
Abstract
Purpose of Review
In addition to the actions of the endocrine hormone, 1alpha,25-dihydroxyvitamin D (1,25(OH)2D) in stimulating intestinal calcium absorption, the regulation of bone mineral metabolism by 1,25(OH)2D is also considered an important contributor to calcium homeostasis. However, recent evidence suggest that 1,25(OH)2D acting either via endocrine or autocrine pathways plays varied roles in bone, which suggests that vitamin D contributes to the maintenance of bone mineral in addition to its catabolic roles. This review highlights the contrasting evidence for the direct action for vitamin D metabolism and activity in bone.
Recent Findings
Numerous cells within bone express vitamin D receptor (VDR), synthesise and catabolise 1,25(OH)2D via 25-hydroxyvitamin D 1alpha-hydroxylase (CYP27B1), and 25-hydroxyvitamin D 24-hydroxylase (CYP24A1) enzymes, respectively. Recent evidence suggests that all three genes are required to regulate processes of bone resorption, mineralization and fracture repair.
Summary
The actions of vitamin D in bone appear to negatively or positively regulate bone mineral depending on the physiological and pathological circumstances, suggesting that vitamin D plays pleiotropic roles in bone.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vitamin D and calcium are important for bone health, aiding in the prevention of osteoporosis and associated fractures. Inadequate calcium and vitamin D nutrition and vitamin D deficiency are common in the elderly [1] which is associated with increased risk of fracture [2, 3]. While vitamin D supplementation is commonly recommended to prevent osteoporosis and reduce fracture risk, current controversies regarding vitamin D supplementation have been difficult to resolve. In particular, the levels of optimal circulating vitamin D (25(OH)D) for preventing fracture remains controversial. This is due, in part, to an incomplete understanding of the physiology by which vitamin D promotes bone structure and strength.
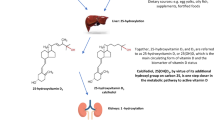
Vitamin D is well known to exert a wide variety of biological effects which extend beyond the traditional role for vitamin D in maintaining calcium homeostasis. This is due to the large number and wide variety of genes that are directly regulated by active vitamin D, 1,25(OH)2D3 [4]. Probably the most commonly known gene that is induced by vitamin D is the enzyme 25-hydroxyvitamin D 24-hydroxylase or CYP24A1. In a classical endocrine negative feedback loop, this vitamin D receptor (VDR)-mediated transcriptional upregulation of CYP24A1 serves to attenuate the activity of 1,25(OH)2D3 in target tissues. The range of target tissues, as determined by the expression of VDR, is wide. However, the number of tissues where the synthesis of active vitamin D occurs, as determined by the mitochondrondrial enzyme 25-hydroxyvitamin D 1alpha-hydroxylase or CYP27B1, is similarly broad as that for VDR [5, 6]. This suggests that many tissues may not only respond to 1,25(OH)2D but may also have a complete autocrine system of vitamin D which involves its synthesis in addition to activity and catabolism. While examples of the importance of vitamin D autocrinology are limited, the bone is one such tissue where advancements in the understanding of VDR, CYP27B1 and CYP24A1 activities have been made as shown in Fig. 1. These advancements have challenged the view that the only role for 1,25(OH)2D is to promote osteoclastic bone resorption.

Autocrine activities of vitamin D metabolism within bone cells. Serum 25(OH)D is utilised by bone cells such as osteoblasts, osteoclasts, osteocytes and chondrocytes by CYP27B1-mediated conversion to active 1,25(OH)2D which regulates processes of bone mineral homeostasis. CYP24A1 inactivates 1,25(OH)2D producing inactive metabolites. 24,25(OH)2D, produced by CYP24A1-stimulated hydroxylation of 25(OH)D, regulates processes of fracture repair
Vitamin D Receptor
Much of what has been determined regarding the role of vitamin D in various systems has been garnered from using gene deletion mouse models. While the global deletion of VDR, CYP27B1 or CYP24A1 in mouse models each demonstrate an impact on skeletal growth and homeostasis, this has been thought to largely do the indirect effects on active intestinal calcium absorption. In weanling Vdr- and Cyp27b1-null mice, a lack of vitamin D activity in the intestine rapidly results in hypocalcaemia, hyperparathyroidism and a rickets-like bone structure [7, 8]. However, although a diet containing high levels of calcium and phosphorus [7, 9,10,11] allows for sufficient passive calcium absorption to allow for normal bone development in young mice, by 17 weeks of age osteopenia develops despite the correction in serum calcium and phosphorus levels [12]. The decline in bone mineral volume in these adult null mice was associated with a deficit in osteoblastic activity and decline in mineral apposition, with no apparent change to bone resorption [12, 13]. A deficit in osteoblast activity has also been shown in osteoblast-specific VDR knockout mice. Using 2.3 kb type 1-collagen (T1-Col) promoter-Cre transgenic mice to delete VDR in osteoblasts, trabecular bone volume increased due to impaired RANKL-mediated osteoclastogenesis [14]. Unlike in the adult global Vdr-null mice, however, no apparent deficit in bone formation was observed at 16 weeks of age, suggesting that the dominant role for VDR in osteoblasts is to regulate bone resorption, at least in growing mice under normal conditions. Curiously, however, two other models of VDR deletion in the osteoblast lineage have yielded contrasting observations. When Osterix-Cre transgenic mice were recently used to delete VDR in osteoblasts, an apparent decrease in femoral trabecular bone volume was observed without an increase in bone resorption [15]. In contrast, when Dentin matrix protein 1 (DMP-1) promoter-Cre transgenic mice were used to delete VDR in osteocytes, no change in trabecular bone was observed [16••]. The lack of an effect of VDR deletion on osteocytic RANKL activity is consistent with other studies showing that RANKL was induced by 1,25(OH)2D only in immature osteoblastic cells but not in mature cells [17]. Thus, while vitamin D-responsive RANKL is expressed at most stages of osteoblastic maturation, the data published to date suggest that the role for VDR in regulating RANKL-mediated osteoclastogenesis has varying levels of importance depending on the stage of osteoblastic maturation and on physiological and pathological circumstances.
Vitamin D promotes anabolic activities of osteoblasts in addition to regulating bone resorption [18, 19••, 20,21,22,23,24]. Yet, few animal studies to date have identified whether 1,25(OH)2D can elicit positive actions on bone mineralization. Indeed, deleting or overexpressing VDR in late osteoblasts and osteocytes has produced contrasting observations on the role of vitamin D on mineralisation. For example, the absence of VDR in osteocytes in the aforementioned DMP-1-Cre/VDRKO mice prevent the effects of sustained high 1,25(OH)2D administration on bone which would otherwise cause hyperosteoidosis [16••]. This suggests that chronically high levels of 1,25(OH)2D in osteocytes inhibits bone mineralization, which could be achieved by the marked induction of genes such as Enpp1 relative to alkaline phosphatase activity [16••], resulting in increased ratio of pyrophosphate (PPi) to inorganic phosphate (Pi), which results in the inhibition of mineralisation [25]. In contrast, transgenic overexpression of osteoblastic VDR in a mouse model exhibit substantially increased cortical and trabecular bone mineral volume and does not exhibit inhibition of mineralisation at least under normal circumstances [26, 27]. Curiously, increased bone in osteoblast-specific VDR-transgenic mice has been clearly demonstrated to be due to enhanced mineral apposition on trabecular and periosteal surfaces and unexpected reduction in RANKL-mediated activity [26, 27]. These mice also are partially resistant to bone loss due to the effects of vitamin D depletion [28]. While these data suggest VDR-mediated activities may lead to anabolism, at least one study demonstrates that the osteoblast VDR-transgenic mice can exhibit lower circulating PTH levels [28], suggesting altered feedback may also contribute to lower bone resorption. However, the anabolic activities of the vitamin D analogue, eldecalcitol, increase bone volume via reducing bone resorption. The actions of eldecalcitol were ameliorated in the Osterix-Cre/VDRKO mouse model, indicating that actions of vitamin D in osteoblasts can result in reduced bone resorption at least under certain circumstances [15]. Whether these varied responses in bone can be explained by the dose and the duration of exposure of vitamin D is not yet clear. In addition, whether these varied observations can be explained by the differences in physiological circumstances in which these models are tested, (i.e. pathological verses optimal circumstances) requires further investigation.
CYP27B1
Adding to the complexity of the roles for vitamin D activity in bone is the evidence that vitamin D can be converted to its active form within osteoblasts, osteoclasts and chondrocytes [29]. With regards to osteoblasts, when 25(OH)D is converted to 1,25(OH)2D, in human or mouse osteoblasts, cell proliferation was inhibited and the degree of matrix mineralisation increased [19••, 22, 30]. In addition, exposure of primary osteoblasts to 25(OH)D increased the expression of mRNA species of genes associated with the mature osteoblast or osteocyte phenotype and stimulated a mature osteocyte morphology and increased mineralisation, but only in the presence of active CYP27B1 [22, 31, 32]. 25(OH)D also promotes the expression of Enpp1 and yet substantially increases the mineralisation of the osteoblastic cell-line MLO-A5 [32]. This is contrast to the effects of exogenous 1,25(OH)2D on bone [16••], possibly suggesting that the expression of Enpp1 can act to promote bone mineralisation with local synthesis of 1,25(OH)2D. We have generated preliminary data demonstrating that the overexpression of CYP27B1 in mature osteoblasts results in increased bone volume in both male and female due to increased bone formation without a change to bone resorption, resulting in thicker trabeculae [33]. These anabolic effects occurred without changes to circulating 1,25(OH)2D, suggesting that local synthesis of 1,25(OH)2D is more critical for bone mineral increases at least under normal growing conditions. These data are consistent the observations that Cyp27b1-null mice developed osteopenia with age due to reduced mineral apposition even though serum calcium and PTH levels were normalised through dietary calcium and phosphate supplementation [34]. Consistent with the notion of local 1,25(OH)2D synthesis in bone, serum 25(OH)D levels are a major positive determinant of bone mineral volume in a long-term trial of varying levels of dietary vitamin D in rats [35,36,37,38]. Peak bone mineral volume was achieved with serum 25(OH)D levels between 60 and 80 nmol/L, by both reducing bone resorption and increased bone formation period (i.e. length of time osteoblasts mineralise bone) [35]. It is noteworthy that these levels of 25(OH)D exceeded the minimum level required to synthesise adequate renal 1,25(OH)2D by approximately seven-fold [39]. Thus, these data collectively suggest that 25(OH)D, an inactive metabolite, is important in ensuring the local synthesis and activity of 1,25(OH)2D in bone in order to regulate osteoblastic bone formation and overall bone remodelling. How these observations relate to humans and the development of osteoporosis remains somewhat unclear. Few studies to date have investigated relationship between 25(OH)D status and local metabolism of vitamin D within bone with respect to bone health. However, one such study has shown that elderly women with subcapital fracture of the femur exhibited five-times lower levels of bone-derived 1,25(OH)2D levels when compared with aged-matched women without fracture, despite comparable circulating levels of 1,25(OH)2D [40]. Whether the changes in local levels of 1,25(OH)2D were due to changes in vitamin D metabolism in fracture patients is unknown. However, these data provides a clinical picture that warrants further investigations.
CYP24A1
Catabolism of 25(OH)D and 1,25(OH)2D occurs exclusively through CYP24A1 activity [41]. The importance of CYP24A1 in regulating vitamin D activity has been demonstrated in numerous reports of CYP24A1 mutations resulting in excessive 1,25(OH)2D activities, resulting in hypercalcaemia, hypercalciuria, nephrocalcinosis and nephrolithiasis [42]. In contrast, CYP24A1 over-activity has also been suggested to be associated with various diseases such as in hyper-proliferative disorders [43, 44] and in chronic kidney disease [45]. While much of the focus of altered CYP24A1 activity in disease is focused on the renal catabolism for vitamin D, there is increasing attention being placed on the putative role that CYP24A1 activity may play in non-renal tissues in the etiology of the disease. Although experimental evidence has expanded on the roles for vitamin D within the bone micro-environment, the question of whether CYP24A1 plays a critical role in directly determining bone mineral homeostasis has, however, largely remained unquestioned.
Cyp24a1 is expressed in numerous cells within the bone. Growth plate chondrocytes, osteoblasts, osteocytes, osteoclasts and precursor cells have been shown through cell culture systems to respond to 1,25(OH)2D by way of rapidly inducing the expression of Cyp24a1 [22, 32, 46,47,48,49]. The characterisation of global Cyp24a1-null mice has provided important experimental evidence for the impact that CYP24A1 has on bone [50]. Half of all Cyp24a1-null mice die before 3 weeks of age due to hypervitaminosis D and hypercalcaemia [50]. While surviving Cyp24a1-null mice compensate by limiting the renal synthesis of 1,25(OH)2D [51], chronic elevation of 1,25(OH)2D in Cyp24a1-null mice results in marked undermineralisation of bone, which is consistent with the effects of excessive exogenous 1,25(OH)2D3 actions on bone, as discussed earlier. The undermineralised bone in Cyp24a1-null mice can be rescued by crossing these mice with Vdr-null mice [50], which confirms that high levels of 1,25(OH)2D, acting through VDR, is responsible for the undermineralised bone. Interestingly, the overexpression of CYP24A1 in a transgenic rat model (Cyp24a1-Tg) also develops a rickets-like undermineralised bone phenotype [52, 53]. However, unlike in the Cyp24a1-null mice, the bone phenotype in Cyp24a1-Tg rats occurred without change to serum calcium, phosphate and 1,25(OH)2D, as renal CYP27B1 activity increased in compensation due to elevated PTH. However, serum 25(OH)D levels were strikingly low in the Cyp24a1-Tg rats, consistent with elevated CYP24A1 catabolism of 25(OH)D. Interestingly, when Cyp24a1-Tg rats were infused with 25(OH)D, the undermineralised bone phenotype was normalised. While the Cyp24a1 activity in bone was not tested directly, these data could theoretically be explained by high Cyp24a1 activity in osteoblasts abolishing the effects of 25(OH)D to 1,25(OH)2D conversion. If so, the infusion of 25(OH)D may have served to improve local 1,25(OH)2D synthesis.
A recent study in a unique mouse model of rickets suggests that targeting CYP24A1 activity may be of key importance to healing the bone disorder. In the rare genetic disorder of X-linked hypophosphatemia (XLH), affected children exhibit growth retardation associated with rickets and osteomalacia [54]. Its genetic basis is a mutation of the PHEX endopeptidase, leading to increased expression of the phosphaturic hormone FGF23, which in turn causes phosphate wasting, and enhancing vitamin D catabolism via CYP24A1 induction [55, 56]. The development of rickets in the mouse homolog model for XLH (HYP) and in other hypophosphataemic disorders is attributable to impaired chondrocyte apoptosis and to disordered function of osteoblasts and osteocytes [57, 58]. XLH was previously known as vitamin D-resistant rickets due to renal impairment of 1,25(OH)2D production. The case for resistance to vitamin D activity at the tissue levels has been less clear until recently when HYP mice were crossed with global Cyp24a1-null mice [59••]. This cross was done with the intention to show the absence of vitamin D catabolism would increase serum 1,25(OH)2D3 levels, improve phosphate absorption and assist in the healing of HYP bone. While the compound HYP x Cyp24a1-null mice demonstrated healing of rickets, the amelioration of the animals’ skeletal defects occurred in the absence of correcting low circulating 1,25(OH)2D levels and severe hypophosphatemia. Furthermore, FGF23 levels were further increased in the compound HYP x Cyp24a1-null mice compared with levels detected in the HYP mice, perhaps reflecting the further stimulation of Fgf23 transcription by the prolonged local effects of 1,25(OH)2D in osteocytes. These paradoxical findings are important as they demonstrate that the hypophosphatemic rickets that occurs in this FGF23 excess disorder is not principally a disorder of phosphate inadequacy or low circulating 1,25(OH)2D3 levels, but rather an intrinsic disorder of mineralisation itself, perhaps characterised by the active prevention of mineral deposition due to CYP24A1 activity. While it is not possible to determine whether the CYP24A1 activity in bone is responsible for the undermineralisation phenotype, this would be consistent with the undermineralisation that occurs in CYP24A1-Tg rats, as discussed earlier. Further studies are required to study whether inactivation of CYP24A1 heals the skeleton in HYP mice by prolonging the local half-life of 1,25(OH)2D in bone, resulting in the positive effects of 1,25(OH)2D on mineralisation [59••].
In addition to its catabolic effects, CYP24A1 produces the intermediate metabolite of 24,25(OH)2D, which has been proposed to exert biological effects (as reviewed in (48)). In particular, the less differentiated cells of the resting zone of the growth plate, respond to 24,25(OH)2D by decreasing cell proliferation and stimulating differentiation and maturation [60]. 24,25(OH)2D–mediated stimulation of resting zone cells produces phospholipase D and promotes the conversion of phosphatidylcholine ultimately to lysophosphatidic acid (LPA) production. LPA in turn stimulates increases in alkaline phosphatase activity and protects resting zone cells from apoptosis [61]. These observations support the hypothesis that 24,25(OH)2D plays a role in cartilage development. It has also been proposed that 24,25(OH)2D plays a role in fracture repair. Circulating levels of 24,25(OH)2D increase during fracture repair in chickens due to an increase in CYP24A1 activity [62] and administration of 24,25(OH)2D, in combination with 1,25(OH)2D, improved bone healing [63]. Furthermore, Cyp24a1-null mice exhibit a delayed bone fracture healing, supporting a role for 24,25(OH)2D in mammalian fracture repair [48]. However, Cyp27b1-null mice did not have impaired direct intramembranous bone regeneration during distraction osteogenesis [64], a process of healing that is distinct from indirect fracture repair which consists of both intramembranous and endochondral bone formation. These data suggest that CYP24A1 and 24,25(OH)2D plays a greater role in callus formation during endochondral fracture repair, rather than intramembranous fracture repair.
Conclusions
The development of novel strategies to treat or prevent bone diseases which involve altered vitamin D activity are currently hampered by an incomplete understanding of the pleiotropic roles that vitamin D plays in bone. A greater understanding of how the local synthesis and activity of vitamin D within the bone micro-environment will contribute to the rationale for vitamin D and calcium supplementation. A clearer understanding of how CYP24A1 activity in bone, as well as in other extra-renal tissues, contributes to the development of rickets in the disorder of XLH, could lead to using inhibitors to therapeutically target CYP24A1 activity. Similarly, examination of the role of CYP24A1 in the process of fracture healing may result in changes to clinical practice in orthopedics. While vitamin D activities in bone provide evidence of actions regulating important aspects of bone mineral metabolism, integration of the evidence from the endocrine and autocrine activities of vitamin D are required to establish these strategies in the future.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Pasco JA, Sanders KM, Henry MJ, Nicholson GC, Seeman E, Kotowicz MA. Calcium intakes among Australian women: Geelong osteoporosis study. Aust NZ J Med. 2000;30(1):21–7.
Tang BM, Eslick GD, Nowson C, Smith C, Bensoussan A. Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: a meta-analysis. Lancet. 2007;370(9588):657–66.
Lai JKC, Lucas RM, Clements MS, Roddam AW, Banks E. Hip fracture risk in relation to vitamin D supplementation and serum 25-hydroxyvitamin D levels: a systematic review and meta-analysis of randomised controlled trials and observational studies. BMC Public Health. 2010;10:331.
Pike JW, Meyer MB, Lee SM, Onal M, Benkusky NA. The vitamin D receptor: contemporary genomic approaches reveal new basic and translational insights. J Clin Invest. 2017.
Anderson PH, Hendrix I, Sawyer RK, Zarrinkalam R, Manavis J, Sarvestani GT, et al. Co-expression of CYP27B1 enzyme with the 1.5 kb CYP27B1 promoter-luciferase transgene in the mouse. Mol Cell Endocrinol. 2008;285(1–2):1–9.
Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, et al. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab. 2001;86(2):888–94.
Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94(18):9831–5.
Dardenne O, Prud'homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142(7):3135–41.
Erben RG, Soegiarto DW, Weber K, Zeitz U, Lieberherr M, Gniadecki R, et al. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol Endocrinol. 2002;16(7):1524–37.
Dardenne O, Prud'homme J, Hacking SA, Glorieux FH, St-Arnaud R. Correction of the abnormal mineral ion homeostasis with a high-calcium, high-phosphorus, high-lactose diet rescues the PDDR phenotype of mice deficient for the 25-hydroxyvitamin D-1alpha-hydroxylase (CYP27B1). Bone. 2003;32(4):332–40.
Amling M, Priemel M, Holzmann T, Chapin K, Rueger JM, Baron R, et al. Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology. 1999;140(11):4982–7.
Panda DK, Miao D, Bolivar I, Li J, Huo R, Hendy GN, et al. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279(16):16754–66.
Ryan JW, Starczak Y, Tsangari H, Sawyer RK, Davey RA, Atkins GJ, et al. Sex-related differences in the skeletal phenotype of aged vitamin D receptor global knockout mice. J Steroid Biochem Mol Biol. 2015;164:361–8.
Yamamoto Y, Yoshizawa T, Fukuda T, Shirode-Fukuda Y, Yu T, Sekine K, et al. Vitamin D receptor in osteoblasts is a negative regulator of bone mass control. Endocrinology. 2013;154(3):1008–20.
Nakamichi Y, Udagawa N, Horibe K, Mizoguchi T, Yamamoto Y, Nakamura T, et al. VDR in osteoblast-lineage cells primarily mediates vitamin D treatment-induced increase in bone mass by suppressing bone resorption. J Bone Miner Res. 2017;32(6):1297–308.
•• Lieben L, Masuyama R, Torrekens S, Van Looveren R, Schrooten J, Baatsen P, et al. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest. 2012;122(5):1803–15. This paper describes the relative importance of VDR-mediated siganlling in osteocytes in the process of regulating mineralisation. Data presented here illustrate that that bone mineralisation is a co-ordinated process which can be inhibited by excessive vitamin D activity.
Atkins GJ, Kostakis P, Pan B, Farrugia A, Gronthos S, Evdokiou A, et al. RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res. 2003;18(6):1088–98.
Owen TA, Aronow MS, Barone LM, Bettencourt B, Stein GS, Lian JB. Pleiotropic effects of vitamin D on osteoblast gene expression are related to the proliferative and differentiated state of the bone cell phenotype: dependency upon basal levels of gene expression, duration of exposure, and bone matrix competency in normal rat osteoblast cultures. Endocrinology. 1991;128(3):1496–504.
•• Geng S, Zhou S, Glowacki J. Effects of 25-hydroxyvitamin D(3) on proliferation and osteoblast differentiation of human marrow stromal cells require CYP27B1/1alpha-hydroxylase. J Bone Miner Res. 2011;26(5):1145–53. This papers contributes important evidence tht local synthesis of vitamin D within osteoblasts-lineage cells plays a key role on the promotion of osteoblastogenesis.
Kato H, Ochiai-Shino H, Onodera S, Saito A, Shibahara T, Azuma T. Promoting effect of 1,25(OH)2 vitamin D3 in osteogenic differentiation from induced pluripotent stem cells to osteocyte-like cells. Open Biol. 2015;5(2):140201.
Xue Y, Karaplis AC, Hendy GN, Goltzman D, Miao D. Exogenous 1,25-dihydroxyvitamin D3 exerts a skeletal anabolic effect and improves mineral ion homeostasis in mice that are homozygous for both the 1alpha-hydroxylase and parathyroid hormone null alleles. Endocrinology. 2006;147(10):4801–10.
Atkins GJ, Anderson PH, Findlay DM, Welldon KJ, Vincent C, Zannettino AC, et al. Metabolism of vitamin D(3) in human osteoblasts: evidence for autocrine and paracrine activities of 1alpha,25-dihydroxyvitamin D(3). Bone. 2007;40(6):1517–28.
Yang D, Turner AG, Wijenayaka AR, Anderson PH, Morris HA, Atkins GJ. 1,25-Dihydroxyvitamin D3 and extracellular calcium promote mineral deposition via NPP1 activity in a mature osteoblast cell line MLO-A5. Mol Cell Endocrinol. 2015;412:140–7.
Yang D, Atkins GJ, Turner AG, Anderson PH, Morris HA. Differential effects of 1,25-dihydroxyvitamin D on mineralisation and differentiation in two different types of osteoblast-like cultures. J Steroid Biochem Mol Biol. 2013;136:166–70.
Mackenzie NC, Huesa C, Rutsch F, MacRae VE. New insights into NPP1 function: lessons from clinical and animal studies. Bone. 2012;51(5):961–8.
Gardiner EM, Baldock PA, Thomas GP, Sims NA, Henderson NK, Hollis B, et al. Increased formation and decreased resorption of bone in mice with elevated vitamin D receptor in mature cells of the osteoblastic lineage. FASEB J. 2000;14(13):1908–16.
Triliana R, Lam NN, Sawyer RK, Atkins GJ, Morris HA, Anderson PH. Skeletal characterization of an osteoblast-specific vitamin D receptor transgenic (ObVDR-B6) mouse model. J Steroid Biochem Mol Biol. 2016;164:331–6.
Lam NN, Triliana R, Sawyer RK, Atkins GJ, Morris HA, O'Loughlin PD, et al. Vitamin D receptor overexpression in osteoblasts and osteocytes prevents bone loss during vitamin D-deficiency. J Steroid Biochem Mol Biol. 2014;144:128–31.
Anderson PH, Atkins GJ. The skeleton as an intracrine organ for vitamin D metabolism. Mol Asp Med. 2008;29(6):397–406.
van Driel M, Koedam M, Buurman CJ, Hewison M, Chiba H, Uitterlinden AG, et al. Evidence for auto/paracrine actions of vitamin D in bone: 1alpha-hydroxylase expression and activity in human bone cells. FASEB J. 2006;20(13):2417–9.
Turner AG, Hanrath MA, Morris HA, Atkins GJ, Anderson PH. The local production of 1, 25 (OH) 2 D 3 promotes osteoblast and osteocyte maturation. J Steroid Biochem Mol Biol. 2014;144:114–8.
Anderson PH, Atkins GJ, Findlay DM, Oloughlin PD, Welldon K, Vincent C, et al. RNAi-mediated silencing of CYP27B1 abolishes 1,25(OH)2D3 synthesis and reduces osteocalcin and CYP24 mRNA expression in human osteosarcoma (HOS) cells. J Steroid Biochem Mol Biol. 2007;103(3–5):601–5.
Turner AG, RK Sawyer, O’Loughlin PD, Atkins GJ, Morris HA, Anderson PH. Transgene expression of CYP27B1 in osteoblasts promotes bone formation without altering bone resorption. J Bone Miner Res. 2012;27(Suppl 1). Available at: http://www.asbmr.org/education/AbstractDetail?aid=05ac9f70-6487-4091-b38f-49cd239a7a29.
Panda DK, Miao DS, Bolivar I, Li JR, Huo RJ, Hendy GN, et al. Inactivation of the 25-hydroxyvitamin D 1 alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279(16):16754–66.
Anderson PH, Sawyer RK, Moore AJ, May BK, O'Loughlin PD, Morris HA. Vitamin D depletion induces RANKL-mediated osteoclastogenesis and bone loss in a rodent model. J Bone Miner Res. 2008;23(11):1789–97.
Lee AM, Anderson PH, Sawyer RK, Moore AJ, Forwood MR, Steck R, et al. Discordant effects of vitamin D deficiency in trabecular and cortical bone architecture and strength in growing rodents. J Steroid Biochem Mol Biol. 2010;121(1–2):284–7.
Lee AM, Sawyer RK, Moore AJ, Morris HA, O'Loughlin PD, Anderson PH. Adequate dietary vitamin D and calcium are both required to reduce bone turnover and increased bone mineral volume. J Steroid Biochem Mol Biol. 2013;144:159–62.
Anderson P, Sawyer R, May B, O’Loughlin P, Morris H. 25-Hydroxyvitamin D requirement for maintaining skeletal health utilising a Sprague-Dawley rat model. J Steroid Biochem Mol Biol. 2007;103(3):592–5.
Anderson PH, Sawyer RK, May BK, O'Loughlin PD, Morris HA. 25-Hydroxyvitamin D requirement for maintaining skeletal health utilising a Sprague-Dawley rat model. J Steroid Biochem Mol Biol. 2007;103(3–5):592–5.
Lidor C, Sagiv P, Amdur B, Gepstein R, Otremski I, Hallel T, et al. Decrease in bone levels of 1,25-dihydroxyvitamin D in women with subcapital fracture of the femur. Calcif Tissue Int. 1993;52(2):146–8.
Masuda S, Byford V, Arabian A, Sakai Y, Demay MB, St-Arnaud R, et al. Altered pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 in the blood and tissues of the 25-hydroxyvitamin D-24-hydroxylase (Cyp24a1) null mouse. Endocrinology. 2005;146(2):825–34.
Dinour D, Beckerman P, Ganon L, Tordjman K, Eisenstein Z, Holtzman EJ. Loss-of-function mutations of CYP24A1, the vitamin D 24-hydroxylase gene, cause long-standing hypercalciuric nephrolithiasis and nephrocalcinosis. J Urol. 2013;190(2):552–7.
Sumantran VN, Mishra P, Bera R, Sudhakar N. Microarray analysis of differentially-expressed genes encoding CYP450 and phase II drug metabolizing enzymes in psoriasis and melanoma. Pharmaceutics. 2016;8(1).
Masuda S, Strugnell SA, Knutson JC, St-Arnaud R, Jones G. Evidence for the activation of 1alpha-hydroxyvitamin D2 by 25-hydroxyvitamin D-24-hydroxylase: delineation of pathways involving 1alpha,24-dihydroxyvitamin D2 and 1alpha,25-dihydroxyvitamin D2. Biochim Biophys Acta. 2006;1761(2):221–34.
Quarles LD. Role of FGF23 in vitamin D and phosphate metabolism: implications in chronic kidney disease. Exp Cell Res. 2012;318(9):1040–8.
Armbrecht HJ, Hodam TL, Boltz MA, Partridge NC, Brown AJ, Kumar VB. Induction of the vitamin D 24-hydroxylase (CYP24) by 1,25-dihydroxyvitamin D3 is regulated by parathyroid hormone in UMR106 osteoblastic cells. Endocrinology. 1998;139(8):3375–81.
Ito N, Findlay DM, Anderson PH, Bonewald LF, Atkins GJ. Extracellular phosphate modulates the effect of 1 alpha,25-dihydroxy vitamin D-3 (1,25D) on osteocyte like cells. J Steroid Biochem Mol Biol. 2013;136:183–6.
St-Arnaud R, Naja RP. Vitamin D metabolism, cartilage and bone fracture repair. Mol Cell Endocrinol. 2011;347(1–2):48–54.
Kogawa M, Anderson PH, Findlay DM, Morris HA, Atkins GJ. The metabolism of 25-(OH)vitamin D-3 by osteoclasts and their precursors regulates the differentiation of osteoclasts. J Steroid Biochem Mol Biol. 2010;121(1–2):277–80.
St-Arnaud R, Arabian A, Travers R, Barletta F, Raval-Pandya M, Chapin K, et al. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology. 2000;141(7):2658–66.
Masuda S, Byford V, Arabian A, Sakai Y, Demay MB, St-Arnaud R, et al. Altered pharmacokinetics of 1a,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 in the blood and tissues of the 25-hydroxyvitamin D-24-hydroxylase (Cyp24a1) null mouse. Endocrinology. 2005;146:825–34.
Hosogane N, Shinki T, Kasuga H, Taketomi S, Toyama Y, Suda T. Mechanisms for the reduction of 24,25-dihydroxyvitamin D3 levels and bone mass in 24-hydroxylase transgenic rats. FASEB J. 2003;17(6):737–9.
Kasuga H, Hosogane N, Matsuoka K, Mori I, Sakura Y, Shimakawa K, et al. Characterization of transgenic rats constitutively expressing vitamin D-24-hydroxylase gene. Biochem Biophys Res Commun. 2002;297(5):1332–8.
Friedman NE, Lobaugh B, Drezner MK. Effects of calcitriol and phosphorus therapy on the growth of patients with X-linked hypophosphatemia. J Clin Endocrinol Metab. 1993;76(4):839–44.
A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11(2):130–6.
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291(1):E38–49.
Miedlich SU, Zalutskaya A, Zhu ED, Demay MB. Phosphate-induced apoptosis of hypertrophic chondrocytes is associated with a decrease in mitochondrial membrane potential and is dependent upon Erk1/2 phosphorylation. J Biol Chem. 2010;285(24):18270–5.
Sabbagh Y, Carpenter TO, Demay MB. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A. 2005;102(27):9637–42.
•• Bai X, Miao D, Xiao S, Qiu D, St-Arnaud R, Petkovich M, et al. CYP24 inhibition as a therapeutic target in FGF23-mediated renal phosphate wasting disorders. J Clin Invest. 2016;126(2):667–80. This paper describes desmostrates that CYP24A1 inhibtion can prevent rickets without the need to restore circulating phosphate or calcitriol levels in the disorder of x-linked hypophosphatemia.
Schwartz Z, Sylvia VL, Del Toro F, Hardin RR, Dean DD, Boyan BD. 24R,25-(OH)(2)D(3) mediates its membrane receptor-dependent effects on protein kinase C and alkaline phosphatase via phospholipase a(2) and cyclooxygenase-1 but not cyclooxygenase-2 in growth plate chondrocytes. J Cell Physiol. 2000;182(3):390–401.
Hurst-Kennedy J, Boyan BD, Schwartz Z. Lysophosphatidic acid signaling promotes proliferation, differentiation, and cell survival in rat growth plate chondrocytes. Biochim Biophys Acta. 2009;1793(5):836–46.
Seo EG, Norman AW. Three-fold induction of renal 25-hydroxyvitamin D3-24-hydroxylase activity and increased serum 24,25-dihydroxyvitamin D3 levels are correlated with the healing process after chick tibial fracture. J Bone Miner Res. 1997;12(4):598–606.
Seo EG, Einhorn TA, Norman AW. 24R,25-dihydroxyvitamin D3: an essential vitamin D3 metabolite for both normal bone integrity and healing of tibial fracture in chicks. Endocrinology. 1997;138(9):3864–72.
Husseini A, St-Arnaud R. CYP24A1-deficiency does not affect bone regeneration in distraction osteogenesis. J Steroid Biochem Mol Biol. 2016; doi:10.1016/j.jsbmb.2016.11.003.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Paul Anderson declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Skeletal Biology and Regulation
Rights and permissions
About this article

Cite this article
Anderson, P.H. Vitamin D Activity and Metabolism in Bone. Curr Osteoporos Rep 15, 443–449 (2017). https://doi.org/10.1007/s11914-017-0394-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-017-0394-8