
Abstract
The classic endogenous somnogen adenosine promotes sleep via A1 and A2A receptors. In this chapter, we present an overview of the current knowledge regarding the regulation of adenosine levels, adenosine receptors, and available pharmacologic and genetic tools to manipulate the adenosine system. This is followed by a summary of current knowledge of the role of adenosine and its receptors in the regulation of sleep and wakefulness. Despite strong data implicating numerous brain areas, including the basal forebrain, the tuberomammillary nucleus, the lateral hypothalamus, and the nucleus accumbens, in the adenosinergic control of sleep, the complete neural circuitry in the brain involved in the sleep-promoting effects of adenosine remains unclear. Moreover, the popular demand for natural sleep aids has led to a search for natural compounds that can promote sleep via adenosine receptor activation. Finally, we discuss the effects of caffeine in man and the possible use of more selective adenosine receptor drugs for the treatment of sleep disorders.
The authors dedicate this chapter to the late Prof. Osamu Hayaishi, whose demise saddened everyone who knew him as a great scientist and extraordinary individual.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Adeno-associated virus
- Astrocytes
- CGS 21680
- DREADD
- Istradefylline
- Modafinil
- Non-rapid eye movement sleep
- Optogenetics
- Prostaglandin D2
- Slow-wave sleep
1 The Concept of a Sleep Substance
The neural and cellular basis of the need for sleep or, alternatively, the “sleep drive” remains unresolved, but can be conceptualized as a homeostatic pressure that builds up during the waking period and dissipates during sleep. One potential mechanism is that the gradual accumulation of one or more endogenous somnogenic factors during wake is the underpinning of sleep homeostatic pressure. Rosenbaum presented the first formal hypothesis that sleep is regulated by humoral factors in 1892 (Rosenbaum 1892), and Ishimori (Ishimori 1909; Kubota 1989) and Pieron (Legendre and Pieron 1913) independently demonstrated the existence of sleep-promoting chemicals a few years later. Both Ishimori and Pieron proposed, and indeed established, the presence of hypnogenic substances or “hypnotoxins” in the cerebrospinal fluid of sleep-deprived dogs (Inoué et al. 1995). Over the past century, several additional putative hypnogenic substances implicated in the sleep homeostatic process have been identified [for review, see Urade and Hayaishi (2011)], including prostaglandin D2 (Qu et al. 2006; Ueno et al. 1982) [for review, see Urade and Lazarus (2013)], cytokines (Krueger et al. 1984) [for review, see Krueger et al. (2011)], adenosine (Porkka-Heiskanen et al. 1997), anandamide (Garcia-Garcia et al. 2009), and the urotensin II peptide (Huitron-Resendiz et al. 2005). Extensive evidence suggests that sleep regulation is interrelated with components of the host defense (immune) system, such as pro-inflammatory cytokines (Krueger and Majde 2003; Krueger et al. 2001; Mullington et al. 2000, 2001) and prostaglandins (Lazarus et al. 2007; Oishi et al. 2015; Urade and Lazarus 2013; Ushikubi et al. 1998). Several excellent reviews of the different theories of how neural switching occurs between sleep and wakefulness are available [for example, Fuller et al. (2015); Saper et al. (2005, 2010)]. In the present chapter, we focus on the possible role of adenosine as a sleep substance.
2 Adenosine in Physiology and Pathophysiology
Adenosine is a neuromodulator and not a neurotransmitter. Although it is released from nerve endings, its formation can be increased by various processes in all types of cells, and in all parts of these cells. Furthermore, the basal level of adenosine depends only on fundamental cell biology and is independent of nerve activity. Adenosine acts on four evolutionarily well-conserved receptors that are present on most if not all cells. Adenosine fulfills physiologic and pathophysiologic functions (Fredholm 2014).
2.1 Regulation of Adenosine Levels
Adenosine is formed by hydrolysis of adenosine monophosphate (AMP) or S-adenosylhomocysteine (Fredholm 2007; Schrader 1983). Adenosine is formed from S-adenosylhomocysteine by the enzyme S-adenosylhomocysteine hydrolase, which can also act to trap adenosine in the presence of excess l-homocysteine. This takes place intracellularly and the fact that the enzyme is bidirectional ensures the constant presence of a finite concentration of adenosine in the cell. The formation of adenosine from 5′-AMP can occur both intracellularly and extracellularly, mediated by different enzymes. The intracellular 5′nucleotidase generates adenosine, which can be used to generate AMP by adenosine kinase. This bidirectional reaction ensures the constant presence of a finite intracellular concentration of adenosine in the range of 10 to a few hundred nanomolar (nM) under physiologic conditions (Ballarin et al. 1991).
The fact that the intracellular concentration of adenosine is not zero ensures that there is also a not insubstantial extracellular concentration of adenosine, because all cells appear to possess one or more equilibrative purine transporters (Geiger and Fyda 1991). Extracellular adenosine is increased when an adenine nucleotide is released. Extracellular adenosine is formed by the conversion of ATP to adenosine by a series of ecto enzymes on the cell surface. Extracellular ATP can be released from various cell types by multiple mechanisms, including co-release from storage vesicles together with other hormones (neurotransmitter), a “kiss-and-run” mechanism (MacDonald et al. 2006), lysosome exocytosis (Zhang et al. 2007), controlled release through pannexin hemichannels (Chekeni et al. 2010; Elliott et al. 2009), release from inflammatory cells or vascular endothelia through connexin hemichannels and channels such as P2X7 receptors (Chen et al. 2006; Faigle et al. 2008; Linden 2006), and uncontrolled leakage from necrotic cells (Eltzschig 2009). Extracellular ATP and adenosine diphosphate (ADP) are broken down to AMP via many different ecto enzymes, especially CD39 (Yegutkin 2008). In the brain, the AMP formed is then broken down to adenosine only via ecto-5′-nucleotidase, CD73 (Resta et al. 1998). Importantly, the levels of extracellular adenine nucleotides are particularly high when cell membranes are broken, which allows the high intracellular content to escape. Thus, trauma in any form is associated with elevated levels of adenine nucleotides, as is blood sampling because most methods for this lead to a breakdown of blood platelets. AMP is also released directly from apoptotic cells (Yamaguchi et al. 2014).
ADP is released by platelets (Hollopeter et al. 2001), and ATP can be released by many different cells, including endothelial cells (Bodin and Burnstock 1998), astrocytes (Guthrie et al. 1999), and neurons (Fields and Stevens 2000), via many different mechanisms (Burnstock and Verkhratsky 2012). In the brain, extracellular adenosine might originate from neurons (both nerve terminals and postsynaptic components) and from surrounding non-neuronal cells such as glial cells (Halassa et al. 2007, 2009). For example, using inducible, astrocyte-specific transgenic dominant negative SNARE mouse approaches, Haydon’s group suggested that astrocytes are an important source of extracellular adenosine via gliotransmission (Halassa et al. 2009). ATP released from neurons (both nerve terminals and postsynaptic components) also contributes to extracellular adenosine production via the CD73 enzyme, the only enzyme that degrades AMP to adenosine in the brain (Lovatt et al. 2012; Wall and Dale 2013). In striatal neurons, extracellular adenosine formed via CD73 may preferentially act at the A2A receptor as extracellular CD73 is selectively co-expressed (Ena et al. 2013) and is physically associated with A2A receptors in striatopallidal neurons (Augusto et al. 2013). Under pathologic conditions, such as cortical seizures, adenosine-mediated synaptic depression is independent of CD73 activity and not a consequence of astrocytic (or neuronal) ATP release, but is due to the activation of postsynaptic neurons, which leads to the release of adenosine, thus constituting an autonomic feedback mechanism that suppresses excitatory transmission during prolonged activity (Lovatt et al. 2012). It may also be that under physiologic conditions, such as those involved in sleep and wakefulness, adenosine is generated in a similar CD73-independent manner.
Adenosine levels are decreased by the enzyme adenosine deaminase (ADA; this enzyme is particularly important when adenosine levels are high) and by uptake into cells other than those that produced it. In these cells, the adenosine taken up is rapidly phosphorylated to AMP by adenosine kinase. The formation and removal of extracellular adenosine determine its levels. Under basal conditions, these levels are low, usually in the order of 30–300 nM (Ballarin et al. 1991). Under more extreme conditions, such as mild hypoxia or strenuous exercise, the levels can approach 1 μM or more, and in severely traumatic situations, including local ischemia, can reach up to several tens of μM (Fredholm 2007).
To link adenosine levels to sleep, one must be able to measure adenosine levels in the brain but adenosine levels rapidly change in both the blood and tissue upon sampling and tissue samples must be frozen within a second or less to preserve in vivo levels. Such rapid inactivation is difficult to achieve, even with focused microwave techniques, and hence measuring regional adenosine brain levels by sampling tissue is highly problematic. Microdialysis could be a feasible method, but as already demonstrated in the first reports using this technique, it takes a long time to overcome the consequences of the initial traumatization required to insert the microdialysis probes (Ballarin et al. 1991; Benveniste et al. 1989; Zetterstrom et al. 1982). Furthermore, if the probe is allowed to remain in the tissue too long it will be covered with glial cells that hamper the exchange of purines (Benveniste et al. 1989). Electrochemical methods were recently used to measure local adenosine levels (Dale and Frenguelli 2012), but this method is also associated with caveats. Thus, reported adenosine levels in sleep and wakefulness should not be uncritically accepted.
2.2 Adenosine Receptors
Extracellular adenosine reacts with one of the four adenosine receptors, A1, A2A, A2B, and A3 (Fredholm et al. 2011). If these receptors are expressed at the same level (~200,000 receptors/cell), adenosine appears to be equally potent at A1, A2A, and A3 receptors (Fig. 1), and the levels of adenosine occurring under basal physiologic conditions are sufficient to activate these receptors. The data suggest that higher concentrations of adenosine are needed to activate A2B receptors. Nevertheless, it is important to remember that the potency of an agonist such as adenosine on its receptor depends on the number of receptors available, i.e., in the presence of only a few receptors higher adenosine concentrations are required to see an effect. Local expression of the A1 and A2A receptors appears to be higher than that of the other two receptors (Fredholm et al. 2005a). Thus, these two receptor types may be primarily involved in sleep regulation.

Schematic illustration of the ability of adenosine to activate the four adenosine receptors. Note that A1, A2A, and A3 receptors are activated by basal levels of adenosine at sites where the receptor number is high. In contrast, A2B receptors are mostly activated in pathologic conditions
First, we briefly discuss how the role(s) of adenosine in physiology and pathophysiology can be determined.
3 How Do We Learn About the Roles of Adenosine?
3.1 Receptor Antagonists Including Caffeine
To assess the in vivo actions of adenosine receptors, selective pharmacologic tools are crucial. Over the last 20 years, medicinal chemistry has generated agonists and antagonists with high affinity (Kd values in the low nM range) and selectivity (>100–200-fold over other adenosine receptor subtypes) for the human variants of each of the four receptors (Fredholm et al. 2011). Most of the known adenosine receptor agonists are derivatives of purine nucleosides, either adenosine or xanthosine, while adenosine receptor antagonists have diverse structures (Müller and Jacobson 2011). Many A2A-selective antagonists from different structural classes have been developed, including 8-(3-chlorostyryl)caffeine, MSX-2, and its water-soluble prodrugs MSX-3, ZM 241385, SCH-58261, and KW6002. In addition, radioactive, and more recently, fluorescent, ligands of adenosine receptors were also developed and introduced for drug screening and monitoring in vivo receptor occupancy in humans.
Caffeine, the most widely consumed arousal-promoting psychostimulant, is a classic nonselective adenosine receptor antagonist, although it is rather weakly potent at adenosine receptors (K i ≈ 10 μM). At doses commonly consumed by humans, caffeine produces its profound arousal effect by partial (estimated to be 25–50%) and nonselective (similar affinity for both A1 and A2A receptors) blockade of adenosine receptors (Fredholm et al. 1999). Caffeine is metabolized to paraxanthine and theophylline (Arnaud 2011). These metabolites are more potent than caffeine as inhibitors of A1 and A2A receptors. Therefore, elimination of caffeine does not predict the elimination of adenosine receptor blockade and hence the effects of caffeine administration.
Importantly, several A2A receptor antagonists are in clinical trials for Parkinson’s disease (PD), including istradefylline (KW6002), SYN-115, and preladenant (SCH-442416). Phase IIB and III clinical trials with A2A receptor antagonists showed a very consistent and excellent safety profile in more than 3,000 patients with advanced PD (Hauser et al. 2011; Jenner et al. 2009). The safety profile of these A2A receptor antagonists is entirely consistent with the widespread use of the nonselective adenosine receptor antagonist caffeine in 70% of the human population. Importantly, this provides an opportunity to rapidly translate A2A receptor antagonists to achieve pharmacologic control of the sleep–wake cycle.
3.2 Receptor Knockouts and Other Genetic Targeting Techniques
Over the past two decades, genetic knockout (KO) models for all four G-protein-coupled adenosine receptors were generated by targeted deletion of critical exons (Fredholm et al. 2005b; Wei et al. 2011). These adenosine receptor KO models have provided insights into the physiologic function of modulation of the sleep–wake cycle by overcoming the limitations of pharmacologic agents with partial specificity and by targeting the adenosine receptor in defined cellular populations. For example, the use of A2A receptor KO models can overcome concerns about the partial specificity of A2A receptor antagonists (particularly after focal injection at relatively high concentrations), and convincingly demonstrated that the sleep-promoting effect of A2A receptor agonists and caffeine-induced arousal effect are mediated by A2A receptors (not A1 receptors). Global A1 and A2A receptor KO approaches, however, have intrinsic limitations of the confounding developmental effect and lack of cell-type specificity (Fredholm et al. 2005b). To overcome these limitations, conditional KO of some adenosine receptor genes in defined brain regions (e.g., forebrain versus striatum) and cell-type (e.g., neurons versus astrocytes) has been achieved using the Cre-loxP system [for review see Fuller et al. (2015); Wei et al. (2011)]. Brain-regional deletion of A2A receptors has been achieved in the forebrain (i.e., striatum, cortex, hippocampus) (Bastia et al. 2005; Yu et al. 2008) and striatum (Shen et al. 2008). Local deletion of A1 receptors in hippocampal CA1 or CA3 neurons and A2A receptors in the nucleus accumbens (NAc) has also been achieved by local injection of adeno-associated virus (AAV) vectors containing the cre transgene into the brains of mice carrying loxP-flanked A1 receptor (Scammell et al. 2003) or A2A receptor (Lazarus et al. 2011) genes. The conditional KO strategy permits a temporal and regional specificity that has uncovered previously under-appreciated functions of adenosine receptors in the basal ganglia for controlling the sleep–wake cycle (see detailed discussion below). In addition, the development of AAV carrying short-hairpin RNA targeted to produce site-specific silencing of the A2A receptor gene allowed for the clear demonstration in rats that the arousal effect of caffeine is mediated by A2A receptors in the NAc shell (Lazarus et al. 2011). Lastly, the recent development of optogenetics based on specific local modulation of neuronal activity using genetically engineered optical switches (e.g., channelrhodopsin) (Boyden et al. 2005; Deisseroth 2014; Yizhar et al. 2011) or chemogenetics to study G-protein signaling in freely behaving animals by the directed molecular evolution of designer receptors exclusively activated by designer dugs (DREADD) (Farrell et al. 2013; Giguere et al. 2014) has refined our understanding of novel brain circuits underlying the sleep–wake cycle (Fuller et al. 2015). Recently, a probe for selective optogenetic control of A2A receptor signaling (optoA2A receptor) was developed (Li et al. 2015).
4 Adenosine and Sleep
4.1 Adenosine Levels During Sleep and Wakefulness
Adenosine has long been known to represent a state of relative energy deficiency: ATP depletion and the elevation of extracellular adenosine levels are positively correlated (Kalinchuk et al. 2003) and positively associated with sleep (Porkka-Heiskanen et al. 1997). Adenosine levels in samples collected from several brain areas of cats during spontaneous sleep–wake cycles by in vivo microdialysis were higher during non-rapid eye movement (non-REM, NREM) sleep than wakefulness for all probed brain areas (Porkka-Heiskanen et al. 1997, 2000). Moreover, in vivo microdialysis experiments in the brain of cats also revealed that adenosine concentrations specifically increase twofold in the basal forebrain (BF) during a prolonged 6-hour period of wakefulness compared with that at the beginning of sleep deprivation (Porkka-Heiskanen et al. 1997, 2000).
Sixty years have passed since the discovery of the hypnotic effect of adenosine in the mammalian brain (Feldberg and Sherwood 1954); however, the brain cell types involved in the sleep-promoting effects of adenosine remain unclear. In principle, adenosine (and ATP, which is rapidly degraded to adenosine) can be released from neurons or glia cells. Genetically engineered mice that selectively express a dominant negative SNARE domain in astrocytes to nonspecifically block the release of ATP exhibit decreased concentrations of extracellular adenosine (Pascual et al. 2005). Although the amount of wakefulness, NREM, and REM sleep in these mice is indistinguishable from that in wild-type mice, these mice exhibit reduced slow-wave activity and recovery sleep after sleep deprivation (Halassa et al. 2009), suggesting that adenosine released from astrocytes is involved in the accumulation of sleep pressure. Direct proof is still lacking, however, and thus the exact sources of adenosine remain unknown.
The late Miodrag Radulovacki and colleagues extensively investigated the effects of adenosine on wakefulness. They used the ADA inhibitor deoxycoformycin to increase the levels of adenosine in the central nervous system of rats and found that REM and NREM sleep were increased (Radulovacki et al. 1983), further supporting a hypnotic role for adenosine.
Adenosine is reported to promote sleep by acting through A1 or A2A receptors, but the relative contribution of these receptors to sleep induction remains controversial (Basheer et al. 2004; Huang et al. 2007). Indirect evidence by comparison of the effects of caffeine, the A1 receptor antagonist 8-cyclopentyltheophylline, and the nonselective A1/A2A receptor antagonist alloxazine on sleep in rats (Virus et al. 1990) might partially account for the prevailing opinion that the A1 receptor is more important in sleep–wake regulation than the A2A receptor. The aforementioned pharmacologic approach and related studies, however, have non-trivial limitations, particularly with respect to data interpretation. For example, receptor antagonists are difficult to compare due to differences in solubility, blood–brain-barrier permeability, and neuropharmacodynamics, and most importantly, have “off-target” effects, especially at higher concentrations. Moreover, the diffuse expression of inhibitory A1 receptors in the brain may have differential effects on sleep and wakefulness in a region-specific manner (Ochiishi et al. 1999a, b; Reppert et al. 1991; Rivkees et al. 1995). The advent of genetically engineered systems, including transgenic animals and recombinant viral vectors, and findings in humans have convincingly established over the last decade a pivotal role of A2A receptors in the regulation of sleep and wakefulness (Holst and Landolt 2015; Lazarus et al. 2012, 2013).
4.2 Effects of A1 Receptors
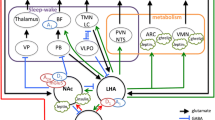
The A1 receptor agonist N6-cyclopentyladenosine produces dose-dependent increases in slow-wave activity in electroencephalography during NREM sleep when administered systemically or intracerebroventricularly in rats (Benington et al. 1995), but lateral ventricle infusions of N6-cyclopentyladenosine in mice do not change the amounts of observed NREM and REM sleep (Urade et al. 2003), which may indicate opposing effects on sleep and wakefulness in different areas of the brain. For example, adenosine acting via A1 receptors induces sleep by inhibiting arousal-related cell groups in the BF, such as the horizontal limb of the diagonal band of Broca and the substantia innominata (Fig. 2a) (Alam et al. 1999; Strecker et al. 2000). Moreover, adenosine may promote sleep by A1 receptor-mediated inhibition of glutamatergic inputs to cortically projecting cholinergic and γ-aminobutyric acid (GABA) neurons of the BF (Yang et al. 2013). Adenosine could also promote sleep by suppressing hypocretin/orexin neurons in the lateral hypothalamus, because an A1 receptor agonist produced NREM and REM sleep and the receptor’s antagonist induced wakefulness (Fig. 2b) (Thakkar et al. 2008). ADA is predominantly localized in the tuberomammillary nucleus (TMN) of the brain and the TMN is enriched in histamine neurons containing A1 receptors, thereby suggesting that the histaminergic arousal system is actively regulated by adenosine in the TMN. In fact, bilateral injections of the A1 receptor agonist N 6 -cyclopentyladenosine into the rat TMN significantly increase the amount of NREM sleep (Fig. 2c) (Oishi et al. 2008). Moreover, bilateral injections of adenosine or the ADA inhibitor coformycin into the rat TMN also increase NREM sleep, which is completely abolished by co-administration of the selective A1 receptor antagonist 1,3-dimethyl-8-cyclopenthylxanthine. These results indicate that endogenous adenosine in the TMN suppresses the histaminergic system via A1 receptors to promote NREM sleep. Interestingly, single-nucleotide polymorphism analyses have identified a human genetic ADA variant with low enzymatic activity that is linked to the enhancement of deep sleep and slow-wave activity during sleep (Bachmann et al. 2012; Mazzotti et al. 2012; Rétey et al. 2005). By contrast, activation of A1 receptors in the lateral preoptic area of the hypothalamus by local infusion of an A1 receptor agonist promotes wakefulness (Methippara et al. 2005).

Circuit basis of sleep–wake regulation. Model 1 (shown in panel a): adenosine inhibits the release of acetylcholine from the basal forebrain (BF) cholinergic neurons to produce slow-wave sleep. Model 2 (shown in panels b–d): a flip–flop switching mechanism involving mutually inhibitory interactions between sleep-promoting neurons in the ventrolateral preoptic area (VLPO) and wake-promoting neurons in the hypothalamus [i.e., histaminergic tuberomammillary nucleus (TMN)], and brainstem [i.e., noradrenergic locus coeruleus (LC), serotonergic dorsal raphe nucleus (DR), and cholinergic laterodorsal tegmental nucleus (LDT)]. The flip-flop switch between the VLPO and hypothalamus and brainstem is stabilized by orexin/hypocretin (OX/Hcrt) inputs from the lateral hypothalamus (LHA). Adenosine acts as an endogenous somnogen and promotes sleep via inhibitory A1 receptors (A1) in the basal forebrain, VLPO, LHA, and TMN and excitatory A2A receptors (A2A) in the nucleus accumbens (NAc) and VLPO (Huang et al. 2007, 2011; Lazarus et al. 2012, 2013). Other abbreviations: Ach acetylcholine, 5-HT serotonin, NE norepinephrine
4.3 Effects of A2A Receptors
CGS 21680, a highly selective A2A receptor agonist, produces profound increases in NREM and REM sleep after infusion into the subarachnoid space underlying the ventral surface region of the rostral BF in rats or into the lateral ventricle of mice (Satoh et al. 1996; Urade et al. 2003). In vivo microdialysis experiments demonstrated that infusions of CGS 21680 into the BF inhibit the release of histamine in both the frontal cortex and medial preoptic area in a dose-dependent manner, and increase the release of GABA in the TMN of the hypothalamus, but not in the frontal cortex (Hong et al. 2005). CGS 21680-induced blocking of histamine release is antagonized when the TMN is perfused with the GABA antagonist picrotoxin, suggesting that the A2A receptor agonist induces sleep by inhibiting the histaminergic system through an increase in GABA release in the TMN. It was previously proposed that sleep is promoted by activating sleep neurons in the ventrolateral preoptic area (VLPO) and reciprocal suppression of histaminergic wake neurons in the TMN through GABAergic and galaninergic inhibitory projections (Sherin et al. 1996, 1998). The existence of two distinct types of VLPO neurons in terms of their responses to serotonin and adenosine was demonstrated by intracellular recordings of VLPO neurons in rat brain slices (Fig. 2b). VLPO neurons are uniformly inhibited by the arousing neurotransmitters noradrenaline and acetylcholine, and primarily inhibited by an A1 receptor agonist. Serotonin inhibits type-1 neurons but excites type-2 neurons, whereas an A2A receptor agonist postsynaptically excites type-2, but not type-1 neurons. These results implicate the involvement of type-2 neurons in the initiation of sleep, whereas type-1 neurons contribute to sleep consolidation as they are only activated in the absence of inhibitory effects from the arousal systems (Gallopin et al. 2005).
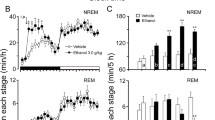
The administration of CGS 21680 into the rostral BF, however, produces c-fos expression not only in the VLPO, but also within the NAc shell and the medial portion of the olfactory tubercle (Satoh et al. 1999; Scammell et al. 2001). Interestingly, direct perfusion of the A2A receptor agonist into the NAc induces NREM and REM sleep that corresponds to about three-quarters of the amount of sleep measured when the A2A receptor agonist is infused into the subarachnoid space (Satoh et al. 1999). These results may indicate that A2A receptors within or close to the NAc predominantly promote sleep (Fig. 2a). Acting opposite to adenosine, caffeine enhances wakefulness because it acts as an antagonist of both A1 and A2A receptor subtypes (Fredholm et al. 1999). Experiments using global genetic KOs of the A1 and A2A receptors revealed that A2A receptors, but not A1 receptors, mediate the arousal-inducing effect of caffeine (Huang et al. 2005). The specific role of A2A receptors in the basal ganglia (BG) was investigated using powerful tools for site-specific gene manipulations, such as conditional KO mice of the A2A receptor based on the Cre/lox technology or local infection with AAV carrying short-hairpin RNA of A2A receptors to silence the expression of the receptor subtype (Lazarus et al. 2011). Deletion of A2A receptors selectively in the NAc shell blocked caffeine-induced wakefulness (Fig. 3). Excitatory A2A receptors within the NAc shell must be tonically activated by adenosine for caffeine to be effective as an A2A receptor antagonist. This tonic activation probably occurs in the NAc shell because sufficient levels of adenosine are available under basal conditions and A2A receptors are abundantly expressed throughout the striatum, including the NAc shell (Rosin et al. 1998; Svenningsson et al. 1999). Thus, activation of A2A receptors in the NAc shell contributes to the restraint of the arousal system, whereby caffeine overrides the “adenosine brake” to promote wakefulness. Interestingly, deletion of the dopamine transporter (DAT), which is responsible for the re-uptake of dopamine (Giros and Caron 1993), in mice reduces NREM sleep, increases wakefulness, and unmasks hypersensitivity to the wake-promoting effects of caffeine (Wisor et al. 2001). The last observation may indicate that the expression of NAc D2 receptors working opposite to A2A receptors is involved in the arousal effect of modafinil, a wakefulness-promoting compound. Despite the fact that stimulating A2A receptors leads to decreased affinity for dopamine at D2 receptors via intramembrane interactions and to a reduction in Gi-protein coupling of the D2 receptor for the inhibition of cAMP production (Fuxe et al. 2003), adenosine and its antagonists, such as caffeine, can modulate the activity of medium spiny projection neurons in the striatum via A2A receptors independently of D2 receptors (Aoyama et al. 2000; Chen et al. 2001). Interestingly, humans with a genetic reduction of striatal DAT show an elevated sensitivity to caffeine stimulation and increased homeostatic response when they are sleep deprived (Holst and Landolt 2015).

The arousal effects of caffeine are abolished in rats with site-specific deletion of A2A receptors (A2AR) in the shell of the nucleus accumbens (NAc). To identify the neurons on which caffeine acts to produce arousal, A2A receptors were focally depleted by bilateral injections of adeno-associated virus carrying short-hairpin RNA for the A2A receptor into the core (dashed green line in the left panel) or shell (dashed red line in the right panel) of the nucleus accumbens of rats (Lazarus et al. 2011). Typical hypnograms that show changes in wakefulness and in rapid eye movement (REM) and non-REM (NREM) sleep after administration of caffeine at a dose of 15 mg/kg indicate that rats with shell, but not core, knockdown of the A2A receptors showed a strongly attenuated caffeine arousal. Green and red areas in the hypnograms represent wakefulness after caffeine administration that correspond to the depletion of A2A receptors in the core and shell of the nucleus accumbens, respectively
The study of NAc adenosine-mediated modulation of the sleep–wake cycle led to a new proposal that the BG represents a key structural element for the control of sleep and wakefulness (Lazarus et al. 2011, 2012). Dysfunction in the BG, such as PD and Huntington’s disease and lesions in the BG, results in a wide range of disorders of movement, cognition, and sleep–wake function (Adler and Thorpy 2005; Dale et al. 2004; Goodman and Barker 2010; Obeso et al. 2000; Wetter et al. 2000). In fact, bilateral lesions using ibotenic acid to kill intrinsic neurons in the striatum (caudoputamen), internal and external globus pallidus (GPe), subthalamic nucleus, substantia nigra (pars reticulata or compacta), or thalamus revealed that bilateral lesions made in the striatum or specifically in the NAc result in a significant reduction in time spent in wakefulness and fragmentation of both sleep and wakefulness (Qiu et al. 2010). Only lesions in the caudate–putamen and globus pallidus increased sleep by 10% and wake by 50%, respectively (Qiu et al. 2010). Moreover, the complete deletion of D2 receptors, which are prominently, albeit not exclusively, expressed in the BG, significantly decreases wakefulness with a concomitant increase in NREM and REM sleep and a drastic decrease in the NREM sleep delta power (Qu et al. 2010). Excessive sleepiness in PD and other sleep disorders are commonly treated with modafinil (Hogl et al. 2002; Minzenberg and Carter 2007) and interestingly, the arousal effect of modafinil is exclusively mediated by D1 and D2 receptors, with D2 receptors being of primary importance (Qu et al. 2008). Based on these findings, it was proposed that activation of A2A receptors leads to enhanced activity of GABAergic output neurons in the striatopallidal pathway and subsequently arousal systems in the thalamus, hypothalamus, brainstem, and ultimately the cerebral cortex are maintained under a tight inhibitory control. In fact, stereotaxic-based brain microinjections of Cre-recombinase-dependent AAV vectors carrying channelrhodopsin or DREADD into the NAc of transgenic mice in which Cre-recombinase is expressed under the A2A-receptor promoter robustly induced NREM sleep during selective activation of striatopallidal neurons by light or the small molecule clozapine-N-oxide (Oishi Y, Xu Q, et al., unpublished).
Moreover, a recent study found that blocking A2A receptors or A2A receptor-expressing neurons in the olfactory bulb of rodents increases REM sleep, suggesting the possibility that the olfactory bulb is a key site for regulating REM sleep by the adenosine/A2A receptor system (Wang et al. 2016). Because olfactory dysfunction can be ameliorated with an A2A receptor antagonist, for example, caffeine or ZM 241385 (Prediger et al. 2005), it is possible that REM sleep and the perception of odors are linked in the olfactory bulb. Interestingly, the ability to smell is reduced in patients with REM sleep behavior disorder (Stiasny-Kolster et al. 2007).
4.4 Effects of Natural Compounds on Sleep and Wakefulness via Adenosine Receptors
Several studies recently demonstrated that a variety of natural compounds promote sleep via adenosine receptor activation. In strong support of the role of A2A receptors in the regulation of sleep, Japanese sake yeast supplementation improves the quality of sleep in humans (Monoi et al. 2016) and sake yeast-induced NREM sleep was abolished in mice by pretreatment with the A2A receptor antagonist ZM 241385 (Nakamura et al. 2016). Because sake yeast, but not other Saccharomyces cerevisiae yeasts (e.g., baker’s and brewer’s yeast), contains a large amount of S-adenosyl-l-methionine and the S-adenosyl-l-methionine metabolite methylthioadenosine, Urade and colleagues suggested that the sleep-inducing effect of sake yeast is likely due to the activation of A2A receptors by S-adenosyl-l-methionine or methylthioadenosine.
In contrast, paeoniflorin, one of the principal active ingredients of Paeonia Radix, shortens sleep latency and increases the amount of NREM sleep exclusively via the activation of A1 receptors, a conclusion based on the finding that paeoniflorin effects can be blocked by treatment with an A1 receptor antagonist and are absent in A1 receptor-KO mice (Chen et al. 2015). In addition, paeoniflorin significantly increases the mechanical pain threshold, prolongs the thermal latency, and increases NREM sleep in partial sciatic nerve ligation mice, a mouse neuropathic pain model characterized by persistent pain and insomnia (Yin et al. 2015). Therefore, the A1 receptor-mediated analgesic and hypnotic effects of paeoniflorin may be of potential use for the treatment of neuropathic pain and associated insomnia.
Moreover, N6-(4-hydroxybenzyl) adenine riboside isolated from Gastrodia elata has hypnotic effects in mice (Zhang et al. 2012) and may dose-dependently increase NREM sleep via mechanisms that involve A1 and A2A receptors. Finally, cordycepin (3-deoxyadenosine), an adenosine analogue isolated from Cordyceps fungi, promotes NREM sleep in rats, but it remains unclear if the sleep-inducing effect is, in fact, mediated by adenosine receptors (Hu et al. 2013).
5 Effects of Caffeine in Man and the Possible Role of More Selective Adenosine Receptor Drugs
Whereas the above data linking adenosine to sleep were obtained from animal experiments, the evidence suggests that this link also holds true in humans. This is largely due to the well-known effects of caffeine on sleep. Caffeine is the most consumed psychoactive compound in the world. It is readily available through dietary products, such as coffee, tea, soft drinks, and chocolate, but it is also added to non-prescription medications, such as pain-relievers and cold remedies. Regardless of the source, worldwide average caffeine consumption is estimated to be just under 80 mg/d, although the levels of intake in countries such as Sweden and Finland are in the range of 400 mg caffeine per day (Fredholm et al. 1999). Caffeine is widely used to promote wakefulness and to counteract fatigue in doses that are well in the range where adenosine antagonism is the dominant effect. Some individuals, however, experience anxiety and panic attacks (Chait 1992; Evans and Griffiths 1991) at normal consumption levels, and this is more common at higher doses. One study found that people with polymorphisms at the A2A-receptor-gene are at risk of experiencing increased anxiety when consuming coffee, tea, energy drinks, or other caffeine-containing products (Alsene et al. 2003). A2A receptor polymorphisms also consistently modulate the objective and subjective effects of caffeine on sleep quality and electroencephalogram (Bodenmann et al. 2012; Byrne et al. 2012; Rétey et al. 2007).
Whether caffeine affects circadian rhythm and thereby alters the timing of sleep is widely unknown; however, recent developments revealed caffeine effects on the mammalian circadian clock. For example, caffeine delays the human circadian melatonin rhythm by blocking A1 receptors (Burke et al. 2015). Chronic treatment with caffeine lengthens the circadian period of molecular oscillations in human osteosarcoma U2OS cells expressing clock gene luciferase reporters. Further, application of pharmacologic tools and small-interfering RNA knockdown revealed that the effect of caffeine on molecular oscillation is attenuated by perturbation of A1 receptor signaling but not ryanodine receptor or phosphodiesterase activity. This finding establishes a possible molecular mechanism for the clinical observation in a double-blind, placebo-controlled, ~49-day long, within-subject study that bedtime caffeine consumption induces a ~40-min phase delay of the circadian melatonin rhythm in humans. Another study revealed that caffeine increases the light sensitivity of the mouse circadian clock (van Diepen et al. 2014).
Society demands the means to bend sleep to the needs of modern lifestyle instead of the other way around and thus sleep-avoidance has become a popular research topic. Scientists and clinicians worldwide are searching for new methods of keeping people alert on limited sleep. Much more about the effects of coffee and other beverages containing caffeine on sleep in man will be covered elsewhere in this book [for review, see Clark and Landolt (2017)]. Moreover, consistent with the fact that the arousal effect of caffeine in mice is exclusively mediated by A2A receptors, emerging evidence supports the modulation of the sleep–wake cycle by A2A receptor antagonists. For example, the newly developed dual adenosine A2A/A1 receptor antagonist JNJ-40255293 enhances wakefulness (Atack et al. 2014). Moreover, since the clinical approval of the A2A receptor antagonist istradefylline (KW-6002) for motor improvement in PD in Japan in 2013, a report of four cases indicated that evening treatment with this antagonist reduces sleep duration in the evening and increases daytime sleepiness in patients (Matsuura and Tomimoto 2015). Thus, A2A receptor antagonists may have considerable potential as eugeroics (wakefulness enhancing drugs) while avoiding some of the aforementioned A2A-independent side effects of caffeine (such as anxiety and disturbance of the circadian rhythm) or negative effects of other psychostimulants, including dependence.
The possibility that stimulation of adenosine receptors could be used to promote sleep should also be considered. Currently, there are 60 million prescriptions for sleeping pills in the USA each year, 43 million of which are for the nonbenzodiazepine zolpidem, also known as Ambien. Benzodiazepines and nonbenzodiazepines, both of which enhance the effect of the neurotransmitter GABA at the GABAA receptor, are used for the treatment of insomnia and poor sleep quality despite their wide range of disadvantages and safety issues, ranging from low sleep quality by increasing light sleep at the expense of physiological deep sleep to side effects (e.g., next-day sedation, cognitive impairment, and amnesic effects) and the development of tolerance and dependence by long-term administration. Because an A2A receptor agonist strongly increases sleep in wild-type mice (Satoh et al. 1996; Urade et al. 2003), pharmacologic A2A receptor activation may be an alternative strategy for the treatment of insomnia. Due to the lack of brain-permeability, however, not all currently existing A2A receptor agonists are suitable for treating the nervous system. If the observation in animals that adenosine levels are elevated during prolonged wakefulness holds also true for humans, a freely penetrating allosteric modulator may effectively enhance the sleep-inducing effect of endogenous adenosine and help people with insomnia to fall asleep.
6 Conclusion
Here we demonstrated that adenosine is indeed one of the several somnogenic substances that act in concert to ensure normal sleep–wake patterns. Adenosine tends to increase sleep, but the source of the adenosine is still poorly understood. Many cells and processes could play a role. Similarly, adenosine promotes sleep by several mechanisms, in several locations, via A1 or A2A receptors. We emphasized A1 receptor-mediated effects on histamine neurons and the A2A receptor-mediated effects in the NAc. Both of these receptors are antagonized by caffeine – to a large extent explaining the awakening effect of this common drug.
References
Adler CH, Thorpy MJ (2005) Sleep issues in Parkinson’s disease. Neurology 64:S12–S20
Alam MN, Szymusiak R, Gong H, King J, McGinty D (1999) Adenosinergic modulation of rat basal forebrain neurons during sleep and waking: neuronal recording with microdialysis. J Physiol 521:679–690. doi:10.1111/j.1469-7793.1999.00679.x
Alsene K, Deckert J, Sand P, de Wit H (2003) Association between A2a receptor gene polymorphisms and caffeine-induced anxiety. Neuropsychopharmacology 28:1694–1702. doi:10.1038/sj.npp.1300232
Aoyama S, Kase H, Borrelli E (2000) Rescue of locomotor impairment in dopamine D2 receptor-deficient mice by an adenosine A2A receptor antagonist. J Neurosci 20:5848–5852
Arnaud MJ (2011) Pharmacokinetics and metabolism of natural methylxanthines in animal and man. Handb Exp Pharmacol 200:33–91. doi:10.1007/978-3-642-13443-2_3
Atack JR et al (2014) JNJ-40255293, a novel adenosine A2A/A1 antagonist with efficacy in preclinical models of Parkinson’s disease. ACS Chem Neurosci 5:1005–1019. doi:10.1021/cn5001606
Augusto E et al (2013) Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J Neurosci 33:11390–11399. doi:10.1523/jneurosci.5817-12.2013
Bachmann V et al (2012) Functional ADA polymorphism increases sleep depth and reduces vigilant attention in humans. Cereb Cortex 22:962–970. doi:10.1093/cercor/bhr173
Ballarin M, Fredholm BB, Ambrosio S, Mahy N (1991) Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand 142:97–103. doi:10.1111/j.1748-1716.1991.tb09133.x
Basheer R, Strecker RE, Thakkar MM, McCarley RW (2004) Adenosine and sleep–wake regulation. Prog Neurobiol 73:379–396. doi:10.1016/j.pneurobio.2004.06.004
Bastia E, Xu YH, Scibelli AC, Day YJ, Linden J, Chen JF, Schwarzschild MA (2005) A crucial role for forebrain adenosine A(2A) receptors in amphetamine sensitization. Neuropsychopharmacology 30:891–900. doi:10.1038/sj.npp.1300630
Benington JH, Kodali SK, Heller HC (1995) Stimulation of A1 adenosine receptors mimics the electroencephalographic effects of sleep deprivation. Brain Res 692:79–85. doi:10.1016/0006-8993(95)00590-m
Benveniste H, Hansen AJ, Ottosen NS (1989) Determination of brain interstitial concentrations by microdialysis. J Neurochem 52:1741–1750. doi:10.1111/j.1471-4159.1989.tb07252.x
Bodenmann S, Hohoff C, Freitag C, Deckert J, Rétey JV, Bachmann V, Landolt HP (2012) Polymorphisms of ADORA2A modulate psychomotor vigilance and the effects of caffeine on neurobehavioural performance and sleep EEG after sleep deprivation. Br J Pharmacol 165:1904–1913. doi:10.1111/j.1476-5381.2011.01689.x
Bodin P, Burnstock G (1998) Increased release of ATP from endothelial cells during acute inflammation. Inflamm Res 47:351–354
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 8:1263–1268. doi:10.1038/nn1525
Burke TM et al (2015) Effects of caffeine on the human circadian clock in vivo and in vitro. Sci Transl Med 7:146–305. doi:10.1126/scitranslmed.aac5125
Burnstock G, Verkhratsky A (2012) Mechanisms of ATP release and inactivation. In: Purinergic signalling and the nervous system. Springer, Berlin Heidelberg, pp 79–118. doi:10.1007/978-3-642-28863-0_4
Byrne EM, Johnson J, McRae AF, Nyholt DR, Medland SE, Gehrman PR, Heath AC, Madden PA, Montgomery GW, Chenevix-Trench G, Martin NG (2012) A genome-wide association study of caffeine-related sleep disturbance: confirmation of a role for a common variant in the adenosine receptor. Sleep 35:967–975. doi:10.5665/sleep.1962
Chait LD (1992) Factors influencing the subjective response to caffeine. Behav Pharmacol 3:219–228
Chekeni FB et al (2010) Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 467:863–867. doi:10.1038/nature09413
Chen J-F et al (2001) The role of the D2 dopamine receptor (D2R) in A2A adenosine receptor (A2AR)-mediated behavioral and cellular responses as revealed by A2A and D2 receptor knockout mice. Proc Natl Acad Sci U S A 98:1970–1975. doi:10.1073/pnas.98.4.1970
Chen Y et al (2006) ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314:1792–1795. doi:10.1126/science.1132559
Chen C et al (2015) Paeoniflorin promotes non-rapid eye movement sleep via adenosine A1 receptors. J Pharmacol Exp Ther 356:64–73. doi:10.1124/jpet.115.227819
Clark I, Landolt HP (2017) Coffee, caffeine, and sleep: a systematic review of epidemiological studies and randomized controlled trials. Sleep Med Rev 31:70–78. doi:10.1016/j.smrv.2016.01.006
Dale N, Frenguelli BG (2012) Measurement of purine release with microelectrode biosensors. Purinergic Signal 8:27–40. doi:10.1007/s11302-011-9273-4
Dale RC et al (2004) Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain 127:21–33. doi:10.1093/brain/awh008
Deisseroth K (2014) Circuit dynamics of adaptive and maladaptive behaviour. Nature 505:309–317. doi:10.1038/nature12982
Elliott MR et al (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461:282–286. doi:10.1038/nature08296
Eltzschig HK (2009) Adenosine: an old drug newly discovered. Anesthesiology 111:904–915. doi:10.1097/ALN.0b013e3181b060f2
Ena SL, De Backer J-F, Schiffmann SN, de Kerchove d’Exaerde A (2013) FACS array profiling identifies Ecto-5′ nucleotidase as a striatopallidal neuron-specific Gene involved in striatal-dependent learning. J Neurosci 33:8794–8809. doi:10.1523/jneurosci.2989-12.2013
Evans SM, Griffiths RR (1991) Dose-related caffeine discrimination in normal volunteers: individual differences in subjective effects and self-reported cues. Behav Pharmacol 2:345–356
Faigle M, Seessle J, Zug S, El Kasmi KC, Eltzschig HK (2008) ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS One 3:e2801. doi:10.1371/journal.pone.0002801
Farrell MS et al (2013) A Galphas DREADD mouse for selective modulation of cAMP production in striatopallidal neurons. Neuropsychopharmacology 38:854–862. doi:10.1038/npp.2012.251
Feldberg W, Sherwood SL (1954) Injections of drugs into the lateral ventricle of the cat. J Physiol 123:148–167
Fields RD, Stevens B (2000) ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci 23:625–633. doi:10.1016/S0166-2236(00)01674-X
Fredholm BB (2007) Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 14:1315–1323
Fredholm BB (2014) Adenosine – a physiological or pathophysiological agent? J Mol Med 92:201–206. doi:10.1007/s00109-013-1101-6
Fredholm BB, Bättig K, Holmén J, Nehlig A, Zvartau EE (1999) Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev 51:83–133
Fredholm BB, Chen J-F, Cunha RA, Svenningsson P, Vaugeois J-M (2005a) Adenosine and brain function. Int Rev Neurobiol 63:191–270. doi:10.1016/S0074-7742(05)63007-3
Fredholm BB, Chen JF, Masino SA, Vaugeois JM (2005b) Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol 45:385–412. doi:10.1146/annurev.pharmtox.45.120403.095731
Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Muller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors – an update. Pharmacol Rev 63:1–34. doi:10.1124/pr.110.003285
Fuller PM, Yamanaka A, Lazarus M (2015) How genetically engineered systems are helping to define, and in some cases redefine, the neurobiological basis of sleep and wake. Temperature 2:406–417. doi:10.1080/23328940.2015.1075095
Fuxe K et al (2003) Receptor heteromerization in adenosine A2A receptor signaling: relevance for striatal function and Parkinson’s disease. Neurology 61:S19–S23
Gallopin T et al (2005) The endogenous somnogen adenosine excites a subset of sleep-promoting neurons via A2A receptors in the ventrolateral preoptic nucleus. Neuroscience 134:1377–1390
Garcia-Garcia F, Acosta-Pena E, Venebra-Munoz A, Murillo-Rodriguez E (2009) Sleep-inducing factors. CNS Neurol Disord Drug Targets 8:235–244
Geiger JD, Fyda DM (1991) Adenosine transport in nervous system tissues. In: Stone TW (ed) Adenosine in the nervous system. Academic Press, London, pp 1–23. doi:10.1016/B978-0-12-672640-4.50007-8
Giguere PM, Kroeze WK, Roth BL (2014) Tuning up the right signal: chemical and genetic approaches to study GPCR functions. Curr Opin Cell Biol 27:51–55. doi:10.1016/j.ceb.2013.11.006
Giros B, Caron MG (1993) Molecular characterization of the dopamine transporter. Trends Pharmacol Sci 14:43–49. doi:10.1016/0165-6147(93)90029-J
Goodman A, Barker R (2010) How vital is sleep in Huntington’s disease? J Neurol 257:882–897. doi:10.1007/s00415-010-5517-4
Guthrie PB, Knappenberger J, Segal M, Bennett MVL, Charles AC, Kater SB (1999) ATP released from astrocytes mediates glial calcium waves. J Neurosci 19:520–528
Halassa MM, Fellin T, Haydon PG (2007) The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med 13:54–63. doi:10.1016/j.molmed.2006.12.005
Halassa MM et al (2009) Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61:213–219
Hauser RA et al (2011) Preladenant in patients with Parkinson’s disease and motor fluctuations: a phase 2, double-blind, randomised trial. Lancet Neurol 10:221–229. doi:10.1016/s1474-4422(11)70012-6
Hogl B et al (2002) Modafinil for the treatment of daytime sleepiness in Parkinson’s disease: a double-blind, randomized, crossover, placebo-controlled polygraphic trial. Sleep 25:905–909
Hollopeter G et al (2001) Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 409:202–207. doi:10.1038/35051599
Holst SC, Landolt H-P (2015) Sleep homeostasis, metabolism, and adenosine. Curr Sleep Med Rep 1:27–37. doi:10.1007/s40675-014-0007-3
Hong Z-Y, Huang Z-L, Qu W-M, Eguchi N, Urade Y, Hayaishi O (2005) An adenosine A2A receptor agonist induces sleep by increasing GABA release in the tuberomammillary nucleus to inhibit histaminergic systems in rats. J Neurochem 92:1542–1549. doi:10.1111/j.1471-4159.2004.02991.x
Hu Z et al (2013) Cordycepin increases nonrapid eye movement sleep via adenosine receptors in rats. Evid Based Complement Alternat Med 2013:8. doi:10.1155/2013/840134
Huang Z-L et al (2005) Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nat Neurosci 8:858–859. http://www.nature.com/neuro/journal/v8/n7/suppinfo/nn1491_S1.html
Huang Z-L, Urade Y, Hayaishi O (2007) Prostaglandins and adenosine in the regulation of sleep and wakefulness. Curr Opin Pharmacol 7:33–38. doi:10.1016/j.coph.2006.09.004
Huang ZL, Urade Y, Hayaishi O (2011) The role of adenosine in the regulation of sleep. Curr Top Med Chem 11:1047–1057
Huitron-Resendiz S et al (2005) Urotensin II modulates rapid eye movement sleep through activation of brainstem cholinergic neurons. J Neurosci 25:5465–5474. doi:10.1523/jneurosci.4501-04.2005
Inoué S, Honda K, Komoda Y (1995) Sleep as neuronal detoxification and restitution. Behav Brain Res 69:91–96. doi:10.1016/0166-4328(95)00014-k
Ishimori K (1909) True cause of sleep: a hypnogenic substance as evidenced in the brain of sleep-deprived animals. Tokyo Igakkai Zasshi 23:429–457
Jenner P, Mori A, Hauser R, Morelli M, Fredholm BB, Chen JF (2009) Adenosine, adenosine A2A antagonists, and Parkinson’s disease. Parkinsonism Relat Disord 15:406–413. doi:10.1016/j.parkreldis.2008.12.006
Kalinchuk AV et al (2003) Local energy depletion in the basal forebrain increases sleep. Eur J Neurosci 17:863–869. doi:10.1046/j.1460-9568.2003.02532.x
Krueger JM, Majde JA (2003) Humoral links between sleep and the immune system: research issues. Ann N Y Acad Sci 992:9–20
Krueger JM, Walter J, Dinarello CA, Wolff SM, Chedid L (1984) Sleep-promoting effects of endogenous pyrogen (interleukin-1). Am J Phys 246:R994–R999
Krueger JM, Obal F Jr, Fang J, Kubota T, Taishi P (2001) The role of cytokines in physiological sleep regulation. Ann N Y Acad Sci 933:211–221
Krueger JM, Clinton JM, Winters BD, Zielinski MR, Taishi P, Jewett KA, Davis CJ (2011) Involvement of cytokines in slow wave sleep. Prog Brain Res 193:39–47. doi:10.1016/b978-0-444-53839-0.00003-x
Kubota K (1989) Kuniomi Ishimori and the first discovery of sleep-inducing substances in the brain. Neurosci Res 6:497–518
Lazarus M, Yoshida K, Coppari R, Bass CE, Mochizuki T, Lowell BB, Saper CB (2007) EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci 10:1131–1133. http://www.nature.com/neuro/journal/v10/n9/suppinfo/nn1949_S1.html
Lazarus M et al (2011) Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. J Neurosci 31:10067–10075. doi:10.1523/jneurosci.6730-10.2011
Lazarus M, Huang Z-L, Lu J, Urade Y, Chen J-F (2012) How do the basal ganglia regulate sleep–wake behavior? Trends Neurosci 35:723–732. doi:10.1016/j.tins.2012.07.001
Lazarus M, Chen J-F, Urade Y, Huang Z-L (2013) Role of the basal ganglia in the control of sleep and wakefulness. Curr Opin Neurobiol 23:780–785. doi:10.1016/j.conb.2013.02.001
Legendre R, Pieron H (1913) Recherches sur le besoin de sommeil consécutif à une veille prolongée. Z Allegem Physiol 14:235–262
Li Y et al (2015) Optogenetic activation of adenosine a receptor signaling in the dorsomedial striatopallidal neurons suppresses goal-directed behavior. Neuropsychopharmacology 41:1003–1013. doi:10.1038/npp.2015.227
Linden J (2006) Purinergic chemotaxis. Science 314:1689–1690. doi:10.1126/science.1137190
Lovatt D et al (2012) Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc Natl Acad Sci U S A 109:6265–6270. doi:10.1073/pnas.1120997109
MacDonald PE, Braun M, Galvanovskis J, Rorsman P (2006) Release of small transmitters through kiss-and-run fusion pores in rat pancreatic beta cells. Cell Metab 4:283–290. doi:10.1016/j.cmet.2006.08.011
Matsuura K, Tomimoto H (2015) Istradefylline is recommended for morning use: a report of 4 cases. Intern Med 54:509–511. doi:10.2169/internalmedicine.54.3522
Mazzotti DR, Guindalini C, de Souza AAL, Sato JR, Santos-Silva R, Bittencourt LRA, Tufik S (2012) Adenosine deaminase polymorphism affects sleep EEG spectral power in a large epidemiological sample. PLoS One 7:e44154. doi:10.1371/journal.pone.0044154
Methippara MM, Kumar S, Alam MN, Szymusiak R, McGinty D (2005) Effects on sleep of microdialysis of adenosine A1 and A2a receptor analogs into the lateral preoptic area of rats. Am J Physiol Regul Integr Comp Physiol 289:R1715–R1723. doi:10.1152/ajpregu.00247.2005
Minzenberg MJ, Carter CS (2007) Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology 33:1477–1502
Monoi N et al (2016) Japanese sake yeast supplementation improves the quality of sleep: a double-blind randomised controlled clinical trial. J Sleep Res 25:116–123. doi:10.1111/jsr.12336
Müller CE, Jacobson KA (2011) Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta 1808:1290–1308. doi:10.1016/j.bbamem.2010.12.017
Mullington J, Korth C, Hermann DM, Orth A, Galanos C, Holsboer F, Pollmächer T (2000) Dose-dependent effects of endotoxin on human sleep. Am J Physiol Regul Integr Comp Physiol 278:R947–R955
Mullington JM, Hinze-Selch D, Pollmacher T (2001) Mediators of inflammation and their interaction with sleep: relevance for chronic fatigue syndrome and related conditions. Ann N Y Acad Sci 933:201–210
Nakamura Y, Midorikawa T, Monoi N, Kimura E, Murata-Matsuno A, Sano T, Oka K, Sugafuji T, Uchiyama A, Murakoshi M, Sugiyama K, Nishino H, Urade Y (2016) Oral administration of Japanese sake yeast (Saccharomyces cerevisiae sake) promotes non-rapid eye movement sleep in mice via adenosine A2A receptors. J Sleep Res 25:746–753. doi:10.1111/jsr.12434
Obeso JA et al (2000) Pathophysiologic basis of surgery for Parkinson’s disease. Neurology 55:S7–12
Ochiishi T et al (1999a) Cellular localization of adenosine A1 receptors in rat forebrain: immunohistochemical analysis using adenosine A1 receptor-specific monoclonal antibody. J Comp Neurol 411:301–316
Ochiishi T et al (1999b) High level of adenosine A1 receptor-like immunoreactivity in the CA2/CA3a region of the adult rat hippocampus. Neuroscience 93:955–967
Oishi Y, Huang Z-L, Fredholm BB, Urade Y, Hayaishi O (2008) Adenosine in the tuberomammillary nucleus inhibits the histaminergic system via A1 receptors and promotes non-rapid eye movement sleep. Proc Natl Acad Sci U S A 105:19992–19997. doi:10.1073/pnas.0810926105
Oishi Y, Yoshida K, Scammell TE, Urade Y, Lazarus M, Saper CB (2015) The roles of prostaglandin E2 and D2 in lipopolysaccharide-mediated changes in sleep. Brain Behav Immun 47:172–177. doi:10.1016/j.bbi.2014.11.019
Pascual O et al (2005) Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–116. doi:10.1126/science.1116916
Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW (1997) Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science 276:1265–1268
Porkka-Heiskanen T, Strecker RE, McCarley RW (2000) Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience 99:507–517. doi:10.1016/S0306-4522(00)00220-7
Prediger RDS, Batista LC, Takahashi RN (2005) Caffeine reverses age-related deficits in olfactory discrimination and social recognition memory in rats: involvement of adenosine A1 and A2A receptors. Neurobiol Aging 26:957–964. doi:10.1016/j.neurobiolaging.2004.08.012
Qiu M-H, Vetrivelan R, Fuller PM, Lu J (2010) Basal ganglia control of sleep–wake behavior and cortical activation. Eur J Neurosci 31:499–507. doi:10.1111/j.1460-9568.2009.07062.x
Qu W-M et al (2006) Lipocalin-type prostaglandin D synthase produces prostaglandin D2 involved in regulation of physiological sleep. Proc Natl Acad Sci U S A 103:17949–17954
Qu W-M, Huang Z-L, Xu X-H, Matsumoto N, Urade Y (2008) Dopaminergic D1 and D2 receptors are essential for the arousal effect of modafinil. J Neurosci 28:8462–8469. doi:10.1523/jneurosci.1819-08.2008
Qu W-M, Xu X-H, Yan M-M, Wang Y-Q, Urade Y, Huang Z-L (2010) Essential role of dopamine D2 receptor in the maintenance of wakefulness, but not in homeostatic regulation of sleep, in mice. J Neurosci 30:4382–4389. doi:10.1523/jneurosci.4936-09.2010
Radulovacki M, Virus RM, Djuricic-Nedelson M, Green RD (1983) Hypnotic effects of deoxycorformycin in rats. Brain Res 271:392–395
Reppert SM, Weaver DR, Stehle JH, Rivkees SA (1991) Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol Endocrinol 5:1037–1048. doi:10.1210/mend-5-8-1037
Resta R, Yamashita Y, Thompson LF (1998) Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev 161:95–109
Rétey JV et al (2005) A functional genetic variation of adenosine deaminase affects the duration and intensity of deep sleep in humans. Proc Natl Acad Sci U S A 102:15676–15681. doi:10.1073/pnas.0505414102
Rétey JV, Adam M, Khatami R, Luhmann UF, Jung HH, Berger W, Landolt HP (2007) A genetic variation in the adenosine A2A receptor gene (ADORA2A) contributes to individual sensitivity to caffeine effects on sleep. Clin Pharmacol Ther 81:692–698
Rivkees SA, Price SL, Zhou FC (1995) Immunohistochemical detection of A1 adenosine receptors in rat brain with emphasis on localization in the hippocampal formation, cerebral cortex, cerebellum, and basal ganglia. Brain Res 677:193–203
Rosenbaum E (1892) Warum müssen wir schlafen?: eine neue Theorie des Schlafes. August Hirschwald, Berlin
Rosin DL, Robeva A, Woodard RL, Guyenet PG, Linden J (1998) Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J Comp Neurol 401:163–186
Saper CB, Scammell TE, Lu J (2005) Hypothalamic regulation of sleep and circadian rhythms. Nature 437:1257–1263
Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE (2010) Sleep state switching. Neuron 68:1023–1042
Satoh S, Matsumura H, Suzuki F, Hayaishi O (1996) Promotion of sleep mediated by the A2a-adenosine receptor and possible involvement of this receptor in the sleep induced by prostaglandin D2 in rats. Proc Natl Acad Sci U S A 93:5980–5984
Satoh S, Matsumura H, Koike N, Tokunaga Y, Maeda T, Hayaishi O (1999) Region-dependent difference in the sleep-promoting potency of an adenosine A2A receptor agonist. Eur J Neurosci 11:1587–1597. doi:10.1046/j.1460-9568.1999.00569.x
Scammell TE et al (2001) An adenosine A2a agonist increases sleep and induces Fos in ventrolateral preoptic neurons. Neuroscience 107:653–663. doi:10.1016/s0306-4522(01)00383-9
Scammell TE, Arrigoni E, Thompson MA, Ronan PJ, Saper CB, Greene RW (2003) Focal deletion of the adenosine A1 receptor in adult mice using an adeno-associated viral vector. J Neurosci 23:5762–5770
Schrader J (1983) Metabolism of adenosine and sites of production in the heart. In: Berne R, Rall T, Rubio R (eds) Regulatory function of adenosine, Developments in pharmacology, vol 2. Springer US, New York, pp 133–156. doi:10.1007/978-1-4613-3909-0_9
Shen HY et al (2008) A critical role of the adenosine A2A receptor in extrastriatal neurons in modulating psychomotor activity as revealed by opposite phenotypes of striatum and forebrain A2A receptor knock-outs. J Neurosci 28:2970–2975
Sherin JE, Shiromani PJ, McCarley RW, Saper CB (1996) Activation of ventrolateral preoptic neurons during sleep. Science 271:216–219
Sherin JE, Elmquist JK, Torrealba F, Saper CB (1998) Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci 18:4705–4721
Stiasny-Kolster K, Clever SC, Moller JC, Oertel WH, Mayer G (2007) Olfactory dysfunction in patients with narcolepsy with and without REM sleep behaviour disorder. Brain 130:442–449. doi:10.1093/brain/awl343
Strecker RE et al (2000) Adenosinergic modulation of basal forebrain and preoptic/anterior hypothalamic neuronal activity in the control of behavioral state. Behav Brain Res 115:183–204. doi:10.1016/s0166-4328(00)00258-8
Svenningsson P, Fourreau L, Bloch B, Fredholm BB, Gonon F, Le Moine C (1999) Opposite tonic modulation of dopamine and adenosine on c-fos gene expression in striatopallidal neurons. Neuroscience 89:827–837
Thakkar MM, Engemann SC, Walsh KM, Sahota PK (2008) Adenosine and the homeostatic control of sleep: effects of A1 receptor blockade in the perifornical lateral hypothalamus on sleep–wakefulness. Neuroscience 153:875–880. doi:10.1016/j.neuroscience.2008.01.017
Ueno R, Ishikawa Y, Nakayama T, Hayaishi O (1982) Prostaglandin D2 induces sleep when microinjected into the preoptic area of conscious rats. Biochem Biophys Res Commun 109:576–582. doi:10.1016/0006-291x(82)91760-0
Urade Y, Hayaishi O (2011) Prostaglandin D2 and sleep/wake regulation. Sleep Med Rev 15:411–418. doi:10.1016/j.smrv.2011.08.003
Urade Y, Lazarus M (2013) Prostaglandin D2 in the regulation of sleep. In: Shaw PJ, Tafti M, Thorpy MJ (eds) The genetic basis of sleep and sleep disorders. Cambridge University Press, New York, pp 73–83
Urade Y et al (2003) Sleep regulation in adenosine A2A receptor-deficient mice. Neurology 61:S94–S96
Ushikubi F et al (1998) Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature 395:281–284
van Diepen HC, Lucassen EA, Yasenkov R, Groenen I, Ijzerman AP, Meijer JH, Deboer T (2014) Caffeine increases light responsiveness of the mouse circadian pacemaker. Eur J Neurosci 40:3504–3511. doi:10.1111/ejn.12715
Virus RM, Ticho S, Pilditch M, Radulovacki M (1990) A comparison of the effects of caffeine, 8-cyclopentyltheophylline, and alloxazine on sleep in rats. Possible roles of central nervous system adenosine receptors. Neuropsychopharmacology 3:243–249
Wall MJ, Dale N (2013) Neuronal transporter and astrocytic ATP exocytosis underlie activity-dependent adenosine release in the hippocampus. J Physiol 591:3853–3871. doi:10.1113/jphysiol.2013.253450
Wang Y-Q et al (2016) Adenosine A2A receptors in the olfactory bulb suppress rapid eye movement sleep in rodents. Brain Struct Funct. doi:10.1007/s00429-016-1281-2
Wei CJ, Li W, Chen JF (2011) Normal and abnormal functions of adenosine receptors in the central nervous system revealed by genetic knockout studies. Biochim Biophys Acta 1808:1358–1379. doi:10.1016/j.bbamem.2010.12.018
Wetter TC, Collado-Seidel V, Pollmacher T, Yassouridis A, Trenkwalder C (2000) Sleep and periodic leg movement patterns in drug-free patients with Parkinson’s disease and multiple system atrophy. Sleep 23:361–367
Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM (2001) Dopaminergic role in stimulant-induced wakefulness. J Neurosci 21:1787–1794
Yamaguchi H, Maruyama T, Urade Y, Nagata S (2014) Immunosuppression via adenosine receptor activation by adenosine monophosphate released from apoptotic cells. Elife 3:e02172. doi:10.7554/eLife.02172
Yang C, Franciosi S, Brown RE (2013) Adenosine inhibits the excitatory synaptic inputs to basal forebrain cholinergic, GABAergic and parvalbumin neurons in mice. Front Neurol 4:77. doi:10.3389/fneur.2013.00077
Yegutkin GG (2008) Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta 1783:673–694. doi:10.1016/j.bbamcr.2008.01.024
Yin D et al (2015) Paeoniflorin exerts analgesic and hypnotic effects via adenosine A1 receptors in a mouse neuropathic pain model. Psychopharmacology 233:281–293. doi:10.1007/s00213-015-4108-6
Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K (2011) Optogenetics in neural systems. Neuron 71:9–34. doi:10.1016/j.neuron.2011.06.004
Yu L et al (2008) Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol 63:338–346. doi:10.1002/ana.21313
Zetterstrom T, Vernet L, Ungerstedt U, Tossman U, Jonzon B, Fredholm BB (1982) Purine levels in the intact rat brain. Studies with an implanted perfused hollow fibre. Neurosci Lett 29:111–115
Zhang Z et al (2007) Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol 9:945–953
Zhang Y, Li M, Kang R-X, Shi J-G, Liu G-T, Zhang J-J (2012) NHBA isolated from Gastrodia elata exerts sedative and hypnotic effects in sodium pentobarbital-treated mice. Pharmacol Biochem Behav 102:450–457. doi:10.1016/j.pbb.2012.06.002
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Lazarus, M., Chen, JF., Huang, ZL., Urade, Y., Fredholm, B.B. (2017). Adenosine and Sleep. In: Landolt, HP., Dijk, DJ. (eds) Sleep-Wake Neurobiology and Pharmacology . Handbook of Experimental Pharmacology, vol 253. Springer, Cham. https://doi.org/10.1007/164_2017_36
Download citation
DOI: https://doi.org/10.1007/164_2017_36
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11270-7
Online ISBN: 978-3-030-11272-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)