
Abstract
Although the fundamental concepts of pharmacokinetics remain the same, ocular pharmacokinetics has its own challenges due to the uniqueness of barrier properties posed by various ocular tissues and its growing complexity with different routes of ocular administration. A thorough understanding of the barrier nature will aid in tailoring a drug or its carrier’s physicochemical properties to its advantage. In order to deliver the right payload of a drug at the target site, various approaches can be taken to leverage the pharmacokinetics that includes molecular design based on desirable physicochemical properties, formulation approaches, and alternative routes of administration. In this chapter, a brief overview of the barrier properties with respect to various routes of administration is presented along with the physicochemical properties that influence the pharmacokinetics of ocular drugs. Recent advances in ocular pharmacokinetics are discussed in addition to new perspectives in interpreting existing data.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
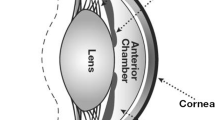
The pharmacokinetic processes of absorption, distribution, metabolism, and elimination determine the time course of a drug in the body and the amount delivered to the site of action. An understanding of these interrelated processes is critical in deciding the dose and dosing frequency of a drug and thereby influences its efficacy and safety. The mode of delivering a drug including the route of administration and design of vehicle or carrier is dependent on its pharmacokinetic properties. The eye is a complex structure composed of several distinct tissues each with a specific function that poses numerous constraints to drug delivery. Due to the unique anatomy and physiology of eye, the pharmacokinetic process of a drug is affected by the ocular tissues and other barriers encountered in the administered route. Since a plethora of literature is available on the ocular structure and barriers to drug delivery, this chapter will provide a brief overview on this topic and will mainly focus on the recent advances in ocular pharmacokinetics with an emphasis on the influence of molecular and physicochemical properties that dictate the ocular fate of a drug. From a drug delivery perspective, anterior segment and posterior segment are the two major routes of ocular drug delivery which are entirely different and have unique properties (Fig. 1). The choice of administration route not only depends on the target ocular tissue, but also on the barriers encountered in the route along with the physicochemical properties of the drug. Since the administration route has an impact on the ocular pharmacokinetics of a drug, this chapter provides an overview of the pharmacokinetic processes associated with the major routes of ocular drug delivery.

Schematic representation of ocular delivery from a pharmacokinetic perspective. Adapted and modified from National Eye Institute, National Institute of Health (NIH)
2 Ocular Pharmacokinetics of the Anterior Segment
The anterior segment of the eye constitutes cornea, conjunctiva, aqueous humor, lens, iris and ciliary body (ICB). The primary routes for drug delivery to the anterior segment include topical administration, subconjunctival, and intracameral injections. Pharmacokinetic processes involved with each of these major routes of administration are discussed below under each section.
2.1 Pharmacokinetics After Topical Administration
Topical administration is the most convenient route of drug delivery to the anterior segment of eye. Following topical instillation, majority of the administered drug is cleared rapidly from the ocular surface resulting in only 1–7% of the dose to reach the aqueous humor (Ghate and Edelhauser 2006). Precorneal clearance mechanisms including tear fluid turnover and blinking, selective permeability of the corneal epithelial barrier, and drug loss through nasolacrimal as well as systemic circulation attribute to the low bioavailability of drugs administered by this route.
2.1.1 Factors Affecting Absorption and Bioavailability
The critical factors that may affect the absorption process and alter the intraocular bioavailability of topical drops consist of physiological factors relevant to ocular tissues and molecular properties unique to drugs. A complete understanding of the interaction between these factors is essential to enhance the pharmacokinetic processes.
2.1.1.1 Loss of Drug from the Precorneal Surface
The tear volume in humans under normal condition is 7–9 μL with a turnover rate of 0.5–2.2 μL/min. Many commercially available eyedroppers deliver a typical volume of 25–56 μL to the precorneal tear film resulting in an increase in the tear volume. Under normal conditions, human palpebral fissure can hold 30 μL without overflowing. This abrupt increase in the volume due to topical instillation causes reflex blinking and rapid drainage from ocular surface. Majority of the applied medication is drained from the surface through the nasolacrimal duct and eventually cleared via systemic circulation.
2.1.1.2 Corneal Barriers of Drug Absorption
Cornea is the primary route for drug penetration to the anterior segments of eye following topical administration. The cornea is composed of epithelium, Bowman’s membrane, stroma, Descemet’s membrane, and endothelium (Grass and Robinson 1988). The relative thickness of corneal epithelium, stroma, and endothelium are around 0.1:1:0.01. These three layers of cornea serve as substantial barriers for absorption. The epithelium is comprised of a basal layer of columnar cells, two to three layers of wing cells, and one or two outermost layers of squamous superficial cells. The superficial cells have tight intercellular junctions while the wing cells and basal cells consist of wider intercellular spaces. The tight junctions of the superficial corneal epithelial cells limit the absorption of hydrophilic drugs and favor transcellular permeation of lipophilic compounds. The paracellular route predominates for hydrophilic compounds of small molecular weight (<350 Da) (Ghate and Edelhauser 2006). Stroma is relatively a hydrophilic environment where drugs can diffuse through with minor resistance. Hydrophilic compounds with optimal molecular radius can easily diffuse through the stroma. Endothelium is a leaky barrier due to the large intercellular junctions between the monolayer of cells that partially resists penetration of lipophilic compounds.
2.1.1.3 Drug Properties Affecting Absorption
In case of topically administered drugs, absorption through cornea may occur via transcellular or paracellular pathways or by active transport. Drug properties that influence these processes such as lipophilicity and aqueous solubility play a key role in the penetration of drugs across cornea. Lipophilicity (LogP) in the range of 2–3 was found to be optimal for corneal permeation of steroids and β-blockers (Schoenwald and Huang 1983). An exploratory analysis of the apparent corneal permeability (P app) values for more than 100 compounds indicated that corneal permeability is dependent on the distribution coefficient (LogD at experimental pH) (Prausnitz and Noonan 1998). As the dataset was mostly comprised of small molecules, no apparent dependency on molecular weight was observed. Further analysis of this permeability data with other molecular descriptors revealed potential correlation of corneal permeability with polar surface area (PSA) (Fig. 2) (unpublished data). PSA is the sum of surfaces of polar atoms, primarily oxygen, nitrogen, and their attached hydrogen atoms. PSA along with lipophilicity and molecular size influence the passive diffusion of molecules.

Relationship between corneal permeability (CP) and polar surface area (PSA). Corneal permeability data comes from Prausnitz and Noonan (1998)
Due to the hydrophilic nature of corneal stroma, highly lipophilic compounds have limited permeability across this tissue. Stromal permeability data from a limited number of molecules (N = 19) indicated a strong dependence on its molecular weight and radius but no apparent relationship with any of the lipophilicity indicators (LogP or LogD) (Prausnitz and Noonan 1998). As indicated earlier, stroma is a thick, fibrous, and hydrophilic tissue where diffusion plays a major role in the transport of molecules. Thus stromal permeability is negatively correlated with molecular weight and radius, the parameters that affect the diffusion of a compound. Permeability of corneal endothelium shows a good correlation with both LogD and molecular radius indicating the role of both lipophilic and hydrophilic pathways. Similar to intact cornea, an increase in the corneal endothelial permeability was observed with a moderate increase in lipophilicity. However, the data was limited by the absence of highly lipophilic compounds to further investigate the barrier properties. Due to the presence of large intercellular junctions and leaky nature of the endothelial layer, as anticipated, strong correlation was observed with molecular radius. In general, taking into consideration the overall data, corneal epithelium serves as main barrier to transport of molecules across cornea. Small molecules with favorable lipophilicity readily cross corneal epithelium but stroma may provide a barrier to macromolecules.
Several formulation approaches are employed to overcome the absorption barriers and improve the ocular bioavailability. More information on these can be found in a recent review article (Ghate and Edelhauser 2006).
2.1.1.4 Non-corneal Routes of Absorption
Apart from the corneal route, topically administered drugs may be absorbed via non-corneal pathways that involve permeation across the conjunctiva and scleral layers. These routes play a major role in the penetration of drugs with poor corneal permeability that includes hydrophilic compounds and macromolecules (Ahmed and Patton 1985). Thus the drug properties determine the relative contribution of the non-corneal routes to absorption.
The conjunctiva is comprised of a stratified columnar epithelium and lamina propria. The superficial conjunctival epithelium has tight junctions with intercellular spaces wider than the corneal epithelium. Thus permeability of hydrophilic molecules is comparatively greater in conjunctiva. Further, large molecules such as inulin and FITC-dextran which are impermeable through cornea have limited permeability across conjunctiva. Based on the limited data available, no significant trend was observed between conjunctival permeability and lipophilicity while a possible dependency was observed with increasing molecular weight (Prausnitz and Noonan 1998). However, more data on large molecules is required to establish its role in conjunctival permeability.
2.1.2 Distribution of Drugs in the Anterior Segment of Eye
Topically administered drugs permeate across the cornea and enter the aqueous humor followed by distribution to the surrounding ocular tissues including iris–ciliary body, lens, choroid–retina, and vitreous (Ghate and Edelhauser 2006). Drugs that may exhibit non-corneal routes of absorption enter the uveal tract and vitreous without entering the aqueous humor. The rate and extent of drug distribution in the anterior segment is determined by a number of factors including permeability, diffusion in the aqueous humor, binding to proteins and surrounding ocular tissue components. Most of these factors are influenced by a drug’s physicochemical properties including lipophilicity, solubility, and molecular weight. The apparent volume of distribution (V d) of drugs can be measured by direct administration into the aqueous humor (intracameral). However, there is a paucity of data on the pharmacokinetics of drugs following intracameral injection. The V d for few ophthalmic drugs administered by intracameral route is summarized along with key physicochemical properties in Table 1.
Based on the volume of aqueous humor in rabbits (0.3 mL), the V d ranged from two- to tenfold larger than the aqueous humor volume. Although no clear trend was observed between V d and molecular weight or LogP, drugs with higher protein binding had a lower V d in the aqueous humor. Drugs that extensively bind to plasma proteins were known to exhibit a low V d and can have a long plasma half-life. Flurbiprofen, a highly protein bound drug, has a longer elimination half-life in aqueous humor when compared to other moderate to weakly bound drugs (Table 1). For topically administered drugs, protein binding occurs first in the tear fluid which has a rapid turnaround time and as a result only the free unbound drug is available for corneal absorption. More binding of the absorbed drug occurs in the cornea and aqueous humor. Protein content of the aqueous humor is different when compared to plasma. Concentration of proteins in the aqueous humor is approximately 200 times less than in plasma. However, these levels may increase in certain disease states that involve inflammatory conditions and subsequently result in increased binding of drugs. The effect of protein binding was investigated by adding increasing amounts of rabbit serum albumin to pilocarpine solution before topical administration (Mikkelson et al. 1973). The results indicated a 75- to 100-fold reduction in response (pupillary diameter) by the addition of 3% albumin indicating a decreased bioavailability as a result of protein binding. Nevertheless, more data comparing the pharmacokinetics of drugs in normal versus diseased state (e.g., inflammation, blood-aqueous barrier breakdown, etc.) is required to understand the effect of protein binding on the disposition of topically administered drugs.
From a therapeutic perspective, distribution of a drug to its target site is essential to achieve the desired efficacy. Although measurement of drug concentration in the aqueous humor provides an estimation of V d, measuring drug levels in the surrounding ocular tissues is required to assess if the drug has reached the site of action. While pharmacokinetic studies with extensive tissue distribution data are scarce, few studies report the drug concentrations in key ocular tissues in addition to the aqueous humor. Given the number of animals required and the destructive nature of tissue sample collection, this is not uncommon in the ophthalmology field. Table 2 summarizes the AUC ratio of tissue:aqueous humor for few topical drugs of interest along with key physicochemical properties.
As expected, the relative exposure was higher in the cornea following topical instillation of drugs. The relative exposure in cornea was several folds higher for high molecular weight compounds (azithromycin and cyclosporin) when compared to other drugs. Also, the relative exposure of drugs in iris–ciliary body is higher than in aqueous humor. With the exception of lomefloxacin, for which data was available from infected rabbit eyes, the relative exposure in ICB decreased with increasing lipophilicity (LogP). Several explanations have been postulated to explain this higher exposure in ICB (Schoenwald 2003). Iris of rabbit eye is a porous and highly vascular tissue with majority of its surface area exposed to aqueous humor thereby allowing extensive distribution from aqueous humor. Further, an increased affinity/capacity for binding to melanin pigment in the iris could enhance the distribution to this tissue. Brimonidine, a drug well known to bind melanin, has higher relative exposure in ICB than in cornea (Table 2). Levobetaxolol, a cardioselective beta-adrenergic receptor blocking agent, has higher affinity to melanin with ICB exposure several folds higher than in aqueous humor.
An alternative explanation for the higher exposure in ICB could be due to potential contribution of non-corneal absorption routes via conjunctival/scleral pathways. Based on their physicochemical properties, certain drugs may be preferentially absorbed by conjunctiva and sclera to reach ICB without entering the aqueous humor. Chien et al. (1990) investigated the ocular absorption via corneal and conjunctival/scleral routes of clonidine, p-aminoclonidine, and AGN 190342 after drug perfusion in vivo. When drug was maintained over the conjunctiva over a period of time, the rank order of drug concentration in the anterior chamber tissues was conjunctiva > cornea > ciliary body > aqueous humor; whereas, when drug solution was in contact with cornea, the rank order for tissues was cornea > aqueous humor > ciliary body > conjunctiva. Besides, the conjunctival/scleral pathway was contributed as the predominant pathway for the least lipophilic (p-aminoclonidine) compound. Further experiments carried out using beta-blocking agents with varying lipophilicity, sucrose and inulin demonstrated that the outer layer of sclera provides less resistance to penetration of hydrophilic drugs when compared to cornea (Ahmed and Patton 1985). Moreover, the estimated permeability of conjunctival and scleral tissues was found to be 15–25 times higher than the cornea and was not affected by molecular size (Hamalainen et al. 1997).
2.1.3 Metabolism and Role of Transporters in Drug Disposition from the Anterior Segment
With the growing body of knowledge and evidence of its expression in various ocular tissues, drug metabolizing enzymes and transporters are gaining more attention from researchers to overcome the barriers for ocular drug delivery. Since there is an abundance of literature that provides a comprehensive overview of the distribution of these enzymes and their role in drug delivery (Attar et al. 2005; Zhang et al. 2008b), this chapter will focus only on key enzymes and transporters of clinical significance where in vivo evidence exists for their role in metabolism or drug–drug interaction (DDI).
Gene expression of aldehyde oxidase, an enzyme involved in oxidative metabolism, was detected in rabbit ocular tissues including ciliary body, iris, and cornea (Attar et al. 2005). Following a single topical administration of brimonidine, aldehyde oxidase mediated brimonidine metabolites were detected in the rabbit conjunctiva, cornea, and ICB (Acheampong et al. 2002). NADPH-dependent ketone reductase activity has been characterized in the corneal epithelium, ICB, conjunctiva, and the lens. After topical instillation, levobunolol undergoes reductive metabolism to dihydrolevobunolol in the corneal epithelium and ICB (Lee et al. 1988). Dihydrolevobunolol is an equally active metabolite with longer half-life than the parent drug and higher exposure in cornea, ICB, and aqueous humor.
Hydrolytic enzymes including esterases have been identified in several ocular tissues. Furthermore, there were recognized differences in their differential expression in various ocular tissues and among species. The major site for metabolism of dipivefrin, an anti-glaucoma agent, was identified as rabbit cornea although higher rates of metabolism were detected in ICB. Conversely, co-administration of an esterase inhibitor, echothiophate iodide in humans did not affect dipivefrin therapy indicating a lack of DDI (Mindel et al. 1981). The authors postulated that arylesterase could be responsible for the metabolism of dipivefrin (a phenol ester) which was not subject to inhibition by echothiophate iodide (a cholinesterase inhibitor). Besides, acetyl-, butyryl-, and carboxylesterases have been identified in the pigmented rabbit eye. Latanoprost, an isopropyl ester prodrug, is hydrolyzed by esterases in the cornea before reaching aqueous humor (Sjoquist and Stjernschantz 2002). Aminopeptidase activity is also determined in various ocular tissues including corneal epithelium, ICB, conjunctiva, and aqueous humor of albino rabbits (Stratford and Lee 1985). Following topical administration of bimatoprost, a prostamide analog, bimatoprost acid levels were detected in aqueous humor and cornea indicating the involvement of aminopeptidase in the metabolism of bimatoprost (Shafiee et al. 2013).
Although the presence of various transporters in several ocular tissues has been characterized, the efflux pump transporter P-glycoprotein (P-gp) has been the most investigated. P-gp has been reported to exist in both corneal and conjunctival tissues (Dey et al. 2003). The corneal exposure of erythromycin, a lipophilic compound, was significantly increased in the presence of testosterone, a P-gp inhibitor indicating its significance in improving corneal bioavailability.
2.1.4 Elimination of Drugs from the Anterior Segment
Majority of the topically administered drug is lost through the nasolacrimal duct followed by systemic absorption. This portion of the drug is metabolized and eliminated by systemic pathways. Remaining drug undergoes intraocular absorption to reach the aqueous humor followed by distribution to surrounding ocular tissues. Elimination of drugs from the aqueous humor occurs by its turnover through the chamber angle and Sclemm’s canal and by the venous blood flow of the anterior uvea (Schoenwald 2003). The turnover rate of aqueous humor in rabbit eye is 1.5% of the anterior chamber volume per minute which translates to a half-life of 46 min. Due to the rapid turnover rate of the aqueous humor, clearance of hydrophilic drugs will be faster than highly lipophilic drugs. This is further evident from the elimination t 1/2 of intracamerally administered drugs, where the t 1/2 ranged from 0.4 to 0.69 h for less lipophilic drugs while the t 1/2 of flurbiprofen (LogP 4.11) was 1.55 h (see Table 1). Table 3 summarizes the half-lives of few ophthalmic drugs of interest in various anterior ocular tissues following topical administration. In case of topically administered highly lipophilic drugs (Log P > 2), elimination t 1/2 was longer in cornea when compared to other anterior chamber tissues. Corneal stroma, being hydrophilic, acts as a depot for highly lipophilic drugs and thereby slows its clearance from cornea.
2.2 Subconjunctival Pharmacokinetics for Drug Delivery to Anterior Segment
Although subconjunctival administration has been demonstrated to deliver drugs to the uvea, this route of administration is not familiar for the delivery of drugs to the anterior segment due to the morbidity of repeated subconjunctival injections. Since the corneal–conjunctival barrier is circumvented after subconjunctival injection, this route of administration is most beneficial for hydrophilic drugs. When gentamicin was administered by the subconjunctival route, sustained effective drug concentration was observed in patients undergoing cataract surgery (Baum and Barza 1983). Similar results were seen for subconjunctival vancomycin where substantially higher concentrations were observed in the aqueous humor in comparison to topical drops. More detail on this route of administration is provided in the later part of this chapter with relevance to posterior segment drug delivery (see Sect. 3.1.1).
2.3 Pharmacokinetics of Intracameral Administration
Direct injection of drug into the anterior chamber bypasses all the corneal and other external barriers to achieve higher drug levels in aqueous humor and surrounding ocular tissues. Results from a recent study indicate the benefits of intracameral injection of antibiotics. After analyzing a large number of cases in cataract surgery, a 22-fold drop in endophthalmitis was seen following the use of intracameral antibiotics (Shorstein et al. 2013). Due to the vicinity of target tissues involved in the regulation of intraocular pressure (IOP), direct injection of a drug or delivery system into the anterior chamber will have a beneficial effect when compared to topical delivery in the treatment of glaucoma. Utilizing this approach, recent research is focused on developing intracameral drug delivery systems mainly for sustained delivery of anti-glaucoma drugs. By administering a single intracameral implant made up of biodegradable polymeric delivery system containing travoprost in beagle dogs, sustained IOP lowering effect was maintained over 8 months with significantly lower aqueous humor concentration of travoprost in comparison to that of topical drops (Navratil et al. 2015). An intracameral implant containing 270 μg bimatoprost was designed to release the drug at a slower rate over 5 months (Hughes 2014). When injected into the anterior chamber of a beagle dog’s eye, a sustained reduction in the IOP was observed for at least 5 months. A desired pharmacokinetic or pharmacodynamic profile can be achieved by controlling the release of drug from delivery system with the right proportion of its constituents. Data collected as part of screening various formulations can be collated to develop an in vitro–in vivo correlation (IVIVC) model to optimize the formulation with desired drug release profile to achieve target concentration or pharmacologic response. These types of model can be developed in the absence of pharmacokinetic data as well by directly linking the in vitro dissolution profile to the pharmacodynamic response of interest.
2.4 Translational Pharmacokinetics for the Anterior Segment
Due to severe limitations in collecting serial pharmacokinetic samples from human eyes, the substantial reliance on data from animal studies is not uncommon in ocular drug development. Unlike the systemic drug development where allometric and physiologically based pharmacokinetic (PBPK) models are well recognized, inter-species scaling is not established in ocular pharmacokinetics. However, anatomical differences in the eye and physiological distinctions across various species can be integrated to augment the predictive capability of PKPD models in human. Utilizing this concept, a semi-mechanistic translational PKPD model was developed using pharmacokinetic and IOP data collected from rabbits and dogs to predict the IOP in human (Durairaj et al. 2014). The pharmacodynamic components of the model included diurnal variation in IOP and physiological parameters representing the turnover rate of aqueous humor in respective species (Fig. 3). Based on the assumption that differences in IOP across species can be attributed to their physiological differences in aqueous humor dynamics, IOP after drug treatment in human was simulated using the preclinical PKPD models. For human simulations, all model parameters representing PK and PD components were fixed and only the aqueous humor dynamics parameters (F in, F us, and C trab) of animals were replaced with values for human. The model was able to predict the IOP in human based on the preclinical PKPD data with reasonable accuracy. Similar scaling approaches can be utilized with an understanding of the mechanistic pathways and physiological differences to extend the predictability of PKPD models to human.

A semi-mechanistic pharmacokinetic–pharmacodynamic model of (a) Brimonidine and (b) Latanoprost to predict IOP in patients. Reproduced with permission from (Durairaj et al. 2014)
3 Ocular Pharmacokinetics of the Posterior Segment
From a pharmacokinetic perspective, drug delivery to the posterior segment tissues is unique and has its own challenges when compared to the anterior segment. Depending on the location of target site in the posterior segment of eye, various delivery routes can be employed to enhance the drug delivery. The most commonly employed routes include transscleral delivery and intravitreal injection.
3.1 Pharmacokinetics of Transscleral Delivery
Transscleral delivery typically comprises subconjunctival, retrobulbar, peribulbar, and sub-tenon injections. These routes are also called as periocular injections and are less invasive when compared with direct intravitreal injection. The relatively larger surface area of sclera and its unique properties in comparison with the cornea make it an attractive means of delivery to posterior segment tissues. Similar to corneal stroma, the permeability of sclera seems to have no dependence on the lipophilicity and a strong dependence on the molecular radius (Prausnitz and Noonan 1998). Based on in vitro experiments, large molecules such as dextran (40 kDa) and albumin (69 kDa) were shown to penetrate the sclera. However, the presence of scleral diseases and scleral thinning may pose additional menace to utilize this mode of drug delivery. In addition to these molecular properties, transscleral route is impeded by static, dynamic, and metabolic barriers (Shah et al. 2010). The static barriers comprise of sclera, Bruch’s – choroid membrane, and retinal pigment epithelium (RPE) which have selective permeability to compounds of distinct physicochemical properties. The high blood and lymphatic flow rates in the conjunctiva and choroid constitute a dynamic barrier leading to higher clearance of drugs. Further, the presence of drug transporter proteins and efflux pumps pose another hurdle for delivery of drugs through this route. Enzymatic activity by cytochrome P450 and lysosomal enzymes may serve as metabolic barriers limiting the fraction available for absorption at this site.
3.1.1 Subconjunctival Pharmacokinetics for Drug Delivery to Posterior Segment
As mentioned earlier, the conjunctival–corneal barrier which is a substantial rate-limiting barrier for hydrophilic drugs is bypassed following subconjunctival injection as drug penetration occurs across sclera. The utility of this route for delivering various therapeutic agents to the posterior segment of the eye has been demonstrated in various studies (Shah et al. 2010). Barza et al. (1993) investigated the drug distribution after a single subconjunctival administration of four antibiotics in rabbits. Significant amount of drug levels were detected in the retina and vitreous humor following subconjunctival injection indicating drug penetration through the scleral route to reach the posterior tissues. Subconjunctival administration of dexamethasone yielded substantially higher drug levels in the vitreous humor when compared to oral and peribulbar routes of administration (Weijtens et al. 2000). More direct evidence on the superiority of this route over topical delivery comes from a study comparing these routes for delivering bevacizumab, a humanized monoclonal antibody in rabbits (Nomoto et al. 2009). Bevacizumab exposure (both AUC and Cmax) after subconjunctival injection was several folds higher in the retina/choroid and vitreous humor when compared to topical drops. As the drug has to permeate across retina/choroid to enter the vitreous, bevacizumab exposure (dose-normalized AUC) was higher in the retina/choroid than vitreous humor (645 vs. 45 ng. wk/g/mg) following subconjunctival administration. Moreover, due to the choroidal blood flow, substantial amount of drug is cleared into the systemic circulation before reaching vitreous humor. Kim et al. (2008) investigated the distribution and clearance of gadolinium-diethylenetriaminopentaacetic acid (Gd-DTPA) infused in the subconjunctival or intrascleral space of rabbits by means of dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI). Results from this study indicated that subconjunctival infusion did not yield detectable levels of Gd-DTPA in the choroid/retina due to rapid clearance by conjunctival blood vessels and lymphatics in addition to the choroidal blood flow. Besides, the presence of RPE and the tight junctions between the endothelial cells of the retinal capillaries restrict the perfusion of drug into the retina and vitreous.
3.2 Pharmacokinetics of Intravitreal Administration
Injection of drug directly into the vitreous is an expedient way of delivering to the posterior tissues. Besides its invasive nature and other complications associated with intravitreal injections, this route of administration remains the pragmatic choice for drug delivery to the posterior segment diseases. Nomoto et al. (2009) compared the pharmacokinetics of bevacizumab after administration of repeated topical drops, a single subconjunctival injection or a single intravitreal injection in pigmented rabbits. Bevacizumab exposure (C max) in the ICB and retina/choroid were 109,192.6 and 93,990 ng/g, respectively, after intravitreal injection, while the levels were 1,418.7 and 295.8 ng/g, respectively, following subconjunctival administration. Topical dosing of bevacizumab resulted in far less exposure than the other two administration routes. Similar trend was observed for AUC as well. The authors concluded that intravitreal injection was the most effective mode of delivering bevacizumab to intraocular tissues. Several articles demonstrate the enhanced delivery of small molecules and macromolecules to posterior tissues following intravitreal injection (Shah et al. 2010).
There are two major routes of elimination for drugs from the vitreous: anterior and posterior. In the anterior route, drugs diffuse across the vitreous to enter the posterior chamber followed by clearance through the aqueous humor turnover or uveal blood flow. In the posterior route, drugs permeate across the retina and eventually cleared by the choroidal blood flow. Due to the relatively large surface area, tissue partitioning, and involvement of active transport mechanisms, molecules eliminated by the posterior (retinal) pathway have typically short half-life in the vitreous. Thus, the molecular and physicochemical properties of a drug play a major role in determining the primary elimination route from the vitreous humor. Table 4 shows the drug exposure (AUC) in vitreous, aqueous, and retina/choroid tissues after a single intravitreal injection of few ophthalmic drugs of interest in rabbit or monkey eyes.
As evident from Fig. 4, an exponential relationship exists between the half-life of drugs in the vitreous and the ratio of AUC (aqueous humor/vitreous humor). The elimination half-life in vitreous is shorter for drugs with lower partitioning ratio from vitreous to aqueous humor. In other words, drugs that are predominantly eliminated from the vitreous through the anterior pathway (aqueous humor turnover) have longer half-lives in the vitreous. Although a clear relationship cannot be elucidated with the limited data, the overall trend indicates a decrease in the vitreal half-life with increasing lipophilicity consistent with the expectation (see Table 4). As indicated earlier, adequate lipophilicity is required to penetrate the tight junctions of RPE barrier which results in large molecular weight and low lipophilic compounds to have prolonged half-life in the vitreous.

Relationship between elimination half-life in vitreous and the ratio of AUC (aqueous humor/vitreous humor) for intravitreally injected drugs
Maurice and Mishima (1984) demonstrated the relationship between molecular weight and aqueous/vitreous ratio indicating that primary route of elimination for high molecular weight compounds is by way of the anterior chamber. Dias and Mitra (2000) showed an inverse relationship between the molecular weight and vitreous elimination rate constant for high molecular weight FITC-dextrans. Using computer generated concentration contours, Maurice (2001) demonstrated that high molecular weight compounds exhibit prolonged half-life in vitreous. Figure 5 shows the relationship between LogP and the AUC ratio of retina-choroid/vitreous humor. Although, only limited data was available, the dependence on lipophilicity is clearly evident for partitioning into the retina/choroid which is in accordance with the previous reports. Similar results were reported by Liu et al. (1998) for a small group of structurally similar antibiotics where an excellent correlation between lipophilicity and vitreous elimination was observed.

Dependence on lipophilicity (LogP) for partitioning into retina from vitreous after intravitreal injection
A comprehensive relationship was developed using a diverse set of compounds to establish the relationship between physicochemical properties and half-life of drugs in the vitreous (Durairaj et al. 2009). A multiple linear regression analysis was conducted to identify the physicochemical properties that are predictors of half-life of a drug in the vitreous. The correlation model developed indicated that molecular weight, lipophilicity (LogP or LogD), and dose number (dose/solubility at pH 7.4) are the significant physicochemical properties that impact the half-life of molecules in the vitreous. The general model developed using the entire dataset (Log t 1/2 = −0.178 + 0.267 Log MW – 0.093 Log D + 0.003 Dose/Solubility7.4 + 0.153 PF) predicted the half-life of drugs in vitreous with good accuracy (R 2 = 0.725). Figure 6 shows the relationship between vitreal half-life and the key physicochemical properties identified in the regression model as significant contributors.

Correlation between vitreal half-life and key physicochemical properties. Reproduced with permission from Durairaj et al. (2009)
Dose number is a derived variable that includes the dose administered and its solubility at pH 7.4. When the injected dose exceeds its solubility in the vitreous, a depot or suspension is formed thereby releasing the drug in a sustained manner. As evident from the Fig. 6, when the dose injected exceeds the solubility limit (Dose/Solubility at pH 7.4 > 1), a steep increase in the apparent elimination half-life of drug was observed due to the slow release of the drug from the suspension or depot. Thus, including the dose and solubility improved the prediction for suspension formulations as well. Besides this general model, a number of submodels were also developed for various subsets depending on the dosage form administered (solution vs. suspension), animal model (albino vs. pigmented), ionization state (acids, base, neutral, and zwitterions), and molecule size (small vs. macromolecules). The models developed for these subsets provided insight into the key molecular properties that are unique for each of those classes. For instance, molecular weight was the major determinant of the half-life for macromolecules while lipophilicity was the main predictor for the acidic, basic, and zwitterionic compounds.
Kidron et al. (2012) used a narrower molecular weight range of compounds (<1,500 Da) to develop a model to predict the intravitreal half-life using 33 physicochemical descriptors relating to lipophilicity, hydrogen bonding, and mass. The final model for whole dataset included LogD at pH 7.4 and the total number of hydrogen bonds as predictors of half-life (log t 1/2, mixed = −0.046 – 0.051 (logD7.4) + 0.640 (LogHtot). Since compounds with molecular weight <1,500 Da were only included in the dataset, the final model did not include MW as a key descriptor. However, the model developed had a good predictive capability with Q 2 of 0.64 using the training set and 0.69 using the test set.
3.3 Effect of Pigmentation on Ocular Pharmacokinetics
In the eye, melanin is primarily distributed in ICB, choroid, and RPE. Moreover, regional differences in the distribution of melanin within RPE have been reported in human eyes (Schmidt and Peisch 1986) and in animals (Durairaj et al. 2012). Despite the route of administration, ocular drugs encounter these pigmented tissues during their pharmacokinetic life cycle as part of distribution or elimination process. Akin to protein binding, binding of drugs to melanin has raised interest in investigating its role in the disposition of drugs from eye. Few studies have investigated the pharmacokinetics of intravitreally injected drugs in pigmented animals. Table 5 summarizes the vitreal half-lives of compounds reported in both albino and pigmented rabbits after intravitreal injection. For at least more than half of the compounds, vitreal half-life is longer in pigmented than in albino rabbits (Table 5). However, with this limited set of data no clear trend can be established with any physicochemical properties.
Melanin is a polyanionic polymer comprised of repeating units of 5,6-dihydroxy indole-2-carboxylic acid and 5,6-dihydroxy indole (Nofsinger et al. 2000). Identification of key molecular properties that influence binding to melanin is more complex and depends on the nature of interaction (reversible vs. irreversible), chemical groups involved in binding, ionization status at the given pH, etc. Besides, the extent to which melanin binding can alter the pharmacokinetics of a drug is also dependent on the predominant route of elimination for that compound from the vitreous.
3.4 Influence of Disease State on Ocular Pharmacokinetics
The effect of disease state on the pharmacokinetics of ocular drugs is one of the less explored areas in ocular drug delivery. Barza et al. (1993) investigated the pharmacokinetics of cephalosporins after subconjunctival and intravitreal injections in the normal and infected eyes of rabbits. Repeated subconjunctival injections in the infected eyes resulted in two- to ninefold higher drug concentration in the vitreous when compared to a single subconjunctival injection in normal eyes. However, these higher levels in the infected eyes were probably related to repeated dosing rather than inflammation. The half-life of ceftizoxime and ceftriaxone were longer in the infected eyes than in normal eyes after intravitreal injection. This is presumably due to the ocular inflammation that generally causes damage to the transport pump and thereby prolonging the half-life of drugs that are eliminated by the posterior (retinal) route. After intravitreal injection in rabbits, the vitreal half-life of ketorolac in normal eyes was 2.28 h (Wang et al. 2012) while the half-life was 4.27 h (Baranano et al. 2009) in eyes with ocular inflammation. Similar results were reported by other authors where the vitreal half-life of ceftriaxone (Jay et al. 1984) and cefazolin (Ficker et al. 1990) was longer in the aphakic eye when compared to phakic eyes. Conversely, a decrease in the vitreal half-life of vancomycin was reported following intravitreal injection in infected rabbit eyes (Coco et al. 1998). The vitreal half-life decreased from 62.3 h in normal eyes to 13.6 h in infected eyes. This increased clearance was attributed to the increased permeability due to the disruption of blood–retinal barrier (BRB).
Cheruvu et al. (2009) investigated the effect of diabetes on transscleral delivery of celecoxib in rats. Following induction of diabetes in albino and pigmented rats, a breakdown in the BRB was observed with 2.4- to 3.5-fold higher leakage than in controls. When a single periocular injection of celecoxib was administered to both the rats with BRB breakdown, celecoxib exposure was 1.5- and 2-folds higher in the retina and vitreous humor of treated eyes as a result of the disruption of the BRB.
Shen et al. (2014) compared the ocular pharmacokinetics of brimonidine and dexamethasone after a single intravitreal injection in rabbits and monkeys with BRB breakdown. In case of rabbits, dexamethasone exposure (AUC) in aqueous humor, retina, and choroid was lower in disease animals than in normal animals. Similar trend was observed for brimonidine as well. In contrary, the central retina/choroid region where choroidal neovascularization (CNV) was established by laser lesions was the only ocular tissue in monkeys with consistent lower drug exposure. The AUC for brimonidine and dexamethasone was significantly higher by 59% and 23%, respectively, in normal animals when compared to CNV monkeys (P < 0.05). In addition to the anatomical and physiological differences, different induction methods were used in these species to disrupt the BRB that could have contributed to this difference. Besides, these results supported the enhanced clearance in animals with BRB breakdown thereby resulting in lower exposure in ocular tissues. Further, this study emphasized the consideration of differences that are compound and disease model specific when extrapolating the data to other species.
4 Summary
Due to various complexities involved in ocular drug delivery that pertains to the uniqueness of ocular barriers, target site for therapy (anterior vs. posterior segment), and different routes of administration, ocular pharmacokinetics is distinct and more intricate than systemic pharmacokinetics. A good understanding of the properties of ocular tissues primarily that act as barriers and targets for drug delivery is essential to understand its interaction with the drug. As most of the ocular diseases afflict anterior or posterior tissues and given the distinct properties of various ocular tissues, ocular pharmacokinetics should be deliberated discretely for these two regions (anterior and posterior segments) in alignment with drug delivery.
Considerable advances have been made to understand the mechanism of drug delivery to anterior segment tissues following various administration routes. Several experiments conducted in various animals and using isolated tissues over these years have advanced the understanding of the barrier properties, identifying the targets, and optimizing the drug and delivery platform to achieve target pharmacokinetic profile. With regard to the anterior segment, investigation on the barrier property of the anterior tissues (cornea, conjunctiva, and anterior sclera) and the establishment of desired molecular properties to circumvent the barriers has resulted in designing smart delivery vehicles and in experimenting novel administration routes. In case of posterior segment, advances in the field of computational sciences and statistical research have resulted in the development of in silico models that predict the half-lives of drugs based on the physicochemical properties. Innovation in the materials science has contributed to the birth of biodegradable implants that prolong the drug release for several months thereby drastically reducing the dosing frequency and improving patient compliance.
Regardless of these advancements, there are still unmet needs to further advance the ocular pharmacokinetics to the next level. For instance, the lack of allometric models to extrapolate the findings from preclinical species to human still exists despite the large number of studies carried out in various species. With the recent advancements in the novel intraocular delivery systems (including intracameral, subconjunctival, and intravitreal), another area that needs pharmacokinetic intervention is the development of IVIVC. Since there will be increasing demand for screening various prototype delivery systems during the development stage in order to identify the ideal delivery system with desired pharmacokinetic profile, an IVIVC model will be of esteem value in minimizing the number of preclinical/clinical studies required. Moreover, one of the main purposes of collecting pharmacokinetic information is to correlate with efficacy or safety data so that an optimal dose and dosing regimen can be established. In the absence of any such correlation, a standalone pharmacokinetic data can serve little purpose as linking with in vitro potency parameters involve assumptions that may not hold true in an in vivo setting. Thus, linking the pharmacokinetics to endpoints of interest (efficacy, safety, or biomarkers) through PKPD models is of great value in establishing the importance of drug exposure and its relevance to successful therapy.
References
Acheampong AA, Shackleton M, Tang-Liu DD, Ding S, Stern ME, Decker R (1999) Distribution of cyclosporin A in ocular tissues after topical administration to albino rabbits and beagle dogs. Curr Eye Res 18(2):91–103
Acheampong AA, Shackleton M, John B, Burke J, Wheeler L, Tang-Liu D (2002) Distribution of brimonidine into anterior and posterior tissues of monkey, rabbit, and rat eyes. Drug Metab Dispos 30(4):421–429
Ahmed I, Patton TF (1985) Importance of the noncorneal absorption route in topical ophthalmic drug delivery. Invest Ophthalmol Vis Sci 26(4):584–587
Akpek EK, Vittitow J, Verhoeven RS, Brubaker K, Amar T, Powell KD, Boyer JL, Crean C (2009) Ocular surface distribution and pharmacokinetics of a novel ophthalmic 1% azithromycin formulation. J Ocul Pharmacol Ther 25(5):433–439. doi:10.1089/jop.2009.0026
Asena L, Akova YA, Goktas MT, Bozkurt A, Yasar U, Karabay G, Demiralay E (2013) Ocular pharmacokinetics, safety and efficacy of intracameral moxifloxacin 0.5% solution in a rabbit model. Curr Eye Res 38(4):472–479. doi:10.3109/02713683.2012.763101
Attar M, Shen J, Ling KH, Tang-Liu D (2005) Ophthalmic drug delivery considerations at the cellular level: drug-metabolising enzymes and transporters. Expert Opin Drug Deliv 2(5):891–908. doi:10.1517/17425247.2.5.891
Baranano DE, Kim SJ, Edelhauser HF, Durairaj C, Kompella UB, Handa JT (2009) Efficacy and pharmacokinetics of intravitreal non-steroidal anti-inflammatory drugs for intraocular inflammation. Br J Ophthalmol 93(10):1387–1390. doi:10.1136/bjo.2009.157297
Barza M, Lynch E, Baum JL (1993) Pharmacokinetics of newer cephalosporins after subconjunctival and intravitreal injection in rabbits. Arch Ophthalmol 111(1):121–125
Baum J, Barza M (1983) Topical vs subconjunctival treatment of bacterial corneal ulcers. Ophthalmology 90(2):162–168
Buitrago E, Hocht C, Chantada G, Fandino A, Navo E, Abramson DH, Schaiquevich P, Bramuglia GF (2010) Pharmacokinetic analysis of topotecan after intra-vitreal injection. Implications for retinoblastoma treatment. Exp Eye Res 91(1):9–14. doi:10.1016/j.exer.2010.03.009
Cheruvu NP, Amrite AC, Kompella UB (2009) Effect of diabetes on transscleral delivery of celecoxib. Pharm Res 26(2):404–414. doi:10.1007/s11095-008-9757-2
Chien DS, Homsy JJ, Gluchowski C, Tang-Liu DD (1990) Corneal and conjunctival/scleral penetration of p-aminoclonidine, AGN 190342, and clonidine in rabbit eyes. Curr Eye Res 9(11):1051–1059. doi:10.3109/02713689008997579
Coco RM, Lopez MI, Pastor JC, Nozal MJ (1998) Pharmacokinetics of intravitreal vancomycin in normal and infected rabbit eyes. J Ocul Pharmacol Ther 14(6):555–563. doi:10.1089/jop.1998.14.555
Conrad JM, Robinson JR (1977) Aqueous chamber drug distribution volume measurement in rabbits. J Pharm Sci 66(2):219–224
Dey S, Patel J, Anand BS, Jain-Vakkalagadda B, Kaliki P, Pal D, Ganapathy V, Mitra AK (2003) Molecular evidence and functional expression of P-glycoprotein (MDR1) in human and rabbit cornea and corneal epithelial cell lines. Invest Ophthalmol Vis Sci 44(7):2909–2918
Dias CS, Mitra AK (2000) Vitreal elimination kinetics of large molecular weight FITC-labeled dextrans in albino rabbits using a novel microsampling technique. J Pharm Sci 89(5):572–578. doi:10.1002/(SICI)1520-6017(200005)89:5<572::AID-JPS2>3.0.CO;2-P
Durairaj C, Shah JC, Senapati S, Kompella UB (2009) Prediction of vitreal half-life based on drug physicochemical properties: quantitative structure-pharmacokinetic relationships (QSPKR). Pharm Res 26(5):1236–1260. doi:10.1007/s11095-008-9728-7
Durairaj C, Kadam RS, Chandler JW, Hutcherson SL, Kompella UB (2010) Nanosized dendritic polyguanidilyated translocators for enhanced solubility, permeability, and delivery of gatifloxacin. Invest Ophthalmol Vis Sci 51(11):5804–5816. doi:10.1167/iovs.10-5388
Durairaj C, Chastain JE, Kompella UB (2012) Intraocular distribution of melanin in human, monkey, rabbit, minipig and dog eyes. Exp Eye Res 98:23–27. doi:10.1016/j.exer.2012.03.004
Durairaj C, Shen J, Cherukury M (2014) Mechanism - based translational pharmacokinetic - pharmacodynamic model to predict intraocular pressure lowering effect of drugs in patients with glaucoma or ocular hypertension. Pharm Res 31(8):2095–2106. doi:10.1007/s11095-014-1311-9
Elena PP, Jauch A (1997) Ocular distribution of lomefloxacin 0.3% after a single instillation in the infected eye of pigmented rabbits. J Ocul Pharmacol Ther 13(6):551–558. doi:10.1089/jop.1997.13.551
Ficker L, Meredith TA, Gardner S, Wilson LA (1990) Cefazolin levels after intravitreal injection. Effects of inflammation and surgery. Invest Ophthalmol Vis Sci 31(3):502–505
Gaudreault J, Fei D, Rusit J, Suboc P, Shiu V (2005) Preclinical pharmacokinetics of Ranibizumab (rhuFabV2) after a single intravitreal administration. Invest Ophthalmol Vis Sci 46(2):726–733. doi:10.1167/iovs.04-0601
Ghate D, Edelhauser HF (2006) Ocular drug delivery. Expert Opin Drug Deliv 3(2):275–287. doi:10.1517/17425247.3.2.275
Grass GM, Robinson JR (1988) Mechanisms of corneal drug penetration. II: ultrastructural analysis of potential pathways for drug movement. J Pharm Sci 77(1):15–23
Hamalainen KM, Kananen K, Auriola S, Kontturi K, Urtti A (1997) Characterization of paracellular and aqueous penetration routes in cornea, conjunctiva, and sclera. Invest Ophthalmol Vis Sci 38(3):627–634
Hughes PM (2014) Intraocular pressure reduction with intracameral bimatoprost implants. Google Patents
Iyer MN, He F, Wensel TG, Mieler WF, Benz MS, Holz ER (2005) Intravitreal clearance of moxifloxacin. Trans Am Ophthalmol Soc 103:76–81, discussion 81–73
Iyer MN, He F, Wensel TG, Mieler WF, Benz MS, Holz ER (2006) Clearance of intravitreal moxifloxacin. Invest Ophthalmol Vis Sci 47(1):317–319. doi:10.1167/iovs.05-1124
Jay WM, Shockley RK, Aziz AM, Aziz MZ, Rissing JP (1984) Ocular pharmacokinetics of ceftriaxone following subconjunctival injection in rabbits. Arch Ophthalmol 102(3):430–432
Kidron H, Del Amo EM, Vellonen KS, Urtti A (2012) Prediction of the vitreal half-life of small molecular drug-like compounds. Pharm Res 29(12):3302–3311. doi:10.1007/s11095-012-0822-5
Kim H, Csaky KG, Chan CC, Bungay PM, Lutz RJ, Dedrick RL, Yuan P, Rosenberg J, Grillo-Lopez AJ, Wilson WH, Robinson MR (2006) The pharmacokinetics of rituximab following an intravitreal injection. Exp Eye Res 82(5):760–766. doi:10.1016/j.exer.2005.09.018
Kim SH, Csaky KG, Wang NS, Lutz RJ (2008) Drug elimination kinetics following subconjunctival injection using dynamic contrast-enhanced magnetic resonance imaging. Pharm Res 25(3):512–520. doi:10.1007/s11095-007-9408-z
Lee VH, Chien DS, Sasaki H (1988) Ocular ketone reductase distribution and its role in the metabolism of ocularly applied levobunolol in the pigmented rabbit. J Pharmacol Exp Ther 246(3):871–878
Liu W, Liu QF, Perkins R, Drusano G, Louie A, Madu A, Mian U, Mayers M, Miller MH (1998) Pharmacokinetics of sparfloxacin in the serum and vitreous humor of rabbits: physicochemical properties that regulate penetration of quinolone antimicrobials. Antimicrob Agents Chemother 42(6):1417–1423
Lopez-Cortes LF, Pastor-Ramos MT, Ruiz-Valderas R, Cordero E, Uceda-Montanes A, Claro-Cala CM, Lucero-Munoz MJ (2001) Intravitreal pharmacokinetics and retinal concentrations of ganciclovir and foscarnet after intravitreal administration in rabbits. Invest Ophthalmol Vis Sci 42(5):1024–1028
Maltese A, Bucolo C (2003) Pharmacokinetic profile of topical flunarizine in rabbit eye and plasma. J Ocul Pharmacol Ther 19(2):171–179. doi:10.1089/108076803321637708
Maurice D (2001) Review: practical issues in intravitreal drug delivery. J Ocul Pharmacol Ther 17(4):393–401. doi:10.1089/108076801753162807
Maurice DM, Mishima S (1984) Ocular pharmacokinetics. In: Sears ML (ed) Pharmacology of the eye. Springer, Heidelberg, pp 19–116. doi:10.1007/978-3-642-69222-2_2
Mayers M, Rush D, Madu A, Motyl M, Miller MH (1991) Pharmacokinetics of amikacin and chloramphenicol in the aqueous humor of rabbits. Antimicrob Agents Chemother 35(9):1791–1798
Mikkelson TJ, Chrai SS, Robinson JR (1973) Altered bioavailability of drugs in the eye due to drug-protein interaction. J Pharm Sci 62(10):1648–1653
Mindel JS, Yablonski ME, Tavitian HO, Podos SM, Orellana J (1981) Dipivefrin and echothiophate. Efficacy of combined use in human beings. Arch Ophthalmol 99(9):1583–1586
Navratil T, Garcia A, Verhoeven RS, Trevino L (2015) Advancing ENV515 (travoprost) intracameral implant into clinical development: nonclinical evaluation of ENV515 in support of first-time-in-human phase 2a clinical study. Paper presented at the ARVO Annual Meeting
Nofsinger JB, Forest SE, Eibest LM, Gold KA, Simon JD (2000) Probing the building blocks of eumelanins using scanning electron microscopy. Pigment Cell Res 13(3):179–184
Nomoto H, Shiraga F, Kuno N, Kimura E, Fujii S, Shinomiya K, Nugent AK, Hirooka K, Baba T (2009) Pharmacokinetics of bevacizumab after topical, subconjunctival, and intravitreal administration in rabbits. Invest Ophthalmol Vis Sci 50(10):4807–4813. doi:10.1167/iovs.08-3148
Prausnitz MR, Noonan JS (1998) Permeability of cornea, sclera, and conjunctiva: a literature analysis for drug delivery to the eye. J Pharm Sci 87(12):1479–1488
Proksch JW, Granvil CP, Siou-Mermet R, Comstock TL, Paterno MR, Ward KW (2009) Ocular pharmacokinetics of besifloxacin following topical administration to rabbits, monkeys, and humans. J Ocul Pharmacol Ther 25(4):335–344. doi:10.1089/jop.2008.0116
Rao CS, Schoenwald RD, Barfknecht CF, Laban SL (1992) Biopharmaceutical evaluation of ibufenac, ibuprofen, and their hydroxyethoxy analogs in the rabbit eye. J Pharmacokinet Biopharm 20(4):357–388
Review and Evaluation of Pharmacology and Toxicology Data (1999) http://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/021114_S000_BETAXON%200.5%25_PHARMR_P1.PDF. Accessed 25 May 2016
Schmidt SY, Peisch RD (1986) Melanin concentration in normal human retinal pigment epithelium. Regional variation and age-related reduction. Invest Ophthalmol Vis Sci 27(7):1063–1067
Schoenwald RD (2003) Ocular pharmacokinetics and pharmacodynamics. In: Mitra AK (ed) Ophthalmic drug delivery systems, 2nd edn. Marcel Dekker, Inc., New York, pp 135–179
Schoenwald RD, Huang HS (1983) Corneal penetration behavior of beta-blocking agents I: physiochemical factors. J Pharm Sci 72(11):1266–1272
Schopf L, Enlow E, Popov A, Bourassa J, Chen H (2014) Ocular pharmacokinetics of a novel loteprednol etabonate 0.4% ophthalmic formulation. Ophthalmol Ther 3(1-2):63–72. doi:10.1007/s40123-014-0021-z
Shafiee A, Bowman LM, Hou E, Hosseini K (2013) Ocular pharmacokinetics of bimatoprost formulated in DuraSite compared to bimatoprost 0.03% ophthalmic solution in pigmented rabbit eyes. Clin Ophthalmol 7:1549–1556. doi:10.2147/OPTH.S48766
Shah SS, Denham LV, Elison JR, Bhattacharjee PS, Clement C, Huq T, Hill JM (2010) Drug delivery to the posterior segment of the eye for pharmacologic therapy. Expert Rev Ophthalmol 5(1):75–93. doi:10.1586/eop.09.70
Shen YC, Wang MY, Wang CY, Tsai TC, Tsai HY, Lee YF, Wei LC (2007) Clearance of intravitreal voriconazole. Invest Ophthalmol Vis Sci 48(5):2238–2241. doi:10.1167/iovs.06-1362
Shen YC, Wang MY, Wang CY, Tsai TC, Tsai HY, Lee HN, Wei LC (2009) Pharmacokinetics of intracameral voriconazole injection. Antimicrob Agents Chemother 53(5):2156–2157. doi:10.1128/AAC.01125-08
Shen J, Durairaj C, Lin T, Liu Y, Burke J (2014) Ocular pharmacokinetics of intravitreally administered brimonidine and dexamethasone in animal models with and without blood-retinal barrier breakdown. Invest Ophthalmol Vis Sci 55(2):1056–1066. doi:10.1167/iovs.13-13650
Shorstein NH, Winthrop KL, Herrinton LJ (2013) Decreased postoperative endophthalmitis rate after institution of intracameral antibiotics in a Northern California eye department. J Cataract Refract Surg 39(1):8–14. doi:10.1016/j.jcrs.2012.07.031
Sjoquist B, Stjernschantz J (2002) Ocular and systemic pharmacokinetics of latanoprost in humans. Surv Ophthalmol 47(Suppl 1):S6–S12
Sjoquist B, Basu S, Byding P, Bergh K, Stjernschantz J (1998) The pharmacokinetics of a new antiglaucoma drug, latanoprost, in the rabbit. Drug Metab Dispos 26(8):745–754
Stratford RE Jr, Lee VH (1985) Ocular aminopeptidase activity and distribution in the albino rabbit. Curr Eye Res 4(9):995–999
Tang-Liu DD, Liu SS, Weinkam RJ (1984) Ocular and systemic bioavailability of ophthalmic flurbiprofen. J Pharmacokinet Biopharm 12(6):611–626
Tang-Liu DD, Liu S, Neff J, Sandri R (1987) Disposition of levobunolol after an ophthalmic dose to rabbits. J Pharm Sci 76(10):780–783
Wang M, Liu W, Lu Q, Zeng H, Liu S, Yue Y, Cheng H, Liu Y, Xue M (2012) Pharmacokinetic comparison of ketorolac after intracameral, intravitreal, and suprachoroidal administration in rabbits. Retina 32(10):2158–2164. doi:10.1097/IAE.0b013e3182576d1d
Weijtens O, Schoemaker RC, Lentjes EG, Romijn FP, Cohen AF, van Meurs JC (2000) Dexamethasone concentration in the subretinal fluid after a subconjunctival injection, a peribulbar injection, or an oral dose. Ophthalmology 107(10):1932–1938
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34(Database issue):D668–D672. doi:10.1093/nar/gkj067
Zhang J, Wang L, Gao C, Zhang L, Xia H (2008a) Ocular pharmacokinetics of topically-applied ketoconazole solution containing hydroxypropyl beta-cyclodextrin to rabbits. J Ocul Pharmacol Ther 24(5):501–506. doi:10.1089/jop.2008.0015
Zhang T, Xiang CD, Gale D, Carreiro S, Wu EY, Zhang EY (2008b) Drug transporter and cytochrome P450 mRNA expression in human ocular barriers: implications for ocular drug disposition. Drug Metab Dispos 36(7):1300–1307. doi:10.1124/dmd.108.021121
Zhang L, Li Y, Zhang C, Wang Y, Song C (2009) Pharmacokinetics and tolerance study of intravitreal injection of dexamethasone-loaded nanoparticles in rabbits. Int J Nanomedicine 4:175–183
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Durairaj, C. (2016). Ocular Pharmacokinetics. In: Whitcup, S., Azar, D. (eds) Pharmacologic Therapy of Ocular Disease. Handbook of Experimental Pharmacology, vol 242. Springer, Cham. https://doi.org/10.1007/164_2016_32
Download citation
DOI: https://doi.org/10.1007/164_2016_32
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58288-7
Online ISBN: 978-3-319-58290-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)