
Abstract
Exoenzyme Y (ExoY) was identified as a component of the Pseudomonas aeruginosa type 3 secretion system secretome in 1998. It is a common contributor to the arsenal of type 3 secretion system effectors, as it is present in approximately 90% of Pseudomonas isolates. ExoY has adenylyl cyclase activity that is dependent upon its association with a host cell cofactor. However, recent evidence indicates that ExoY is not just an adenylyl cyclase; rather, it is a promiscuous cyclase capable of generating purine and pyrimidine cyclic nucleotide monophosphates. ExoY’s enzymatic activity causes a characteristic rounding of mammalian cells, due to microtubule breakdown. In endothelium, this cell rounding disrupts cell-to-cell junctions, leading to loss of barrier integrity and an increase in tissue edema. Microtubule breakdown seems to depend upon tau phosphorylation, where the elevation of cyclic nucleotide monophosphates activates protein kinases A and G and causes phosphorylation of endothelial microtubule associated protein tau. Phosphorylation is a stimulus for tau release from microtubules, leading to microtubule instability. Phosphorylated tau accumulates inside endothelium as a high molecular weight, oligomeric form, and is then released from the cell. Extracellular high molecular weight tau causes a transmissible cytotoxicity that significantly hinders cellular repair following infection. Thus, ExoY may contribute to bacterial virulence in at least two ways; first, by microtubule breakdown leading to loss of endothelial cell barrier integrity, and second, by promoting release of a high molecular weight tau cytotoxin that impairs cellular recovery following infection.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Endothelium lines blood and lymphatic vessels throughout all organ systems, and in this capacity, serves as a semi-permeable barrier controlling the flux of solutes, macromolecules, and cells between the circulation and the underlying tissue (Townsley and Stevens 2015). This barrier property is highly dynamic, and is modulated according to the demands of the local environment. Adenosine 3′,5′-cyclic monophosphate (cAMP) is a ubiquitous second messenger that greatly influences the strength of endothelial cell adhesion (Sayner 2011). Circulating epinephrine (Sayner 2011) and prostacyclin (Birukova et al. 2007, 2012, 2013, 2015) activate transmembrane adenylyl cyclases, which increase endothelial cell cAMP that promotes cell-to-cell adhesion and decreases permeability (Fig. 1a). In contrast, neurohumoral inflammatory calcium agonists inhibit endothelial transmembrane adenylyl cyclase activity, which decreases cAMP leading to loss of cell-to-cell adhesion and an increase in permeability (Cioffi et al. 2002; Stevens et al. 1995). Approaches to elevate endothelial cell cAMP concentration would therefore appear to represent an appropriate anti-inflammatory strategy (Sayner 2011; Moore et al. 1998). However, this is not the whole story.

Signaling through transmembrane and soluble adenylyl cyclases elicit different physiological outcomes in endothelium. (a) Circulating first messengers, such as epinephrine and prostacyclin, bind endothelial cell G protein coupled receptors that activate transmembrane adenylyl cyclases. Production of cAMP by transmembrane adenylyl cyclases strengthens the cortical actin rim, which stabilizes adherens junctions. This cAMP signal promotes microtubule interaction with peripheral actin. (b) ExoY is a promiscuous nucleotidyl cyclase that produces cGMP, cAMP, cUMP, and cCMP in endothelium. The cAMP signal is responsible for microtubule breakdown, leading to cell rounding and endothelial cell barrier disruption. cAMP, and to a lesser extent cGMP, activates PKA, which phosphorylates the microtubule associated protein tau. As tau becomes hyperphosphorylated, it is released from microtubules and forms apparent high molecular weight oligomers. These oligomers are released from the endothelium, and are transmissible among cells causing hyperpermeability and cytotoxicity. The stimulus for tau oligomer release is unknown. However, its release parallels the rise in cUMP, and emerging evidence suggests that cUMP may act as a second messenger important for stimulating tau oligomer release (unpublished). Other promiscuous nucleotidyl cyclases are used as part of the virulence arsenal of bacteria, including cyaA of Bordetella pertussis, edema factor of Bacillus anthracis, and MARTX of Vibrio vulnificus. These nucleotidyl cyclases have not yet been systematically studied in endothelium, although they are each soluble cylases with activity resembling ExoY
Bacteria utilize soluble adenylyl cyclases as mammalian cell toxins, and these toxins contribute significantly to bacterial virulence (Sayner 2011). Such enzymatic activity was first described with the Bacillus anthracis edema factor (Leppla 1982) and Bordetella pertussis cyaA (Glaser et al. 1988), and more recently with the biotype 3 variant Vibrio vulnificus multifunctional autoprocessing RTX (MARTX) toxin (Ziolo et al. 2014) and Pseudomonas aeruginosa exoenzyme Y (ExoY) (Yahr et al. 1998). Because P. aeruginosa is a principal cause of pneumonia that can progress to sepsis and acute lung injury, our group studied the contribution of ExoY to endothelial hyperpermeability (Fig. 1b). We have seen that ExoY functions not only as an adenylyl cyclase, but rather, as a promiscuous purine and pyrimidine nucleotidyl cyclase in vascular endothelium, generating cGMP, cAMP, cUMP and, to a lesser extent, cCMP (Morrow et al. 2015). The elevation of these cyclic nucleotide monophosphates causes endothelial tau hyperphosphorylation, which leads to microtubule breakdown and endothelial hyperpermeability (Balczon et al. 2013; Morrow et al. 2016; Ochoa et al. 2012; Prasain et al. 2009; Sayner et al. 2011). Hyperphosphorylation appears to trigger tau oligomerization and release, contributing to a transmissible proteinopathy that perpetuates cytotoxicity during infection (Morrow et al. 2016).
It is now apparent that cyclic nucleotide monophosphate signal transduction is far more complex than initially conceived. In endothelium, the location of cAMP production is a critical determinant of its end effect, where transmembrane adenylyl cyclases produce a cAMP signal that is barrier protective and cytosolic adenylyl cyclases produce a cAMP signal that is barrier disruptive (Sayner 2011; Moore et al. 1998). Studies exploiting the actions of ExoY have revealed that endothelium constitutively possesses both purine and pyrimidine (i.e., non-canonical) cyclic nucleotide monophosphates, yet little is known about the non-canonical cyclic nucleotide monophosphates. This chapter highlights key discoveries that led to our current understanding of cyclic nucleotide monophosphate signaling in endothelium, especially as it relates to control of barrier function.
2 The Endothelial cAMP Signal as a Barrier Protective Mechanism
Epinephrine has been a long-standing therapy for acute management of anaphylaxis. Among its many physiological effects, epinephrine binds endothelial β2 receptors that are functionally coupled to an elevation in cAMP (Sayner 2011). This increase in intracellular cAMP activates protein kinase A (PKA), resulting in phosphorylation of effectors such as the actin binding protein filamin (Sayner et al. 2011). Phosphorylated filamin promotes F-actin alignment in a cortical rim that increases attachments among junctional complexes, strengthening the endothelial cell barrier. Thus, it is evident that cAMP arising from transmembrane adenylyl cyclases protects endothelial cell barrier integrity, and serves as a dominant intracellular signal controlling adhesion (Cioffi et al. 2002; Stevens et al. 1995).
It is curious then how circulating inflammatory agonists disrupt the endothelial cell barrier. Many of these agonists, such as substance P, histamine, bradykinin, and thrombin, elevate cytosolic calcium, which leads to myosin light chain kinase phosphorylation and realignment of the cortical actin rim into centripetally directed stress fibers (Dudek and Garcia 2001). This increase in tension is accompanied by loss of cell-to-cell adhesions, enabling formation of transient inter-endothelial cell gaps that allow fluid, solute, and protein leak into the tissue (Mehta and Malik 2006).
The strength of this calcium signal is amplified by calcium inhibition of transmembrane adenylyl cyclases (Stevens et al. 1995). Endothelial cells express the type 6 adenylyl cyclase (AC6), which is inhibited by submicromolar increases in calcium, especially by calcium influx through store operated calcium entry channels (Cioffi et al. 2002; Creighton et al. 2003; Stevens et al. 1999). The sensitivity of calcium inhibition is most evident when measuring the ATP-to-cAMP turnover rates in plasma membrane fractions, although it can be detected by radioimmunoassay in whole cell lysates as well. Development of Fluorescence Resonance Energy Transfer (FRET) approaches to measure cAMP also enables visualization of AC6 calcium inhibition in single living cells (Werthmann et al. 2009). Depending upon the endothelial cell phenotype studied, the magnitude of calcium inhibition can range from 20 to nearly 90% of the membrane cyclase activity (Stevens et al. 1999), bringing into question the importance of this cross-talk mechanism in controlling endothelial cell barrier integrity.
To address this issue, a calcium stimulated adenylyl cyclase (AC8) was expressed in endothelium, and its impact on permeability assessed (Cioffi et al. 2002). AC8 was correctly targeted to the plasma membrane where it produced cAMP in apparent lipid raft domains, as appropriate. Expression of AC8 did not change baseline cAMP concentrations, yet it converted calcium inhibition into modest calcium stimulation of cAMP. Under these conditions, even high thrombin concentrations failed to disrupt the endothelial cell barrier, providing direct evidence that AC6 mediates physiologically relevant crosstalk between calcium and cAMP.
3 Edema Factor: A Soluble Adenylyl Cyclase
It seemed clear that elevations in cAMP protect the endothelial cell barrier, and further, that calcium influx lowers cAMP as a necessary prerequisite to the barrier disruption that causes edema. However, an apparent paradox to this understanding was lurking in the microbiology literature, and had not been considered by the vascular biology community. Studies in the 1950s from microbiologists examining Bacillus anthracis revealed that the death of infected animals was accompanied by tissue edema (Smith et al. 1955). Exudate obtained from an admixture of plasma and peritoneal and thoracic exudates was sufficient to cause small edematous lesions following intradermal injection. Thus, it appeared that B. anthrax produced some edema-causing agent; an agent that we would later learn is an adenylyl cyclase.
Over the ensuing 30 years, the molecular basis for anthrax toxicity was determined to depend upon three components: protective antigen, lethal factor, and edema factor (Keppie et al. 1963; Stanley and Smith 1961, 1963). Protective antigen binds to the mammalian cell surface to enable introduction of either lethal factor or edema factor. In the early 1980s, the molecular basis of edema factor came into focus (Leppla 1982). In what has become a citation classic, Leppla reported that edema factor increases mammalian cell cAMP. He considered the possibility that edema factor increases cAMP because of the known actions of cholera toxin, which like edema factor, increases vascular permeability upon its introduction into the skin of animals (Craig 1965; Johnson et al. 1971). Cholera toxin induces ADP ribosylation of Gsα proteins, inhibiting GTPase catalytic activity, which allows transmembrane adenylyl cyclases to remain active, causing an increase in cAMP. Coincident with increased cAMP, cholera toxin also produces a characteristic CHO cell shape change. This change in cell shape could be reproduced using active edema factor, leading Leppla to consider edema factor was in some way generating intracellular cAMP. Leppla examined whether edema factor acts like cholera toxin, causing ADP ribosylation of a Gsα protein (referred to as ADP-ribosylation of the cyclase, since its Gsα target had not been determined). However, his evidence did not support this assertion, and rather, suggested that edema factor itself directly contributes to an adenylyl cyclase activity. We now know that this latter conclusion is correct. Edema factor is introduced into mammalian cells where it is localized within the cell’s cytosolic fraction. In this fraction, edema factor interacts with calmodulin to confer enzymatic activity. As discussed below, evidence that edema factor activity is resolved within the cytosol is essential to the biological function of such “soluble” enzymes.
Edema factor is just one example of a broader family of soluble (class II) adenylyl cyclases (Linder and Schultz 2003). These enzymes are produced by multiple different bacterial species, including B. anthracis (e.g., edema factor), Bordetella pertussis (e.g., cyaA), Vibrio vulnificus (e.g., MARTX toxin), and Pseudomonas aeruginosa (e.g., ExoY). Our group became increasingly interested in the enzymatic activity of these soluble enzymes following discovery of ExoY. P. aeruginosa infection is a common cause of pneumonia that can progress to sepsis and acute lung injury, especially in immunocompromised patients. Moreover, P. aeruginosa pneumonia is described to cause vasculitis and coagulative necrosis, indicating interaction of the bacterium with the lung microvascular endothelium (Winn et al. 2008).
4 Studies on the Discovery of ExoY and Its Enzymatic Activity
Pseudomonas aeruginosa maintains a large genome that encodes the regulatory and structural genes required to infect a variety of hosts. Significant resources are dedicated to making use of a type 3 secretion system (T3SS), through which P. aeruginosa injects four known effector proteins (ExoS, ExoT, ExoU, and ExoY) (Engel and Balachandran 2009; Hauser 2009). Transcription of these four exoenzymes is regulated by the proximal transcription factor, ExsA. Prior to 1998, three of the four effector proteins had been identified and their activities during eukaryotic cellular infections described (Engel and Balachandran 2009; Hauser 2009). The functional roles of these effectors were postulated to facilitate infection of epithelial cells followed by dissemination of the bacterium to the bloodstream.
The fourth potential effector – now known to be ExoY – had been observed from early work comparing the secretome of ExsA(+) and ExsA(−) P. aeruginosa strains (Yahr et al. 1997). The effector was identified as a 42-kDa protein that was detectable, albeit at much lower levels than ExoS and ExoT, in strains 388 and PAK but not in the extracellular supernatants of strain PA103. Amino-terminal sequences obtained in these analyses were subsequently used in a search of the newly sequenced (at that time) PAO1 genome (Stover et al. 2000). After adjusting for a frameshift and searching for the most probable open reading frame, a hypothetical protein corresponding to 42-kDa was identified that shared significant homology with the extracellular adenylyl cyclases of Bordetella pertussis (e.g., CyaA) and Bacillus anthracis (e.g., edema factor). The absence of signal sequence processing and possession of a consensus ExsA binding site suggested that this protein – ExoY – was a T3SS effector that may possess adenylyl cyclase activity. Yahr et al. (1998) determined that ExoY purified from E. coli possessed adenylyl cyclase activity and that amino acid residues important for ATP-binding were conserved in ExoY, CyaA, and edema factor. Although ExoY shares significant homology in the catalytic domain with CyaA and edema factor, the previously characterized calmodulin-binding domain was absent from ExoY. Calmodulin is required to measure edema factor catalytic activity, and it stimulates CyaA activity over 500-fold (Karst et al. 2010; Schuler et al. 2012; Selwa et al. 2012, 2014). ExoY, however, was not stimulated or activated by calmodulin in the Yahr studies (Yahr et al. 1998). Stimulation of adenylyl cyclase activity was, however, detectable after the addition of a post-nuclear extract from Chinese hamster ovary cells (CHO). This complex mixture of cytosolic proteins stimulated ExoY activity at least 500-fold in a dose–response relationship. Heated extracts were inactive, suggesting that the cofactor for ExoY was likely a protein. The identity of the eukaryotic cofactor(s) for ExoY remains elusive, although just recently, filamentous actin (F-actin) has been reported to be sufficient to confer ExoY activity (Belyy et al. 2016). To study the biological activity of ExoY, an effector-less strain of P. aeruginosa was constructed (PA103ΔexoUexoT::Tc) and rExoY was coordinately expressed with the T3SS machinery for injection into CHO cells (Yahr et al. 1998). In contrast to the biological activity of heat-labile enterotoxin and pertussis toxin, which caused an elongated or clustering of CHO cells, ExoY injected by the T3SS rounded CHO cells. This differential regulation of cell morphology was hypothesized to either be due to the magnitude of cAMP generation or to differences in subcellular distribution of the toxin. Two key studies by Sayner et al. confirmed the latter hypothesis. In the first of these studies, Sayner and colleagues utilized a P. aeruginosa strain (PA103ΔexoUexoT::Tc pUCPexoY) capable of introducing only ExoY into mammalian cells through the T3SS (Sayner et al. 2004). Using this bacterial strain, and a second strain introducing inactive ExoY (PA103ΔexoUexoT::Tc pUCPexoY K81M), they determined that ExoY localized to the cytosol of pulmonary microvascular endothelial cells, and found that the ExoY-derived cAMP signal caused endothelial cell rounding leading to disruption of cell-to-cell adhesions and increased permeability (Sayner et al. 2004). This study provided direct evidence that ExoY acts like an “edema factor,” directly contributing to permeability edema. The findings of this study were in stark contrast to the widely held belief that cAMP is barrier protective. However, they were taken to mean that the activation of soluble adenylyl cyclases, that is, enzymes located in the cytosol, is barrier disruptive, whereas activation of transmembrane adenylyl cyclases is barrier protective.
In a second study, Sayner and colleagues (Sayner et al. 2006) evaluated whether transmembrane or soluble adenylyl cyclase activity dominates in control of endothelial barrier integrity. To test this idea, a soluble mammalian adenylyl cyclase chimera, the sACI/II enzyme, was expressed in endothelium. This chimeric enzyme mimics the localization of ExoY in the cytosol. The enzyme is constitutively inactive, and is directly stimulated by forskolin. Forskolin, however, also activates transmembrane adenylyl cyclases, and so it is capable of simultaneously producing cAMP from both transmembrane and soluble enzymes. In this case, forskolin acutely disrupted the endothelial cell barrier, demonstrating that soluble adenylyl cyclase activity dominates in control of the endothelial cell barrier (Sayner et al. 2006). An important facet of this work relates to the magnitude of increase in cAMP that is necessary to disrupt the endothelial barrier. sACI/II cytosolic activity is less than one-tenth of the transmembrane adenylyl cyclase activity, indicating that it takes very little cytosolic cAMP to disrupt the endothelial barrier. This issue is important to the effector function of ExoY, as ExoY will need to produce only low amounts of cAMP in order to cause edema.
5 ExoY and Disruption of the Endothelial Cytoskeleton
ExoS and ExoT possess RhoGAP activity, which remodels F-actin leading to cell rounding. Cowell and colleagues (Cowell et al. 2005) examined whether the adenylyl cyclase activity of ExoY was similarly associated with F-actin remodeling. ExoY+ and ExoYK81M infections both led to apparent F-actin disruption in epithelial cells within 2-hours, suggesting the ExoY protein but not its enzymatic activity interferes with F-actin alignment. Just recently, the Mechold group (Belyy et al. 2016) revealed an interaction between ExoY and F-actin. F-actin was found to bind directly to ExoY where it served as a putative enzymatic cofactor. ExoY interaction with F-actin competitively dislodged other F-actin binding proteins, such as the Arp2/3 complex, providing a mechanism for remodeling the actin cytoskeleton. Consistent with these results, soluble adenylyl cyclase activity decreased phosphorylation of myosin light chain 20, indicating impaired actomyosin interaction. It therefore appears that ExoY intimately interacts with F-actin, perhaps necessary to confer its enzymatic activity, yet ExoY activity is not required for remodeling of the F-actin cytoskeleton.
Although ExoY intoxication does not promote F-actin contraction, it leads to progressive breakdown of peripheral microtubules, coincident with cell rounding, and inter-endothelial cell gap formation (Fig. 2). Balczon and colleagues examined the mechanism underlying this microtubule breakdown (Balczon et al. 2013). ExoY production of cAMP activates protein kinase A, which phosphorylates the microtubule associated protein tau. This endothelial cell tau stabilizes dynamic microtubule remodeling. Following protein kinase A phosphorylation, however, tau dissociates from microtubules, which promotes dynamic instability. Microtubule breakdown could result from decreased centrosome nucleation, increased rate of disassembly, or decreased rate of assembly. ExoY intoxication did not impact the rate of centrosome nucleation and it did not impact the rate at which cold exposure disassembled microtubules. Rather, ExoY decreased the rate at which microtubules assembled. Thus, ExoY catalytic activity targets endothelial cell tau for phosphorylation, causing microtubule breakdown by impairing its rate of assembly.

Loss of peripheral microtubules following ExoY+ intoxication. Pulmonary microvascular endothelial cells were infected with either ExoY+ or ExoYK81M at a multiplicity of infection (MOI) of 20:1, and microtubule abundance was assessed by anti-tubulin fluorescence microscopy. Microtubules were also assessed in uninfected (Control) cells. Scale bar = 10 μm. Adapted from Balczon et al. (2013)
6 Discovery That ExoY Is a Guanylyl Cyclase
During the initial biochemical characterization of ExoY, GTP was added to a standard reaction mix to assess whether ExoY possessed guanylyl cyclase activity (Yahr et al. 1998). However, cGMP synthesis was not detected under these conditions and the assay for ExoY-mediated guanylyl cyclase activity was not revisited with assay conditions containing eukaryotic cytosolic cofactors. Thus, ExoY had been considered an adenylyl cyclase for approximately 12 years when Gottle et al. (2010) demonstrated that edema factor and CyaA could generate multiple cyclic nucleotides (namely cCMP, cUMP, and to a lesser extent cIMP (Gottle et al. 2010)). Given the shared enzymatic properties between ExoY, CyaA, and edema factor, it was reasonable to consider that ExoY generates multiple cyclic nucleotide monophosphates. Coupling this possibility with the fact that cAMP and cGMP both influence endothelial barrier integrity, Ochoa et al. (2012) hypothesized that ExoY possessed both adenylyl and guanylyl cyclase activity when introduced into pulmonary microvascular endothelial cells (PMVECs). When PMVECs were infected, ExoY intoxication generated a significantly higher (~10-fold) cGMP than cAMP signal, suggesting ExoY is principally a guanylyl cyclase (Ochoa et al. 2012). Yahr et al. (1998) previously demonstrated that substituting a single lysine residue within the ATP-binding domain at position 81 with methionine (e.g., ExoYK81M) abolished adenylyl cyclase activity. ExoYK81M also abolished ExoY’s guanylyl cyclase activity. ExoY was coined a “promiscuous cyclase,” as multiple lines of evidence now clearly illustrate ExoY’s multifunctional enzymatic activity. The nature of the enzyme catalytic pocket enabling recognition of multiple substrates has not yet been determined, although we consider that mammalian cofactor(s) may modulate substrate specificity, or alternatively, enzyme intracellular location may be a determinant of which substrate ExoY hydrolyzes.
7 ExoY as a Purine and Pyrimidine Cyclase
Given the structural similarities between P. aeruginosa ExoY, edema factor, and CyaA, coupled with evidence that CyaA and edema factor possess cytidylyl and uridylyl cyclase activity (Gottle et al. 2010), the possibility that ExoY might have broad substrate specificity, and therefore be able to generate purine and pyrimidine cyclic nucleotides, was reasonable. Beckert et al. (2014a) tested this idea in B103 neuroblastoma and A549 lung carcinoma cells. B103 cells were transfected with ExoY and purine and pyrimidine cyclic nucleotide monophosphates were measured by mass spectrometry in the same samples over a 3-day time course. They observed a long-lasting cyclic nucleotide monophosphate increase in the order cGMP ~ cUMP > cAMP ~ cCMP. Next, to evaluate the effects of a T3SS-delivered ExoY, they infected B103 and A549 cells at a multiplicity of infection (MOI) of 5:1 for 0–4 h and assessed cyclic nucleotide monophosphate levels. Four hours post-infection in B103 cells they observed a cyclic nucleotide monophosphate increase in the order cUMP ~ cGMP > > cCMP > cAMP. They observed a similar pattern in the A549 cells following infection. Interestingly, cUMP and cGMP increased first between 1–2 h post-infection, followed by increases in both cAMP and cCMP between 2 and 3 h post-infection. From these studies it is clear that ExoY: (1) generates both purine and pyrimidine cyclic nucleotide monophosphates in mammalian cells, and (2) its capacity to do so depends upon the cellular phenotype. To determine whether ExoY similarly increases purine and pyrimidine cyclic nucleotide monophosphates in vivo, Bahre et al. (2015) infected mice with P. aeruginosa ExoY+. cUMP levels rapidly increased in lung tissue, and remained elevated 2–3 days post-infection. They also reported that cGMP levels exhibited a slow, gradual increase over a 24-hour time course post-infection (Bahre et al. 2015). The precise cNMP signal that is responsible for sustained vascular damage following ExoY+ intoxication remains to be elucidated. Although these studies further supported the notion that ExoY is a promiscuous cyclase, the cell type(s) responsible for increased cyclic nucleotide monophosphates were unclear; it was suggested that both epithelial and endothelial cells contribute to this increase in cyclic nucleotide monophosphates. We examined whether ExoY+ produces both purine and pyrimidine cyclic nucleotide monophosphates in lung endothelium (Morrow et al. 2015). Intoxication of pulmonary endothelium at an MOI of 20:1 with ExoY+ produces cGMP first, followed by cUMP, and then cAMP; cCMP concentrations increase last, and this increase is of a relatively low magnitude. Although the temporal relationship among cyclic nucleotide monophosphates is similar between cell types, the magnitude of their increase is not. ExoY increases all cyclic nucleotide monophosphates to a greater extent in pulmonary artery than in pulmonary microvascular endothelial cells. This difference in cyclic nucleotide monophosphate metabolism has important implications regarding the physiological response to infection, mechanisms regulating ExoY catalytic activity, e.g., enzyme cofactors, and potentially, cellular mechanisms responsible for cyclic nucleotide monophosphate turnover, e.g., phosphodiesterase activity and extrusion. Perhaps most provocatively, however, this work establishes a temporal relationship among cyclic nucleotide monophosphates as they are synthesized in endothelium, where cGMP is synthesized first, followed by cUMP and then cAMP. In future studies it will be important to resolve the signaling function of these cyclic nucleotides in time and space.
8 The Intracellular Function of Purine and Pyrimidine Cyclic Nucleotide Monophosphates
The function(s) of purine and pyrimidine cyclic nucleotide monophosphates in endothelium is complex and poorly understood. cGMP is sufficient to activate protein kinases G and A, which may contribute to tau phosphorylation. However, this cGMP signal does not appear to be the principal cause of tau phosphorylation (Ochoa et al. 2012). Other signaling roles played by cGMP in this context have not been explored, although the issue is important, as cGMP is not only the first, it is the most prominent signal produced by ExoY. cUMP increases second, along with cAMP. At present there is no known role for cUMP in endothelium. Evidence from the Seifert group (Wolter et al. 2011) indicates that cUMP can activate protein kinase A, and be hydrolyzed by phosphodiesterases 3a, 3b, and 9 (Reinecke et al. 2011), suggesting it serves as an important second messenger in mammalian cells. The cAMP signal best parallels endothelial cell barrier disruption, an effect that has been replicated by activation of other soluble adenylyl cyclases that produce only cAMP (Sayner et al. 2006). The ExoY-induced cAMP signal activates protein kinase A that clearly phosphorylates tau. Other intracellular targets of this cAMP response have not been thoroughly studied. Most of what we know about ExoY signaling is based upon its ability to increase cAMP and cause tau hyperphosphorylation.
Tau hyperphosphorylation has important physiological consequences, as this signal causes tau dissociation from microtubules leading to their breakdown. Tau accumulates in the endothelial cytosol for several hours following infection, and then is released in a high molecular weight, injurious form (Morrow et al. 2016) (see below). Interestingly, tau oligomerization and release closely parallels the increase in cUMP levels. Thus, it is possible that cUMP contributes to the release of oligomerized tau. This mechanism is of particular interest because, as has been recently established within the neurological field, extracellular tau oligomers are transmissible among cells as a mechanism of cytotoxicity (Frost et al. 2009; Kfoury et al. 2012).
9 Is ExoY Relevant to Disease?
Whether ExoY is relevant to the virulence of infection has been a topic of debate since its initial discovery in 1998. The definition of “virulence” varies among investigators, especially in light of the variety of cellular endpoints used to assess it. Virulence is broadly defined as “the capability of a microorganism to cause disease.” In the context of ExoY then, the relevant matter is whether or not introduction of ExoY through the T3SS contributes to pathology. The majority of studies addressing this issue have measured the: (1) severity of acute or initial infection, determined by host survival within the first 24–48 h post-infection; (2) bacterial dissemination or spread from the initial site of infection; (3) pathogen survival; or (4) cytotoxicity using in vitro culture systems. Multiple bacterial strains have been tested, including PAO1, PA14, PA103, and PAK (Bjorn et al. 1979; Holloway 1955; Liu 1966a, b; Rahme et al. 1995; Takeya and Amako 1966) (Table 1). The broad conclusion from this work has been that ExoY does not feature prominently in the virulence of infection. However, the role that ExoY plays in infection is more complex than initially appreciated.
10 Evidence That ExoY Does Not Contribute to Virulence
Early studies addressing the contribution of ExoY to infection severity uniformly concluded it played little role in virulence. For example, deletion of exoY from the PAK strain (Fig. 3) did not impact CHO cell cytotoxicity, as measured by LDH release (Lee et al. 2005), and similarly, exoY deletion did not impact CHO cell survival, as assessed by a cell plating efficiency assay. In the latter case, cells were infected at an MOI of 10 for 3 h and then trypsinized, serially diluted, and seeded in culture medium containing antibiotics. Under these conditions, CHO cell plating efficiency was similar after infection by PAKΔY and PAK wild-type strains. Bacterial survival and dissemination were also assessed using a mouse pneumonia model. Here, the deletion of exoY (PAKΔY strain) did not impact either bacterial survival in the lung or bacterial dissemination to the spleen and liver when compared to control strains. In separate studies, A549 cells were infected with individual clinical P. aeruginosa isolates at an MOI of ~80 for 3 h and LDH release measured, and J774 cells were infected at an MOI of ~200 for 6 h and apoptosis assessed. Each clinical isolate was instilled into the nares of BALB/c mice, animal survival was monitored over the course of seven days, and each strain’s LD50 was determined. In the background of ExoU, ExoY did not further increase cytotoxicity, apoptosis-like cell death, or virulence (Schulert et al. 2003), as assessed by each strain’s LD50. Overall, these studies were taken to mean that ExoY plays a limited pathogenic role.

Secretion profiles of P. aeruginosa strain PAK and isogenic derivatives induced for type 3 secretion. Secreted proteins were separated by 12% PAGE and revealed by staining with Coomassie. The positions of ExoS, ExoT, and ExoY on the gel are indicated by arrowheads. WT, wild type. Genetic deletion of exoY from PAK produces little to no change in ExoY protein levels. Adapted from Lee et al. (2005)
11 Evidence That ExoY Contributes to Virulence
Although early studies suggested that ExoY is functionally dispensable, emerging evidence reveals previously unappreciated ways in which ExoY contributes to virulence. ExoY initially causes cell rounding, due in part to a non-enzymatic regulation of the F-actin cytoskeleton and an enzymatic control of microtubule architecture. As cell rounding progresses to cellular death, the enzymatic activity of ExoY is responsible for production and release of amyloid proteins, like non-neuronal tau, that cause transmissible cytotoxicity.
The interaction between ExoY and the F-actin cytoskeleton remains poorly understood and represents an important area of study. Fleiszig and colleagues hypothesized that the adenylyl cyclase activity of ExoY may contribute to actin cytoskeleton disruption. As such, immortalized rabbit corneal epithelial cells were inoculated with either ExoY+ or ExoYK81M at an MOI of 10, and F-actin microfilaments were observed by fluorescence microscopy 2 and 4 h post-infection. ExoY+ and ExoYK81M both caused F-actin redistribution at 2 and 4 h. However, only cells infected with ExoY+ displayed significant cell rounding, and this cell rounding was observed at 4 h (Cowell et al. 2005). These data are consistent with recent evidence implicating a role for F-actin as an ExoY cofactor, enabling production of cyclic nucleotide monophosphates that are responsible for cell rounding.
In a follow-up study, Hritonenko and colleagues hypothesized that ExoY may contribute to corneal epithelial cell virulence, an effect mediated by bacterial trafficking to plasma membrane blebs with subsequent bacterial survival and replication. In cultured human corneal epithelial cells, ExoY adenylyl cyclase activity was sufficient to allow epithelial cell bleb-niche formation. However, whereas bleb-niche formation had previously been shown to correlate with bacterial survival and replication (Angus et al. 2010), there was no evidence that ExoY contributed to either survival or replication in vitro (Hritonenko et al. 2011). Interestingly, when applied to Black Swiss Surfactant Protein D knockout mice, ExoY+ significantly increased the corneal disease severity score (used to assess virulence) at both 24 and 48 h post-inoculation, when compared with ExoYK81M. Taken together, these results indicate that ExoY contributes to virulence in vivo without increasing bacterial survival or replication in vitro.
Beckert and colleagues determined that ExoY transfection into B103 neuroblastoma cells results in significant (~40% of total cell population) cell death 72 h post-transfection (Beckert et al. 2014a). This effect was hypothesized to be dependent upon cGMP and cUMP, due to ExoY nucleotidyl cyclase activity. As such, this group wanted to test whether the cytotoxic activity of ExoY was due to cGMP and cUMP generation. In a follow-up study, B103 cells treated with a combination of cGMP/cUMP-AM for 48 h displayed rounded cell morphology and apoptotic/necrotic cell death that was similar to cells transfected with ExoY (Beckert et al. 2014b). Although these studies were conducted in B103 neuroblastoma cells and were based upon a transfection approach rather than bacterial infection, they raise the possibility that ExoY in certain environments may do more than simply cause F-actin redistribution and cell rounding, as previously shown in CHO cells.
ExoY synthesizes cyclic nucleotide monophosphates; elevation of these cyclic nucleotide monophosphates results in phosphorylation of the microtubule associated protein tau. Tau hyperphosphorylation leads to its release from microtubules into the supernatant in an oligomeric form where it functions as a cytotoxic agent. Enrichment of purified oligomeric tau from cellular supernatant is sufficient to cause cell rounding and propagate injury in the absence of infection. Indeed, generation and release of oligomeric tau is an unequivocal mechanism of ExoY-induced virulence that has not been previously considered. The production of oligomeric tau is not just an in vitro phenomenon, but it is a part of the natural response to infection in vivo. ExoY+ airway instillation causes pneumonia, which progresses to acute respiratory distress syndrome. Histological assessment of this infection reveals perivascular cuff formation and alveolar edema with hemorrhage (Stevens et al. 2014). ExoY+ infection has long-standing consequences, as animals recovering from infection retain evidence for vascular dysfunction at least one week later, and preliminary studies reveal that oligomeric tau can be recovered from the bronchoalveolar lavage of infected animals 24 and 72 h post-infection (data not shown). Future studies will be required to assess the long-term impact of oligomeric tau exposure in the lung and determine the biodistribution of oligomeric tau among physiological compartments following infection.
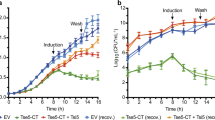
There is no question that production and release of oligomeric tau contributes to ExoY-induced virulence. Interestingly, ExoU activity is also sufficient to induce oligomeric tau production and release. ExoU visibly damages endothelial cells within 3–4 h, whereas ExoY disrupts cell morphology within 6–8 h (Morrow et al. 2016) (Fig. 4). We have directly compared cytotoxicity due to ExoY+- and PA103-generated supernatant. Supernatant collected from ExoY+-infected cells causes cytotoxicity that is equal to or greater than supernatant collected from PA103-infected cells (Fig. 5). In addition, ExoY+- and PA103-generated cytotoxic supernatant containing high molecular weight tau causes endothelial barrier disruption in the isolated perfused lung. In considering the direct effect of ExoY on cellular morphology, its ability to cause necrosis, and its ability to generate cytotoxic tau oligomers, ExoY’s contribution to bacterial virulence and pathogenesis is likely more extensive than initially thought.

Gap formation following P. aeruginosa infection. Pulmonary microvascular endothelial cells were inoculated with either PA103 or with ExoY+ bacterial strain in Hanks’ balanced salt solution (HBSS) at an MOI of 20:1, and gap formation was visually assessed by phase-contrast microscopy. Gap formation at 7 h post-infection with ExoY+ was comparable to that observed at 4 h post-infection with PA103; overall cell health was worse in PA103-infected cells with prominent sunken nuclei observed. Images were captured at 20× magnification with scale bar = 10 μm. Adapted from Morrow et al. (2016)

Cytotoxic supernatant from PA103 and ExoY+-infected cells applied to naïve endothelium. Filtered supernatants collected 4.5 h post-PA103 and 7 h post-ExoY+ infection were transferred to naïve PMVECs, and images of injurious effects were captured 6 and 16 h post-transfer. Supernatant from ExoY+-infected cells produced comparable damage to supernatant from PA103-infected cells. Images were captured at 20× magnification with scale bar = 10 μm. Adapted from Morrow et al. (2016)
12 Summary
Study of ExoY in vascular endothelium has led to a number of novel observations. Importantly, these experiments have given greater insight as to the importance of cAMP compartmentalization in control of endothelial cell barrier integrity, where cAMP generated by membrane adenylyl cyclases is barrier protective and cAMP generated by soluble adenylyl cyclases is barrier disruptive. The presence of pyrimidine cyclic nucleotide monophosphates in endothelium has now been reported. Further the intracellular milieu in pulmonary artery endothelial cells is more conducive to the production of all cyclic nucleotides by ExoY intoxication than is the environment in pulmonary microvascular endothelial cells. Evidence for expression of an endothelial tau isoform that stabilizes microtubules was obtained studying ExoY, and these observations led to discovery that ExoY enzymatic activity promotes tau oligomerization and release. In this latter case, tau oligomers are sufficient to cause cytotoxicity that perpetuates organ dysfunction even after the infection has been cleared. It appears increasingly evident that ExoY figures prominently among the arsenal of P. aeruginosa virulence factors. It will be important moving forward to address how the bacterium utilizes the complexity of T3SS effectors to interact with host cell microenvironment, especially among phenotypically diverse host cell populations.
References
Angus AA, Evans DJ, Barbieri JT, Fleiszig SM (2010) The ADP-ribosylation domain of Pseudomonas aeruginosa ExoS is required for membrane bleb niche formation and bacterial survival within epithelial cells. Infect Immun 78:4500–4510
Bahre H, Hartwig C, Munder A, Wolter S, Stelzer T, Schirmer B, Beckert U, Frank DW, Tummler B, Kaever V, Seifert R (2015) cCMP and cUMP occur in vivo. Biochem Biophys Res Commun 460:909–914
Balczon R, Prasain N, Ochoa C, Prater J, Zhu B, Alexeyev M, Sayner S, Frank DW, Stevens T (2013) Pseudomonas aeruginosa exotoxin Y-mediated tau hyperphosphorylation impairs microtubule assembly in pulmonary microvascular endothelial cells. PLoS One 8, e74343
Beckert U, Wolter S, Hartwig C, Bahre H, Kaever V, Ladant D, Frank DW, Seifert R (2014a) ExoY from Pseudomonas aeruginosa is a nucleotidyl cyclase with preference for cGMP and cUMP formation. Biochem Biophys Res Commun 450:870–874
Beckert U, Grundmann M, Wolter S, Schwede F, Rehmann H, Kaever V, Kostenis E, Seifert R (2014b) cNMP-AMs mimic and dissect bacterial nucleotidyl cyclase toxin effects. Biochem Biophys Res Commun 451:497–502
Belyy A, Raoux-Barbot D, Saveanu C, Namane A, Ogryzko V, Worpenberg L, David V, Henriot V, Fellous S, Merrifield C, Assayag E, Ladant F, Renault L, Mechold U (2016) Actin activates Pseudomonas aeruginosa ExoY nucleotidyl cyclase toxin and ExoY-like effector domains from MARTX toxins. Nat Comm 7:13582
Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, Birukov KG (2007) Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res 313:2504–2520
Birukova AA, Tian Y, Dubrovskyi O, Zebda N, Sarich N, Tian X, Wang Y, Birukov KG (2012) VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J Cell Physiol 227:3405–3416
Birukova AA, Wu T, Tian Y, Meliton A, Sarich N, Tian X, Leff A, Birukov KG (2013) Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur Respir J 41:165–176
Birukova AA, Meng F, Tian Y, Meliton A, Sarich N, Quilliam LA, Birukov KG (2015) Prostacyclin post-treatment improves LPS-induced acute lung injury and endothelial barrier recovery via Rap1. Biochim Biophys Acta 1852:778–791
Bjorn MJ, Pavlovskis OR, Thompson MR, Iglewski BH (1979) Production of exoenzyme S during Pseudomonas aeruginosa infections of burned mice. Infect Immun 24:837–842
Cioffi DL, Moore TM, Schaack J, Creighton JR, Cooper DM, Stevens T (2002) Dominant regulation of interendothelial cell gap formation by calcium-inhibited type 6 adenylyl cyclase. J Cell Biol 157:1267–1278
Cowell BA, Evans DJ, Fleiszig SM (2005) Actin cytoskeleton disruption by ExoY and its effects on Pseudomonas aeruginosa invasion. FEMS Microbiol Lett 250:71–76
Craig JP (1965) A permeability factor (toxin) found in cholera stools and culture filtrates and its neutralization by convalescent cholera sera. Nature 207:614–616
Creighton JR, Masada N, Cooper DM, Stevens T (2003) Coordinate regulation of membrane cAMP by Ca2+-inhibited adenylyl cyclase and phosphodiesterase activities. Am J Physiol Lung Cell Mol Physiol 284:L100–L107
Dudek SM, Garcia JG (2001) Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) 91:1487–1500
Engel J, Balachandran P (2009) Role of Pseudomonas aeruginosa type III effectors in disease. Curr Opin Microbiol 12:61–66
Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852
Glaser P, Sakamoto H, Bellalou J, Ullmann A, Danchin A (1988) Secretion of cyclolysin, the calmodulin-sensitive adenylate cyclase-haemolysin bifunctional protein of Bordetella pertussis. EMBO J 7:3997–4004
Gottle M, Dove S, Kees F, Schlossmann J, Geduhn J, Konig B, Shen Y, Tang WJ, Kaever V, Seifert R (2010) Cytidylyl and uridylyl cyclase activity of Bacillus anthracis edema factor and Bordetella pertussis CyaA. Biochemistry 49:5494–5503
Hauser AR (2009) The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7:654–665
Holloway BW (1955) Genetic recombination in Pseudomonas aeruginosa. J Gen Microbiol 13:572–581
Hritonenko V, Mun JJ, Tam C, Simon NC, Barbieri JT, Evans DJ, Fleiszig SM (2011) Adenylate cyclase activity of Pseudomonas aeruginosa ExoY can mediate bleb-niche formation in epithelial cells and contributes to virulence. Microb Pathog 51:305–312
Johnson GS, Friedman RM, Pastan I (1971) Restoration of several morphological characteristics of normal fibroblasts in sarcoma cells treated with adenosine-3′:5′-cyclic monophosphate and its derivatives. Proc Natl Acad Sci U S A 68:425–429
Karst JC, Sotomayor Perez AC, Guijarro JI, Raynal B, Chenal A, Ladant D (2010) Calmodulin-induced conformational and hydrodynamic changes in the catalytic domain of Bordetella pertussis adenylate cyclase toxin. Biochemistry 49:318–328
Keppie J, Harris-Smith PW, Smith H (1963) The chemical basis of the virulence of Bacillus anthracis. IX. Its aggressins and their mode of action. Br J Exp Pathol 44:446–453
Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI (2012) Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 287:19440–19451
Lee VT, Smith RS, Tummler B, Lory S (2005) Activities of Pseudomonas aeruginosa effectors secreted by the type III secretion system in vitro and during infection. Infect Immun 73:1695–1705
Leppla SH (1982) Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci U S A 79:3162–3166
Linder JU, Schultz JE (2003) The class III adenylyl cyclases: multi-purpose signalling modules. Cell Signalling 15:1081–1089
Liu PV (1966a) The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis. III. Identity of the lethal toxins produced in vitro and in vivo. J Infect Dis 116:481–489
Liu PV (1966b) The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis. II. Effects of lecithinase and protease. J Infect Dis 116(1):112–116
Mehta D, Malik AB (2006) Signaling mechanisms regulating endothelial permeability. Physiol Rev 86:279–367
Moore TM, Chetham PM, Kelly JJ, Stevens T (1998) Signal transduction and regulation of lung endothelial cell permeability. Interaction between calcium and cAMP. Am J Physiol 275:L203–L222
Morrow KA, Seifert R, Kaever V, Britain AL, Sayner SL, Ochoa CD, Cioffi EA, Frank DW, Rich TC, Stevens T (2015) Heterogeneity of pulmonary endothelial cyclic nucleotide response to Pseudomonas aeruginosa ExoY infection. Am J Physiol Lung Cell Mol Physiol 309:L1199–L1207
Morrow KA, Ochoa CD, Balczon R, Zhou C, Cauthen L, Alexeyev M, Schmalzer KM, Frank DW, Stevens T (2016) Pseudomonas aeruginosa exoenzymes U and Y induce a transmissible endothelial proteinopathy. Am J Physiol Lung Cell Mol Physiol 310:L337–L353
Ochoa CD, Alexeyev M, Pastukh V, Balczon R, Stevens T (2012) Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability. J Biol Chem 287:25407–25418
Prasain N, Alexeyev M, Balczon R, Stevens T (2009) Soluble adenylyl cyclase-dependent microtubule disassembly reveals a novel mechanism of endothelial cell retraction. Am J Physiol Lung Cell Mol Physiol 297:L73–L83
Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM (1995) Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902
Reinecke D, Burhenne H, Sandner P, Kaever V, Seifert R (2011) Human cyclic nucleotide phosphodiesterases possess a much broader substrate-specificity than previously appreciated. FEBS Lett 585:3259–3262
Sayner SL (2011) Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am J Physiol Lung Cell Mol Physiol 300:L667–L678
Sayner SL, Frank DW, King J, Chen H, VandeWaa J, Stevens T (2004) Paradoxical cAMP- induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ Res 95:196–203
Sayner SL, Alexeyev M, Dessauer CW, Stevens T (2006) Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ Res 98:675–681
Sayner SL, Balczon R, Frank DW, Cooper DM, Stevens T (2011) Filamin A is a phosphorylation target of membrane but not cytosolic adenylyl cyclase activity. Am J Physiol Lung Cell Mol Physiol 301:L117–L124
Schuler D, Lubker C, Lushington GH, Tang WJ, Shen Y, Richter M, Seifert R (2012) Interactions of Bordetella pertussis adenylyl cyclase toxin CyaA with calmodulin mutants and calmodulin antagonists: comparison with membranous adenylyl cyclase I. Biochem Pharmacol 83:839–848
Schulert GS, Feltman H, Rabin SD, Martin CG, Battle SE, Rello J, Hauser AR (2003) Secretion of the toxin ExoU is a marker for highly virulent Pseudomonas aeruginosa isolates obtained from patients with hospital-acquired pneumonia. J Infect Dis 188:1695–1706
Selwa E, Laine E, Malliavin TE (2012) Differential role of calmodulin and calcium ions in the stabilization of the catalytic domain of adenyl cyclase CyaA from Bordetella pertussis. Proteins 80:1028–1040
Selwa E, Davi M, Chenal A, Sotomayor-Perez AC, Ladant D, Malliavin TE (2014) Allosteric activation of Bordetella pertussis adenylyl cyclase by calmodulin: molecular dynamics and mutagenesis studies. J Biol Chem 289:21131–21141
Smith H, Keppie J, Stanley JL (1955) The chemical basis of the virulence of Bacillus anthracis. V. The specific toxin produced by B. Anthracis in vivo. Br J Exp Pathol 36:460–472
Stanley JL, Smith H (1961) Purification of factor I and recognition of a third factor of the anthrax toxin. J Gen Microbiol 26:49–63
Stanley JL, Smith H (1963) The three factors of anthrax toxin: their immunogenicity and lack of demonstrable enzymic activity. J Gen Microbiol 31:329–337
Stevens T, Nakahashi Y, Cornfield DN, McMurtry IF, Cooper DM, Rodman DM (1995) Ca2+-inhibitable adenylyl cyclase modulates pulmonary artery endothelial cell cAMP content and barrier function. Proc Natl Acad Sci U S A 92:2696–2700
Stevens T, Creighton J, Thompson WJ (1999) Control of cAMP in lung endothelial cell phenotypes. Implications for control of barrier function. Am J Physiol 277:L119–L126
Stevens TC, Ochoa CD, Morrow KA, Robson MJ, Prasain N, Zhou C, Alvarez DF, Frank DW, Balczon R, Stevens T (2014) The Pseudomonas aeruginosa exoenzyme Y impairs endothelial cell proliferation and vascular repair following lung injury. Am J Physiol Lung Cell Mol Physiol 306:L915–L924
Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964
Takeya K, Amako K (1966) A rod-shaped Pseudomonas phage. Virology 28:163–165
Townsley M, Stevens T (2015) Lung Endothelium. Morgan and Claypool Life Sciences
Werthmann RC, von Hayn K, Nikolaev VO, Lohse MJ, Bunemann M (2009) Real-time monitoring of cAMP levels in living endothelial cells: thrombin transiently inhibits adenylyl cyclase 6. J Physiol 587:4091–4104
Winn WC, LaSala PR, Leslie KO (2008) Bacterial Infections. In: Cagle PT, Farver CF, Fraire AE, Tomashefski JF (eds) Pulmonary Pathology. Springer, New York, pp 228–315
Wolter S, Golombek M, Seifert R (2011) Differential activation of cAMP- and cGMP-dependent protein kinases by cyclic purine and pyrimidine nucleotides. Biochem Biophys Res Commun 415:563–566
Yahr TL, Mende-Mueller LM, Friese MB, Frank DW (1997) Identification of type III secreted products of the Pseudomonas aeruginosa exoenzyme S regulon. J Bacteriol 179:7165–7168
Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW (1998) ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci U S A 95:13899–13904
Ziolo KJ, Jeong HG, Kwak JS, Yang S, Lavker RM, Satchell KJ (2014) Vibrio vulnificus biotype 3 multifunctional autoprocessing RTX toxin is an adenylate cyclase toxin essential for virulence in mice. Infect Immun 82:2148–2157
Acknowledgments
This work is supported by National Institutes of Health grants HL60024 (T. Stevens) and HL66299 (T. Stevens, R. Balczon, and D. W. Frank), and an American Heart Association postdoctoral fellowship 14POST18080004 (K. A. Morrow).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Morrow, K.A., Frank, D.W., Balczon, R., Stevens, T. (2016). The Pseudomonas aeruginosa Exoenzyme Y: A Promiscuous Nucleotidyl Cyclase Edema Factor and Virulence Determinant. In: Seifert, R. (eds) Non-canonical Cyclic Nucleotides. Handbook of Experimental Pharmacology, vol 238. Springer, Cham. https://doi.org/10.1007/164_2016_5003
Download citation
DOI: https://doi.org/10.1007/164_2016_5003
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52671-3
Online ISBN: 978-3-319-52673-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)