
Abstract
Targeted therapies are revolutionizing the treatment of advanced non-small cell lung cancer (NSCLC). The discovery of key oncogenic events mainly in lung adenocarcinoma, like EGFR mutations or ALK rearrangements, has changed the treatment landscape while improving the prognosis of lung cancer patients. Inevitably, virtually all patients initially treated with targeted therapies develop resistance because of the emergence of an insensitive cellular population, selected by pharmacologic pressure. Diverse mechanisms of resistance, in particular to EGFR, ALK and ROS1 tyrosine-kinase inhibitors (TKIs), have now been discovered and may be classified in three different groups: (1) alterations in the target (such as EGFR T790M and ALK or ROS1 mutations); (2) activation of alternative pathways (i.e. MET amplification, KRAS mutations); (3) phenotype transformation (to small cell lung cancer, epithelial–mesenchymal transition). These basic mechanisms are informing the development of novel therapeutic strategies to overcome resistance in the clinic. Novel-generation molecules include osimertinib, for EGFR-T790M-positive patients, and new ALK-TKIs. Nevertheless, the possible concomitant presence of multiple resistance mechanisms, as well as their heterogeneity among cells and disease localizations, makes research in this field particularly arduous. In this chapter, available evidence and perspectives concerning precise mechanisms of escape to pharmacological inhibition in oncogene-addicted NSCLC are reported for single targets, including but not limited to EGFR and ALK.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Lung cancers currently figure among the most frequent tumor diagnoses and are the most relevant in terms of mortality worldwide (Siegel et al. 2016).
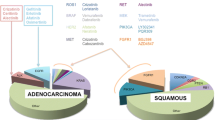
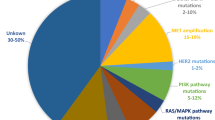
Before the year 2000 the dichotomy between small cell and non-small cell lung cancer (SCLC and NSCLC, respectively) was sufficient to address treatment strategies. Further histologic definition within NSCLC (squamous cell carcinoma and adenocarcinoma) was therefore recognized as clinically meaningful (Scagliotti et al. 2008). Since the last decades, molecular sub-typing of NSCLC (with an almost exclusive regard to adenocarcinoma) is providing a drastic refinement in the detection of alterations suitable of specific inhibition, generating a dramatic evolution in patients’ management. Such aberrations (whose incidence in western population is showed in Fig. 1), in general mutually exclusive, normally represent the very funding oncogenic event (Gainor et al. 2013). The targeting of such altered tyrosine-kinase (TK) receptors by means of specific inhibitors (TKIs, actively competing against ATP-binding) usually generates extremely rapid and profound tumor responses, defining thus far the scenario of oncogene addiction (Lynch et al. 2004; Paez et al. 2004; Kwak et al. 2010). In this field, the superiority of targeted agents over standard chemotherapy, in the advanced setting, is at this point evident (Mok et al. 2009; Solomon et al. 2014).

Distribution of molecular aberrations responsible for oncogene addiction in lung adenocarcinoma affecting Western populations
Albeit targeted therapies are revolutionizing the treatment of advanced NSCLC, sooner or later resistance appears in virtually every patient. Molecular treatment exhaustion denotes the emergence of a cellular population insensitive and selected by the pharmacologic pressure. In parallel to the crucial recognition of specific mechanisms on the diagnostic samples, the detection of molecular reasons explaining treatment resistance at the moment of disease progression, obtaining of novel tumor material (re-biopsy) harbours a similar pivotal importance. The role of re-biopsies in the clinical setting is currently gaining more and more relevance due to the development of novel-generations Epidermal Growth Factor Receptor (EGFR)-TKIs, like osimertinib (AZD9291), active against T790M EGFR mutation, whose emergence is the most common mechanism of resistance to first-and second-generation anti-EGFR compounds (Kobayashi et al. 2005; Cross et al. 2014; Mok et al. 2016) (see next paragraphs).
Patterns of resistance to tailored therapies are shared among different activating aberration, and lessons regarding targets rare in lung cancers can be driven from other tumors. The general molecular ways lung cancer cells find to escape directed targeting are resumed in Table 1. The possible concomitant presence of multiple mechanisms, as well as their heterogeneity among cells and disease localizations (Suda et al. 2016; Hata et al. 2015), makes research in this field particularly arduous. Available evidence and perspectives concerning precise mechanisms of escape to pharmacological inhibition in oncogene-addicted NSCLC are reported for single targets in the next paragraphs.
2 Resistance Mechanisms to EGFR-Driven NSCLC
Mutations in the EGFR gene are the most frequent oncogenic drivers in NSCLC, reported in approximately 10–15% of Caucasian NSCLC patients (Rosell et al. 2009) and 30–50% of Asians ones (Mok et al. 2009). The development of EGFR-TKIs, such as erlotinib, gefitinib (belonging to the first generation) and afatinib (second generation), shaped a great shift in the therapeutic management of EGFR-mutated NSCLC patients resulting in improved response rate (RR), progression free survival (PFS) and quality of life compared to first-line platinum-based chemotherapy (Mok et al. 2009; Rosell et al. 2012; Yang et al. 2015).
Unfortunately, prognosis remains unfavourable because of the occurrence of treatment resistance.
However, the identification of some mechanisms of resistance improved the therapeutic chances of these patients. In particular, the point mutation p.Thr790Met (T790M) occurring in EGFR exon 20 is responsible of resistance in about 50–60% of the patients when progression occurs (Sequist et al. 2011). Recently, the third-generation TKI osimertinib improved outcomes in patients harbouring this new mutation (Mok et al. 2016). Some other molecular resistance mechanisms have already been identified, but other information are needed to better understand and effectively overcome resistance to EGFR-TKIs in the remaining 40–50% lacking T790M mutation. Although exciting survival data and response rates have been registered in patients treated with osimertinib, acquired resistance unfortunately still occurs also during this therapy (Minari et al. 2016). Here, we will review principle mechanisms of resistance described during NSCLC treatment with both first-/second- and third-generation EGFR-TKIs.
2.1 Resistance to First- and Second-Generation EGFR-TKIs
Today erlotinib and gefitinib, together with the second-generation afatinib, are recognized as the standard first-line therapy in NSCLC patients with activating EGFR mutations (Mok et al. 2009; Rosell et al. 2012; Yang et al. 2015). Despite these important results, some patients with confirmed mutations in the EGFR-TK domain do not respond to EGFR-TKIs at all (de novo/intrinsic resistance). The remaining EGFR-mutated patients, after favourable and prolonged responses, inevitably exhibit disease progression (acquired resistance), usually after 10–14 months of treatment. Although the large majority of evidence concerns tumor evasion of targeted treatments represented by erlotinib and gefitinib, afatinib exhaustion seems to share the same molecular mechanisms (Campo et al. 2016). Several mechanisms of resistance have been identified and they may be classified in three different groups, as indicated in the introduction: (1) EGFR mutations; (2) activation of alternative pathways; (3) phenotype transformation (Table 1 and Fig. 2).

Mechanisms of resistance to first/second (yellow) and third (red)-generation EGFR-TKIs; shared mechanisms among the three TKI generations are depicted in white. NSCLC non-small cell lung cancer, SCLC small cell lung cancer, SCC squamous cell carcinoma
2.1.1 Preclinical Evidence and Clinical Relevance of Resistance Mechanisms
Mechanisms of primary resistance are still not fully understood, but several cases of de novo inefficacy of EGFR-TKIs are the consequence of the presence of non-sensitive EGFR mutations. Exon 20 insertions, which represent the 1–10% of the total number of EGFR mutations, adding residues at the N-lobe of EGFR (M766 to C775) in particular in the C-helix (A767 to C775), frequently reduce affinity for EGFR-TKIs (Yasuda et al. 2013). New sequencing technologies are able to detect cases of concomitant (double or multiple) EGFR mutations. Patients with a combination of typical and atypical mutations reported less favourable outcomes compared to patients with a single typical mutation (Wu et al. 2011). Also the coexistence of different driver alterations in other genes, such as ALK rearrangements and KRAS mutations, resulted associated with worse prognosis after EGFR-TKI treatment in EGFR-mutated NSCLC (Ulivi et al. 2016).
The most common mechanism of resistance is the development of acquired T790M EGFR gene mutation (Sequist et al. 2011), a secondary point mutation in exon 20, engendering the substitution of methionine (T) for threonine (M) at codon position 790, that sterically prevents the EGFR-TKI binding in the TK domain (TKD), allowing the ATP-mediated activation of the receptor (Kobayashi et al. 2005). Nevertheless, T790M mutation has been also identified as a de novo mutation (Inukai et al. 2006). In this case of primary resistance, it is predictive for poor survival outcomes under EGFR-TKI treatment (Su et al. 2012). Moreover, the T790M impact on responsiveness to EGFR-TKI therapy may depend on the proportion of pre-treatment EGFR T790M-positive clones (Hata et al. 2016).
Third-generation EGFR-mutant selective inhibitors (such as osimertinib and rociletinib) have been developed for patients whose cancers acquire the T790M mutation. These third-generation EGFR-TKIs realize selective inhibition of activating as well as T790M alterations, by means of an irreversible covalent binding to the target while sparing wild-type EGFR (Cross et al. 2014), with important efficacy results and reduced toxic effects (Mok et al. 2016; Jänne et al. 2015; Sequist et al. 2015).
Other rare resistance EGFR point mutations including D761Y, T854A and L747S have been reported in less than 10% of mutated NSCLC patients. The mechanism underlying resistance conferred by these mutations is still unclear (Nguyen et al. 2009).
The activation of alternative pathways is now recognized as a different mechanism of resistance (Niederst and Engelman, 2013; Yu et al. 2013a).
The MET gene amplification is the second most common mechanisms of acquired resistance, affecting about 5–20% of NSCLC patients during EGFR-TKI treatment, irrespective of the T790M mutation status (Sequist et al. 2011; Engelman et al. 2007). MET amplification, accompanied by HGF (MET ligand) autocrine signalling, drives resistance to EGFR-TKIs acting upon molecular elements regulating critical intracellular pathways (Engelman et al. 2007; Turke et al. 2010). MET inhibition has proven to be effective in cell lines with MET gene amplification and many preclinical and clinical data demonstrate that contemporary inhibition of MET and EGFR may be a strategy to overcoming resistance (Engelman et al. 2007; Bahcall et al. 2016; Gainor et al. 2016a).
HER2 amplification is a rare event in lung adenocarcinoma at diagnosis, accounting for about 1–2% of cases, but it has been reported in up to 13% of NSCLC with acquired resistance to EGFR-TKIs (Yu et al. 2013a; Takezawa et al. 2012), whereas resulting absent in other series (Sequist et al. 2011). Mutated EGFR has the tendency to heterodimerize with HER2, the resulting heterodimers being resistant to degradation (Takezawa et al. 2012). Therefore, HER2 heterodimerization could support EGFR-TKIs resistance in presence of both T790M mutation and HER2 amplification itself as acquired mechanisms of drug exhaustion.
Boosting of cell signalling pathways due to activation of BRAF, PIK3CA and AXL has been proposed as mechanism of drug resistance in cancer cells and in EGFR-mutated NSCLC patients, in which their emergence can be overall detected in up to 20–25% of cases (Sequist et al. 2011; Ohashi et al. 2012; Wang et al. 2014; Zhang et al. 2012).
A third resistance mechanism is the phenotypic transformation of lung cancer cells. Histological transformation in SCLC has been observed after the development of acquired resistance to EGFR-TKI in about 3–14% of patients (Sequist et al. 2011; Yu et al. 2013a). The mechanism underlying this histological modification is still not completely known: minor pre-existent cells under the selection pressure of EGFR-TKIs could originate SCLC cells or adenocarcinoma cells could trans-differentiate in SCLC cells (Oser et al. 2015); alternatively, SCLC cells could develop from multi-potent pre-existing stem cells (Oser et al. 2015). Whatever the funding cellular evolution, the loss of Rb protein seems a common and necessary event for this kind of transformation (Niederst et al. 2015a).
Loss of E-cadherin expression and upregulation of mesenchymal proteins such as vimentin, fibronectin and N-cadherin are the main features of epithelial–mesenchymal transition (EMT). In the EMT setting, AXL upregulation and alterations in the Hedgehog pathway have been recently recognized as mechanisms of resistance to targeted agents in EGFR-mutated NSCLC (Zhang et al. 2012; Thomson et al. 2005).
Moreover, transformation from adenocarcinomas to squamous cell carcinomas during the administration of anti-EGFR molecules has been reported as a mechanism of acquired drug resistance (Haratani et al. 2016).
Anyway, the cause of resistance remains still unknown in 18–30% of NSCLC patients resistant to anti-EGFR targeted therapy (Sequist et al. 2011; Yu et al. 2013a).
2.1.2 Detection of T790M Mutation
According to current guidelines, after progression to first-line EGFR-TKI treatment, carrying out a new biopsy to identify the molecular mechanism of acquired resistance and to select patients for targeted therapies is a reasonable procedure (Novello et al. 2016). The feasibility and utility of re-biopsies have been evaluated in several clinical experiences (Mok et al. 2016; Campo et al. 2016; Arcila et al. 2011).
However, serial tumor sampling to monitor cancer evolution is not always feasible in clinical practice. An alternative approach in NSCLC patients may be indeed the use of the so-called liquid biopsy, whereby circulating cell-free tumor DNA (ctDNA), DNA fragments passively released into the blood by primary cancer cells, or circulating tumor cells (CTCs), viable or apoptotic cells released from the primary tumor, can be analysed in the peripheral blood to detect EGFR mutations (Crowley et al. 2013; Douillard et al. 2014). Dynamic changes of EGFR mutational status in ctDNA seem to predict the clinical outcome to EGFR-TKI treatment (Tseng et al. 2015). A meta-analysis showed a sensitivity of 61% and a specificity of 90% for blood (plasma and serum) analysis compared to tissue evaluation in identifying EGFR mutations with a concordance rate of 79% (Mao et al. 2015).
Many studies confirmed the utility and validity of plasma DNA in detection T790M mutation in patients with NSCLC who progressed under EGFR-TKI therapy (Mok et al. 2016; Sundaresan et al. 2016; Remon et al. 2017). According to results of many studies, this method it is today approved by the FDA (http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm519922). Considering the level of sensitivity, when a liquid biopsy is negative for the detection of EGFR T790M, this result should be confirmed on tissue biopsy specimen (Oxnard et al. 2016). In addition, a study recently demonstrated satisfying agreements in T790M status definition between urine, plasma and tissue (Reckamp et al. 2016); further urine-based tests are indeed under study.
As mentioned before, T790M mutations account for up to 60% of resistant cases to first- and second-generation EGFR-TKIs (Sequist et al. 2011; Yu et al. 2013a). The remaining T90M-negative cases, for which, in addition, molecular treatment strategies are less developed, can be less characterized by liquid biopsy, with special regard to morpho-phenotypic changes. Recently nevertheless, a high quote of KRAS activating mutations has been uncovered in ctDNA in EGFR-mutant NSCLC patients progressing to first or second-generation TKIs (Del Re et al. 2016).
2.1.3 Potential Strategies to Overcome Resistance
Some clinical strategies have indeed been developed in order to deal with or overcome resistance to first- and second-EGFR-TKIs.
Because of cancer heterogeneity, once the onset of resistance is manifest, some clones may continue to remain sensitive to EGFR-TKIs, whose continuation can slow down disease progression. For these reasons, in selected patients with slow-growing and low-volume disease, progression in non-critical or asymptomatic sites, no clinical deterioration or intolerable toxicity, first-line EGFR-TKI treatment can be continued beyond progression, as several retrospective studies and some prospective experience showed (Park et al. 2016; Yap et al. 2017).
In the case of clinical progression in circumscribed localization, disease behaviour reflects the spatial heterogeneity of resistance. Retention of the targeted treatment with the addition of local approaches (including surgical resection or radiotherapy) to the dimensionally increasing lesions results a suitable option in order to achieve long-term disease control, acting directly against the resistant counterparts while maintaining active EGFR suppression (Weickhardt et al. 2012; Yu et al. 2013b).
Brain metastases interest around 20% of EGFR-mutated NSCLC patients at diagnosis, while 30–60% of them, during an effective EGFR-TKI administration, develop central nervous system lesions, often representing the isolated site of disease recurrence (Heon et al. 2010; Khalifa et al. 2016). If the progression after first-line EGFR-TKI therapy is characterized by the development of isolated brain metastases, stereotactic radiotherapy or surgery, when possible, is recommended, while multiple lesions require whole-brain radiotherapy. In order to keep extra-cerebral disease control, EGFR-TKI treatment should be continued (Khalifa et al. 2016).
These reported are effective clinical ways to delay the requirement of novel (cytotoxic or targeted) treatment. In virtually every patient indeed, disease progression not allowing such approaches sooner or later occurs. The research of T790M mutation on tumor specimens or ctDNA is crucial, both for the quote of patients harbouring it and for the actual possibility to overcome it with the novel molecules of third generation, such as osimertinib, rociletinib, HM61713 (olmutinib), ASP8273, EGF816 and PF-06747775. The clinical development of rociletinib and olmutinib has been recently interrupted and ASP8273, EGF816 and PF-06747775 are under early investigation.
Osimertinib is an oral, irreversible EGFR-TKI that is selective for both activating and T790M-resistance mutations (Cross et al. 2014), with significant activity against central nervous system metastases too (Mok et al. 2016; Ballard et al. 2016). In the phase 1 trial (AURA) the RR for osimertinib in patients with T790M-positive tumors was 61%, with a median PFS of 9.6 months (Jänne et al. 2015). These findings were confirmed in a pooled analysis of two subsequent phase 2 studies (Yang et al. 2016), one of which recently published (AURA2) (Goss et al. 2016). On the basis of these results, FDA approved osimertinib in T790M-positive NSCLC patients. A confirmatory, randomized, open-label, international phase 3 trial (AURA3) was conducted and osimertinib showed significantly greater efficacy than platinum plus pemetrexed chemotherapy in patients with T790M-positive cancers after progression under first- or second-generation EGFR-TKIs (Mok et al. 2016). Median PFS for osimertinib was 10.1 months, compared to 4.4 months for chemotherapy (HR: 0.30; 95% CI 0.23–0.41; p < 0.001) (Mok et al. 2016). Under the AURA development, osimertinib became the standard of care in second-line treatment for EGFR-mutated patients harbouring the T790M mutation.
At the time of progression for patients who are not candidate to osimertinib due to the absence of EGFR T790M resistance mutation, different therapeutic options have been or are under investigation. The randomized phase III IMPRESS trial compared gefitinib with versus chemotherapy alone in 265 EGFR-mutated NSCLC resistant to first-line gefitinib (Soria et al. 2015). The underlying objective was to sound out the potential contribution of maintaining inhibition of the driver molecule in addition to standard cytotoxic treatment. No benefit in survival was observed when gefitinib was associated with chemotherapy, suggesting that the EGFR-TKI should be discontinued in resistant patients when the switch to chemotherapy is required (Soria et al. 2015, 2016).
In order to overcome the resistance mediated by specific bypass mechanisms, targeting the detected drivers of resistance itself in combination with EGFR-TKIs may be a sound therapeutic possibility. In particular, the use of MET inhibitors in combination with EGFR-TKI recently revealed as a promising strategy for EGFR-mutated and MET amplified NSCLC. Cabozantinib, capmatinib and tepotinib reported significant results in terms of disease response if associated with anti-EGFR agents in this subgroup of NSCLC (Bahcall et al. 2016; Wu et al. 2016; Soo et al. 2015).
In cases of rapid systemic progression, performing a new biopsy is recommended because in presence of a phenotypic transformation to SCLC, to squamous cell carcinoma or when EMT is evident, the use of the chemotherapy could be more beneficial than the use of target therapies.
2.2 Resistance to Third-Generation EGFR-TKIs
The introduction of third-generation EGFR-TKIs resulted in a further outcome improvement for a selected group of NSCLC patients. Nevertheless, despite the high RR and the significant prolongation of survival, after 9–13 months, unfortunately, acquired resistance occurs again (Mok et al. 2016; Jänne et al. 2015; Goss et al. 2016). Several (and not fully recognized) underlying molecular mechanisms have been described (Minari et al. 2016).
2.2.1 Preclinical Evidence and Clinical Relevance of Resistance Mechanisms
In the case of third-generation EGFR-TKIs too, we can classify the mechanisms of resistance in three different categories: (1) EGFR-dependent mechanisms; (2) activation of alternative pathways; (3) phenotypic transformation (Table 1 and Fig. 2).
The emergence of tertiary EGFR mutations has been repeatedly reported in the presence of acquired resistance to third-generation TKIs and it has been well characterized in cell lines models (Minari et al. 2016). The EGFR p.Cys797Ser (C797S) mutation in the exon 20 is the most common mutation responsible for resistance to osimertinib. Firstly, C797S was identified in ctDNA of 6 out of 15 (40%) patients progressing to osimertinib in the AURA phase I/II study (Thress et al. 2015). It seems also responsible of acquired resistance to other third-generation EGFR-TKIs such as HM61713 and WZ4002, but it is rare after rociletinib (Niederst et al. 2015b; Ercan et al. 2015; Chabon et al. 2016). The substitution of a cysteine with a serine in the position 797 of the tyrosine kinase domain reduces the inhibitory effect of third-generation TKIs by interfering with their covalent binding to EGFR (Thress et al. 2015). Interestingly, according to preclinical evidence, the location of C797S mutation among other EGFR alleles (in cis vs. in trans) may affect the efficacy of subsequent treatments (Niederst et al. 2015b).
After the report of this first mutation responsible of resistance third-generation EGFR-TKIs, several other single-site alterations, such as L718Q and L844V, have been reported in patients and in cellular models treated with osimertinib or other third-generation TKIs (Minari et al. 2016). Importantly, liquid biopsy confirmed its value in detecting such mutations in ctDNA, reinforcing its importance as a clinical tool (Thress et al. 2015; Ercan et al. 2015; Chabon et al. 2016; Piotrowska et al. 2015).
Again, EGFR-independent mechanisms of resistance during third-generation TKI treatment can emerge. HER2 amplification was discovered in a NSCLC patient with disease progression after 12 months of osimertinib in the AURA trial (Planchard et al. 2015). It appeared to be mutually exclusive with EGFR T790M mutation, as described for first-generation TKIs (Takezawa et al. 2012), and not associated with C797S. Similar findings were reported in other patients treated with osimertinib (Minari et al. 2016), while in the case of resistance to rociletinib, HER2 amplification was associated with T790M persistence (Chabon et al. 2016). MET amplification was first reported in a single case of NSCLC after 10 months of osimertinib treatment, in the absence of T790M or C797S mutations (Planchard et al. 2015), and it was documented too as a mechanism of acquired resistance, both in preclinical in vitro models and clinical cases (Ortiz-Cuaran et al. 2016; Ou et al. 2016a).
In preclinical studies of acquired resistance to osimertinib, an increased dependence on RAS signalling was reported. NRAS mutations, including a novel E63K mutation, and NRAS or KRAS amplification have been described as mechanisms of acquired resistance to osimertinib (Eberlein et al. 2015). The emergence of three KRAS activating mutations (p.G12A, p.Q61H and p.A146T), alone or in combination with other resistance mechanisms, has been reported after rociletinib (Chabon et al. 2016). Moreover, at the time of progression, p.E542K and p.E545K mutations in PIK3CA gene have been described in five patients treated with rociletinib (Chabon et al. 2016). Other reported resistance mechanisms include BRAF p.V600E mutation (Oxnard et al. 2015) and EGFR amplification (Chabon et al. 2016; Piotrowska et al. 2015).
Finally, after third-generation TKIs too, in some cases resistant tumors showed phenotypic changes, such as SCLC transformation or EMT (Piotrowska et al. 2015; Kim et al. 2015; Ham et al. 2016).
2.2.2 Potential Strategies to Overcome Resistance
Currently, different therapeutic strategies to overcome the above-described heterogeneous resistance mechanisms to third-generation TKIs are under development.
A new era of fourth-generation TKIs is coming (Minari et al. 2016). EAI045 is the first of a new class of inhibitors, able to overcome T790M and C797S mutations, being selective against mutant-EGFR while sparing the wild-type forms. The combination of EAI045 and cetuximab showed efficacy in mouse models of lung cancer carrying EGFR L858R/T790M/C797S mutations (Jia et al. 2016).
Combinations with third-generation TKIs are being investigated in several studies to avoid the occurrence or overcome resistance (Minari et al. 2016). The association of the MEK inhibitor trametinib with the third-generation EGFR-TKI WZ4002 was able to prevent the emergence of resistance in EGFR-mutant lung cancer models (Tricker et al. 2015). The association of osimertinib and another MEK inhibitor, selumetinib, prevented the onset of resistance in cellular lines and reported in vivo cancer regression in an EGFR-mutated, T790M-positive, osimertinib-resistant transgenic model (Eberlein et al. 2015).
Some evidences suggest that patients with C797S and T790M mutations in trans could be sensitive again to the association of first/second-generation TKIs with third-generation ones, while the in cis disposition results in resistance to all molecules, both alone and in combination (Niederst et al. 2015b). Moreover, the occurrence of C797S in T790M wild-type cells is responsible of resistance to third-generation TKIs, despite the sensitivity to first-generation TKIs (Niederst et al. 2015b). Patients progressing on rociletinib achieved response with osimertinib (Sequist et al. 2016), suggesting a slight different activity of the molecules developed against the T790M mutation.
For patients whose tumors undergo SCLC transformation or EMT, switching platinum-based chemotherapy could be recommended.
Surely, other escape mechanisms are likely to emerge, highlighting the importance of molecular characterization at the time of progression, aiming at the definition of the most correct therapeutic strategy.
3 Resistance Mechanisms to ALK- and ROS1-Driven NSCLC
ALK and ROS1 rearrangements are present in approximately 4–7% and 1–2% of NSCLC, respectively (Barlesi et al. 2016; Bergethon et al. 2012). These two oncogenes share profound similarities in phylogenesis, biology, genomic sequences, profiles of pharmacological inhibition and tumor clinical features (Ou et al. 2012). Importantly, tumors driven by either ALK or ROS1 manifest similar mechanisms of resistance to targeted agents, which will be approached in parallel. Several mechanisms of drug escape have been identified and they may be classified, similarly to EGFR-TKIs, in three different groups, as indicated in the introduction: (1) involving the target (ALK or ROS1); (2) activation of alternative pathways; (3) phenotype transformation (Table 1 and Fig. 3).

Thorough representation of mechanisms of resistance to crizotinib and novel-generation inhibitors in ALK-rearranged non-small cell lung cancer. See Gainor et al. Cancer Discovery 2016 (Gainor et al. 2016b) to distinguish the differential mechanisms for single molecules
3.1 Mechanisms of Crizotinib Resistance
3.1.1 Mechanisms of Crizotinib Resistance Involving ALK and ROS1
Crizotinib, firstly developed as a MET inhibitor (Kwak et al. 2010), is currently registered by FDA and EMA for patients suffering from ALK- and ROS1-rearranged NSCLC. Similarly to EGFR-driven tumors, mutations in the target have been reported as the first mechanism of resistance to crizotinib for both oncogenes (Choi et al. 2010; Awad et al. 2013). The number of reported ALK mutations responsible of acquired resistance is high, whereas altogether their detection is present in around 30% of clinical samples (Gainor et al. 2016b).
Concomitantly with the first report of crizotinib clinical activity (Kwak et al. 2010), Choi and colleagues described ALK C1156Y and L1196M mutations as responsible of acquired resistance to the drug (Choi et al. 2010). L1196 corresponds to the gatekeeper residue in ALK tyrosine kinase domain and this substitution is among the most frequently reported single-nucleotide alteration responsible of crizotinib resistance in NSCLC, together with G1269A (Gainor et al. 2016b). Biopsies obtained in ALK-positive NSCLC patients at the moment of disease progression to crizotinib (Katayama et al. 2012; Doebele et al. 2012) allowed the detection a conspicuous number of mutations in ALK TKD. These latter engender crizotinib resistance by means of two main mechanisms: by increasing enzymatic activity at a level not suitable of crizotinib inhibition or interfering with its binding (Gainor et al. 2016b; Friboulet et al. 2014). Considering also isolated case reports, single-nucleotide substitutions reported thus far include 1151Tins, L1152R, C1156Y, I1171N/S/T, F1174C/V, L1196M, G1202R, D1203N, S1206Y, F1245C and G1269A (Gainor et al. 2016b; Facchinetti et al. 2016a) (Fig. 3).
Due to the more limited rate of patients harbouring the oncogenic aberration and to the particularly recent introduction of crizotinib for ROS1-positive advanced NSCLC, information concerning resistance emerging in this setting is barely less abundant. Given the homology between ALK and ROS1 TKD (which share >80% sequence identity within their ATP-binding sites), every mutation reported so far to negatively affect crizotinib activity in ROS1-positive patients find its corresponding with comparison to ALK (Facchinetti et al. 2016b). G2032R (Awad et al. 2013), D2033N (Drilon et al. 2016a) and S1986Y/F (Facchinetti et al. 2016b), here reported in order of discovery, can be indeed aligned with the corresponding ALK G1202R, D1203N and C1156Y, respectively. Moreover, the gatekeeper L2026M substitution, together with L1951R, has been recently reported in a patient biopsy after crizotinib resistance (McCoach et al. 2016). The clinical relevance of other mutations (L2155S, K2003I, L1951R and M2128V) reported in vitro only has yet to be established (Katayama et al. 2015; Song et al. 2015).
ALK activation in NSCLC is a consequence of gene fusions and the most frequently partner gene is represented by EML4. Several fusion variants are possible, showing slight differential sensitivity to crizotinib in cellular models (Heuckmann et al. 2012) and potentially accounting to the variable durations of disease control in the clinics (Yoshida et al. 2016; Woo et al. 2016).
Crizotinib exhaustion can depend from the amplification/copy number gain of the ALK gene itself (Doebele et al. 2012; Shaw et al. 2014) and the possibility of loss of the driver alteration under selective pressure has been proposed in the clinics (Doebele et al. 2012).
These mechanisms have not yet been reported in ROS1-postive NSCLC, neither in preclinical studies nor in the clinics.
3.1.2 Activation of Bypass Pathways Explaining Crizotinib Resistance
Occurrence of either ALK or ROS1 rearrangements together with KRAS mutations in NSCLC can explain primary (Mengoli et al. 2016; Schmid et al. 2016) or acquired (Doebele et al. 2012) resistance to crizotinib. Moreover, KRAS and NRAS activation through mutations or amplifications leads to the exhaustion of first-generation inhibitors activity in ROS1-positive cellular models (Cargnelutti et al. 2015).
EGFR signalling has been reported as the responsible of crizotinib resistance in a non-negligible rate of patients-derived biopsies and in cellular models (Katayama et al. 2012; Doebele et al. 2012). Similarly to what observed for RAS alterations, EGFR signalling could augment, with or without the evidence of classical activating mutations (Katayama et al. 2012; Doebele et al. 2012; Kim et al. 2013). Several experiences indeed shown the amplification of EGFR gene and implementation of EGFR phosphorylation have been functionally related to acquired resistance to crizotinib, in both ALK- and ROS1-dependent NSCLC (Katayama et al. 2012; Song et al. 2015; Kim et al. 2013; Davies et al. 2013).
KIT gene amplification or activating mutations in ALK and ROS1-rearranged NSCLC patients can, respectively, explain the acquisition of crizotinib resistance (Katayama et al. 2012; Dziadziuszko et al. 2016).
Moreover, IGF1R, SRC and MEK/ERK activation can mediate resistance to specific inhibitors in ALK-dependent cell models, suggesting thus far the potential of combinatorial strategies in the clinics (Lovly et al. 2014; Crystal et al. 2014; Hrustanovic et al. 2015).
3.1.3 Further Mechanisms of Crizotinib Resistance
The largest part of ALK-positive NSCLC patients exposed to crizotinib experiences intracranial disease progression in spite of extra-cerebral disease control. Pharmacokinetics issues concerning the low blood–brain barrier penetration of the compound account for its reduced, albeit present, activity in CNS lesions (Costa et al. 2011). Data concerning ROS1-driven disease are still too limited to infer such similar conclusions.
Morpho-phenotypic tumor changes in ALK-positive disease are more limited than EGFR mutated, while in ROS1-rearranged cells they are limited to preclinical reports (Song et al. 2015). The neuroendocrine phenotypes of NSCLC can be responsible of either de novo crizotinib resistance (Omachi et al. 2014) or for the exhaustion of drug efficacy after initial responses, as transformation from adenocarcinoma to SCLC (Caumont et al. 2016).
In vitro data suggests EMT contribution to the establishing of resistance to ALK inhibitors (Kogita et al. 2014). Experimental data are sustained by clinical reports (Kobayashi et al. 2013).
3.1.4 Overcoming Crizotinib Resistance
In the very recent years, several other molecules have been developed aiming to maintain specific inhibition of ALK and ROS1 when crizotinib runs out of activity. Second-generation compounds include ceritinib, alectinib and brigatinib, while lorlatinib belongs to the third-generation of drugs. If ceritinib, brigatinib and lorlatinib are characterized by half maximal inhibitory concentration (IC50) values suitable of clinical application in both ALK- and ROS1-driven diseases, according to cellular assays alectinib exclusively hits ALK (Gainor et al. 2016b; Facchinetti et al. 2016b; Davare et al. 2015).
The mentioned inhibitors shown crucial properties, such as the ability to inhibit the target more potently than crizotinib, the activity against multiple mutant forms of ALK and ROS1 and the good brain penetration, confirming their relevant activity in the setting of crizotinib resistance (Facchinetti et al. 2016a, 2017). Data from clinical trials and patients’ cohorts suggests the clear advantage of administering new-generation compounds after crizotinib (Facchinetti et al. 2016a, 2017). There is moreover an efficacy gradient from the less recent to the newest inhibitors, as the latest are more potent and active against a larger number of mutations in the targets conferring resistance, as long as the brain penetration and the selectivity increase (Gainor et al. 2016b; Zou et al. 2015).
Among the cited novel inhibitors, only ceritinib and lorlatinib ostensibly harbour a significant role as ROS1 TKIs, considering the reduced potency of brigatinib against the wild-type and mutated forms of the enzyme (Chong et al. 2017), together with the inefficacity of alectinib.
Other wide-spectrum tyrosine-kinase inhibitors (e.g. cabozantinib, foretinib, entrectinib) shown activity against ROS1 and/or ALK (Facchinetti et al. 2017). The current availability of the mentioned specific inhibitors with important anti-ALK activity makes questionable the utilization of the latter ones in ALK-driven diseases. Among these less-specific molecules, cabozantinib only could have a role, as the sole drug potentially active against the G2032R ROS1 mutation, the most frequent mechanism of crizotinib resistance reported so far, albeit in very small series (Katayama et al. 2015; Gainor et al. 2016c). Nevertheless, the toxicity spectrum of cabozantinib at the systemic concentrations required to inhibit mutant ROS1 forms, still leave doubts about its potential clinical proposition; beside, all other crizotinib-resistance mutations seem to be overcome by the more manageable drug lorlatinib (Facchinetti et al. 2017).
3.2 Resistance to Next-Generation ALK and ROS1 Inhibitors
Recently, Gainor and colleagues reported the results from a wide series of ALK-positive tumor biopsies obtained at progression to crizotinib or second-generation inhibitors (Gainor et al. 2016b). Some of the codons involved in TKD mutations were previously unreported in the clinics, as E1210K, conferring crizotinib resistance, and V1180L, occurring after alectinib administration and already approached in in vitro studies (Gainor et al. 2016b; Katayama et al. 2014).
After ceritinib and alectinib, amino-acidic substitutions mediating resistance were observed in more than 50% of the samples, compared with the 20–30% target alterations responsible of crizotinib exhaustion (Gainor et al. 2016b). Although post-brigatinib biopsies were limited in the series, an enrichment in ALK G1202R mutation was reported after the onset of resistance for all the three new inhibitors compared to post-crizotinib samples. Other ALK mutations responsible of new-generation inhibitors resistance, unreported after crizotinib, are emerging, as reported in a patient developing ceritinib exhaustion due to the G1123S substitution (Toyokawa et al. 2015), underlying the differential selective pressure exerted by the inhibitors.
PIK3CA G106V activating mutation was detected in an alectinib-resistant specimen, thus allowing to envisaging the involvement of the AKT-mTOR pathway in resistance (Redaelli et al. 2016), as seen in EGFR-mutant NSCLC (Sequist et al. 2011).
Bypass signalling activation mediated by IGF1R, HER3 (with the concomitant overexpression of its ligand neuregulin-1) and MET has been proven as mechanism of resistance to alectinib in cellular models (Isozaki et al. 2016).
Morpho-phenotypic changes driving EMT were clearly depicted in one ceritinib-resistant sample and present, with different levels of intensity, in up to 42% (five out of 12) specimens, often in the presence of ALK mutations (Gainor et al. 2016b). Two cases of SCLC transformation were reported in alectinib-resistant tumors (Fujita et al. 2016; Takegawa et al. 2016).
Great interest was raised by the first and, to date, the only report of specific lorlatinib resistance in an ALK-rearranged NSCLC patient. Tumor cells, already harbouring the crizotinib and ceritinib resistant mutation C1156Y, developed the previously unknown L1198F substitution, determining in vitro resistance to all available ALK inhibitors except for crizotinib (Shaw et al. 2015). Administration of the first-generation compound led indeed to disease response. Data concerning resistance to new-generations inhibitors in ROS1-positive NSCLC models or patients are still lacking.
4 Resistance Mechanisms to Targeted Drugs in NSCLC Driven by Other Oncogenes
As several other targets and corresponding pharmacological compounds are emerging in lung cancer, mechanisms of resistance in this new field are rising and lessons can be inferred from tumor models other than NSCLC, sharing driving molecular aberrations and specific inhibition.
4.1 MET
Beside its role in mediating resistance to EGFR inhibition (Engelman et al. 2007), MET is known as a meaningful driver oncogene in NSCLC since around a decade. Nevertheless, its precise mechanisms of activation, harbouring biological and clinical relevance, have been profitably elucidated in the last 2 years (Drilon et al. 2017). MET gene amplification needs a precise definition for achieving a meaningful role in predicting response to specific inhibitors, the most relevant one to date again represented by crizotinib (Drilon et al. 2017). Moreover, MET can be biologically activated by a newly recognized mechanism represented by the loss of its exon 14 (exon 14 skipping), coding for the juxtamembrane domain, that leads to a meaningful increase in MET signalling by means of the decrease of its degradation (Awad 2016). As in this case MET TKD remains intact, crizotinib is indeed active (Drilon et al. 2017). Nevertheless, mutations occurring in MET TKD (D1228N, D1228V, Y1230C, the two latter founded in ctDNA too) have been recently reported as putative responsible of resistance to MET inhibitors in the clinics (Bahcall et al. 2016; Heist et al. 2016; Ou et al. 2016b). Strategies to overcome resistance to type I MET inhibitors (which preferentially bind to the active conformation of the protein, e.g. crizotinib and savolitinib) with type II compounds (which preferentially bind to the inactive molecule conformation, e.g. cabozantinib and capmatinib) yield an in vitro and clinical crucial meaning (Bahcall et al. 2016). If MET activation can explain resistance to EGFR inhibitors, as seen above, the reverse situation has been reported, with the onset of crizotinib resistance in a MET-driven NSCLC associated to the appearance to the activating EGFR L861A mutation (Benderra et al. 2016).
4.2 BRAF
BRAF-mutated NSCLC is similar in biology (in terms of role of oncogenic agent), clinical and therapeutic approaches to melanomas harbouring BRAF activating mutations (Nguyen-Ngoc et al. 2015). According to BRAF-mutant melanomas, the co-inhibition of BRAF and MEK in NSCLC generates better outcomes compared to BRAF blockade alone (Planchard et al. 2016a, b). With regard to BRAF-mutated lung adenocarcinoma, the onset of mutations in KRAS, TP53 and CDKN2A has been proposed as a resistance mechanism to the BRAF inhibitor dabrafenib in the clinics (Rudin et al. 2013).
Clear evidence concerning resistance to BRAF and MEK inhibitors in NSCLC is yet to be provided, but mechanisms could be the same observed in melanoma. Nevertheless, if BRAF mutations in melanoma occur for the largest part in codon V600 (of which V600E is the archetypal), in NSCLC the involvement of different BRAF activating sites in up to 50% of the cases (Nguyen-Ngoc et al. 2015). Non-V600 BRAF mutants are globally less potently inhibited by available anti-BRAF molecules (Gatalica et al. 2015; Noeparast et al. 2016), making the association with MEK inhibitors even more recommended.
4.3 RET
RET rearrangements drive oncogenesis in 1–2% of lung adenocarcinomas (Kohno et al. 2012). Responses to cabozantinib and vandetanib have currently been systematically recognized in phase II clinical trials (Drilon et al. 2016b; Yoh et al. 2017). Moreover, clinical activity of sunitinib (Wu et al. 2015) and alectinib (Lin et al. 2016) has been documented in RET-driven NSCLC. No clinical demonstration of molecular mechanisms of targeted treatment exhaustion is available thus far. An extensive preclinical study recently identified RET mutations conferring differential resistance to cabozantinib and vandetanib, while overcome by ponatinib, the most potent RET inhibitor (Huang et al. 2016), whose activity in patients is currently under study. Another experience revealed the hyperactivation of SRC, a central gene in focal adhesion, as a suitable mechanism of acquired resistance to dovitinib (Kang et al. 2015); specific inhibition of SRC with sarcatinib allowed the re-sensitization to RET inhibition, as robustly seen in ALK-rearranged models (Crystal et al. 2014).
5 Conclusions
The obtaining of the most prolonged disease control with targeted therapies represents nowadays the primary goal in oncogene-driven advanced NSCLC. The profound knowledge of the molecular mechanisms driving resistance to specific inhibitors is basilar in order to develop further treatment strategies. Nevertheless, in experiences when re-biopsies are performed once treatment exhaustion manifests, mechanistic reasons for this clinical behaviour remain biologically uncovered in up to 30–50% of the cases.
Adaptive strategies with novel inhibitors are showing outstanding results both after and in comparison with first-generation molecules when administered upfront, suggesting a scenario in which the most potent drugs would be given immediately. Nevertheless, combinatorial strategies aiming at bypass collapse, achievable with the fruitful blocking of both the primary molecular alteration and the alternative signalling tracks responsible of resistance, are still lacking. Given the relevant potency of novel molecules against their respective targets, together with the emergence of novel resistance mutations thus far uncommon (EGFR C797S, ALK L1198F), activation of bypass pathways are expected to arise in a significant quote of cases and this therapeutic gap would need to be filled.
Inner tumor heterogeneity manifests in this field with the various mechanisms adopted by tumors to find escapes under specific therapeutic pressure, in different individuals as well as in the diverse lesions of the same patient. This represents one of the greatest issues to deal with, hampering the potential of pharmacologic developments. Strategies to face the limits imposed by biologic tumor variability are lately emerging (Suda et al. 2017).
The detection of mutations responsible of resistance to old and novel EGFR inhibitors in the blood represents a major improvement (hopefully applicable to other drivers) as a proof of principle and for practical reasons. Nevertheless, its application still requires additional adjustments.
Taken together, the evidence contained in this chapter depicts a scenario in continuous evolution, in which the search for the best-targeted treatment option in lung cancer strictly relies upon the digging towards the deeper and widest knowledge concerning biologic resistance. Questions to be solved are still copious and complex; nevertheless, the recent advances in both clinical and preclinical research, allowed by the impressive developments in experimental methods and techniques, generate enthusiasm and hope for the very next future.
References
Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF et al (2011) Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 17:1169–1180
Awad MM (2016) Impaired c-Met receptor degradation mediated by MET exon 14 mutations in non-small-cell lung cancer. J Clin Oncol 34:879–881
Awad MM, Katayama R, McTigue M, Liu W, Deng Y-L, Brooun A et al (2013) Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med 368:2395–2401
Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS, Kuang Y et al (2016) Acquired METD1228V mutation and resistance to MET inhibition in lung cancer. Cancer Discov 6:1334–1341
Ballard P, Yates JWT, Yang Z, Kim DW, Yang JCH, Cantarini M et al (2016) Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res 22:5130–5140
Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H et al (2016) Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French cooperative thoracic intergroup (IFCT). Lancet 387:1415–1426
Benderra MA, Aspeslagh S, Postel-Vinay S, Bigot L, De Baere T, Loriot Y et al (2016) Acquired EGFR mutation as the potential resistance driver to crizotinib in a MET-mutated tumor. J Thorac Oncol 11:e21–e23
Bergethon K, Shaw AT, Ou S-HI, Katayama R, Lovly CM, McDonald NT et al (2012) ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 30:863–870
Campo M, Gerber D, Gainor JF, Heist RS, Temel JS, Shaw AT et al (2016) Acquired resistance to first-line afatinib and the challenges of prearranged progression biopsies. J Thorac Oncol 11:2022–2026
Cargnelutti M, Corso S, Pergolizzi M, Mévellec L, Aisner DL, Dziadziuszko R et al (2015) Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget 6:5182–5194
Caumont C, Veillon R, Gros A, Laharanne E, Bégueret H, Merlio JP (2016) Neuroendocrine phenotype as an acquired resistance mechanism in ALK-rearranged lung adenocarcinoma. Lung Cancer 92:15–18
Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ et al (2016) Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 7:11815
Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T et al (2010) EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 363:1734–1739
Chong CR, Bahcall M, Capelletti M, Kosaka T, Ercan D, Sim T et al (2017) Identification of existing drugs that effectively target NTRK1- and ROS1-rearrangements in lung cancer. Clin Cancer Res 23:204–213
Costa DB, Kobayashi S, Pandya SS, Yeo WL, Shen Z, Tan W et al (2011) CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 29:e443–e445
Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ et al (2014) AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 4:1046–1061
Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A (2013) Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol 10:472–484
Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL et al (2014) Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346:1480–1486
Davare MA, Vellore NA, Wagner JP, Eide CA, Goodman JR, Drilon A et al (2015) Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc Natl Acad Sci 112:E5381–E5390
Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK et al (2013) Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One 8:1–13
Del Re M, Tiseo M, Bordi P, D’Incecco A, Camerini A, Petrini I et al (2016) Contribution of KRAS mutations and c.2369C > T (p.T790M) EGFR to acquired resistance to EGFR-TKIs in EGFR mutant NSCLC: a study on circulating tumor DNA. Oncotarget [Epub ahead of print]
Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ et al (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 18:1472–1482
Douillard JJ-Y, Ostoros G, Cobo M, Ciuleanu T, Cole R, McWalter G et al (2014) Gefitinib treatment in EGFR mutated caucasian NSCLC: circulating-free tumor DNA as a surrogate for determination of EGFR status. J Thorac Oncol 9:1345–1353
Drilon A, Somwar R, Wagner JP, Vellore NA, Eide CA, Zabriskie MS et al (2016a) A novel crizotinib-resistant solvent-front mutation responsive to cabozantinib therapy in a patient with ROS1-rearranged lung cancer. Clin Cancer Res 22:2351–2358
Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M et al (2016b) Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 17:1653–1660
Drilon A, Cappuzzo F, Ou SICD (2017) Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol 12:15–26
Dziadziuszko R, Le AT, Wrona A, Jassem J, Camidge DR, Varella-Garcia M et al (2016) An activating KIT mutation induces crizotinib resistance in ROS1-positive lung cancer. J Thorac Oncol 11:1273–1281
Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR et al (2015) Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res 75:2489–2500
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043
Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ et al (2015) EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res 21:3913–3923
Facchinetti F, Tiseo M, Di Maio M, Graziano P, Novello S (2016a) Tackling ALK in non-small cell lung cancer: the role of novel inhibitors. Transl Lung Cancer Res 5:301–321
Facchinetti F, Loriot Y, Kuo MS, Mahjoubi L, Lacroix L, Planchard D et al (2016b) Crizotinib-resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1- and ALK-rearranged lung cancers. Clin Cancer Res 15:5983–5991
Facchinetti F, Rossi G, Bria E, Soria JC, Besse B, Minari R et al (2017) Oncogene addiction in non-small cell lung cancer: focus on ROS1 inhibition. Cancer Treat Rev. doi:10.1016/j.ctrv.2017.02.010
Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS et al (2014) The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov 4:662–673
Fujita S, Masago K, Katakami N, Yatabe Y (2016) Transformation to SCLC after treatment with the ALK inhibitor alectinib. J Thorac Oncol 11:e67–e72
Gainor JF, Varghese AM, Ou SHI, Kabraji S, Awad MM, Katayama R et al (2013) ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res 19:4273–4281
Gainor JF, Niederst MJ, Lennerz JK, Dagogo-Jack I, Stevens S, Shaw AT et al (2016a) Dramatic response to combination erlotinib and crizotinib in a patient with advanced, EGFR-mutant lung cancer harboring de novo MET amplification. J Thorac Oncol 11:e83–e85
Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R et al (2016b) Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov 6:1118–1133
Gainor JF, Friboulet L, Yoda S, Dardaei Alghalandis L, Farago AF, Logan J et al (2016c) Frequency and spectrum of ROS1 resistance mutations in ROS1-positive lung cancer patients progressing on crizotinib. J Clin Oncol 34:(suppl; abstr 9072)
Gatalica Z, Burnett K, Bender R, Feldman R, Vranic S, Reddy S (2015) BRAF mutations are potentially targetable alterations in a wide variety of solid cancers. Eur J Cancer 51(suppl 3):S31
Goss G, Tsai C-M, Shepherd FA, Bazhenova L, Lee JS, Chang G-C et al (2016) Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 17:1643–1652
Ham JS, Kim S, Kim HK, Byeon S, Sun JM, Lee SH et al (2016) Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J Thorac Oncol 11:e1–e4
Haratani K, Hayashi H, Watanabe S, Kaneda H, Yoshida T, Takeda M et al (2016) Two cases of EGFR mutation-positive lung adenocarcinoma that transformed into squamous cell carcinoma: successful treatment of one case with rociletinib. Ann Oncol 27:200–202
Hata A, Katakami N, Yoshioka H, Kaji R, Masago K, Fujita S et al (2015) Spatiotemporal T790M heterogeneity in individual patients with EGFR-mutant non–small-cell lung cancer after acquired resistance to EGFR-TKI. J Thorac Oncol 10:1553–1559
Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE et al (2016) Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 22:262–269
Heist RS, Sequist LV, Borger D, Gainor JF, Arellano RS, Le LP et al (2016) Acquired resistance to crizotinib in NSCLC with MET exon 14 skipping. J Thorac Oncol 11:1242–1245
Heon S, Yeap BY, Britt GJ, Costa DB, Rabin MS, Jackman DM et al (2010) Development of central nervous system metastases in patients with advanced non-small cell lung cancer and somatic EGFR mutations treated with gefitinib or erlotinib. Clin Cancer Res 16:5873–5882
Heuckmann JM, Balke-Want H, Malchers F, Peifer M, Sos ML, Koker M et al (2012) Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res 18:4682–4690
Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM et al (2015) RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med 21:1038–1047
Huang Q, Schneeberger VE, Luetteke N, Jin C, Afzal R, Budzevich MM et al (2016) Preclinical modeling of KIF5B-RET fusion lung adenocarcinoma. Mol Cancer Ther 15(10):2521–2529
Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J et al (2006) Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res 66:7854–7858
Isozaki H, Ichihara E, Takigawa N, Ohashi K, Ochi N, Yasugi M et al (2016) Non-small cell lung cancer cells acquire resistance to the ALK inhibitor alectinib by activating alternative receptor tyrosine kinases. Cancer Res 76:1506–1516
Jänne PA, Yang JC-H, Kim D-W, Planchard D, Ohe Y, Ramalingam SS et al (2015) AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 372:1689–1699
Jia Y, Yun C-H, Park E, Ercan D, Manuia M, Juarez J et al (2016) Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 534:129–132
Kang CW, Jang KW, Sohn J, Kim S-M, Pyo K-H, Kim H et al (2015) Antitumor activity and acquired resistance mechanism of dovitinib (TKI258) in RET-rearranged lung adenocarcinoma. Mol Cancer Ther 14:2238–2248
Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B et al (2012) Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med 4:120ra17
Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF et al (2014) Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res 20:5686–5696
Katayama R, Kobayashi Y, Friboulet L, Lockerman EL, Koike S, Shaw AT et al (2015) Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clin Cancer Res 21:166–174
Khalifa J, Amini A, Popat S, Gaspar LE, Faivre-Finn C (2016) Brain metastases from NSCLC: radiation therapy in the era of targeted therapies. J Thorac Oncol 11:1627–1643
Kim S, Kim TM, Kim D-W, Go H, Keam B, Lee S-H et al (2013) Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol 8:415–422
Kim TM, Song A, Kim D-W, Kim S, Ahn Y-O, Keam B et al (2015) Mechanisms of acquired resistance to AZD9291, a mutation-selective, irreversible EGFR inhibitor. J Thorac Oncol 10:1736–1744
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M et al (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352:786–792
Kobayashi Y, Sakao Y, Ito S, Sakakura N, Usami N, Mitsudomi T et al (2013) Transformation to sarcomatoid carcinoma in ALK-rearranged adenocarcinoma, which developed acquired resistance to crizotinib and received subsequent chemotherapies. J Thorac Oncol 8:75–78
Kogita A, Togashi Y, Hayashi H, Sogabe S, Terashima M, De Velasco MA et al (2014) Hypoxia induces resistance to ALK inhibitors in the H3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition. Int J Oncol 45:1430–1436
Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T et al (2012) KIF5B-RET fusions in lung adenocarcinoma. Nat Med 18:375–357
Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG et al (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363:1693–1703
Lin JJ, Kennedy E, Sequist LV, Brastianos PK, Goodwin KE, Stevens S et al (2016) Clinical activity of alectinib in advanced RET-rearranged non-small-cell lung cancer. J Thorac Oncol 11:2027–2032
Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y et al (2014) Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med 20:1027–1034
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW et al (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139
Mao C, Yuan J-Q, Yang Z-Y, Fu X-H, Wu X-Y, Tang J-L (2015) Blood as a substitute for tumor tissue in detecting EGFR mutations for guiding EGFR TKIs treatment of nonsmall cell lung cancer: a systematic review and meta-analysis. Medicine (Baltimore) 94:e775
McCoach CE, Le AT, Aisner D, Gowan K, Jones KL, Merrick D et al (2016) Resistance mechanisms to targeted therapies in ROS1+ and ALK+ non-small cell lung cancer. J Clin Oncol 34:(suppl; abstr 9065)
Mengoli MC, Barbieri F, Bertolini F, Tiseo M, Rossi G (2016) K-RAS mutations indicating primary resistance to crizotinib in ALK-rearranged adenocarcinomas of the lung: report of two cases and review of the literature. Lung Cancer 93:55–58
Minari R, Bordi P, Tiseo M (2016) Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: review on emerged mechanisms of resistance. Transl Lung Cancer Res 5:695–708
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N et al (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361:947–957
Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS et al (2016) Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med [Epub ahead of print]
Nguyen K-SH, Kobayashi S, Costa DB (2009) Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer 10:281–289
Nguyen-Ngoc T, Bouchaab H, Adjei AA, Peters S (2015) BRAF alterations as therapeutic targets in non-small cell lung cancer. J Thorac Oncol 10:1396–1403
Niederst MJ, Engelman JA (2013) Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal 6:re6
Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR et al (2015a) RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 6:6377
Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z et al (2015b) The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res 21:3924–3933
Noeparast A, Teugels E, Giron P, Verschelden G, De Brakeleer S, Decoster L et al (2016) Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of trametinib and dabrafenib. Oncotarget [Epub ahead of print]
Novello S, Barlesi F, Califano R, Cufer T, Ekman S, Levra MG et al (2016) Metastatic non-small-cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 27:v1–27
Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL et al (2012) Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A 109:E2127–E2133
Omachi N, Shimizu S, Kawaguchi T, Tezuka K, Kanazu M, Tamiya A et al (2014) A case of large-cell neuroendocrine carcinoma harboring an EML4-ALK rearrangement with resistance to the ALK inhibitor crizotinib. J Thorac Oncol 9:e40–e42
Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L et al (2016) Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res 22:4837–4847
Oser MG, Niederst MJ, Sequist LV, Engelman JA (2015) Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 16:e165–e172
Ou S-HI, Tan J, Yen Y, Soo RA (2012) ROS1 as a “druggable” receptor tyrosine kinase: lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther 12:447–456
Ou SHI, Agarwal N, Ali SM (2016a) High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer 98:59–61
Ou S-HI, Young L, Schrock AB, Johnson A, Klempner SJ, Zhu VW et al (2016b) Emergence of pre-existing MET Y1230C mutation as a resistance mechanism to crizotinib in NSCLC with MET exon 14 skipping. J Thorac Oncol 12:137–140
Oxnard GR, Thress K, Paweletz CP, Stetson D, Dougherty B, Lai Z et al (2015) Mechanisms of acquired resistance to AZD9291 in EGFRT790M positive lung cancer. J Thorac Oncol 9(suppl 2):S207
Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M et al (2016) Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J Clin Oncol 34:3375–3382
Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S et al (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500
Park K, Yu C-J, Kim S-W, Lin M-C, Sriuranpong V, Tsai C-M et al (2016) First-line erlotinib therapy until and beyond response evaluation criteria in solid tumors progression in Asian patients with epidermal growth factor receptor mutation-positive non-small-cell lung cancer: the ASPIRATION study. JAMA Oncol 2:305–312
Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M et al (2015) Heterogeneity underlies the emergence of EGFRT790wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov 5:713–723
Planchard D, Loriot Y, André F, Gobert A, Auger N, Lacroix L et al (2015) EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol 26:2073–2208
Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F et al (2016a) Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 17:642–650
Planchard D, Besse B, Groen HJ, Souquet PJ, Quoix E, Baik CS et al (2016b) Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 17:984–993
Reckamp KL, Melnikova VO, Karlovich C, Sequist LV, Camidge DR, Wakelee H et al (2016) A highly sensitive and quantitative test platform for detection of NSCLC EGFR mutations in urine and plasma. J Thorac Oncol 11:1690–1700
Redaelli S, Ceccon M, Antolini L, Rigolio R, Pirola A, Peronaci M et al (2016) Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget 7:72886–72897
Remon J, Caramella C, Jovelet C, Lacroix L, Lawson A, Smalley S et al (2017) Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Ann Oncol [Epub ahead of print]
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C et al (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361:958–967
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E et al (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13:239–246
Rudin CM, Hong K, Streit M (2013) Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with BRAF-mutant non-small-cell lung cancer. J Thorac Oncol 8:e41–e42
Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C et al (2008) Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol 26:3543–3551
Schmid S, Gautschi O, Rothschild S, Mark M, Froesch P, Klingbiel D et al (2016) Clinical outcome of ALK-positive non-small cell lung cancer (NSCLC) patients with de novo EGFR or KRAS co-mutations receiving tyrosine kinase inhibitors (TKI). J Thorac Oncol [Epub ahead of print]
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P et al (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3:75ra26
Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A et al (2015) Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med 372:1700–1709
Sequist LV, Piotrowska Z, Niederst MJ, Heist RS, Digumarthy S, Shaw AT et al (2016) Osimertinib responses after disease progression in patients who had been receiving rociletinib. JAMA Oncol 2:541–543
Shaw AT, Kim D-W, Mehra R, Tan DSW, Felip E, Chow LQM et al (2014) Ceritinib in ALK-rearranged non–small-cell lung cancer. N Engl J Med 370:1189–1197
Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A et al (2015) Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med 374:54–61
Siegel RL, Miller KD, Jemal A (2016) Cancer statistics 2016. CA Cancer J Clin 66:7–30
Solomon BJ, Mok T, Kim D-W, Wu Y-L, Nakagawa K, Mekhail T et al (2014) First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 371:2167–2177
Song A, Kim TM, Kim D-W, Kim S, Keam B, Lee S-H et al (2015) Molecular changes associated with acquired resistance to crizotinib in ROS1-rearranged non-small cell lung cancer. Clin Cancer Res 21:2379–2387
Soo R, Kim D-W, Yang JC-H, Park K, Stammberger U, Xiong H et al (2015) Highly selective c-Met inhibitor tepotinib plus gefitinib is active in Asian patients with c-Met+ NSCLC. Ann Oncol 26(suppl 9):ix143
Soria JC, Wu YL, Nakagawa K, Kim SW, Yang JJ, Ahn MJ et al (2015) Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol 16:990–998
Soria JC, Kim SW, Wu YL, Nakagawa K, Yang JJ, Ahn MJ et al (2016) Gefitinib/chemotherapy vs chemotherapy in EGFR mutation-positive NSCLC after progression on 1st line gefitinib (IMPRESS study): final overall survival (OS) analysis. Ann Oncol 27(suppl 6):1201O
Su KY, Chen HY, Li KC, Kuo ML, Yang JCH, Chan WK et al (2012) Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol 30:433–440
Suda K, Murakami I, Sakai K, Tomizawa K, Mizuuchi H, Sato K et al (2016) Heterogeneity in resistance mechanisms causes shorter duration of epidermal growth factor receptor kinase inhibitor treatment in lung cancer. Lung Cancer 91:36–40
Suda K, Bunn PA, Rivard CJ, Mitsudomi T, Hirsch FR (2017) Primary double-strike therapy for cancers to overcome EGFR kinase inhibitor resistance: proposal from the bench. J Thorac Oncol 12:27–35
Sundaresan TK, Sequist LV, Heymach JV, Riely GJ, Jänne PA, Koch WH et al (2016) Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin Cancer Res 22:1103–1110
Takegawa N, Hayashi H, Iizuka N, Takahama T, Ueda H, Tanaka K et al (2016) Transformation of ALK rearrangement-positive adenocarcinoma to small-cell lung cancer in association with acquired resistance to alectinib. Ann Oncol 27:953–955
Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E et al (2012) HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFR T790M mutation. Cancer Discov 2:922–933
Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N et al (2005) Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 65:9455–9462
Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B et al (2015) Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 21:560–562
Toyokawa G, Inamasu E, Shimamatsu S, Yoshida T, Nosaki K, Hirai F et al (2015) Identification of a novel ALK G1123S mutation in a patient with ALK-rearranged non-small-cell lung cancer exhibiting resistance to ceritinib. J Thorac Oncol 10:e55–e57
Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A et al (2015) Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discov 5:960–971
Tseng JS, Yang TY, Tsai CR, Chen KC, Hsu KH, Tsai MH et al (2015) Dynamic plasma EGFR mutation status as a predictor of EGFR-TKI efficacy in patients with EGFR-mutant lung adenocarcinoma. J Thorac Oncol 10:603–610
Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E et al (2010) Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 17:77–88
Ulivi P, Chiadini E, Dazzi C, Dubini A, Costantini M, Medri L et al (2016) Nonsquamous, non-small-cell lung cancer patients who carry a double mutation of EGFR, EML4-ALK or KRAS: frequency, clinical-pathological characteristics, and response to therapy. Clin Lung Cancer 17:384–390
Wang L, Hu H, Pan Y, Wang R, Li Y, Shen L et al (2014) PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS One 9:e0088291
Weickhardt AJ, Scheier B, Burke JM, Gan G, Lu X, Bunn PA et al (2012) Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogene-addicted non-small-cell lung cancer. J Thorac Oncol 7:1807–1814
Woo CG, Seo S, Kim SW, Jang SJ, Park KS, Song JY et al (2016) Differential protein stability and clinical responses of EML4-ALK fusion variants to various ALK inhibitors in advanced ALK-rearranged non-small cell lung cancer. Ann Oncol [Epub ahead of print]
Wu JY, Yu CJ, Chang YC, Yang CH, Shih JY, Yang PC (2011) Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res 17:3812–3821
Wu H, Shih JY, Yang JC (2015) Rapid response to sunitinib in a patient with lung adenocarcinoma harboring KIF5B-RET fusion gene. J Thorac Oncol 10:e95–e96
Wu YL, Kim DW, Felip E, Zhang L, Liu X, Zhou CC et al (2016) Phase (Ph) II safety and efficacy results of a single-arm ph ib/II study of capmatinib (INC280) + gefitinib in patients (pts) with EGFR-mutated (mut), cMET-positive (cMET + ) non-small cell lung cancer (NSCLC). J Clin Oncol 34:(suppl;abstr 9020)
Yang JCH, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N et al (2015) Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-lung 3 and LUX-lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 16:141–151
Yang J, Ramalingam SS, Jänne PA, Cantarini M, Mitsudomi T (2016) Osimertinib (AZD9291) in pre-treated pts with T790M-positive advanced NSCLC: updated phase 1 (P1) and pooled phase 2 (P2) results. J Thorac Oncol 11(suppl 4):S152–S153
Yap TA, Macklin-Doherty A, Popat S (2017) Continuing EGFR inhibition beyond progression in advanced non-small cell lung cancer. Eur J Cancer 70:12–21
Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL et al (2013) Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 5:216ra177
Yoh K, Seto T, Satouchi M, Nishio M, Yamamoto N, Murakami H et al (2017) Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med 5:42–50
Yoshida T, Oya Y, Tanaka K, Shimizu J, Horio Y, Kuroda H et al (2016) Differential crizotinib response duration among ALK fusion variants in ALK-positive non-small-cell lung cancer. J Clin Oncol 34:3383–3389
Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W et al (2013a) Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 19:2240–2247
Yu HA, Sima CS, Huang J, Solomon SB, Rimner A, Paik P et al (2013b) Local therapy with continued EGFR tyrosine kinase inhibitor therapy as a treatment strategy in EGFR-mutant advanced lung cancers that have developed acquired resistance to EGFR tyrosine kinase inhibitors. J Thorac Oncol 8:346–351
Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T et al (2012) Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 44:852–860
Zou H, Friboulet L, Kodack D, Engstrom L, Li Q, West M et al (2015) PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical odels. Cancer Cell 28:70–81
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Facchinetti, F., Proto, C., Minari, R., Garassino, M., Tiseo, M. (2017). Mechanisms of Resistance to Target Therapies in Non-small Cell Lung Cancer. In: Mandalà, M., Romano, E. (eds) Mechanisms of Drug Resistance in Cancer Therapy. Handbook of Experimental Pharmacology, vol 249. Springer, Cham. https://doi.org/10.1007/164_2017_16
Download citation
DOI: https://doi.org/10.1007/164_2017_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-10506-8
Online ISBN: 978-3-030-10507-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)