian Ministry of Health (No. GR-2016-02364133), and Italian Ministry of Research (No. 2017HTKLRF) to L.E.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hall, I.F., Climent, M., Farina, F.M., Elia, L. (2022). Epigenetics and Vascular Disease. In: Michels, K.B. (eds) Epigenetic Epidemiology. Springer, Cham. https://doi.org/10.1007/978-3-030-94475-9_20
Download citation
DOI: https://doi.org/10.1007/978-3-030-94475-9_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94474-2
Online ISBN: 978-3-030-94475-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)