
Abstract
It is well known that the density is the basic character of a substance. Density measurement is an important characterization method for both static and dynamic analysis and for inferences of chemical composition in specific experiments. For example, the density of polymers depends not only on their chemical composition, but also on how the chains are arranged in space (i.e. crystalline or amorphous arrangements). Therefore, the minute difference in density can be used to analyze polymers with similar atomic compositions. For certain classes of polymers (formed by polymerization of small monomers, such as acrylic esters or other vinyl monomers), the kinetics of the polymerizations can be monitored through the measurement of the change in density associated with polymerization.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


8.1 Introduction
It is well known that the density is the basic character of a substance. Density measurement is an important characterization method for both static and dynamic analysis and for inferences of chemical composition in specific experiments. For example, the density of polymers depends not only on their chemical composition, but also on how the chains are arranged in space (i.e. crystalline or amorphous arrangements). Therefore, the minute difference in density can be used to analyze polymers with similar atomic compositions. For certain classes of polymers (formed by polymerization of small monomers, such as acrylic esters or other vinyl monomers), the kinetics of the polymerizations can be monitored through the measurement of the change in density associated with polymerization. In fact, many types of chemical/biochemical reactions, such as cycloaddition reactions (dimerization) of cyclopentadiene [1], chemical reactions (or binding) on a solid support (e.g., covalent attachments of low-molecular-weight organic molecules to 4-benzyloxybenzaldehyde polystyrene beads [2], and binding of carbonic anhydrase to porous hydrogel particles presenting aryl sulfonamides—a class of inhibitors specific to carbonic anhydrase [3]), and biochemical reactions between antigens and antibodies, can be monitored by analyze the dynamic change in density over time. The density of minerals can also be used to assess their purity, and the difference in density provides the basis for mineral separations [4]. Differently sized carbon nanotubes, coated with structure-discriminating surfactants, show different densities, and thus provide a basis for simple separation.
8.2 Identification of Substances
The type and composition of a matter reflect on its density. Density measurement can be used as a primary method for separating and identifying substances in specific occasions such as analyzing forensic evidence. Common contact trace objects are used in forensic investigations to establish an association (i.e., link criminals to crime scenes and to victims). The objects may be the hairs, fibers, paint chips, and fragments of broken glass that are left at the crime scene. In addition, glitter is another contact trace material that has been used as associative evidence [5]. It is worth noting that, usually, these objects are diamagnetic. The tests of these contact trace objects are sophisticated and expensive, from visual or microscopic inspection to spectroscopic analysis. As introduced above, the density of an object could in principle be used to characterize a trace object. For common density measurement methods, it requires precise determination of the volume of an irregularly shaped objects to measure the density. However, volume measurements are difficult for small objects such as glitter particles, and thus, there exists a need for a method that makes it possible to determine the density of trace objects quickly and easily. Kirk used density columns with the mixture of two organic liquids (e.g., bromoform and bromobenzene) in a specific ratio to analyze glass fragments (naming “sink/float method”) [6]. Questioned and known glass fragments are placed together in the mixture, and the ratio of the organic liquids is adjusted until the two glass fragments are separated and distinguished according to their differences in densities. Otherwise, if the glass fragments cannot be separated (sank or levitated together) in a large range of liquid ratio, it can be concluded that the fragments are originated from the same source. Predictably, this method also requires long testing process, for the adjustment of the mixture contains time consuming trials. Considering the advantages over the common methods, MagLev is applied to identify these small and irregular-shaped forensic evidences.
8.2.1 Method
The objects, such as smokeless gunpowder, glass pieces, fibers, hair, glitter, etc., commonly encountered in crime scenes are diamagnetic and are possible for MagLev. The container is chosen as glass or transparent polymer (such as PMMA) cuvettes for direct observation. Various paramagnetic solutes are available for preparing appropriate solutions (e.g., MnCl2, MnSO4, GdCl3, FeCl3, CuSO4, etc.). Chelated paramagnetic ions, such as Gd(DTPA) and Mn(EDTA), are other choices for aqueous and nonaqueous solvents. The samples immersed in the paramagnetic medium need pretreatment to remove the bubbles adhere to them. These bubbles will significantly influence the measuring result (the density of air is 0.00118 g/cm3 at 25 °C at standard pressure). The repetitive invert of the cuvette can remove bubbles that may have formed on the samples. Washing the samples with ethanol/methanol can also greatly reduce the generation of the bubbles. However, it is not a suitable way for powders and small particles, for these samples may bring too much ethanol/methanol to the medium, which will change the concentration of the medium and finally cause large inaccuracy in testing. Adding a small amount of detergent (e.g., Tween 20, 0.1% (vol./vol.)) to the MnCl2 solution can lower the surface tension of the medium and reduce the interactions between hydrophobic objects, as well as their interactions with the walls of the cuvette. Combined with sonication treatment, the bubbles that form on the walls of the cuvette and on the sample itself can be removed. It has been proved that this treatment can result in the most precise measurements. For the forensic samples are in very small scales, the levitation require a much longer time (several minutes) to reach the equilibrium position.
8.2.2 Identification of Forensic Evidences
The identification of glitters with same appearance shows the advantages of MagLev method [7]. The properties of a series of glitters are shown in Table 8.1. According to their nominal densities, the medium is chosen as 3.0 M MnCl2 aqueous solution. Figure 8.1a exhibits an example of levitating different amount of Crystalina #321 after 6 min. Two standard density beads (1.350 and 1.450 g/cm3) are levitated together for comparison. The densities measured through MagLev and provided by the manufacturer are listed in Fig. 8.1b. The results are obtained by the levitation of 20 pieces of each glitter. Their standard deviation of each result is ± 0.001 g/cm3 based on n = 7 independent measurements.

Reproduced with permission from Ref. [7]. Copyright 2012 Wiley
Identification of glitters using MagLev. a Levitation results of different amount of Crystalina #321 (upper cluster) after 6 min. Two standards (lower spheres) are levitated together of comparison. b Identification results of different glitters.
According to the comparison between the measuring results and nominal densities provided by the manufacturer, the measured densities for the 11 glitters can be categorized into three groups. (i) High agreement: average density difference below 0.05 g/cm3, (ii) Moderate agreement: average density difference between 0.05 and 0.10 g/cm3. (iii) Low agreement: average density difference above 0.10 g/cm3. Most of the samples (6 in 11) are in high agreement level. Three samples are in moderate agreement. Noticing that there are two samples (Alpha Jewels I and II) differed by more than 1.0 g/cm3 from the nominal densities. These samples may be fake or from other unknown sources.
The determination of the density of the glitters have potential to identify the samples extract from a mixture like commercial nail polish. A Sally Hansen diamond strength nail polish sample with Alpha Jewels I glitter (1.394 ± 0.002 g/cm3) inside was applied to demonstrate the idea. 0.5 g of the sample is in 5 mL acetone, and the glitters are then collected by straining the solution via a piece of quantitative filter. The glitters then measured by the MagLev method. Comparing the Alpha Jewels I glitter extract from the nail polish and previous levitation results, it can be found that the levitation height of the extracted glitters is within the standard deviation of the measurement. This means MagLev method is accurate in obtaining density of unknown target objects from complex cosmetic matrices.
Except for the particles, powders are another type of evidence that is always found in crime scenes. For example, smokeless gunpowder is of great vital for shooting scene investigation. The MagLev method is applied to determine the density of six smokeless gunpowder samples. For the powders have a relatively large density range, two media, 3.0 M and 4.0 M MnCl2 aqueous solutions, are used in the testing. The testing result of the powders are shown in Table 8.2. Normally, gunpowder is susceptible to dampness and will not be effective for ignition. The dampness may also affect the density measurement via MagLev. However, the experiments shows different conclusion. The repeated exposure and prolonged exposure to an aqueous MnCl2 solution are considered in experiments. The Hercules Blue Dot samples immersed in a 4.0 M MnCl2 solution for 7 days, and the density was measured every 24 h. Considering the effect on the concentration of the medium caused by the evaporation of the water, standard density beads are introduced to the levitation to calibrate the results. The result shows a maximum change of 0.012 g/cm3 on the change in the average density of the gunpowder. It is within the standard deviation of the initial measurements, which means the dampness will not affect the density of the gunpowder that it can be accurately measure via MagLev. The same conclusion can also be drawn by the experiments on Hercules Blue Dot gunpowder after repeated exposures to MnCl2 solution. The samples are tested for 10 consecutive measurements. The gunpowder is levitated in MagLev device, then removed, rinsed by water, and dried in nitrogen atmosphere for next test. The change of the results is also within the standard deviation. In addition, the MnCl2 solution has a pH of ~3.0. The results also proved that the density of the gunpowder will not affected by little change of the environment.
8.2.3 Identification of Drugs
Drug abuse is a major public health problem. The overdoses cause over 100,000 death around the world. In order to identify a drug according to the forensic standards of analysis, multiple analytical methods have to be used to obtain results that correspond to each other. Common analytical methods are listed in Table 8.3.
The techniques that are able to parse molecular configurations has the highest ranking for forensic analysis, such as X-ray diffractometry and IR, NMR, Raman spectrum, and mass spectrometry [8]. However, their cost and inconvenience in use make these methods available only under laboratory conditions. The less-molecularly-specific separation methods are considered to be the med-level for identification. The methods that cannot or limitedly provide molecular structure information are in the lowest category. In some cases, although the lowest ranking method (e.g. immunoassays [9, 10] and colorimetric tests [11]) can only provide weak evidence of molecular information, but have advantage that they can be easily used in resource-limited circumstances. Therefore, MagLev also have the potential to be used as a presumptive identification method that based on its ability to sensitively test the compound density [12].
Mixtures of illicit drugs can contain a wide range of compounds. These compounds, including hydrochloride salts of fentanyl, acetyl fentanyl, cocaine, heroin, and methamphetamine, can be separated and measured by MagLev method. Noting that most of these compounds are water soluble, the Gd(DPM)3TOPO (450 mM) dissolved in a mixture of 23 vol.% hexane and 77 vol.% tetrachloroethylene is use as the medium. Typical separation results are shown in Fig. 8.2. In fact, some drugs like synthetic opioids requires high dilution (~5 wt%) of the active components, and thus, makes the analysis of these opioids from small amount samples a big challenge. For example, the fentanyl may be present only as a few crystals in a 50 mg mixture.

Reproduced with permission from Ref. [12]. Copyright 2019 Wiley
Separation process of mixtures of powdered illicit drugs, adulterants, and dilutants (2.5–9.5 mg of each compound) using MagLev. Gd(DPM)3TOPO (450 mM) dissolved in a mixture of 23 vol.% hexane and 77 vol.% tetrachloroethylene is used as paramagnetic medium.
A series of separation experiments of lidocaine HCl and caffeine mixture are carried out to demonstrate the advanced ability of MagLev to screen out minute amounts of target matter. The lidocaine HCl and caffeine mixed in different proportions are significantly separated in MagLev device, as shown in Fig. 8.3. It is worth noticing that very small proportion of fentanyl HCl in the mixture (e.g. 1 wt%) can not be detected by FTIR-ATR, but can be clearly identified by FTIR-ATR after separation (Fig. 8.3b). Ulteriorly, the limit of concentration of separable mixture can be even less than 1 wt%. The MagLev method can screen out as few as five 100–200 μm crystals, from 50 mg mixture that contains hundreds to thousands of particles of other compounds. The image analysis (Fig. 8.3d) also verifies that the amount of separated compounds well agrees quantitatively with the known compositions of the samples.

Reproduced with permission from Ref. [12]. Copyright 2019 Wiley
The investigation of MagLev separation of powdered mixtures and the following characterization with spectroscopic techniques. a MagLev separation (30 min) of a mixture of lidocaine HCl and caffeine (95:5 wt%; 50 mg) in a cuvette filled with the paramagnetic solution, and extraction using a Pasteur pipette. b MagLev separation (20 min) of powdered mixtures (50 mg) of lidocaine HCl (top clouds) and caffeine (bottom clouds) in different proportions (wt%). c Scanning electron micrographs of crystals of lidocaine HCl and caffeine (pure compounds). d The projected, two-dimensional areas of the levitating fractions of lidocaine HCl (green line) and caffeine (red line), and their combined area (black line), are plotted against the chemical composition of mixtures. e 1H NMR (600 MHz) characterization of a mixture (50 mg) of lidocaine HCl and caffeine (50:50 wt%) and the fractions after separation (30 min) in the MagLev. f FTIR-ATR characterization (normalized to highest peak) of the samples purified in e except that the residue was characterized as a dry powder.
The separation after MagLev can be further characterized by high ranking method such as NMR (Fig. 8.3e) and FTIR-ATR (Fig. 8.3f). The spectra of the extracted compounds well coincided with the spectra of pure compounds, which suggests excellent separation of crystals of these two compounds using MagLev. Although there should be minute amount of crystals remaining in the medium, the separation can be assumed as totally separated. The separation also enabled clear identification of target compound with FTIR-ATR, which cannot be identified in the mixture before separation (Fig. 8.3f).
8.3 Monitoring the Chemical Reaction
Currently, analytical techniques to monitor the chemical reaction can be classified into two categories: (i) techniques with high-end instruments, such as rheometry, calorimetry, gel-permeation chromatography, various forms of UV/vis spectroscopy, mass spectrometry, and NMR spectroscopy. These methods can provide detailed information (e.g. online monitoring of polymerization reactions [13]), but are expensive for use and maintain. (ii) Unsophisticated tools and methods such as balances, refractometers, colorimetry, and dilatometers [14, 15]. These tools are readily available in research labs and quality control stations.
The change of density can be used to characterize the reaction process. For example, solid-state support chemistry is widely used in the preparation of peptides, nucleic acids, small molecule libraries, affinity purification, and protein target identification of capture reagents [16]. However, a major disadvantage of solid phase chemistry is that there is no cheap and fast way to quantitatively monitor the reaction process on insoluble polymer carriers [17]. Colorimetric is a common method for rapid and qualitative test. Nevertheless, it is affected by artifacts from competing side effects (false positives) and incomplete reactions (false negatives) [18]. Spectroscopic instrument can provide much more information than colorimetric tests, while the method is time-consuming and require expensive instruments (>$10,000). A favorable addition to solid-supported chemistry, especially in the developmental phase of solid-supported synthesis, would be a quick and quantitative method to track the progress of reactions without the need for specialized or expensive equipment. In fact, what is needed is the equivalent of thin layer chromatography (TLC) in solid phase chemistry.
For free-radical polymerization, density variation, which is usually measured by volume dilatometry, is a key character for characterizing the kinetics of free-radical polymerization. The method measures the volumetric shrinkage of a sample of polymer and monomer (the polymerization makes the monomer moves from the van der Waals distance in the liquid monomer to the covalent and shorter distance). It is widely applicable to the shrink (or expand) in volume of the polymer system during polymerization (e.g. bulk, suspension, emulsion polymerization with vinyl monomers). The method could also be used to study photopolymerization after proper modifications. Similarly, volume dilatometry has its shortcomings: (i) It normally requires relatively large volumes of samples (1−10 mL). Thus, it is not applicable to monomers available in limited quantities (<100 μL). (ii) The dilatometer can only monitor the polymerization in the device. It cannot be used as a stand-alone device to monitor polymerization in other reaction vessels. (iii) It is a time-limited method for monitor the early stages of the polymerization. As long as the products, or the mixture, become thick, solidified, or stick to the boundary of the vessel along with the progress of the reaction, the error in reading the positions of the meniscus become larger.
8.3.1 Monitor Chemical Reactions on Solid Supports
It is evident that MagLev method can test minute density change through obvious change in levitation height, by which the chemical composition of polymeric beads can be detected [19]. Figure 8.4 shows a series of examples of derivatives of 4-benzyloxybenzaldehydepolystyrene (diameters from 35 to 75 μm, loading level = 3.5 mmol –CHO/g resin, ∼350 pmol –CHO/bead). The derivatives are prepared through reductive amination reactions.

Reductive amination reactions for preparing different series of 4-benzyloxybenzaldehyde polystyrene
Each reaction (dissolved in 5% CH3COOH-DMF) agitates the beads with 10 equivalent of amine and NaBH3(CN) for 24 h to ensure complete conversion. The beads are sequentially washed by DMF, CH2Cl2, CH3OH, and H2O to remove excess reagent from the polymer. The beads are stained by 2,4-dinitrophenylhydrazine to confirm that the aldehyde is completely consumed. Approximate 100 beads after each reaction are levitated by the MagLev method with the aid of 650 mM GdCl3. The dispersed beads in the solution become coalesced after 5 min levitation and finally get concentrated within 15 min. The height of the center point of the concentrated beads are used to calculate the average density of the beads.
Each bead contains ~350 pmol of small molecule. Benefit from the high sensitivity of the MagLev method, the difference in chemical composition of a single atom will lead to a distinguishable change in levitation height. For example, the b, g, and j subjects shown in Fig. 8.5b has the differences of only one fluorine atom, but reflect obvious derivations in levitation heights.

Reproduced with permission from Ref. [19]. Copyright 2008 American Chemical Society
a Levitation results of 10 different derivatives of the 4-benzyloxybenzaldehyde polystyrene beads in a 650 mM GdCl3 aqueous solution. b Chemical structures for each derivative in (A).
The MagLev method is then proved to be also sensitive to the change in chemical composition (through change of density) of a polymer during the course of a chemical reaction. It is demonstrated by monitoring the reaction of 2,5-diiodobenzoic acid to leucinederivatized Wang polystyrene (diameter) 75–150 μm, 1.8 mmol –NH2/g resin, ∼1 nmol –NH2/bead) at 0 °C using 5 equiv of O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) and 5 equiv of diisopropylethyl amine (DIEA) in MF, as shown in Fig. 8.6.

Reproduced with permission from Ref. [19]. Copyright 2008 American Chemical Society
Schematic of the process for monitoring conversion for a condensation reaction of a solid-supported amine with a carboxylic acid.
Aliquots of beads are withdrawn from the reaction process at different times and immediately washed to eliminate the adhered reagents. About 100 beads from each aliquot are levitated by MagLev device using medium DMF with 650 mM GdCl3 as the medium. The results are shown in Fig. 8.7. The levitation height of the beads decreases which means the average density of the beads increases as the reaction goes on. The beads form tight clusters at the beginning and end of the reaction, but their dispersity increases as the reaction approaches 50% completion. According to the theory of MagLev method, although the sizes of the beads vary from 75 to 150 μm, it does not affect levitation height of the beads. Therefore, the dispersion of the beads means the incomplete reaction of the beads. This reflects in the aggregation of the beads in the MagLev experiment.

Reproduced with permission from Ref. [19]. Copyright 2008 American Chemical Society
a Levitation of the polymer beads cluster (leucinederivatized Wang polystyrene, ∼100 beads/cluster) at different times throughout the course of the reaction. b The correlation of density of the polymer beads with their levitation height. c Pseudo-first-order kinetics plots for the rate of consumption of polymer-bound amine determined by 1H NMR (solid dot) and by levitation (hollow dot) from three independent measurements.
Since the amount of reagent used in the reaction is five times greater than the amount of polymer-bound –NH2, the reaction follows pseudo-first-order kinetics. The verification is carried out by 1H NMR. Figure 8.7c compares 3 independent results from 1H NMR and MagLev. It is obvious the results of MagLev method show high coincidence with 1H NMR results. Both MagLev method and NMR yield similar rates (T1/2 = 23 ± 4 min (NMR) and T1/2 = 18 ± 2 min (MagLev)) for the pseudo-first-order reaction.
8.3.2 Characterize the Kinetics of Free-Radical Polymerization
The polymerization of vinyl monomers presents an obvious change in density. For example, methyl methacrylate (MMA) and poly(methyl methacrylate) has the density of 0.936 and 1.188 g/mL, respectively, which means a 27% change in density in free-radical chain growth polymerization. The MagLev is applied to this polymerization system (through thermal polymerization and photopolymerization) to demonstrate the application to characterize the kinetics of free-radical polymerization [20].
Similar to the chemical reactions on solid supports, the change in density is primarily a direct result of the reduction in volume \(\Delta V\) as the monomer covalently binds to the grown polymer chain and thus the distance between the monomers changes from the van der Waals distance to the shorter covalent distance. For polymerization of methacrylate esters, \(\Delta V\) originates from the addition reaction of the double bonds. Its contribution to the reduction of the density of the product is highly correlated with its side chains. Larger side chains results in the smaller change in density between the polymer and the monomer. Larger molecule polymer can be synthesized by the cross-linking of prepolymer. In this kind of polymerization, the prepolymer has a large volume, which lead to an inconspicuous \(\Delta V\) during polymerization. Thus, the density change of the system could be approximately 0.
Thermal polymerization of MMA, hexyl methacrylate, and octadecyl methacrylate are investigated to experimentally confirmed the prediction that the density change should be smaller for monomer polymerization with larger molecular weight (or volume) but with common polymerizable groups. Small polymer beads (with diameter of ~1 mm) are obtained by suspension polymerization of the monomers. The densities of the monomers and polymers are determined using MagLev and listed in Table 8.4. The estimation of the density of the polymer can be calculated by Eq. 8.2,
where, m (kg) is the mass of a monomer molecule, \(V_m\) (m3) is the average volume of a single molecule in a liquid monomer (including real volume a monomer and the void space averagely occupied by each monomer), \(V_p\) (m3) is the volume that monomeric unit in the polymer occupies, and \(\Delta V\) (m3) is the difference between \(V_m\) and \(V_p\).
As shown in Table 8.4, the data clearly demonstrates an obvious decrease in \(\frac{\rho_p }{{\rho_m }}\) with the increase of molecular weight.
It is known that the polymerization of n-alkyl methacrylate can maintain a reasonably consistent reduction in the molar volume consumed per vinyl methacrylate group when this series of monomers are polymerized to produce a polymer. This consistent reduction in volume results from the shared chemical moiety (i.e., vinyl group) involved in the polymerization. The densities of the monomers can be obtained from the vendors and literatures. The reduction in molar volume is reported as 22.5 mL/mol [21]. Hence, the estimation of the density of some members in this series can calculated (Fig. 8.8). The experimental values are also listed with the estimations and show satisfactory agreement between them.

Reproduced with permission from Ref. [20]. Copyright 2017 American Chemical Society
Density measurement results of methacrylate esters and their polymers via MagLev.
On the other hand, the prepolymer of vinyl-containing siloxane (for synthesizing PDMS (Dow Corning Sylgard 184)) is used to validate the prediction that the cross-linking of large “monomer” will not significantly affect the density throughout the polymerization. The Pt-catalyzed hydrosilylation (addition of Si–H bonds to olefin bonds) between vinyl-ending prepolymers and crosslinkers with Si–H groups results in the polymerization of the prepolymers and produces the final cross-linked (and cured) polymer [22].
Because the ΔV is much smaller than the volume of the prepolymers in hydrosilylation reaction (22.5 cm3/mol, under the assumption that it is the same as observed in n-alkyl methacrylate polymerization, while the molar volume is estimated to be ∼7300 cm3/mol). Therefore, the \(\frac{\rho_p }{{\rho_m }}\) can be calculated as 1.003. The experiment of the polymerization presents a good verification of the discussion. Although the morphological changes are obvious, the levitation height of the droplet of un-cross-linked prepolymers is the same as that of the cross-linked irregular PDMS piece.
Based on the density changes, the kinetics of the polymerization can be characterized. The radical polymerization of MMA is chosen as the standard polymer system for an example (in particular, the polymerization is thermally initiated by 2,2′-azo (isobutyronitrile) or AIBN). The pure monomer of MMA (using inhibitors of 4-methoxyphenol and O2) and AIBN to perform the thermal polymerization. The polymerization of MMA is a first-order reaction for MMA. Its rate equation can be expressed as Eq. 8.3.
where, \(c_M\) (mol/L) is the concentration of the monomer, \(k_P\) (L mol−1 s−1) is the rate constant of radical propagation, \(f\) (unitless) is the efficiency of initiation (the proportion of free radicals produced by the homolysis reaction of the initiator that successfully initiates the polymer chain), \(k_d\) (s − 1) is the rate constant of thermal decomposition of the initiator into radicals, \(c_I\) (mol/L) is the concentration of the initiator, and \(k_t\) (L mol−1 s−1) is the rate constant of radical termination.
The reactions are carried out at four temperatures. The density of the reacting mixture is measured at 2-min intervals, and convert the density to the concentration by Eq. 8.4.
where, \(M_w\) (g/mol) is the molecular weight of the monomer, \(k_I\) is the mass ratio of the initiator to the monomer in the mixture, \(\rho_I\) (g/cm3) is the density of the initiator, and \(\rho\) (g/cm3) is the density of the reacting mixture.
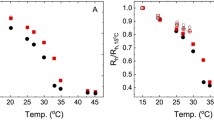
Utilizing MagLev method, the initial rates of polymerization over the first 10 min are determined, as shown in Fig. 8.9. The Arrhenius activation energy of this polymerization can be estimated as 79 kJ/mol, which is coincident with the reported results (range from 62.0 to 84.9 kJ/mol).

Reproduced with permission from Ref. [20]. Copyright 2017 American Chemical Society
Determination of the Arrhenius activation energy of thermally initiated polymerization of MMA using AIBN. a The changes in density of the polymerizing mixture (pure MMA with 1.3 wt% AIBN,) over time by transferring small (∼2 μL), cooled aliquots of the reacting mixture to the MagLev device. b The changes in the concentrations of the remaining monomer in the reacting mixture. c The initial rates of polymerizations with respect to the temperatures.
Different from the thermal polymerization, photopolymerization of MMA allows direct monitor through MagLev device by levitatin a single drop of monomer mixture. The monomer contains a hydrophobic photoinitiator of 2,2-dimethoxy-2-phenylacetophenone. The photopolymerization stimulates by irradiating the drop by a 365 nm UV light. The change in density \({\Delta }\rho\) is measured continuously as reaction proceeded. The verification of the expected behavior of pure MMA photopolymerization is carried out by continuous or periodic UV irradiation. The measured density can be converted to the fractional conversion of the monomer \(x\) using Eq. 8.5
The continuous photopolymerization with the UV light keep irradiating is drawn as the solid black spots in Fig. 8.10a. In the first 20 min, the rate of conversion increases slowly. Then the rate starts accelerating after 30 min, and finally reaches the equilibrium statue at ∼60 min. This autoacceleration behavior in polymerization of MMA is a well-known effect: the Trommsdorff effect, or “gel effect” in short. This is due to the slowing down of the diffusion-limiting termination rate of radical—radical binding as the viscosity of the reaction mixture increases. When irradiating the monomer periodically (the UV switch on/off every 10 min), it can be seen that the polymerization is almost halted when the UV light is off. The residual polymerization during the dark period in gel region (50–60 and 70–80 min) suggests that the free radicals persist during the reaction. When doubling the dark period (20 min), the residual polymerization can still be observed in gel region (100–120 min and 130–150 min), as shown in Fig. 8.10b.

Reproduced with permission from Ref. [20]. Copyright 2017 American Chemical Society
Monitoring the progress of photopolymerization through MagLev. a The pure MMA with photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (5 wt%) irradiated by a continuous (solid dots) or periodic (hollow dots, 10 min of dark period for every 10 min of irradiation) UV light (365 nm from a Hg lamp). b The photopolymerization irradiated using a periodic UV light with longer dark periods (20 min for every 10 min of irradiation).
The average rate of polymerization can be described by the theoretical model as Eq. 8.6. The model combines three key steps of radical polymerization: radical initiation, propagation, and termination. The profile of UV absorption (i.e. Beer’s Law) as light passes through the mixture (sphere droplet) is also considered in the model.
In Eq. 8.6, \(k_p\) (L/mol s) is the rate constant for propagation, \(k_t\)(L/mol s) is the rate constant for radical termination, \(c_A\) (mol/L) is the concentration of initiator, \(R\) (m) is the radius of the mixture droplet, \(\alpha = \varepsilon \ln 10\) (\(\varepsilon\) is molar absorptivity) is the absorption coefficient of A (L/mol cm), \(\phi\) (unitless) is the quantum yield for initiation (the number of propagating chains initiated per light photon absorbed), and \(I_0\) (mol/cm2 s) is the incident light intensity at the surface of the drop. \(K^{\prime}\) is the correction term that corrects the shape effect in absorbing UV light.
Under the condition of the same concentration of photoinitiator, the reaction order of monomers can be determined by measuring the initial polymerization rate of monomers with different concentration. The diluent is chosen as anisole. The density of the droplets is converted to concentrations of the monomer in the drops using Eq. 8.4. The result are plotted in Fig. 8.11. It can be observed that the monomer concentration changes with time are approximately linear in the initial 40 min. Therefore, the data in this range can be used to estimate the initial rate of polymerization. The slope of the linear fitted log–log curve of the initial rate and monomer concentration was 0.993, which indicated that the reaction order of MMA in the photopolymerization was ~1.0.

Reproduced with permission from Ref. [20]. Copyright 2017 American Chemical Society
Determination of the order of reaction for MMA in photopolymerization using MagLev. a The increase in the density of droplet contains MMA and photoinitiator (PI, 2,2-dimethoxy-2-phenylacetophenone, 0.27 wt%). b Calculated concentration of the monomer during the reaction. c Estimated initial rates of polymerization using the slopes of the linear fits in (b).
References
Turnbull AG, Hull HS. A thermodynamic study of the dimerization of cyclopentadiene. Aust J Chem. 1968;21(7):1789–97.
Mirica KA, Shevkoplyas SS, Phillips ST, et al. Measuring densities of solids and liquids using magnetic levitation: fundamentals. J Am Chem Soc. 2009;131(29):10049–58.
Shapiro ND, Mirica KA, Soh S, et al. Measuring binding of protein to gel-bound ligands using magnetic levitation. J Am Chem Soc. 2012;134:5637–46.
Koroznikova L, Klutke C, McKnight S, et al. The use of low-toxic heavy suspensions in mineral sands evaluation and zircon fractionation. J South Afr Inst Min Metall. 2008;108(1):25–33.
Grieve MC. Glitter particles—an unusual source of trace evidence? J Forensic Sci Soc. 1987;27(6):405–12.
Kirk PL. Density and refractive index. Their application in criminal identification. 1951.
Lockett MR, Mirica KA, Mace CR, et al. Analyzing forensic evidence based on density with magnetic levitation. J Forensic Sci. 2013;58(1):40–5.
Ash J, Hickey L, Goodpaster J. Formation and identification of novel derivatives of primary amine and zwitterionic drugs. Forensic Chem. 2018;10:37–47.
Amlani A, McKee G, Khamis N, et al. Why the FUSS (fentanyl urine screen study)? A cross-sectional survey to characterize an emerging threat to people who use drugs in British Columbia, Canada. Harm Reduct J. 2015;12(1):1–7.
Wang G, Huynh K, Barhate R, et al. Development of a homogeneous immunoassay for the detection of fentanyl in urine. Forensic Sci Int. 2011;206(1–3):127–31.
Philp M, Fu S. A review of chemical ‘spot’tests: a presumptive illicit drug identification technique. Drug Test Anal. 2018;10(1):95–108.
Abrahamsson CK, Nagarkar A, Fink MJ, et al. Analysis of powders containing illicit drugs using magnetic levitation. Angew Chem Int Ed. 2020;59(2):874–81.
Hoogenboom R, Fijten MWM, Abeln CH, et al. High-throughput investigation of polymerization kinetics by online monitoring of GPC and GC. Macromol Rapid Commun. 2004;25(1):237–42.
Zoller P. Dilatometry. Encyclopedia of Polymer Science and Technology; 2002.
Martín O, Mendicuti F, Tarazona MP. Use of a reliable homemade dilatometer to study the kinetics of the radical chain polymerization of PMMA: an undergraduate polymer chemistry laboratory kinetics experiment. J Chem Educ. 1998;75(11):1479.
Dörwald Z. Organic synthesis on solid phase. Wiley VCH; 2002.
Cironi P, Álvarez M, Albericio F. A combination of different spectroscopic techniques to monitor the “in situ” solid-phase synthesis of organic molecules. QSAR Comb Sci. 2004;23(1):61–8.
Vazquez J, Qushair G, Albericio F. Qualitative colorimetric tests for solid phase synthesis. In: Methods in enzymology. Vol. 369. Academic Press; 2003, p. 21–35.
Mirica KA, Phillips ST, Shevkoplyas SS, et al. Using magnetic levitation to distinguish atomic-level differences in chemical composition of polymers, and to monitor chemical reactions on solid supports. J Am Chem Soc. 2008;130(52):17678–80.
Ge S, Semenov SN, Nagarkar AA, et al. Magnetic levitation to characterize the kinetics of free-radical polymerization. J Am Chem Soc. 2017;139(51):18688–97.
Patel MP, Braden M, Davy KWM. Polymerization shrinkage of methacrylate esters. Biomaterials. 1987;8(1):53–6.
Lisensky GC, Campbell DJ, Beckman KJ, et al. Replication and compression of surface structures with polydimethylsiloxane elastomer. J Chem Educ. 1999;76(4):537.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Xie, J., Zhang, B., Li, X., Zhao, P. (2024). Magnetic Levitation in Chemistry. In: Zhao, P. (eds) Magnetic Levitation. Springer, Singapore. https://doi.org/10.1007/978-981-99-8314-8_8
Download citation
DOI: https://doi.org/10.1007/978-981-99-8314-8_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-8313-1
Online ISBN: 978-981-99-8314-8
eBook Packages: EngineeringEngineering (R0)