
Abstract
Nature relies on a unique and intricate biochemical setup to achieve sunlight-driven water splitting. Combined experimental and computational efforts have produced significant insights into the structural and functional principles governing the operation of the water-oxidizing enzyme Photosystem II in general, and of the oxygen-evolving manganese–calcium cluster at its active site in particular. Here we review the most important aspects of biological water oxidation, emphasizing current knowledge on the organization of the enzyme, the geometric and electronic structure of the catalyst, and the role of calcium and chloride cofactors. The combination of recent experimental work on the identification of possible substrate sites with computational modeling have considerably limited the possible mechanistic pathways for the critical O–O bond formation step. Taken together, the key features and principles of natural photosynthesis may serve as inspiration for the design, development, and implementation of artificial systems.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The biological conversion of light energy into chemical energy sustains practically all life on our planet [1]. Although not all photoautotrophs perform the same type of chemistry or have the same biochemical machinery, the fundamental functions are essentially the same: light-induced charge separation that drives electron transfer to eventually create energy-rich reduced compounds. A major differentiating factor among photosynthetic organisms is the source of electrons. In oxygenic photosynthesis performed by plants, algae and cyanobacteria, the source of electrons is water:
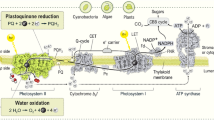
The light-driven oxidation of water takes place in the membrane-embedded pigment–protein supercomplex Photosystem II (PS-II), at an active site (the oxygen-evolving complex, or OEC) which contains an oxo-bridged tetramanganese–calcium cluster (Mn4CaO5) [2–7]. Dioxygen is a waste product and is simply discarded by photosynthetic organisms. However, it is through this biochemical process that the Earth’s atmosphere obtained its high oxygen content and the ozone layer, while life exploited oxygen’s oxidizing power in the evolution of respiration that can sustain more complex life forms. Protons are used for the creation of a membrane gradient driving the chemiosmotic synthesis of ATP (adenosine triphosphate), an energy carrier molecule in metabolic processes. The electrons are transported along other components of the photosynthetic chain (see Fig. 1) to be utilized eventually in the synthesis of NADPH (reduced nicotinamide adenine dinucleotide phosphate). Through NADPH, a carrier of reducing equivalents, the electrons extracted from water are employed in the light-independent carbon fixation reactions that reduce CO2 to carbohydrates:

Components of the light-dependent reactions and electron transfer in oxygenic photosynthesis. Blue arrows indicate the flow of electrons from H2O to NADPH. Q plastoquinone, Pc plastocyanin, Fd ferredoxin, FNR ferredoxin-NADP+ reductase
Among the distinctive features of biological oxygenic photosynthesis are the highly efficient light conversion, utilization of the ultimately abundant “electron source” on our planet (water), and a water oxidation catalyst composed of a readily available first-row transition metal, manganese. Faced by the current and near-future energy challenges of our civilization, and by the need to control the adverse effects of fossil fuel use, it is precisely these features of biological photosynthesis that we are trying today to reproduce in synthetic light-driven systems. The aim is to split water on a large scale to produce molecular hydrogen or other reduced compounds as energy carriers (solar fuels) [8–15]. Although the highly complicated biological system cannot be viewed as a blueprint to be faithfully reproduced in artificial systems, it remains a uniquely successful example of this type of chemistry – and a unique source of chemical insight. Several aspects of natural photosynthesis remain insufficiently understood despite decades of research, but many questions have also been answered and many fundamental principles have been clarified. Instead of attempting a broad and exhaustive overview of all facets of natural photosynthesis, in this chapter we focus on the most relevant aspects of water oxidation performed by PS-II and discuss the structural and functional principles that have emerged from recent research on biological water splitting.
2 Photosystem II Structure and Function
2.1 Overall Structure and Electron Transfer
The refinement of the PS-II structure has been a laborious and lengthy undertaking for X-ray crystallography. The first crystallographic model, at 3.8 Å resolution, was obtained by Zouni and coworkers in 2001 [16] and it took 10 years of effort [17–21] until a resolution of 1.9 Å was achieved [22]. Even more recently, a structure was obtained at 1.95 Å resolution using X-ray free electron laser pulses instead of synchrotron radiation [23], potentially avoiding the effects of extensive radiation damage which compromised earlier crystallographic models. PS-II has a dimeric structure (Fig. 2). Each monomer consists of more than 20 proteins arranged in a pseudo-dimeric fashion. A large number of cofactors are accommodated in the protein, including chlorophylls, pheophytins, carotenes, plastoquinones, lipids, bicarbonate, heme and non-heme iron centers, and an Mn4CaO5 complex at the OEC active site.

Left: structure of the PS-II dimer. Right: pigments, other cofactors and important residues involved in charge separation, electron transfer, and catalytic activity within a PS-II monomer
The core of each PS-II monomer is formed by the two homologous proteins D1 and D2, which contain most of the cofactors involved in catalysis and electron transfer (Fig. 2). Both proteins are highly conserved and very few variations exist among photosynthetic organisms. This is particularly true for D1, which harbors the oxygen-evolving manganese cofactor. Two proteins that contain several chlorophyll molecules, CP43 and CP47, surround the D1 and D2 proteins and are involved in light harvesting and transfer of excitation energy to the charge-separation site (P680), a set of four chlorophylls and two pheophytins located symmetrically around an intersection of the D1 and D2 polypeptide chains. Several other proteins surround the D1–D2 and CP43–CP47 pairs. These include “extrinsic” proteins on the lumenal side of PS-II, i.e., the side facing the interior of the thylakoid. These proteins may be necessary for catalytic function or they may bind only transiently to stabilize specific states or facilitate large-scale transformations such as active site reconstitution [24–26].
Photosystem II converts light energy into electrochemical potential which drives the oxidation of water and the reduction of plastoquinone [1, 27] according to the overall reaction
The dioxygen thus generated is released as a byproduct into the atmosphere, whereas plastoquinol (QH2) diffuses into the membrane to cytochrome b6f. Through the series of electron transport steps depicted in Fig. 1, the chain terminates with the synthesis of NADPH, the biological equivalent of reduced hydrogen. It is conceptually convenient to view the function of PS-II as the sum of four processes: (1) light harvesting and excitation energy transfer to the reaction center, (2) electronic excitation of the reaction center and charge separation, (3) transfer of the electron from the reaction center to plastoquinone, and (4) reduction of the reaction center by the Mn4CaO5 cluster, which in turn oxidizes water.
Following the absorption of visible light by chlorophyll molecules and their subsequent excitation, the excitation energy is transferred to the reaction center, a directed process facilitated by the spatial arrangement of the chromophores. The reaction center consists of chlorophyll (Chl a) and pheophytin (Pheo a) molecules bound to the D1 and D2 proteins (see Fig. 2). Excitation of the reaction center results in charge separation within picoseconds, generating the radical cation, P680•+, and a radical anion, PheoD1 •−. P680•+ has an estimated reduction potential of +1.2 V (vs SHE), making it one of the most oxidizing species known in biology [28, 29]. Following the formation of the initial charge-separated state, the electron is transferred from PheoD1 •− to a tightly bound plastoquinone molecule (QA) and then through a non-heme Fe(II) to the final acceptor QB. The rapid and efficient multi-step electron transfer over this distance of more than 30 Å is critical for suppressing charge recombination and results in the high quantum efficiency of biological water splitting. After two reduction and protonation steps the formed plastoquinol QBH2 diffuses into the thylakoid membrane and is replaced by another plastoquinone.
On the opposite side of the enzyme, the electron hole at P680•+ is filled by an electron donated by the redox-active Tyr161 residue (or YZ) of the D1 protein. Oxidation of Tyr161 is coupled to its deprotonation; the nearby His190 is the residue accommodating the proton on its imidazole ring upon formation of the tyrosyl radical. The YZ • subsequently oxidizes the manganese cluster of the OEC, which can accumulate up to four oxidizing equivalents before it eventually claims back these four electrons by oxidizing two water molecules, forming molecular dioxygen.
2.2 Channel Architecture
In addition to satisfying the requirement of precise spatial organization of redox-active components involved in excitation energy and electron transfer, the folding and structural arrangement of the PS-II proteins must also serve the need for tight control of accessibility and water delivery at the OEC, proton transfer, and product release. Although precise understanding of this type of regulation is still missing, analysis of crystallographic models [18, 20, 30–32], noble gas studies [33], pK a calculations [34], and, most importantly, molecular dynamics studies that take into account the dynamic nature of the channel architecture [35, 36] have already identified possible channels within PS-II that may be involved in water or dioxygen transport and proton transfer.
In this respect, it is important to realize that water plays at least four distinct roles: as the surrounding medium on each side of the thylakoid membrane, as structural water, as ordered chains involved in proton transfer, and as the reactant to be oxidized at the active site of the enzyme. Existing information indicates the presence of five solvent-accessible channels originating at the OEC. It is not currently possible to assign the role of each individual channel (water delivery, proton transfer, or dioxygen release), but it appears likely that at least one of the channels associated with a terminal Mn ion and one channel associated with the Ca2+ ion may serve as water delivery channels, another channel that involves calcium may be a dioxygen release pathway [36], and two channels associated with a proximal chloride ion may facilitate proton transfer away from the OEC [37].
2.3 Photoprotection and Repair
A critical aspect of natural photosynthesis and in particular PS-II is its efficient protection and repair machinery, an aspect that may not lend itself easily to biomimetic approaches in synthetic systems [38]. Being tasked with catalyzing one of the most challenging reactions in biology using sunlight, PS-II has to achieve tight control on how to regulate light absorption and how to use the absorbed energy to drive the electron transport reactions. Excess light energy that cannot be put to use would result in damage through unwanted side reactions and generation of deleterious reactive oxygen species. PS-II has a series of photoprotective mechanisms that serve to dissipate excess energy or quench photoexcited states under such conditions. Without going into detail, these mechanisms may be distinguished in those that involve recirculating electrons within the components of the reaction center, redirecting the electrons to alternative acceptors, or disposing of the absorbed light energy as heat by reconfiguration of the antenna system [39–42].
Nevertheless, photoinhibition cannot be entirely avoided. The D1 chain is the part of PS-II most vulnerable to oxidative damage – of the electron transfer components, amino acid residues, or the Mn cluster itself. There is still controversy regarding how D1 exchange is triggered but it is known that the protein has to be replaced approximately every half hour of operation. A process intimately related to self-repair of PS-II is the assembly of the Mn4Ca cluster: remarkably, the biosynthesis of the inorganic cluster does not rely on chaperones but is a light-driven process which builds the oxo-bridged metal cluster in place by sequential oxidations of Mn2+ ions by YZ •.
3 Oxygen Evolving Complex
3.1 Catalytic Cycle and Redox Leveling
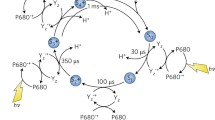
As described above, the tetramanganese cluster of the OEC couples the one-electron process of photoexcitation and charge separation at the reaction center with the four-electron chemistry of water oxidation by storing oxidizing equivalents. At a phenomenological level, four photo-driven charge separation events lead to successive removal of four electrons from the Mn ions of the OEC, via the redox-active Tyr161 (YZ), at which point O2 is evolved and the OEC is reset to its most reduced state. These observations [43] have been encoded in a model of the catalytic cycle consisting of five intermediates (Fig. 3) [44], known as the S i states, where the subscript indicates the number of stored oxidizing equivalents (i = 0–4). The S0 is the most reduced state of the cluster, but S1 is the dark-stable resting state [4]. The S4 is a transient and so far unobserved state that decays spontaneously to S0 by releasing O2.

Left: dioxygen evolved by PS-II with a series of flashes, demonstrating the four-flash periodicity in the maximum yield. Right: the catalytic cycle of five S i states, including YZ radical intermediates
As indicated in Fig. 3, the removal of electrons and protons follows a strictly alternating pattern [45–48], with all transitions involving proton loss except S1 → S2 [2, 28, 49, 50]. Considerable information from electron paramagnetic resonance (EPR) spectroscopy is available on many of the intermediate S i YZ • states, and evidence also exists regarding additional transient intermediates in the S0 → S1 and S2 → S3 transitions [46, 51], and a deprotonated intermediate immediately prior to the final electron transfer after formation of the S3YZ • state [52–54].
The critical importance of the coupled removal of protons and electrons from the OEC is that it avoids the build-up of excess charge which would quickly render the Mn cluster impossible to oxidize by the YZ radical. This is known as a “redox-leveling” effect and allows for four successive oxidations of the cluster to take place within a narrow potential range. This becomes apparent in the reduction potentials of the transitions, which are ca. 0.85, 1.10, 1.15, and 1.0 eV for states S0–S3 [4]. It should be noted that although the electron and proton transfers are coupled, they are not necessarily concerted. Obviously, the highly organized and intricately balanced hydrogen bonding network around the OEC is of fundamental importance in regulating the transfer of protons [55–58].
3.2 Structural Information
The Mn–Mn distances obtained from Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy represented the first insight into the geometry of the Mn4CaO5 core of the OEC. The 1981 study by Klein and coworkers was also the first direct observation of the Mn ions contained in chloroplasts [59]. Already at that time, even before the number of Mn ions per active site was known, the authors concluded that the Mn ions should be connected in a similar way to the bis-μ-oxo-bridged model dimers they also investigated. EXAFS remains a powerful tool and has provided metal–metal and metal–ligand distances for all stable catalytic intermediates [60–63], but it is unlikely that it can decipher structurally heterogeneous states of the OEC. The relative orientation of the metal–metal vectors can in principle be revealed by the use of polarized EXAFS [64], but the extraction of a unique three-dimensional structure remains challenging.
X-Ray diffraction studies of improving resolution have progressively refined our view of the three-dimensional topology and connectivity of the OEC up to the point where an atomic-resolution model (1.9 Å resolution) was reported in 2011 [22]. These studies are typically carried out on dark-adapted samples that have accumulated in the S1 state. However, the employed X-ray doses resulted in radiation damage in the form of partial reduction of the Mn ions [65–67], leading to significant amounts of Mn(II), as opposed to Mn(III) and Mn(IV) which are present in the S1 state. This also explains the significant differences in Mn–Mn distances between the XRD models and those observed by EXAFS or predicted computationally [61, 68, 69]. Most recently, by employing X-ray Free Electron Laser (XFEL) pulses, the three-dimensional structure of the inorganic cluster in the S1 state was obtained at 1.95 Å resolution with probably no radiation damage [23]. Comparison of the XFEL Mn–Mn distances with those of the previous XRD structure [22] confirms the consensus reached by most researchers in the field, that a significant elongation of up to 0.20 Å for the Mn–Mn distances in the XRD structure can result from radiation-induced reduction of Mn ions in the cluster. Importantly, this XFEL structural model has now largely converged to EXAFS with respect to Mn–Mn distances and is in agreement with detailed computational models for the S1 state (see Table 1), although some inconsistencies remain with respect to oxo-bridge positions [70, 71].
The inorganic core of the OEC has been described as taking the shape of a “distorted chair” [22], with the base formed by a heterometallic Mn3CaO4 cuboidal unit and the backrest by an Mn–O linkage connected to one of the Mn ions and one of the oxo bridges of the cubane (see Fig. 4). Four water molecules, or water-derived ligands, are directly ligated to metal ions of the cluster, two at Mn4 and two at Ca. This unit is embedded in the D1 protein and connected to the CP43 protein by one direct ligand. Carboxylate residues bridge Mn1–Mn2 (Asp342), Mn2–Mn3 (CP43-Glu354), Mn3–Mn4 (Glu333), and Ca2+ with Mn2 (Ala344) and Mn4 (Asp170). Only one N-donor, His332, is coordinating the cluster, at Mn1, which is also mono-coordinated by Glu189. Important residues in the second coordination sphere include the Tyr161–His190 couple (electron transfer gate to the special pair), Asp61 (probably important for proton transfer), CP43-Arg357, and His337 (hydrogen bond to O3).

A view of the OEC with the Mn4CaO5 cluster and its immediate environment (crystallographic coordinates are taken from Umena et al. [22]); a common labeling scheme for the inorganic core is indicated in the inset
Remarkably, the precise nuclearity and bonding connectivity of the inorganic cluster and its immediate ligand environment had already been predicted quite accurately in computational studies by Siegbahn, based on energetic considerations, before the advent of the first atomic-resolution XRD structure [72, 73]. It should be noted that XRD as yet reports, ideally if not in practice, on the dark-adapted S1 state only. The structural changes occurring as the catalytic cycle progresses can be traced from EXAFS data as follows. In the S1 and S2 states, there exist three short Mn–Mn distances of 2.7–2.8 Å and a longer Mn–Mn distance of 3.28 Å [62]. The very small structural changes between these two states are consistent with the fact that this transition represents only an oxidation event [45, 74, 75]. The S0 state appears to be characterized by an elongation of one of the short Mn–Mn distances to 2.85 Å, which may arise from the protonation of one of the oxo bridges. The reduction by ca. 0.1 Å between S0 and S1 is in line with observations of geometrical changes in model systems upon oxo bridge protonation [76, 77]. Oxidation of the S2 to the S3 state also leads to elongation of at least one Mn–Mn distance by ca. 0.1 Å. Before the unequivocal assignment of the Mn oxidation states in the S3 state by coupled EPR and theoretical studies [78], this elongation upon oxidation had given weight to arguments in favor of a ligand-centered rather than a metal-centered oxidation in the S2 → S3 transition.
Despite this valuable structural information, the three-dimensional topology and the precise connectivity of the OEC cannot be directly deduced experimentally at the atomic level in each S-state, and information on the protonation states of bridging and terminal oxygen ligands is out of reach. However, all these aspects and a detailed view of the orientation of Mn(III) Jahn–Teller axes in each state can be obtained by experimentally constrained computational models, as described in the following.
3.3 Mn Oxidation States and Detailed Structural Models
Although the relative oxidation levels of the OEC are defined by the Kok cycle (Fig. 3), the absolute oxidation states of the Mn ions are not. Two schemes had historically evolved, which accommodated several structural constraints and spectroscopic observations [79–90]. Since the early 1980s it has been known that the four Mn ions in oxidation states III and IV composing the active site gave rise to an EPR signal with a spin ground state of S GS = 1/2 in the S2 state [79]. This finding could be explained equally well with the oxidation state combinations Mn(III)3Mn(IV) and Mn(III)Mn(IV)3, where in both cases one Mn(III) and one Mn(IV) magnetically couple to S dimer = 1/2 and the remaining two Mn(III) or two Mn(IV) couple to yield S dimer = 0, so that in both cases S GS = 1/2 can be obtained. These two proposals are known as the “low oxidation state” and “high oxidation state” schemes, or equivalently, “low valent” (LV) and “high valent” (HV) schemes, and they differ by two in the total number of electrons in all states. Specifically, the LV scheme would correspond to Mn oxidation states of (II, III, III, III) in S0 up to (III, III, IV, IV) in S3, whereas the HV scheme requires the S0 state to be (III, III, III, IV) instead. Answering this question is central to understanding the principles of biological water oxidation and places immediate restrictions on the mechanism of O–O bond formation, because, for example, in the HV scheme either oxo-oxyl coupling or nucleophilic attack mechanisms can be imagined, whereas in the LV scheme only the latter appears to be a chemically reasonable option.
Attempts to “count” the number of redox equivalents needed to photoactivate the OEC [91, 92] or to disassemble a specific S state [93], as well as experiments that probe the super-reduced S states [94] are promising, as they both involve measurable quantities of Mn(II). However, these experiments as well as H2 16O/H2 18O substrate water exchange experiments [95–97] have been interpreted controversially [7, 98–100]. EPR and electron nuclear double resonance (ENDOR) studies have contributed a wealth of information on the water oxidizing cluster and have also addressed the question of overall oxidation level. Most importantly, the ground state spin multiplicities [101] of all stable intermediates have been established: S GS = 1/2 for the S0 state [102–105], S GS = 0 for the S1 state [106, 107], S GS = 1/2 and ≥5/2 for the S2 state (g = 2 and g ≥ 4.1 signals) [79, 108–110], and S GS = 3 for the S3 state [78, 111, 112]. All of these are compatible with the two oxidation state schemes introduced above. The same is true for the 55Mn hyperfine coupling constants in the S2 state, which have been used both to support the low valent scheme [81] and to rule it out [113, 114], particularly because ENDOR studies of the S0 state were interpreted as indicating the absence of Mn(II) in that state [114], which would exclude the LV scenario.
X-Ray absorption and emission spectroscopies [115, 116] have probed the relative changes in Mn oxidation states, but the limits of interpretation in terms of absolute oxidation states are still being debated. Although in principle the absolute oxidation state of a metal can be extracted from the edge position, the situation for a multinuclear complex in which only one out of four ions changes valence upon oxidation of the cluster is more complicated: the shift of ca. 1 eV per oxidation event for a mononuclear site is diminished to ca. 0.25 eV for a tetranuclear cluster. Additionally, rearrangements of the coordination sphere, which are plausible upon oxidation of a multinuclear system, are known to influence the edge position. XANES data has been interpreted as consistent with both the HV [82–85, 117] and the LV [118–120] schemes, the latter mostly based on comparison of experimental with calculated spectra.
Computational studies have typically been carried out under the assumption of either the LV [119–123] or the HV scheme [72, 124–140]. The first comparative study of the two schemes was recently presented, including all stable intermediates (S0–S3) and combining the widest possible collection of experimental information as evaluation criteria, ranging from EPR properties and EXAFS or XFEL structural information to Mn K pre-edge spectroscopy. After construction of computational models valid for the S0–S3 states in either the LV or the HV schemes, i.e., ranging from Mn(II)Mn(III)3 to Mn(IV)4, and systematic variation of the overall protonation level, protonation sites, orientation of Jahn–Teller axes for Mn(III) and other such permutations, the relative energies, geometric characteristics, spin ground states (see next section for details), 55Mn hyperfine coupling constants and Mn K pre-edge signatures of the models were compared with experimental data. The study relied in large part on a series of methods developed over the years for the accurate calculation of magnetic and spectroscopic properties [130, 141–146]. Although agreement with isolated experimental observations could be reached for some low-valent models, S0 and S1 models based on the LV scheme were found to be incompatible with all experimental constraints. Therefore, this study established beyond doubt that a consistent catalytic cycle is only possible with the high-valent scheme, i.e., Mn(III)2(IV)2 in the S0 state to Mn(IV)4 in the S3 state. Simultaneously, the protonation state and pattern in each S-state were uniquely determined. Figure 5 summarizes the sequence of all stable S-state catalytic intermediates resulting from this comprehensive study.

Computationally determined and experimentally consistent models of the inorganic core of the OEC for all S-state intermediates. The distribution of Mn oxidation states and the ground state spin S are shown for each model. Coordinating amino acid ligands are not shown for clarity
A somewhat different debate, independent of the overall oxidation level of the cluster, concerned the correct interpretation of XANES data with respect to the nature of the S2 to S3 transition [83, 147–149]. The two suggestions were (1) ligand-centered oxidation, as implied by the absence of a K-edge shift in the S2 to S3 transition and (2) metal-centered oxidation with concomitant binding of a ligand to a five-coordinate Mn(III) in S2 to a six-coordinate Mn(IV) in S3, such that the change in coordination environment compensates for the K-edge shift produced by metal center oxidation [83, 149]. A recent combined EPR/ENDOR and DFT study of the S3 state conclusively supports the latter interpretation [78], showing that all four Mn centers are present as octahedrally coordinated Mn(IV) ions before the final oxidation event leading to spontaneous formation and release of dioxygen. Proposed mechanisms for the O–O bond formation step are discussed in a later section.
3.4 Spin States and Structural Flexibility
As noted above, EPR spectroscopy has been central in determining the spin states of the OEC cluster in the S0–S3 states of the catalytic cycle, but also in probing the local electronic properties of the Mn ions, especially through 55Mn-ENDOR [113, 114, 150–152]. In terms of spin states, the data suggest that the Mn cluster evolves from low-spin states in the early stable intermediates (S0 and S1) to coexistence of low-spin and higher-spin states in S2, progressing to a high-spin state in S3, in preparation for the final O–O bond formation step (see Fig. 5).
The S2 state is the catalytic intermediate studied the most by EPR/ENDOR, not only because of its easy accessibility through a single light flash from dark-adapted samples, but also because of the intriguing phenomenology observed for this state. It has also been the state targeted in pioneering studies aimed at providing theoretically supported topological interpretations of spin state data [153, 154] before the availability of crystallographic information. The S2 state is associated with two EPR signals that are quantitatively interconvertible, implying that the structural changes must be small. The two signals correspond to spin ground states of S GS = 1/2 at g = 2.0 (“multiline signal”) and S GS = 5/2 at g ≥ 4.1 (“g4 signal”). Building on the first atomic-resolution XRD structure, Pantazis et al. constructed two models in the HV scheme that fully explained the phenomenology [137]. The structure giving rise to the g4 signal can be viewed as an Mn(IV)3CaO4 cubane, to which the fourth Mn is attached via a μ-oxo bridge providing a link to an Mn ion of the cubane (see Figs. 5 and 6). This structure is termed the “closed cubane” form, similar to a structure proposed by Barber and Murray [155]. It is known that Mn(IV)3Ca cubanes are associated with spin ground states of S GS = 9/2 [156]; therefore, the coupling of the trimeric S cubane = 9/2 unit with the outer Mn(III) with S Mn(III) = 2 leads to the observed spin ground state of S GS = 5/2. The conversion into the almost isoenergetic structure associated with the multiline signal is achieved through the low-barrier translocation of a μ-oxo bridge of the cluster and a valence exchange so that the Mn(III) in the closed cubane structure becomes Mn(IV) in the open cubane structure (see Fig. 5). The open cubane form is similar to that put forward originally by Siegbahn [72, 73]. The thus altered connectivity of the Mn ions in the core leads to a magnetic coupling topology drastically different from that in the closed cubane structure, ultimately rendering the S GS = 1/2 ground state. Two antiferromagnetic coupling pathways (Mn1–Mn2 and Mn3–Mn4) and one ferromagnetic coupling (Mn2–Mn3) dominate the magnetic interactions in the tetranuclear cluster. Importantly, the magnetic coupling topology places restrictions on the pattern of 55Mn hyperfine coupling constants, which have been used as a powerful criterion to discriminate between different electronic configurations, structural possibilities, and protonation states of terminal and bridging ligands [70, 134, 136].

Exchange coupling constants (in cm−1) for the two interconvertible structural models of the S2 state shown in Fig. 5, along with a first-derivative EPR simulation of the two configurations, the open cubane (S = 1/2) g = 2.0 multiline and the closed cubane (S = 5/2) g = 4.1 S2-state OEC signals [137]
These studies represent important showcases of how a consistent picture of spectroscopic observations and structural information is yielded through the combination of experimental facts and a theoretical approach oriented towards prediction of observable properties. Structural polymorphism had previously been invoked for the S1 state by Kusunoki [128], motivated by the quest to rationalize the radiation-damaged crystal structure using a superposition of structural forms, but it now appears that no such drastic rearrangement is present in the S1 state. Several studies suggest that heterogeneity of the geometric and electronic structures of the S2 state [132, 137, 157] may be absent in the S3 state [70, 78, 124], but this point requires further investigation. It is not clear whether structural flexibility in S2 serves a fundamental mechanistic purpose, such as facilitating or controlling water binding [132, 158]. Nevertheless, it suggests that the manganese cluster in PS-II is not merely a small rigid piece of rock embedded in a polypeptide. This structural flexibility, and the progression from low-spin to high-spin states along the catalytic cycle, are two intriguing aspects of the OEC with potential mechanistic implications requiring further experimental and theoretical work to be understood fully.
3.5 The Role of Calcium
Calcium is an integral part of the water-splitting catalyst, demonstrated, for instance, by the fact that the only ion that can replace it while maintaining the water-splitting capability of the OEC is Sr2+, although the efficiency is diminished [159–162]. As other metals of the same charge and similar ionic radii are easily available in nature, the role of Ca2+ must be more than purely structural [163]. In line with the early, redox-equivalent accumulating S-states displaying structural heterogeneity, which the later S-states do not, the role of Ca2+ has also been suggested to change as the catalytic cycle progresses. Although Dy3+ and Cd2+ cause a loss of the O2-evolution capability of the cluster, the OEC substituted with these cations can advance from S1 to S2, giving weight to a dominantly structural role of Ca2+ in the early S states [164]. Ca2+ has also been suggested to bind substrate water [3, 165] and to be involved in maintaining a hydrogen-bond network for proton transfer [166]. The possible involvement of calcium in the delivery of water to the Mn cluster has also been suggested on the basis of ab initio molecular dynamics studies [132].
The similar Lewis acidity of Sr2+ and Ca2+ has been considered to be an important factor for the fully functional substitution [159, 163, 164, 167]. Synthetic studies on MMn3O4 and MMn3O2 model complexes that are structural subunits of the OEC have established a linear correlation between the pK a of M(aquo)n+ ions of redox-inactive metals as a measure of their Lewis acidity and their reduction potentials [168]. Although the two series investigated differ in Mn oxidation states and ancillary ligands, the slopes of the linear correlations are similar. Thus, more negative reduction potentials are achieved by incorporation of redox-inactive metals with high Lewis acidities and by incorporation of more oxo-bridges into the metal-oxo core. The redox potentials of these models shifted distinctively relative to the Ca2+-containing model for ions such as Zn2+, Y3+, Sc3+, and Mn3+, but not for Sr2+, which is attributed to the similar Lewis acidities of Sr2+ and Ca2+ [168, 169]. Arguing by analogy, it might be expected that one reason for Nature’s selectivity for Ca2+ would be the optimization of redox potential range for the Mn4CaO5 cluster, an effect that was also implied by DFT calculations on an OEC model [170].
Additionally, experimental evidence in the form of peptide carbonyl frequency shifts upon Ca2+ substitution by other cations imply that hydrogen bonding interactions are affected by this type of modification of the OEC cluster [171]. Moreover, recent work on the higher states of the OEC suggests that Ca/Sr substitution perturbs the distribution of the conformational microstates in the S3–S0 transition, highlighting the role of calcium in structuring the environment of YZ in the S3 state [54]. Given the proximity of Ca2+ to the redox-active tyrosine (Tyr161, YZ), and because a sequence of balanced redox potentials is necessary for efficient energy conversion, it is pertinent to ask how precisely Ca2+ influences the properties of YZ, besides its effect on the Mn cluster itself. A recent computational study examined the effect of Ca2+ on the acidity, arrangement, and ordering of the water molecules between Ca2+ and YZ [140]. The two major factors are: (1) influence of Ca2+ on the electronic structure and therefore the redox potential of YZ by the organization of hydrogen bonds towards the tyrosine and (2) modulation of the communication between YZ and the cluster through these water molecules. The absence or presence of water molecules, or equivalently the “structured polarity” of the hydrogen-bonding surrounding the phenolic oxygen, leads to significant changes in predicted spectroscopic parameters (g tensor of the tyrosyl radical) and large shifts in the electron affinity of the residue of ca. 0.46/0.28 eV (vertical/adiabatic values). This demonstrates that Ca2+ plays a combined structural and electronic role by controlling the hydration environment of YZ, orienting the tyrosine ring, ordering the hydrogen-bonding network, and regulating the acidities of bound waters and the redox potential of the YZ residue in both its reduced and oxidized forms (Fig. 7) [140].

Depiction of the Ca2+ ion of the OEC and the water molecules involved in the interaction of the ion with the redox-active tyrosine. The structure is taken from an optimized model of the S2YZ • state. The diagram shows the computed change in the electron affinity (squares: adiabatic; circles: vertical) of the tyrosyl radical upon successive loss of interaction with each ordered water molecule (adapted from Retegan et al. [140])
3.6 Role of Chloride
Two sites of Cl− ions in close proximity to the active site have been identified in numerous crystal structures, again by substitution with the heavier and thus easier to identify Br− and I− ions [21, 172]. The role of Cl− can be partially substituted by other negatively charged ions such as Br−, NO3 −, and NO2 − at the cost of a lower efficiency, but probably not with iodide [172]. The binding sites are ca. 6.55 Å and 7.45 Å from the closest Mn ion of the OEC. Most studies have focused on the closer binding site, known as “binding site 1” (see Fig. 4). It is surrounded by Lys317, Asn181, and the backbone N of Glu333. Two water molecules (PDB IDs 517 and 518 in 4UB8) fill the space between this binding site, Asp61 and the W2 ligand to Mn4. A QM cluster study assigned the role of Cl− mainly to its charge, with the effect that the last steps before product release are lowered in energy relative to Cl−-containing models [173]. In molecular dynamics studies, structural flexibility of the Cl− ion and the amino acid residues in its vicinity was observed, with a salt bridge forming between Asp61 and Lys317 upon Cl− depletion [37]. Because Asp61 is considered essential for proton removal from the catalytic site, chloride would serve to stabilize this function and would therefore be essential for the function of the proton relay network [174, 175]. Furthermore, a role of chloride in fine tuning the pK a of Asp61 has been suggested [176].
The close connection to the Mn4 ligand W2 via one water molecule and the position in the immediate vicinity of the Mn1–Mn4 bridging entity Glu332-His333 are reminiscent of the organizational role that Ca2+ plays. Glu333, the D1 residue bridging Mn3 and Mn4, is suggested to interact with D2-Lys317 in the absence of Cl−, so that Cl− depletion would lead to a distorted geometry and hence altered magnetic interactions in the Mn4Ca cluster [176–178].
4 Biological Water Oxidation Mechanism
4.1 Substrate Identification
The origin of the substrates and the evolution of their protonation states in the consecutive steps of the catalytic cycle determine the details of the water oxidation mechanism at the atomic level. Because the substrate waters are present close to the OEC and water molecules play structural roles or participate in proton channels, it is impossible to identify the substrates directly from any source of structural information.
As complex as the OEC is to study experimentally and computationally, one immediate advantage over other bioinorganic catalysts is that the product it forms is gaseous. By exposing the active site to isotopically labeled water (H2 18O) the composition of the final O2 (16O16O, 16O18O, 18O18O) can be analyzed by mass spectrometry. Invaluable advances have been achieved by membrane-inlet mass spectrometry (MIMS) studies [2, 100], in which the exposure time of the protein sample to the labeled water is varied, from which the exchange rates of the substrate waters can be determined [95, 97]. Two substrate binding sites with different exchange rates have thus been identified. Their exchangeability varies as the catalytic cycle progresses: the slowly exchanging water, “Wslow”, exchanges fastest in S0 and slowest in S1, and accelerates its exchange rate again as the cluster is oxidized to the S2 state. Although no molecular model studies exist for direct comparison of H2O/OH−/O2− exchange rates to the natural system, it is unexpected to observe an increase in exchange rate by several orders of magnitude upon metal oxidation. The other water, “Wfast”, cannot be detected in S0 and S1 [179], either because of kinetic limitations in the setup or because it is not yet bound to the cluster. As shown in Table 2, Wfast has a slower exchange rate in the S3 state although still exchanging much more rapidly than Wslow in S3. Combining the MIMS experiment with Ca2+/Sr2+ exchange, it was found that Wslow is accelerated by substitution of Ca2+ with Sr2+, whereas Wfast is unaffected [180]. It should be noted that MIMS experiments are not sensitive to the precise location of the water exchange sites and the protonation state of the exchanging O-entity. This requires input from other experimental techniques and theoretical modeling.
To observe changes that affect water molecules between S states, FTIR difference spectra in the frequency region of OH stretching vibrations are a valuable tool. Weakly H-bonded OH groups are easier to detect than strongly H-bonded ones, as the latter overlap with NH stretching vibrations [181]. By subtracting the spectra of two S states, the protein background is removed, revealing those changes associated with the S-state transition, e.g., the hydration of the OEC. The effect of dehydration of OEC samples was found to be largest for the S2–S3 and S3–S0 transitions, which was interpreted as caused by water binding to the active site during these transitions [181, 182]. The difference in one vibrational mode seen in the S2–S1 difference spectrum upon suspension of PS-II in an H2 18O buffer and the effect on the mode of Ca/Sr substitution also suggested that a candidate for Wslow could be an Mn–O–Ca bridge of the inorganic core [183, 184].
A unique contribution to the effort towards substrate identification at the atomic level comes from EPR techniques. Specifically, high-field 17O-ELDOR-detected NMR (EDNMR) and 17O-ENDOR has been used to probe exchangeable 17O species [184, 185]. Three types of 17O nuclei were observed (μ-oxo bridge, terminal Mn–OH or Mn–OH2, and Ca-bound or second-shell water). Experimentally, either O4 or O5 could correspond to the exchangeable μ-oxo bridge, but DFT models favored the assignments shown in Fig. 8, that is, with O5 as the exchangeable bridge. Further studies relied on the use of ammonia, a substrate analogue that can access the OEC and binds directly to the Mn4Ca cluster by ligand substitution. Although the replacement of a μ-oxo bridge by a deprotonated form of ammonia is still considered as a viable scenario [186], experimental and theoretical studies have come to the conclusion that NH3 most likely binds to the cluster by displacing W1 [139, 187, 188], stabilized at this position by hydrogen bonding (Fig. 8). By observing the response of the EDNMR signal of the exchangeable bridge to the presence of both NH3 binding and Ca/Sr replacement, these studies strongly support the assignment of O5 as the slowly exchanging water binding site.

(a) S2-state 17O-ELDOR-detected NMR spectra (94 GHz) of the OEC exchanged in H2 17O and color-coded assignments of exchangeable H2O/OH− ligands (adapted with permission from Cox et al. [5], copyright 2013 American Chemical Society); (b) DFT model for the binding of NH3 by W1 displacement in the S2 state (adapted from Lohmiller et al. [139])
An exchange rate similar to that of Wslow is still much faster than exchange rates observed for μ-oxo bridges in high-valent Mn systems [185, 189]. This may be because there exists so far no synthetic model that mimics all structural features of the OEC and its environment. The structural flexibility observed in the S2 state, enabled through the dynamic nature of the bonding between O5 and the terminal Mn ions, may be relevant in this respect [137], and recent DFT simulations of water exchange have produced computed barriers consistent with observed exchange rates [190].
4.2 O–O Bond Formation
It would be impossible to review here all the different mechanistic proposals that have appeared over the years and all the variants of strictly hypothetical or computationally defined models for the critical steps in biological water oxidation. We refer the interested reader to the numerous reviews dealing with parts of this enormous subject, e.g., [3, 15, 99]. Here we focus exclusively on some of the possibilities regarding the O–O bond formation step, as constrained by the information presented in previous sections. The assignment of O5 as a substrate considerably limits the number of models that can be imagined, as shown in Fig. 9. The four models depicted here do not exclude variations in the bonding topology for each scheme, for example, in whether a terminal OH group is associated with Mn1 or Mn4. However, mechanisms that can be excluded if the O5 bridge is a substrate are those that would involve nucleophilic attack of a Ca-bound OH/H2O on a terminal Mn4-oxo, and those that would involve direct coupling of terminal Mn4-oxo groups.

(a–d) Possible schemes for O–O bond formation in PS-II
The models in Fig. 9a, b involve nucleophilic attack on O5. Nucleophilic attack of a Ca-bound OH or H2O on a terminal high-valent oxo group, Mn(V)–O (oxo) or Mn(IV)–O• (oxyl) [191] has been discussed extensively in the past [127, 165, 192, 193], but the assignment of O5 as a substrate is not consistent with this idea as it requires this bridge and not a terminal oxo group to play the role of electrophile. A variation of the O–O bond formation scenario depicted in Fig. 9a, b would involve attack on O5 not by an Mn or Ca-bound OH/H2O ligand but by a second-sphere water molecule positioned appropriately by hydrogen bonding interactions. We are not aware of any theoretical studies that explicitly evaluate and compare all these proposals in terms of their electronic, thermodynamic, and kinetic feasibility using realistic OEC models. Of these possibilities, an Mn-bound attacking group (Fig. 9b) would appear to be more consistent with the exchange kinetics described above.
The models in Fig. 9c, d involve radical mechanisms: in the first case the O–O bond is formed between a terminal Mn4(IV)–O• radical and the O5 bridge that is supposed to acquire radical character in the S4 state [194]. The last scheme (Fig. 9d) corresponds to a model elaborated by Siegbahn in a long process of mechanistic refinement aimed at yielding the lowest possible energy barriers. After examining several possible models and O–O bond formation pathways, the most favorable scenario was identified as a direct coupling of an Mn1(IV)–O• radical [195] with the μ-oxo bridge (O5) bound to Mn4 (Fig. 9d) [72, 73, 196]. This structure is entirely consistent with the S3 structure shown to be a best match with experimental observations (see Fig. 5) by independent calculations of spectroscopic properties [70, 78]. In this scenario, the oxyl radical on Mn1 originates from water that binds as OH− upon formation of the S3 state. An important requirement for the O–O bond formation to proceed with a low barrier is an alternate-spin alignment of the two oxygen atoms and the two terminal Mn ions, as shown in Fig. 9d.
The above coupling model represents a convincing and self-consistent hypothesis. However, several aspects of the mechanism of water oxidation at the later stages of the catalytic cycle still require more extensive studies to be sufficiently understood and, most important, to receive solid experimental support.
5 Summary
Water oxidation in biological photosynthesis relies on a complex arrangement of specialized components and finely orchestrated processes. Photosystem II is the “engine of life” that couples sunlight collection with charge separation and water oxidation. The catalytic oxygen evolving complex is composed of four oxo-bridged high-valent Mn ions. Although the cluster resembles a piece of Mn oxide “rock,” it features structural flexibility which might be important for mechanistic regulation in the form of substrate inclusion or electronic control. The catalytic cluster self-assembles from Mn(II) ions by a photo-driven process, and the PS-II enzyme itself has a number of mechanisms both to avoid damage as long as possible and to repair its sensitive components when damage does occur. The Mn ions have localized unpaired electrons and local high-spin configurations in all catalytic states that have been probed experimentally, and the magnetic coupling between the Mn ions leads to a characteristic evolution of ground spin states from low-spin early in the cycle to high-spin at the later stages. The protein matrix plays an important functional role by optimizing accessibility of reactants and removal of products, but also by maintaining an efficient proton-removal network crucial for achieving an almost constant redox potential for four consecutive oxidations of the cluster. Integral (calcium) and peripheral (chloride) cofactors optimize the electronic properties and control the reactivity of the cluster. An all-Mn(IV) state is reached before the final catalytic step, implying formation of an Mn(V) oxo or an Mn(IV) oxyl species in the final step of the catalytic cycle leading to evolution of dioxygen. According to quantum chemical calculations, O–O bond formation most likely proceeds by a radical process. However, experimental proof of the final stages of biological water oxidation is still hard to obtain. Structural aspects of the biological water oxidizing complex, such as the cuboidal Mn3Ca motif, are already partly realized in heterogeneous Mn/Ca oxides which show water oxidizing behavior [197], but without displaying catalytic performance comparable to that of the biological system. It remains to be seen whether features of the natural catalyst such as the structural flexibility, the switching from low-spin to activated high-spin states, and the exquisite control of proton transfer can be, or indeed need to be, implemented in synthetic manganese-based systems for efficient water oxidation.
References
Blankenship RE (2001) Molecular mechanisms of photosynthesis. Blackwell, Oxford
Hillier W, Messinger J (2005) Mechanism of photosynthetic oxygen production. In: Wydrzynski T, Satoh K (eds) Photosystem II. The light-driven water:plastoquinone oxidoreductase, vol 22, Advances in photosynthesis and respiration. Springer, Dordrecht, pp 567–608
McEvoy JP, Brudvig GW (2006) Chem Rev 106:4455–4483
Messinger J, Renger G (2008) Photosynthetic water splitting. In: Renger G (ed) Primary processes of photosynthesis, part 2: principles and apparatus, vol 9. The Royal Society of Chemistry, Cambridge, pp 291–349
Cox N, Pantazis DA, Neese F, Lubitz W (2013) Acc Chem Res 46:1588–1596
Pantazis DA, Cox N, Lubitz W, Neese F (2014) Oxygen-evolving photosystem II. In: Scott RA (ed) Encyclopedia of inorganic and bioinorganic chemistry. Wiley. doi:10.1002/9781119951438.eibc2166
Vinyard DJ, Ananyev GM, Dismukes GC (2013) Annu Rev Biochem 82:577–606
Lubitz W, Reijerse EJ, Messinger J (2008) Energy Environ Sci 1:15–31
Faunce TA, Lubitz W, Rutherford AW, MacFarlane D, Moore GF, Yang P, Nocera DG, Moore TA, Gregory DH, Fukuzumi S, Yoon KB, Armstrong FA, Wasielewski MR, Styring S (2013) Energy Environ Sci 6:695–698
Lewis NS, Nocera DG (2006) Proc Natl Acad Sci U S A 103:15729–15735
Nocera DG (2012) Acc Chem Res 45:767–776
Schlögl R (2010) ChemSusChem 3:209–222
Cogdell RJ, Gardiner AT, Cronin L (2012) Philos Trans R Soc A 370:3819–3826
Dau H, Zaharieva I (2009) Acc Chem Res 42:1861–1870
Dau H, Limberg C, Reier T, Risch M, Roggan S, Strasser P (2010) ChemCatChem 2:724–761
Zouni A, Witt HT, Kern J, Fromme P, Krauss N, Saenger W, Orth P (2001) Nature 409:739–743
Kamiya N, Shen J-R (2003) Proc Natl Acad Sci U S A 100:98–103
Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S (2004) Science 303:1831–1838
Biesiadka J, Loll B, Kern J, Irrgang K-D, Zouni A (2004) Phys Chem Chem Phys 6:4733–4736
Loll B, Kern J, Saenger W, Zouni A, Biesiadka J (2005) Nature 438:1040–1044
Guskov A, Kern J, Gabdulkhakov A, Broser M, Zouni A, Saenger W (2009) Nat Struct Mol Biol 16:334–342
Umena Y, Kawakami K, Shen J-R, Kamiya N (2011) Nature 473:55–60
Suga M, Akita F, Hirata K, Ueno G, Murakami H, Nakajima Y, Shimizu T, Yamashita K, Yamamoto M, Ago H, Shen J-R (2014) Nature 517:99–103
Becker K, Cormann KU, Nowaczyk MM (2011) J Photochem Photobiol B 104:204–211
Shi L-X, Hall M, Funk C, Schröder WP (2012) Biochim Biophys Acta Bioenerg 1817:13–25
Fagerlund RD, Eaton-Rye JJ (2011) J Photochem Photobiol B 104:191–203
Wydrzynski T, Satoh K (eds) (2005) Photosystem II. The light-driven water:plastoquinone oxidoreductase, vol 22. Springer, Dordrecht
Rappaport F, Diner BA (2008) Coord Chem Rev 252:259–272
Diner BA, Rappaport F (2002) Annu Rev Plant Biol 53:551–580
Murray JW, Barber J (2007) J Struct Biol 159:228–237
Ho FM, Styring S (2008) Biochim Biophys Acta Bioenerg 1777:140–153
Ho FM (2008) Photosynth Res 98:503–522
Gabdulkhakov A, Guskov A, Broser M, Kern J, Müh F, Saenger W, Zouni A (2009) Structure 17:1223–1234
Ishikita H, Saenger W, Loll B, Biesiadka J, Knapp E-W (2006) Biochemistry 45:2063–2071
Vassiliev S, Comte P, Mahboob A, Bruce D (2010) Biochemistry 49:1873–1881
Vassiliev S, Zaraiskaya T, Bruce D (2012) Biochim Biophys Acta Bioenerg 1817:1671–1678
Rivalta I, Amin M, Luber S, Vassiliev S, Pokhrel R, Umena Y, Kawakami K, Shen JR, Kamiya N, Bruce D, Brudvig GW, Gunner MR, Batista VS (2011) Biochemistry 50:6312–6315
Najafpour MM, Fekete M, Sedigh DJ, Aro E-M, Carpentier R, Eaton-Rye JJ, Nishihara H, Shen J-R, Allakhverdiev SI, Spiccia L (2015) ACS Catal 1499–1512
Cardona T, Sedoud A, Cox N, Rutherford AW (2012) Biochim Biophys Acta Bioenerg 1817:26–43
Niyogi KK (2000) Curr Opin Plant Biol 3:455–460
Pascal AA, Liu Z, Broess K, van Oort B, van Amerongen H, Wang C, Horton P, Robert B, Chang W, Ruban A (2005) Nature 436:134–137
Derks A, Schaven K, Bruce D (2015) Biochim Biophys Acta Bioenerg 1847:468–485
Joliot P, Barbieri G, Chabaud R (1969) Photochem Photobiol 10:309–329
Kok B, Forbush B, McGloin M (1970) Photochem Photobiol 11:457–475
Dau H, Haumann M (2008) Coord Chem Rev 252:273–295
Klauss A, Haumann M, Dau H (2012) Proc Natl Acad Sci U S A 109:16035–16040
Dau H, Haumann M (2007) Biochim Biophys Acta Bioenerg 1767:472–483
Klauss A, Haumann M, Dau H (2015) J Phys Chem B 119:2677–2689
Lavergne J, Junge W (1993) Photosynth Res 38:279–296
Rappaport F, Lavergne J (2001) Biochim Biophys Acta Bioenerg 1503:246–259
Klauss A, Krivanek R, Dau H, Haumann M (2009) Photosynth Res 102:499–509
Haumann M, Liebisch P, Müller C, Barra M, Grabolle M, Dau H (2005) Science 310:1019–1021
Gerencsér L, Dau H (2010) Biochemistry 49:10098–10106
Rappaport F, Ishida N, Sugiura M, Boussac A (2011) Energy Environ Sci 4:2520–2524
Bao H, Dilbeck P, Burnap R (2013) Photosynth Res 116:215–229
Service RJ, Hillier W, Debus RJ (2014) Biochemistry 53:1001–1017
Bondar A-N, Dau H (2012) Biochim Biophys Acta Bioenerg 1817:1177–1190
Vogt L, Vinyard DJ, Khan S, Brudvig GW (2015) Curr Opin Chem Biol 25:152–158
Kirby JA, Robertson AS, Smith JP, Thompson AC, Cooper SR, Klein MP (1981) J Am Chem Soc 103:5529–5537
Yano J, Pushkar Y, Glatzel P, Lewis A, Sauer K, Messinger J, Bergmann U, Yachandra V (2005) J Am Chem Soc 127:14974–14975
Grundmeier A, Dau H (2012) Biochim Biophys Acta Bioenerg 1817:88–105
Glöckner C, Kern J, Broser M, Zouni A, Yachandra V, Yano J (2013) J Biol Chem 288:22607–22620
Yano J, Yachandra V (2014) Chem Rev 114:4175–4205
Yano J, Kern J, Sauer K, Latimer MJ, Pushkar Y, Biesiadka J, Loll B, Saenger W, Messinger J, Zouni A, Yachandra VK (2006) Science 314:821–825
Dau H, Liebisch P, Haumann M (2004) Phys Chem Chem Phys 6:4781–4792
Grabolle M, Haumann M, Müller C, Liebisch P, Dau H (2006) J Biol Chem 281:4580–4588
Yano J, Kern J, Irrgang K-D, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, Messinger J, Zouni A, Yachandra VK (2005) Proc Natl Acad Sci U S A 102:12047–12052
Luber S, Rivalta I, Umena Y, Kawakami K, Shen JR, Kamiya N, Brudvig GW, Batista VS (2011) Biochemistry 50:6308–6311
Galstyan A, Robertazzi A, Knapp EW (2012) J Am Chem Soc 134:7442–7449
Krewald V, Retegan M, Cox N, Messinger J, Lubitz W, DeBeer S, Neese F, Pantazis DA (2015) Chem Sci 6:1676–1695
Askerka M, Vinyard DJ, Wang J, Brudvig GW, Batista VS (2015) Biochemistry 54:1713–1716
Siegbahn PEM (2008) Chem Eur J 14:8290–8302
Siegbahn PEM (2009) Acc Chem Res 42:1871–1880
Zimmermann JL, Rutherford AW (1984) Biochim Biophys Acta Bioenerg 767:160–167
Brudvig GW, Casey JL, Sauer K (1983) Biochim Biophys Acta Bioenerg 723:366–371
Baldwin MJ, Stemmler TL, Riggs-Gelasco PJ, Kirk ML, Penner-Hahn JE, Pecoraro VL (1994) J Am Chem Soc 116:11349–11356
Krewald V, Lassalle-Kaiser B, Boron TT, Pollock CJ, Kern J, Beckwith MA, Yachandra VK, Pecoraro VL, Yano J, Neese F, DeBeer S (2013) Inorg Chem 52:12904–12914
Cox N, Retegan M, Neese F, Pantazis DA, Boussac A, Lubitz W (2014) Science 345:804–808
Dismukes GC, Siderer Y (1981) Proc Natl Acad Sci U S A 78:274–278
Penner-Hahn JE, Fronko RM, Pecoraro VL, Yocum CF, Betts SD, Bowlby NR (1990) J Am Chem Soc 112:2549–2557
Zheng M, Dismukes GC (1996) Inorg Chem 35:3307–3319
Roelofs TA, Liang W, Latimer MJ, Cinco RM, Rompel A, Andrews JC, Sauer K, Yachandra VK, Klein MP (1996) Proc Natl Acad Sci U S A 93:3335–3340
Iuzzolino L, Dittmer J, Dörner W, Meyer-Klaucke W, Dau H (1998) Biochemistry 37:17112–17119
Dau H, Liebisch P, Haumann M (2003) Anal Bioanal Chem 376:562–583
Ono T-A, Noguchi T, Inoue Y, Kusunoki M, Matsushita T, Oyanagi H (1992) Science 258:1335–1337
Hansson O, Andreasson LE (1982) Biochim Biophys Acta 679:261–268
De Paula JC, Brudvig GW (1985) J Am Chem Soc 107:2643–2648
De Paula JC, Beck WF, Brudvig GW (1986) J Am Chem Soc 108:4002–4009
de Paula JC, Beck WF, Miller A-F, Wilson RB, Brudvig GW (1987) J Chem Soc Faraday Trans 83:3635–3651
Pace RJ, Jin L, Stranger R (2012) Dalton Trans 41:11145–11160
Dasgupta J, Ananyev GM, Dismukes GC (2008) Coord Chem Rev 252:347–360
Kolling DRJ, Cox N, Ananyev GM, Pace RJ, Dismukes GC (2012) Biophys J 103:313–322
Kuntzleman T, Yocum CF (2005) Biochemistry 44:2129–2142
Messinger J, Seaton G, Wydrzynski T, Wacker U, Renger G (1997) Biochemistry 36:6862–6873
Messinger J, Badger M, Wydrzynski T (1995) Proc Natl Acad Sci U S A 92:3209–3213
Beckmann K, Messinger J, Badger M, Wydrzynski T, Hillier W (2009) Photosynth Res 102:511–522
Cox N, Messinger J (2013) Biochim Biophys Acta Bioenerg 1827:1020–1030
Hillier W, Wydrzynski T (2001) Biochim Biophys Acta Bioenerg 1503:197–209
Messinger J (2004) Phys Chem Chem Phys 6:4764–4771
Hillier W, Wydrzynski T (2008) Coord Chem Rev 252:306–317
Haddy A (2007) Photosynth Res 92:357–368
Åhrling KA, Peterson S, Styring S (1997) Biochemistry 36:13148–13152
Messinger J, Nugent JHA, Evans MCW (1997) Biochemistry 36:11055–11060
Messinger J, Robblee JH, Yu WO, Sauer K, Yachandra VK, Klein MP (1997) J Am Chem Soc 119:11349–11350
Åhrling KA, Peterson S, Styring S (1998) Biochemistry 37:8115–8120
Koulougliotis D, Hirsh DJ, Brudvig GW (1992) J Am Chem Soc 114:8322–8323
Hsieh W-Y, Campbell KA, Gregor W, Britt RD, Yoder DW, Penner-Hahn JE, Pecoraro VL (2004) Biochim Biophys Acta Bioenerg 1655:149–157
Zimmermann JL, Rutherford AW (1986) Biochemistry 25:4609–4615
Haddy A, Lakshmi KV, Brudvig GW, Frank HA (2004) Biophys J 87:2885–2896
Horner O, Rivière E, Blondin G, Un S, Rutherford AW, Girerd J-J, Boussac A (1998) J Am Chem Soc 120:7924–7928
Ioannidis N, Petrouleas V (2000) Biochemistry 39:5246–5254
Boussac A, Sugiura M, Rutherford AW, Dorlet P (2009) J Am Chem Soc 131:5050–5051
Kulik LV, Epel B, Lubitz W, Messinger J (2005) J Am Chem Soc 127:2392–2393
Kulik LV, Epel B, Lubitz W, Messinger J (2007) J Am Chem Soc 129:13421–13435
Sauer K, Yano J, Yachandra VK (2005) Photosynth Res 85:73–86
Sauer K, Yano J, Yachandra VK (2008) Coord Chem Rev 252:318–335
Yachandra VK, DeRose VJ, Latimer MJ, Mukerji I, Sauer K, Klein MP (1993) Science 260:675–679
Gatt P, Stranger R, Pace RJ (2011) J Photochem Photobiol B Biol 104:80–93
Jaszewski AR, Petrie S, Pace RJ, Stranger R (2011) Chem Eur J 17:5699–5713
Jaszewski AR, Stranger R, Pace RJ (2011) J Phys Chem B 115:4484–4499
Petrie S, Stranger R, Pace RL (2008) Chem Eur J 14:5482–5494
Gatt P, Petrie S, Stranger R, Pace RJ (2012) Angew Chem Int Ed 51:12025–12028
Petrie S, Gatt P, Stranger R, Pace RJ (2012) Phys Chem Chem Phys 14:11333–11343
Siegbahn PEM (2013) Biochim Biophys Acta Bioenerg 1827:1003–1019
Kusunoki M (2007) Biochim Biophys Acta Bioenerg 1767:484–492
Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS (2008) J Am Chem Soc 130:6728–6730
Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS (2008) J Am Chem Soc 130:3428–3442
Kusunoki M (2011) Photochem Photobiol B 104:100–110
Saito T, Yamanaka S, Kanda K, Isobe H, Takano Y, Shigeta Y, Umena Y, Kawakami K, Shen JR, Kamiya N, Okumura M, Shoji M, Yoshioka Y, Yamaguchi K (2012) Int J Quantum Chem 112:253–276
Pantazis DA, Orio M, Petrenko T, Zein S, Lubitz W, Messinger J, Neese F (2009) Phys Chem Chem Phys 11:6788–6798
Yamaguchi K, Isobe H, Yamanaka S, Saito T, Kanda K, Shoji M, Umena Y, Kawakami K, Shen JR, Kamiya N, Okumura M (2012) Int J Quantum Chem 113:525–541
Bovi D, Narzi D, Guidoni L (2013) Angew Chem Int Ed 52:11744–11749
Schinzel S, Schraut J, Arbuznikov AV, Siegbahn PEM, Kaupp M (2010) Chem Eur J 16:10424–10438
Ames W, Pantazis DA, Krewald V, Cox N, Messinger J, Lubitz W, Neese F (2011) J Am Chem Soc 133:19743–19757
Cox N, Rapatskiy L, Su J-H, Pantazis DA, Sugiura M, Kulik L, Dorlet P, Rutherford AW, Neese F, Boussac A, Lubitz W, Messinger J (2011) J Am Chem Soc 133:3635–3648
Su J-H, Cox N, Ames W, Pantazis DA, Rapatskiy L, Lohmiller T, Kulik LV, Dorlet P, Rutherford AW, Neese F, Boussac A, Lubitz W, Messinger J (2011) Biochim Biophys Acta Bioenerg 1807:829–840
Pantazis DA, Ames W, Cox N, Lubitz W, Neese F (2012) Angew Chem Int Ed 51:9935–9940
Retegan M, Neese F, Pantazis DA (2013) J Chem Theory Comput 9:3832–3842
Lohmiller T, Krewald V, Pérez Navarro M, Retegan M, Rapatskiy L, Nowaczyk MM, Boussac A, Neese F, Lubitz W, Pantazis DA, Cox N (2014) Phys Chem Chem Phys 16:11877–11892
Retegan M, Cox N, Lubitz W, Neese F, Pantazis DA (2014) Phys Chem Chem Phys 16:11901–11910
Zein S, Kulik LV, Yano J, Kern J, Pushkar Y, Zouni A, Yachandra VK, Lubitz W, Neese F, Messinger J (2008) Philos Trans R Soc B 363:1167–1177
Orio M, Pantazis DA, Neese F (2009) Photosynth Res 102:443–453
Pantazis DA, Orio M, Petrenko T, Zein S, Bill E, Lubitz W, Messinger J, Neese F (2009) Chem Eur J 15:5108–5123
Neese F, Ames W, Christian G, Kampa M, Liakos DG, Pantazis DA, Roemelt M, Surawatanawong P, Ye SF (2010) Adv Inorg Chem 62:301–349
Neese F (2009) Coord Chem Rev 253:526–563
DeBeer George S, Petrenko T, Neese F (2008) J Phys Chem A 112:12936–12943
Kusunoki M, Ono T, Noguchi T, Inoue Y, Oyanagi H (1993) Photosynth Res 38:331–339
Messinger J, Robblee JH, Bergmann U, Fernandez C, Glatzel P, Visser H, Cinco RM, McFarlane KL, Bellacchio E, Pizarro SA, Cramer SP, Sauer K, Klein MP, Yachandra VK (2001) J Am Chem Soc 123:7804–7820
Haumann M, Müller C, Liebisch P, Iuzzolino L, Dittmer J, Grabolle M, Neisius T, Meyer-Klaucke W, Dau H (2005) Biochemistry 44:1894–1908
Peloquin JM, Campbell KA, Randall DW, Evanchik MA, Pecoraro VL, Armstrong WH, Britt RD (2000) J Am Chem Soc 122:10926–10942
Peloquin JM, Britt RD (2001) Biochim Biophys Acta Bioenerg 1503:96–111
Kulik L, Lubitz W (2009) Photosynth Res 102:391–401
Hasegawa K, Ono T, Inoue Y, Kusunoki M (1999) Bull Chem Soc Jpn 72:1013–1023
Hasegawa K, Ono T-A, Inoue Y, Kusunoki M (1999) Chem Phys Lett 300:9–19
Barber J, Murray JW (2008) Philos Trans R Soc B 363:1129–1137
Krewald V, Neese F, Pantazis DA (2013) J Am Chem Soc 135:5726–5739
Isobe H, Shoji M, Yamanaka S, Umena Y, Kawakami K, Kamiya N, Shen JR, Yamaguchi K (2012) Dalton Trans 41:13727–13740
Narzi D, Bovi D, Guidoni L (2014) Proc Natl Acad Sci U S A 111:8723–8728
Ghanotakis DF, Babcock GT, Yocum CF (1984) FEBS Lett 167:127–130
Boussac A, Rutherford AW (1988) Biochemistry 27:3476–3483
Ishida N, Sugiura M, Rappaport F, Lai T-L, Rutherford AW, Boussac A (2008) J Biol Chem 283:13330–13340
Boussac A, Rappaport F, Carrier P, Verbavatz J-M, Gobin R, Kirilovsky D, Rutherford AW, Sugiura M (2004) J Biol Chem 279:22809–22819
Brudvig GW (2008) Philos Trans R Soc B 363:1211–1219
Lee C-I, Lakshmi KV, Brudvig GW (2007) Biochemistry 46:3211–3223
Vrettos JS, Limburg J, Brudvig GW (2001) Biochim Biophys Acta Bioenerg 1503:229–245
McEvoy JP, Brudvig GW (2004) Phys Chem Chem Phys 6:4754–4763
Vrettos JS, Stone DA, Brudvig GW (2001) Biochemistry 40:7937–7945
Tsui EY, Agapie T (2013) Proc Natl Acad Sci U S A 110:10084–10088
Tsui EY, Kanady JS, Agapie T (2013) Inorg Chem 52:13833–13848
Siegbahn PEM (2014) Phys Chem Chem Phys 16:11893–11900
Polander BC, Barry BA (2013) J Phys Chem Lett 786–791
Kawakami K, Umena Y, Kamiya N, Shen J-R (2009) Proc Natl Acad Sci U S A 106:8567–8572
Siegbahn PEM (2009) Dalton Trans 10063–10068
Olesen K, Andréasson L-E (2003) Biochemistry 42:2025–2035
Amin M, Vogt L, Szejgis W, Vassiliev S, Brudvig GW, Bruce D, Gunner MR (2015) J Phys Chem B 119:7366–7377
Pokhrel R, McConnell IL, Brudvig GW (2011) Biochemistry 50:2725–2734
Pokhrel R, Service RJ, Debus RJ, Brudvig GW (2013) Biochemistry 52:4758–4773
van Vliet P, Rutherford AW (1996) Biochemistry 35:1829–1839
Hillier W, Wydrzynski T (2004) Phys Chem Chem Phys 6:4882–4889
Hendry G, Wydrzynski T (2003) Biochemistry 42:6209–6217
Noguchi T (2008) Philos Trans R Soc B 363:1189–1195
Noguchi T, Sugiura M (2002) Biochemistry 41:15706–15712
Chu H-A, Sackett H, Babcock GT (2000) Biochemistry 39:14371–14376
Rapatskiy L, Cox N, Savitsky A, Ames WM, Sander J, Nowaczyk MM, Rögner M, Boussac A, Neese F, Messinger J, Lubitz W (2012) J Am Chem Soc 134:16619–16634
McConnell IL, Grigoryants VM, Scholes CP, Myers WK, Chen P-Y, Whittaker JW, Brudvig GW (2012) J Am Chem Soc 134:1504–1512
Pokhrel R, Brudvig G (2014) Phys Chem Chem Phys 16:11812–11821
Pérez Navarro M, Ames WM, Nilsson H, Lohmiller T, Pantazis DA, Rapatskiy L, Nowaczyk MM, Neese F, Boussac A, Messinger J, Lubitz W, Cox N (2013) Proc Natl Acad Sci U S A 110:15561–15566
Schraut J, Kaupp M (2014) Chem Eur J 20:7300–7308
Tagore R, Chen H, Crabtree RH, Brudvig GW (2006) J Am Chem Soc 128:9457–9465
Siegbahn PEM (2013) J Am Chem Soc 135:9442–9449
Yamaguchi K, Takahara Y, Fueno T (1986) Ab-initio molecular orbital studies of structure and reactivity of transition metal-oxo compounds. In: Smith VH Jr, Scheafer HF III, Morokuma K (eds) Applied quantum chemistry. Reidel, Boston, pp 155–184
Pecoraro VL, Baldwin MJ, Caudle MT, Hsieh W-Y, Law NA (1998) Pure Appl Chem 70:925–929
Barber J, Ferreira K, Maghlaoui K, Iwata S (2004) Phys Chem Chem Phys 6:4737–4742
Yamaguchi K, Yamanaka S, Isobe H, Tanaka K, Ueyama N (2012) Int J Quantum Chem 112:3849–3866
Siegbahn PEM, Crabtree RH (1999) J Am Chem Soc 121:117–127
Siegbahn PEM (2006) Chem Eur J 12:9217–9227
Shevela D, Koroidov S, Najafpour MM, Messinger J, Kurz P (2011) Chem Eur J 17:5415–5423
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Krewald, V., Retegan, M., Pantazis, D.A. (2015). Principles of Natural Photosynthesis. In: Tüysüz, H., Chan, C. (eds) Solar Energy for Fuels. Topics in Current Chemistry, vol 371. Springer, Cham. https://doi.org/10.1007/128_2015_645
Download citation
DOI: https://doi.org/10.1007/128_2015_645
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23098-6
Online ISBN: 978-3-319-23099-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)