
Abstract
Vitamin D is a principal factor required for mineral and skeletal homeostasis. Vitamin D deficiency during development causes rickets and in adults can result in osteomalacia and increased risk of fracture. 1,25-Dihydroxyvitamin D3 (1,25(OH)2D3), the hormonally active form of vitamin D, is responsible for the biological actions of vitamin D which are mediated by the vitamin D receptor (VDR). Mutations in the VDR result in early-onset rickets and low calcium and phosphate, indicating the essential role of 1,25(OH)2D3/VDR signaling in the regulation of mineral homeostasis and skeletal health. This chapter summarizes our current understanding of the production of the vitamin D endocrine hormone, 1,25(OH)2D3, and the actions of 1,25(OH)2D3 which result in the maintenance of skeletal homeostasis. The primary role of 1,25(OH)2D3 is to increase calcium absorption from the intestine and thus to increase the availability of calcium for bone mineralization. Specific actions of 1,25(OH)2D3 on the intestine, kidney, and bone needed to maintain calcium homeostasis are summarized, and the impact of vitamin D status on bone health is discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 The Vitamin D Endocrine System: Metabolism and Molecular Mechanism of Action
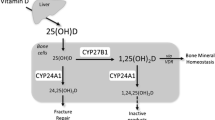
Vitamin D is a principal factor required for the development and maintenance of bone as well as for maintaining normal calcium and phosphorus homeostasis. Vitamin D deficiency during bone development causes rickets, and in adults, vitamin D deficiency, which is common in the elderly, causes secondary hyperparathyroidism that can result in osteomalacia and increased risk of fracture (Weaver et al. 2016; Bouillon and Carmeliet 2018). For vitamin D to affect mineral metabolism, it must first be metabolized to its active form. Vitamin D which is synthesized in the skin from 7-dehydrocholesterol in a reaction catalyzed by ultraviolet irradiation or taken in the diet (few foods, which include fish oils and fortified dairy products, contain appreciable amounts of vitamin D) is first hydroxylated in the liver to 25-hydroxyvitamin D3 [25(OH)D3], the major circulating form of vitamin D and the most reliable index of vitamin D status (Christakos et al. 2019). CYP2R1, a 25-hydroxylase, is likely the key vitamin D based in part on genetic evidence that patients with a mutation in CYP2R1 are deficient in 25(OH)D3 and develop vitamin D-dependent rickets (Cheng et al. 2003, 2004; Thacher et al. 2015). Since in Cyp2r1 null mice levels of 25(OH)D3 are reduced but not abolished, it has been suggested that other hydroxylases are also involved in the conversion in the liver of vitamin D to 25(OH)D (Zhu et al. 2013). The second hydroxylation occurs in the proximal renal tubule through the action of mitochondrial 25(OH)D 1 α hydroxylase (CYP27B1) resulting in the conversion of 25(OH)D to the principal hormonal form of vitamin D, 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), which is responsible for the biological actions of vitamin D (Christakos et al. 2016). Vitamin D and its metabolites are transported in the blood by vitamin D binding protein (DBP) (Christakos et al. 2016, 2019). Mutation in the CYP27B1 gene results in vitamin D dependency rickets type 1 (VDDR-1), characterized by decreased mineralization, hypocalcemia, and low circulating 1,25(OH)2D3 (Kitanaka et al. 1998). The activity of renal CYP27B1 is under stringent control. Parathyroid hormone (PTH) induced in response to hypocalcemia stimulates CYP27B1 (Jones et al. 2014). FGF23, which promotes renal phosphate excretion and requires klotho, a transmembrane protein, suppresses the expression of CYP27B1 (Shimada et al. 2004; Hu et al. 2013). 1,25(OH)2D3, as a feedback mechanism, regulates its own production by inhibiting CYP27B1, downregulating PTH synthesis by the parathyroid gland and upregulating FGF23 production in bone (Jones et al. 2014; Christakos et al. 2016). Recent mouse genomic studies by Meyer et al. (2017, 2019a) identified a kidney specific enhancer module that mediates basal and PTH-induced expression of Cyp27b1 and FGF23 and 1,25(OH)2D3-mediated repression. Studies using human kidney suggest that a kidney-specific module similar to that observed in the mouse exists in humans (Meyer et al. 2019a). These findings represent an important advance in the vitamin D field since they provide insight for the first time at the genomic level on the mechanisms that control Cyp27b1 expression. Additional factors including sex hormones and prolactin have been reported to stimulate CYP27B1 (Tanaka et al. 1978; Ajibade et al. 2010). In addition to the kidney, CYP27B1 is also expressed in the placenta (Zehnder et al. 2002) and in small amounts in a number of different tissues, including bone (Bikle 2010). However, the role of CYP27B1 under normal physiological conditions in tissues other than kidney and placenta remains to be determined.
As an additional autoregulatory mechanism, 1,25(OH)2D3 induces CYP24A1 (25-hydroxyvitamin D3 24-hydroxylase) which accelerates the catabolism of 1,25(OH)2D3 by catalyzing the conversion of 1,25(OH)2D3 into 24-hydroxylated products targeted for excretion (Jones et al. 2014; Christakos et al. 2016). Mutation in the CYP24A1 gene results in hypercalcemia, hypercalciuria, decreased PTH, and normal to high 1,25(OH)2D3 levels (Schlingmann et al. 2011). The levels of 25(OH)D3 have also been reported to be regulated through the catabolic activity of renal CYP24A1 (Jones et al. 2014; Christakos et al. 2016; Meyer et al. 2019b). CYP24A1 is present not only in kidney but also in all cells that contain VDR (Jones et al. 2014; Christakos et al. 2016). Thus, CYP24A1 not only regulates circulating 1,25(OH)2D3 protecting against hypercalcemia but also may modulate the amount of 1,25(OH)2D3 in target cells and control the cellular response. In the kidney CYP24A1 is reciprocally regulated when compared to CYP27B1 (suppressed by low calcium and PTH and induced by FGF23 and 1,25(OH)2D3). This reciprocal regulation occurs only in the kidney. In nonrenal target cells, CYP24A1 is solely regulated by 1,25(OH)2D3 (Meyer et al. 2019b). Recent studies have identified genomic mechanisms resulting in differential regulation of Cyp24a1 in the kidney and nonrenal target tissues (e.g., intestine and bone) (Meyer et al. 2019b).
The biological activities of 1,25(OH)2D3 are mediated by the vitamin D receptor (VDR), a nuclear receptor which is a member of the steroid receptor family. 1,25(OH)2D3-occupied VDR heterodimerizes with the retinoid X receptor (RXR) and together with chromatin active co-regulatory proteins interacts with specific DNA sequences (vitamin D response elements) in and around target genes resulting in activation or repression of transcription (Christakos et al. 2016; Pike and Christakos 2017). VDR-binding sites are located at proximal promoters and also many kilobases upstream and downstream and in intronic and exonic sites. Mutations in VDR result in early-onset rickets, low calcium and phosphate, and high PTH, indicating the essential role of VDR in mediating 1,25(OH)2D3 regulation of mineral homeostasis and skeletal health (Malloy et al. 2014).
2 Effect of Vitamin D on Calcium Homeostasis
2.1 Intestine
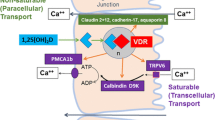
The principal action of vitamin D in maintenance of calcium homeostasis is increased intestinal calcium absorption and thus increased availability of calcium for mineralization of bone. This conclusion is based in part on studies in Vdr null mice. Feeding Vdr null mice a diet which includes high calcium prevents rickets and osteomalacia and results in the normalization of serum calcium and PTH (Amling et al. 1999; Masuyama et al. 2003). In addition, in humans with a mutation in VDR characterized by resistance to 1,25(OH)2D3 (hereditary vitamin D-resistant rickets; HVDRR), administration of calcium alone has been reported to normalize bone and result in normal mineralization (al-Aqeel et al. 1993). Active intestinal calcium absorption occurs when there is an increased need for calcium (during growth, pregnancy, lactation, and under low dietary calcium conditions). Active calcium absorption is mediated at least in part by a transcellular process involving calcium entry via the apical epithelial calcium channel TRPV6, calcium binding by the intracellular calcium-binding protein calbindin-D9k, and calcium extrusion via the basolateral membrane calcium ATPase PMCA1b (Christakos et al. 2014). TRPV6, calbindin, and PMCA1b are regulated by 1,25(OH)2D3/VDR (Van Cromphaut et al. 2001; Song et al. 2003; Lee et al. 2015). Abnormalities in the Vdr null mice develop only after weaning, consistent with studies showing that intestinal VDR, calbindin, and TRPV6 are induced at weaning (Yoshizawa et al. 1997; Song et al. 2003). Overexpression of TRPV6 in the intestine results in hypercalcemia and soft tissue calcification indicating a significant role for TRPV6 in intestinal calcium absorption and suggesting that the inability to transport calcium into the enterocyte may be a primary defect in VDR-dependent rickets (Cui et al. 2012). Direct evidence for a critical role of 1,25(OH)2D3/VDR-mediated intestinal calcium absorption in bone homeostasis was shown in studies in which VDR expression specifically in the intestine of Vdr null mice prevented rickets and normalized serum calcium (Xue and Fleet 2009). These findings indicate that intestinal VDR is essential for controlling bone formation. In addition, when VDR is deleted specifically from mouse intestine, there is calcium malabsorption, bone resorption, increased bone fractures, and normal serum calcium (Lieben et al. 2012). Thus when calcium homeostasis cannot be maintained by 1,25(OH)2D3/VDR-mediated intestinal calcium absorption, serum calcium will be maintained at the expense of skeletal integrity.
2.2 Kidney
When serum calcium cannot be maintained by intestinal calcium absorption, in addition to stimulation of osteoclastogenesis by PTH and 1,25(OH)2D3 (see Sect. 2.3), calcium reabsorption occurs in the distal convoluted tubule and collecting tubule and is regulated by 1,25(OH)2D3 and PTH (Christakos et al. 2016). Similar to the intestine, calcium is reabsorbed in the distal tubule by a transcellular process involving calcium entry through calcium channel TRPV5 (75% sequence homology with TRPV6), calcium binding to the calcium-binding protein calbindin [calbindin-D9k (9,000 Mr; only in mouse kidney) and calbindin-D28k (28,000 Mr; in mouse, rat, and human kidney)], and calcium extrusion via the calcium ATPase PMCA1b and the Na+/Ca2+ exchanger (NCX1 or SLC8A1). 1,25(OH)2D3 induces the expression in the kidney of TRPV5 and the calbindins (Song et al. 2003). PTH has been reported to activate TRPV5 via protein kinase A phosphorylation (de Groot et al. 2009). Studies in Trpv5 null mice show that inactivation of Trpv5 results in diminished calcium reabsorption in the distal tubule, severe hypercalciuria, and significant changes in bone structure (Hoenderop et al. 2003). It has been suggested that calcium uptake by TRPV5 is a rate-limiting step in renal calcium reabsorption and thus in the maintenance of calcium and bone homeostasis (Hoenderop et al. 2003). The kidney is also a major site of production of 1,25(OH)2D3 and its regulation (Christakos et al. 2010, 2016; Pike and Christakos 2017).
2.3 Bone
As outlined above, loss-of-function mutations in the human VDR lead to resistance toward the actions of 1,25(OH)2D3 and result in the development of hereditary vitamin D-resistant rickets type II (Malloy et al. 2014). This condition is mimicked in systemic Vdr null mice that develop hypocalcemia, hypophosphatemia, secondary hyperparathyroidism, and rickets (Li et al. 1997; Yoshizawa et al. 1997; Van Cromphaut et al. 2001). The rickets phenotype is characterized by progressive widening of the epiphyseal growth plates, due to a significant enlargement and disorganization of the zone of hypertrophic chondrocytes. In addition, systemic Vdr null mice develop hyperosteoidosis and osteomalacia, due to a delay in bone mineralization that develops when Vdr null mice become hypocalcemic after weaning (Li et al. 1997; Yoshizawa et al. 1997; Van Cromphaut et al. 2001). This increase in unmineralized osteoid is accompanied by an increased number of osteoblasts, whereas osteoclast numbers are not altered (Amling et al. 1999). Importantly, the bone and mineral phenotype of systemic Vdr null mice is fully corrected by supplementation with a high-calcium/high-lactose diet, underscoring the importance of VDR-mediated intestinal calcium absorption (Li et al. 1998; Amling et al. 1999; Van Cromphaut et al. 2001). Yet, multiple cell types within bone express the VDR, and the highest levels of VDR are present in osteoblasts and osteocytes, which are thus considered as the main mediators of 1,25(OH)2D3 action in bone homeostasis. VDR expression is also present in chondrocytes and in osteoclasts, although much less abundant. As bone homeostasis is regulated by timely controlled interactions between osteoblasts and osteoclasts (Wang et al. 2014; Nakamichi et al. 2017) and between chondrocytes and osteoclasts (Masuyama et al. 2006), VDR expression in all these cell types enables 1,25(OH)2D3 to affect bone development and remodeling. Here, we aim to delineate the role of 1,25(OH)2D3 in bone based on the knowledge obtained from different transgenic mouse models in which Vdr expression is either deleted or overexpressed in one of the different bone cell types. First, we will discuss the bone effects of 1,25(OH)2D3 in conditions of a positive calcium balance, where the amount of (re)absorbed calcium equals or exceeds fecal and renal calcium losses. In this condition, serum calcium levels remain normal and allow calcium deposition in bone. Thereafter, we will focus on the effects of 1,25(OH)2D3 in bone when the calcium balance is negative, as caused, for example, by insufficient intestinal calcium absorption.
2.3.1 1,25(OH)2D3 and Bone Metabolism During a Positive Calcium Balance
When dietary calcium intake is normal, 1,25(OH)2D3 indirectly regulates bone homeostasis and mineralization by guaranteeing adequate calcium supply through stimulation of intestinal and renal calcium (re)absorption (Christakos et al. 2016; Goltzman 2018). In addition, VDR expression in different osteogenic cells enables 1,25(OH)2D3 to directly and locally impact on bone metabolism in a paracrine or autocrine manner. Yet, these latter effects are still not fully elucidated and are likely dependent on the differentiation stage of the osteogenic cells.
2.3.1.1 Stage-Dependent Inactivation of Vdr Expression in Osteoblast-Lineage Cells Points to a Minor Role of Osteoblastic Vdr Expression in Bone Homeostasis
Osteoblasts together with chondrocytes differentiate from a common skeletal progenitor cell. Differentiation along the osteoblast lineage is governed by multiple transcription factors including Runx2 and Osterix (Osx) (Huang et al. 2007). When osteoprogenitor cells differentiate to immature osteoblasts, they start to express genes that encode for proteins of the extracellular matrix (ECM) such as type 1 collagen α1 (Col1a1), whereas mature osteoblasts are typified by the production of osteocalcin, a secreted protein with numerous endocrine functions including the regulation of glucose and energy metabolism (Tangseefa et al. 2018; Dirckx et al. 2019). Once osteoblasts become embedded in the bone matrix, they differentiate toward osteocytes, which fulfill a function in coordinating bone formation and resorption, and these cells are characterized by a high expression of dentin matrix protein 1 (Dmp-1) and the Wnt inhibitor sclerostin (Atkins and Findlay 2012).
During the last decades, several transgenic mouse models have been established in which Vdr expression was either deleted or overexpressed at a specific stage of osteoblast differentiation (Gardiner et al. 2000; Lieben et al. 2012; Yamamoto et al. 2013; Triliana et al. 2016; Nakamichi et al. 2017) (Fig. 1). All studies were performed in mice that received adequate supply of dietary calcium (ranging from 0.8 to 1.18% dietary calcium). Deletion of Vdr expression in osteoblast precursors, by Osx-driven Cre recombination (Osx-Vdr-cKO mice), does not affect calcium or bone homeostasis (Nakamichi et al. 2017) (Fig. 2). Indeed, serum concentrations of calcium, phosphate, PTH, and 1,25(OH)2D3 are normal in 14-week-old Osx-Vdr-cKO mice, while serum FGF23 levels are slightly reduced. Trabecular bone mass in Osx-Vdr-cKO mice is similar to control littermates with no overt changes in bone resorption or in bone mineralization. When Vdr expression is deleted in immature osteoblasts, under the control of the Col1a1 promoter (Col1a1-Vdr-cKO mice), no differences in bone mass are observed in 4- and 9-week-old animals (Yamamoto et al. 2013). However, in 16-week-old animals, a small – but significant – increase in trabecular bone mass is detected, while no significant changes in cortical bone are present. This increased bone mass is attributed to decreased bone resorption rather than to increased bone formation and is accompanied by reduced expression of the receptor activator of nuclear factor κβ ligand (RANKL), a major stimulator of osteoclastic differentiation. This bone phenotype fits with the in vitro observations showing that 1,25(OH)2D3 signaling increases RANKL expression in osteoblasts (Yasuda et al. 1998), although it is not clear why the increased bone mass is only observed in older mice. Serum levels of calcium and phosphate as well as those of PTH and 1,25(OH)2D3 are normal in Col1a1-Vdr-cKO mice.

Overview of different bone cell types in which Vdr expression was targeted

Cell- and stage-specific effects of Vdr expression in osteogenic cells on bone homeostasis during a positive calcium balance. During a positive serum balance, normal serum calcium levels allow calcium deposition in bone and proper mineralization. Vdr expression in osteoprogenitors (characterized by Osx expression (Nakamichi et al. 2017)) or in mature osteoblasts and osteocytes (Dmp1 (Lieben et al. 2012)) does not affect bone homeostasis. However, Vdr expression in immature osteoblasts (Col1a1 (Yamamoto et al. 2013)) and in mature osteoblasts (osteocalcin (Gardiner et al. 2000; Triliana et al. 2016)) is reported to induce or inhibit bone resorption, respectively. Vdr expression in osteoclast progenitors (LysM (Verlinden et al. 2019)) does not influence bone resorption, whereas Vdr signaling in mature osteoclasts (Ctsk) is shown to have no (Nakamichi et al. 2017) or inhibitory effects (Starczak et al. 2018) on osteoclast activity
Deletion of Vdr expression in mature osteoblasts and osteocytes under control of the Dmp-1 promoter (Dmp1-Vdr-cKO mice) showed that VDR signaling in mature osteoblasts and osteocytes is redundant for bone homeostasis (Lieben et al. 2012). Trabecular and cortical bone mass are indistinguishable between 8-week-old Dmp1-Vdr-cKO mice and their wild-type littermates, as are bone resorption and mineralization. Together, these three different conditional knockout models, in which Vdr expression is deleted at different stages of osteoblastic differentiation, suggest that the vitamin D system does not have major direct effects on bone homeostasis in conditions of a positive calcium balance.
On the other hand, studies in mice in which Vdr is overexpressed in mature osteoblasts, under the control of the osteocalcin promoter (osteocalcin-Vdr-cOE mice), point toward a positive effect of VDR signaling on bone mass (Gardiner et al. 2000). Indeed, 4- and 9-month-old osteocalcin-Vdr-cOE mice have increased cortical and trabecular bone mass, which is accompanied by enhanced bone formation and reduced bone resorption. Mechanistically, the decrease in bone resorption in osteocalcin-Vdr-cOE mice is attributed to reduced RANKL and increased OPG, a decoy receptor for RANKL (Misof et al. 2003; Baldock et al. 2006). This decrease in RANKL expression is rather surprising and not fully understood, as VDR signaling has been described to stimulate RANKL expression in osteogenic cells (Yasuda et al. 1998). To investigate whether the observed bone phenotype was dependent on the FVB/N background, these osteocalcin-Vdr-cOE mice were backcrossed and studied in a C57Bl6 background (Triliana et al. 2016). In parallel to the previous study, an increase in cortical and trabecular bone mass is observed in 9-week-old male and female mice and is associated with increased bone formation and reduced bone resorption. However, whereas this phenotype is recapitulated in 20-week-old male mice, no differences are present between 20-week-old female osteocalcin-Vdr-cOE mice and their wild-type littermates.
Taken together, changes in Vdr signaling in osteogenic cells do not manifestly affect bone homeostasis, although some mutant Vdr mice develop a bone phenotype. However, both inactivation and overexpression of Vdr result in an elevated bone mass (Gardiner et al. 2000; Yamamoto et al. 2013; Triliana et al. 2016), and it is at present not clear how to reconcile these apparently contradictory findings. A possible explanation is that the divergent experimental settings, such as diet, age, genetic background of the animals, and gene dosage effects (overexpression versus deletion), intervene with the effect of VDR on bone homeostasis. Alternatively, the effect of 1,25(OH)2D3 on bone metabolism might depend on the osteoblastic differentiation stage, as is suggested by the differential response of in vitro cultured osteoblasts and osteocytes to 1,25(OH)2D3 (St John et al. 2014). Therefore, a direct comparison under controlled and identical circumstances (diet, age, background, analysis method) of different transgenic models with Vdr inactivation at specific stages of osteoblast differentiation is required to enhance our understanding of the direct VDR effects in osteoblasts under a positive calcium balance.
2.3.1.2 Vdr Signaling in Growth Plate Chondrocytes Transiently Regulates Bone and Phosphate Homeostasis
As outlined above, chondrocytes differentiate from the same skeletal progenitor cells as osteoblasts and express low levels of Vdr. Systemic Vdr null mice have expanded epiphyseal growth plates with a widened zone of hypertrophic chondrocytes, due to a reduced apoptosis rate. This impaired induction of caspase-mediated apoptosis of hypertrophic chondrocytes is caused by the low serum phosphate levels in the systemic Vdr null mice (Donohue and Demay 2002; Sabbagh et al. 2005). Correction of the hypophosphatemia by a high-calcium/high-lactose diet leads to normalization of the growth plate phenotype (Li et al. 1998; Amling et al. 1999). Accordingly, inactivation of Vdr expression by Col2-Cre-driven excision does not affect chondrocyte development (Col2-Vdr-cKO mice) (Masuyama et al. 2006). However, early in life (postnatal days 3 and 15), Col2-Vdr-cKO mice have transiently reduced trabecular bone mass, which is associated with decreased vascular invasion and reduced osteoclast number at the growth plate. Mechanistically, chondrocytic VDR signaling can stimulate osteoclast formation directly by induction of RANKL expression as evidenced in chondrocyte/splenocyte cocultures. Surprisingly, serum phosphate and 1,25(OH)2D3 levels are increased in young Col2-Vdr-cKO mice, whereas calcium and PTH levels are normal. These changes can be explained by the lower serum levels of FGF23 in mutant mice, and in vitro cultures confirmed that Vdr inactivation in chondrocytes results in reduced osteoblastic expression of Fgf23. These decreased circulating FGF23 levels in Col2-Vdr-cKO mice lead to elevated renal expression of Cyp27b1 and of the sodium phosphate cotransporter type IIa, which then cause increased serum levels of 1,25(OH)2D3 and phosphate. Together these data demonstrate that VDR signaling in chondrocytes has endocrine actions and is able to affect bone mass.
2.3.1.3 Stage-Specific Deletion of Osteoclastic Vdr Expression Does Not Manifestly Affect Bone Homeostasis
It is well established that 1,25(OH)2D3 can enhance osteoclast formation in vitro in cocultures of osteoblasts and hematopoietic cells by inducing osteoblastic RANKL production (Nakamichi et al. 2018). However, as osteoclasts express Cyp27b1 as well as low levels of Vdr, it is plausible that osteoclastic VDR expression affects bone resorption in an autocrine or paracrine manner. Recent studies addressed this question by deleting Vdr expression either in myeloid cells by use of M lysozyme-driven Cre expression (LysM-Vdr-cKO mice) (Verlinden et al. 2019) or in mature osteoclasts by Cathepsin K-driven Cre recombination (Ctsk-Vdr-cKO mice) (Fig. 2) (Nakamichi et al. 2017; Starczak et al. 2018). VDR inactivation in myeloid cells results in reduced osteoclastic Vdr expression and lower induction of Cyp24a1 in response to 1,25(OH)2D3. However, calcium and bone metabolism are normal in 8-week-old LysM-Vdr-cKO mice, and no changes in bone resorption parameters are observed. Correspondingly, in vitro osteoclast formation with hematopoietic cells from LysM-Vdr-cKO mice occurs normally, suggesting that osteoclastic Vdr expression does not affect osteoclast formation and function (Verlinden et al. 2019). These findings are in agreement with observations in mature osteoclasts. Indeed, Nakamichi et al. reported that 14-week-old Ctsk-Vdr-cKO mice have a normal trabecular bone mass and that osteoclast surface is not different from control littermates (Nakamichi et al. 2017). In addition, osteoblast surface and dynamic bone parameters are unaltered arguing against a paracrine role of osteoclastic VDR expression on osteoblast function. However, Starczak et al. described that trabecular bone mass is slightly, but significantly, reduced in 6-week-old Ctsk-Vdr-cKO mice (Starczak et al. 2018). Yet, no differences are observed in osteoclast surface, number, or size, and dynamic bone parameters are also similar between Ctsk-Vdr-cKO mice and control littermates. After ovariectomy, Ctsk-Vdr-cKO mice experience an exacerbated bone loss, which is associated with enhanced osteoclastic activity but not with increased osteoclast formation. Collectively, their data suggest that osteoclastic VDR expression is a positive determinant of bone mass. This apparent discrepancy between the latter studies may point toward a transient role of osteoclastic VDR expression as Starczak et al. studied the bone phenotype of 6-week-old mice, whereas Nakamichi et al. used 14-week-old animals. Alternatively, the observed difference may be gender-related as Starczak et al. used female mice versus male mice in the study of Nakamichi et al. In analogy with the osteoblast-specific Vdr knockout models, controlled side-by-side analysis of transgenic models with osteoclastic Vdr inactivation will be required to definitively exclude a role for osteoclastic VDR expression in bone homeostasis.
2.3.2 1,25(OH)2D3 and Bone Metabolism During a Negative Calcium Balance
A negative calcium balance occurs when intestinal calcium absorption does not meet the daily calcium demand to maintain stable serum calcium concentrations and to ensure proper calcium deposition within bone. An interesting model to mimic such a negative calcium balance is the intestine-specific Vdr null model where intestinal Vdr expression is inactivated by villin-driven Cre expression (Villin-Vdr-cKO mice) (Lieben et al. 2012). Intestinal calcium absorption is decreased in Villin-Vdr-cKO mice, and in response serum levels of PTH and 1,25(OH)2D3 increase, but calcium and phosphate levels remain within the normal range (Fig. 3). The normal serum calcium levels are in contrast with the hypocalcemia that develops in systemic Vdr null mice, suggesting that in Villin-Vdr-cKO mice, compensation mechanisms are installed in the kidney and bone to ensure normal serum calcium levels. Indeed, urinary calcium loss is decreased in Villin-Vdr-cKO mice due to elevated renal calcium reabsorption. More importantly, Villin-Vdr-cKO mice are characterized by a major reduction in bone mass, by enhanced cortical thinning and porosity, by a manifest increase in the amount of unmineralized matrix (hyperosteoidosis) and, finally, by a reduced mineral content of the mineralized bone (hypomineralization). Together these findings indicate a mass transfer of calcium from bone to serum. Increased bone resorption, in response to the high circulating levels of PTH and 1,25(OH)2D3, contributes to the trabecular and cortical bone loss and to the preservation of normal serum calcium levels. Indeed, suppression of osteoclastic bone resorption by administration of bisphosphonates to Villin-Vdr-cKO mice leads to better preservation of bone mass but concurrently reduces serum calcium levels.

Effects of a negative calcium balance on bone and the contribution of Vdr expression in osteoblasts. In case of intestinal calcium malabsorption, serum levels of PTH and 1,25(OH)2D3 increase and stimulate the osteoblastic production of RANKL, which on its turn enhances osteoclastic bone resorption. In addition, high circulating levels of 1,25(OH)2D3 decrease bone mineralization by transcriptional induction of mineralization inhibitors through Vdr-mediated signaling in late osteoblasts and osteocytes. Elevated bone resorption and decreased mineralization contribute both to the transfer of calcium from the bone to the blood and ensure stable serum calcium concentrations
In addition to the enhanced bone resorption, also impaired bone mineralization in response to the high serum levels of 1,25(OH)2D3 contributes to this calcium transfer. Osteoblastic bone matrix mineralization is a multistep process, characterized by an initial formation of hydroxyapatite crystals in small extracellular matrix vesicles and subsequent deposition of hydroxyapatite minerals outside the vesicles and accumulation of minerals in the extracellular matrix (Van Driel and Van Leeuwen 2017; Goltzman 2018). This mineralization process is inhibited by high pyrophosphate levels and matrix proteins such as osteopontin (Opn). Pyrophosphate levels are regulated by pyrophosphatase phosphodiesterase enzymes (Enpp1, Enpp3), which generate pyrophosphate, and by the transmembrane ankylosis protein (encoded by Ank), which mediates pyrophosphate transport from intracellular to the extracellular matrix (Goltzman 2018). Interestingly, femoral transcript levels of Ennp3 and Ank are significantly elevated in Villin-Vdr-cKO mice (Lieben et al. 2012). In addition, chromatin immunoprecipitation reactions revealed that Enpp1, Enpp3, and Ank are direct transcriptional target genes of 1,25(OH)2D3 signaling. Together these data indicate that 1,25(OH)2D3 inhibits bone mineralization by elevating pyrophosphate levels. In accordance with these findings, lowering the 1,25(OH)2D3-induced pyrophosphate levels by cotreatment with tissue-nonspecific alkaline phosphatase, which reduces pyrophosphate levels, restores mineralization in the presence of 1,25(OH)2D3. In addition, Opn gene expression is increased in Villin-Vdr-cKO mice, and it is known that 1,25(OH)2D3 induces Opn gene expression via VDR-mediated transactivation of the Opn gene (Staal et al. 1996). Further study in Dmp1-Vdr-cKO mice, with Vdr inactivation in mature osteoblasts and osteocytes, revealed that osteocytic Vdr expression is involved in the inhibitory effect of 1,25(OH)2D3 on matrix mineralization. Indeed, Dmp1-Vdr-cKO mice treated with high doses of 1,25(OH)2D3 do not develop hyperosteoidosis, and the hypercalcemia is less pronounced in comparison with wild-type littermates. In conclusion, these findings revealed that during a negative calcium balance, normocalcemia is maintained at the expense of skeletal integrity. Transfer of bone calcium to serum is ensured by enhanced bone resorption as well as by reduced bone mineralization. Elevated bone resorption in response to PTH and 1,25(OH)2D3 is likely due to enhanced osteoblastic expression of RANKL and not to VDR-mediated signaling in osteoclasts. Indeed, bone loss in LysM-Vdr-cKO mice during a negative calcium balance is not different from bone loss in control littermates and suggests that osteoclastic Vdr expression does not play an important role in bone homeostasis (Verlinden et al. 2019).
3 Vitamin D and Bone Health
Vitamin D deficiency, as defined by low serum 25(OH)D levels, is common in the elderly and causes secondary hyperparathyroidism which can result in decreased bone density, accelerated bone loss, osteoporosis, and increased risk of fracture (Carmeliet et al. 2015). Risk factors for vitamin D deficiency include inadequate exposure to sunshine, obesity, dark skin tone, and older age (Holick et al. 2011). With older age there is a decline in the ability of the intestine to absorb calcium, a decline in the ability of the kidney to synthesize 1,25(OH)2D3, and an increase in the catabolism of 1,25(OH)2D3 by CYP24A1 which may contribute to the age-related bone loss (Veldurthy et al. 2016). Although controversy exists between the association of vitamin D supplementation and protection against fracture, it is generally recognized that there is a positive relationship between vitamin D sufficiency and a reduction in the risk of fracture (Bouillon and Carmeliet 2018). Several meta-analyses of randomized controlled clinical trials have reported that vitamin D supplementation in conjunction with sufficient calcium intake has clear benefit in protection against fracture, particularly in the elderly who are vitamin D deficient (Chapuy et al. 2002; Larsen et al. 2004; Gallagher et al. 2012; Weaver et al. 2016; Bouillon and Carmeliet 2018; Macdonald et al. 2018). Vitamin D deficiency is defined by the National Institute of Medicine [now known as the National Academy of Medicine (NAM)] as serum 25(OH)D levels below 50 nM (20 ng/mL) (Ross et al. 2011). However, the Endocrine Society guidelines suggest that a threshold of 75 nM (30 ng/mL) is necessary to maintain bone health (Gallagher et al. 2012). It is not clear, however, that optimal vitamin D levels are the same for Caucasians, Black Africans, and Asians. One problem has been that assays for measuring 25(OH)D vary. Efforts are currently being made to standardize results from different laboratories and more laboratories are implementing liquid chromatography/mass spectrometry (LC/MS) to measure vitamin D metabolites (Christakos et al. 2019). Thus, in the future there may be more of a consensus for optimal 25(OH)D levels for different groups. For vitamin D supplementation, the NAM recommends 600 IU/day for ages 1–70 and 800 IU/day for those over 70 as well as 700–1,300 mg calcium/day for adults (Ross et al. 2011). A combination of vitamin D and calcium together with pharmacological intervention [bisphosphonates and RANKL inhibitor (antiresorptive compounds) and PTH (anabolic drug, teriparatide)] has been recommended for the treatment of osteoporotic patients (Rizzoli 2018). Thus correcting vitamin D deficiency together with sufficient calcium intake may optimize pharmacological treatment of osteoporosis.
4 Conclusion
Vitamin D-regulated intestinal calcium absorption is essential in order to maintain calcium homeostasis and skeletal integrity. When calcium cannot be maintained by intestinal calcium absorption (e.g., under conditions of inadequate calcium intake, vitamin D deficiency, or diminished absorption), 1,25(OH)2D3 together with PTH can stimulate osteoclastogenesis and mobilize calcium from bone and can also enhance calcium reabsorption from the renal distal tubule in order to maintain circulating calcium levels and bone homeostasis. These findings emphasize the need to use a combination of calcium and vitamin D in order to prevent or treat osteoporosis, since vitamin D alone may negatively affect bone during a negative calcium balance. During a positive calcium balance, although the mechanisms are not clearly defined, 1,25(OH)2D3 can modulate bone formation by acting on certain stages of osteoblasts. Thus, through direct and indirect effects, 1,25(OH)2D3 is a key factor in bone mineralization. Although the mechanisms by which vitamin D deficiency contributes to osteoporosis remain to be defined, it is generally recognized that vitamin D supplementation together with sufficient calcium intake has clear benefit in protection against fracture, particularly in the elderly who are vitamin D deficient. Future studies related to mechanisms by which inadequate vitamin D contributes to osteoporosis and the identification of novel targets of 1,25(OH)2D3 action in intestine, kidney and bone involved in calcium homeostasis and changes that occur with aging will provide an increased understanding of age-related dysregulation of calcium homeostasis and may suggest candidates for targeted therapies to sustain calcium balance in the elderly.
References
Ajibade DV, Dhawan P, Fechner AJ et al (2010) Evidence for a role of prolactin in calcium homeostasis: regulation of intestinal transient receptor potential vanilloid type 6, intestinal calcium absorption, and the 25-hydroxyvitamin D(3) 1alpha hydroxylase gene by prolactin. Endocrinology 151(7):2974–2984
Al-Aqeel A, Ozand P, Sobki S et al (1993) The combined use of intravenous and oral calcium for the treatment of vitamin D dependent rickets type II (VDDRII). Clin Endocrinol 39(2):229–237
Amling M, Priemel M, Holzmann T et al (1999) Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology 140(11):4982–4987
Atkins GJ, Findlay DM (2012) Osteocyte regulation of bone mineral: a little give and take. Osteoporos Int 23(8):2067–2079
Baldock PA, Thomas GP, Hodge JM et al (2006) Vitamin D action and regulation of bone remodeling: suppression of osteoclastogenesis by the mature osteoblast. J Bone Miner Res 21(10):1618–1626
Bikle DD (2010) Extrarenal synthesis of 1,25-dihydroxyvitamin D and its health implications. In: Holick MF (ed) Vitamin D: physiology, molecular biology, and clinical applications. Humana Press, New York, pp 277–295
Bouillon R, Carmeliet G (2018) Vitamin D insufficiency: definition, diagnosis and management. Best Pract Res Clin Endocrinol Metab 32(5):669–684
Carmeliet G, Dermauw V, Bouillon R (2015) Vitamin D signaling in calcium and bone homeostasis: a delicate balance. Best Pract Res Clin Endocrinol Metab 29(4):621–631
Chapuy MC, Pamphile R, Paris E et al (2002) Combined calcium and vitamin D3 supplementation in elderly women: confirmation of reversal of secondary hyperparathyroidism and hip fracture risk: the Decalyos II study. Osteoporos Int 13(3):257–264
Cheng JB, Motola DL, Mangelsdorf DJ et al (2003) De-orphanization of cytochrome P450 2R1 - a microsomal vitamin D 25-hydroxylase. J Biol Chem 278(39):38084–38093
Cheng JB, Levine MA, Bell NH et al (2004) Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A 101(20):7711–7715
Christakos S, Ajibade DV, Dhawan P et al (2010) Vitamin D: metabolism. Endocrinol Metab Clin N Am 39(2):243–253. table of contents
Christakos S, Lieben L, Masuyama R et al (2014) Vitamin D endocrine system and the intestine. Bonekey Rep 3:496
Christakos S, Dhawan P, Verstuyf A et al (2016) Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev 96(1):365–408
Christakos S, Li S, De La Cruz J et al (2019) New developments in our understanding of vitamin metabolism, action and treatment. Metabolism 98:112–120
Cui M, Li Q, Johnson R et al (2012) Villin promoter-mediated transgenic expression of transient receptor potential cation channel, subfamily V, member 6 (TRPV6) increases intestinal calcium absorption in wild-type and vitamin D receptor knockout mice. J Bone Miner Res 27(10):2097–2107
De Groot T, Lee K, Langeslag M et al (2009) Parathyroid hormone activates TRPV5 via PKA-dependent phosphorylation. J Am Soc Nephrol 20(8):1693–1704
Dirckx N, Moorer MC, Clemens TL et al (2019) The role of osteoblasts in energy homeostasis. Nat Rev Endocrinol 15:651–665
Donohue MM, Demay MB (2002) Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology 143(9):3691–3694
Gallagher JC, Sai A, Templin T 2nd et al (2012) Dose response to vitamin D supplementation in postmenopausal women: a randomized trial. Ann Intern Med 156(6):425–437
Gardiner EM, Baldock PA, Thomas GP et al (2000) Increased formation and decreased resorption of bone in mice with elevated vitamin D receptor in mature cells of the osteoblastic lineage. FASEB J 14(13):1908–1916
Goltzman D (2018) Functions of vitamin D in bone. Histochem Cell Biol 149(4):305–312
Hoenderop JG, Van Leeuwen JP, Van Der Eerden BC et al (2003) Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest 112(12):1906–1914
Holick MF, Binkley NC, Bischoff-Ferrari HA et al (2011) Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 96(7):1911–1930
Hu MC, Shiizaki K, Kuro-O M et al (2013) Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol 75:503–533
Huang W, Yang S, Shao J et al (2007) Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front Biosci 12:3068–3092
Jones G, Prosser DE, Kaufmann M (2014) Cytochrome P450-mediated metabolism of vitamin D. J Lipid Res 55(1):13–31
Kitanaka S, Takeyama K, Murayama A et al (1998) Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med 338(10):653–661
Larsen ER, Mosekilde L, Foldspang A (2004) Vitamin D and calcium supplementation prevents osteoporotic fractures in elderly community dwelling residents: a pragmatic population-based 3-year intervention study. J Bone Miner Res 19(3):370–378
Lee SM, Riley EM, Meyer MB et al (2015) 1,25-Dihydroxyvitamin D3 controls a cohort of vitamin D receptor target genes in the proximal intestine that is enriched for calcium-regulating components. J Biol Chem 290(29):18199–18215
Li YC, Pirro AE, Amling M et al (1997) Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A 94(18):9831–9835
Li YC, Amling M, Pirro AE et al (1998) Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology 139(10):4391–4396
Lieben L, Masuyama R, Torrekens S et al (2012) Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest 122(5):1803–1815
Macdonald HM, Reid IR, Gamble GD et al (2018) 25-Hydroxyvitamin D threshold for the effects of vitamin D supplements on bone density: secondary analysis of a randomized controlled trial. J Bone Miner Res 33(8):1464–1469
Malloy PJ, Tasic V, Taha D et al (2014) Vitamin D receptor mutations in patients with hereditary 1,25-dihydroxyvitamin D-resistant rickets. Mol Genet Metab 111(1):33–40
Masuyama R, Nakaya Y, Katsumata S et al (2003) Dietary calcium and phosphorus ratio regulates bone mineralization and turnover in vitamin D receptor knockout mice by affecting intestinal calcium and phosphorus absorption. J Bone Miner Res 18(7):1217–1226
Masuyama R, Stockmans I, Torrekens S et al (2006) Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest 116(12):3150–3159
Meyer MB, Benkusky NA, Kaufmann M et al (2017) A kidney-specific genetic control module in mice governs endocrine regulation of the cytochrome P450 gene Cyp27b1 essential for vitamin D3 activation. J Biol Chem 292(42):17541–17558
Meyer MB, Benkusky NA, Kaufmann M et al (2019a) Targeted genomic deletions identify diverse enhancer functions and generate a kidney-specific, endocrine-deficient Cyp27b1 pseudo-null mouse. J Biol Chem 294(24):9518–9535
Meyer MB, Lee SM, Carlson AH et al (2019b) A chromatin-based mechanism controls differential regulation of the cytochrome P450 gene Cyp24a1 in renal and non-renal tissues. J Biol Chem. https://doi.org/10.1074/jbc.RA119.010173
Misof BM, Roschger P, Tesch W et al (2003) Targeted overexpression of vitamin D receptor in osteoblasts increases calcium concentration without affecting structural properties of bone mineral crystals. Calcif Tissue Int 73(3):251–257
Nakamichi Y, Udagawa N, Horibe K et al (2017) VDR in osteoblast-lineage cells primarily mediates vitamin D treatment-induced increase in bone mass by suppressing bone resorption. J Bone Miner Res 32(6):1297–1308
Nakamichi Y, Udagawa N, Suda T et al (2018) Mechanisms involved in bone resorption regulated by vitamin D. J Steroid Biochem Mol Biol 177:70–76
Pike JW, Christakos S (2017) Biology and mechanisms of action of the vitamin D hormone. Endocrinol Metab Clin N Am 46(4):815–843
Rizzoli R (2018) Postmenopausal osteoporosis: assessment and management. Best Pract Res Clin Endocrinol Metab 32(5):739–757
Ross AC, Manson JE, Abrams SA et al (2011) The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 96(1):53–58
Sabbagh Y, Carpenter TO, Demay MB (2005) Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A 102(27):9637–9642
Schlingmann KP, Kaufmann M, Weber S et al (2011) Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med 365(5):410–421
Shimada T, Hasegawa H, Yamazaki Y et al (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19(3):429–435
Song Y, Peng X, Porta A et al (2003) Calcium transporter 1 and epithelial calcium channel messenger ribonucleic acid are differentially regulated by 1,25 dihydroxyvitamin D3 in the intestine and kidney of mice. Endocrinology 144(9):3885–3894
St John HC, Bishop KA, Meyer MB et al (2014) The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Mol Endocrinol 28(7):1150–1165
Staal A, Van Wijnen AJ, Birkenhäger JC et al (1996) Distinct conformations of vitamin D receptor/retinoid X receptor-alpha heterodimers are specified by dinucleotide differences in the vitamin D-responsive elements of the osteocalcin and osteopontin genes. Mol Endocrinol 10(11):1444–1456
Starczak Y, Reinke DC, Barratt KR et al (2018) Absence of vitamin D receptor in mature osteoclasts results in altered osteoclastic activity and bone loss. J Steroid Biochem Mol Biol 177:77–82
Tanaka Y, Castillo L, Wineland MJ et al (1978) Synergistic effect of progesterone, testosterone, and estradiol in the stimulation of chick renal 25-hydroxyvitamin D3-1alpha-hydroxylase. Endocrinology 103(6):2035–2039
Tangseefa P, Martin SK, Fitter S et al (2018) Osteocalcin-dependent regulation of glucose metabolism and fertility: skeletal implications for the development of insulin resistance. J Cell Physiol 233(5):3769–3783
Thacher TD, Fischer PR, Singh RJ et al (2015) CYP2R1 mutations impair generation of 25-hydroxyvitamin D and cause an atypical form of vitamin D deficiency. J Clin Endocrinol Metab 100(7):E1005–E1013
Triliana R, Lam NN, Sawyer RK et al (2016) Skeletal characterization of an osteoblast-specific vitamin D receptor transgenic (ObVDR-B6) mouse model. J Steroid Biochem Mol Biol 164:331–336
Van Cromphaut SJ, Dewerchin M, Hoenderop JG et al (2001) Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci U S A 98(23):13324–13329
Van Driel M, Van Leeuwen JPTM (2017) Vitamin D endocrinology of bone mineralization. Mol Cell Endocrinol 453:46–51
Veldurthy V, Wei R, Oz L et al (2016) Vitamin D, calcium homeostasis and aging. Bone Res 4:16041
Verlinden L, Janssens I, Doms S et al (2019) Vdr expression in osteoclast precursors is not critical in bone homeostasis. J Steroid Biochem Mol Biol 195:105478
Wang Y, Zhu J, Deluca HF (2014) Identification of the vitamin D receptor in osteoblasts and chondrocytes but not osteoclasts in mouse bone. J Bone Miner Res 29(3):685–692
Weaver CM, Alexander DD, Boushey CJ et al (2016) Calcium plus vitamin D supplementation and risk of fractures: an updated meta-analysis from the National Osteoporosis Foundation. Osteoporos Int 27(1):367–376
Xue Y, Fleet JC (2009) Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology 136(4):1317–1327. e1311–1312
Yamamoto Y, Yoshizawa T, Fukuda T et al (2013) Vitamin D receptor in osteoblasts is a negative regulator of bone mass control. Endocrinology 154(3):1008–1020
Yasuda H, Shima N, Nakagawa N et al (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A 95(7):3597–3602
Yoshizawa T, Handa Y, Uematsu Y et al (1997) Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet 16(4):391–396
Zehnder D, Evans KN, Kilby MD et al (2002) The ontogeny of 25-hydroxyvitamin D3 1α-hydroxylase expression in human placenta and decidua. Am J Pathol 161(1):105–114
Zhu JG, Ochalek JT, Kaufmann M et al (2013) CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc Natl Acad Sci U S A 110(39):15650–15655
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Christakos, S., Li, S., DeLa Cruz, J., Verlinden, L., Carmeliet, G. (2019). Vitamin D and Bone. In: Stern, P.H. (eds) Bone Regulators and Osteoporosis Therapy. Handbook of Experimental Pharmacology, vol 262. Springer, Cham. https://doi.org/10.1007/164_2019_338
Download citation
DOI: https://doi.org/10.1007/164_2019_338
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-57377-5
Online ISBN: 978-3-030-57378-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)