
Abstract
Idiopathic pulmonary fibrosis (IPF) is a fatal variation of the interstitial pulmonary illness characterised by extracellular matrix deposition that leads to secretion of inflammatory cytokines and causes fibrosis in the lungs. Further progression of fibrosis leads to cancerous stage of lungs and death. IPF is the worst pathological condition to be focused on and explored because of its rising prevalence, poor prognosis, and inadequate treatment. Even though the disease’s origin is still unknown, several genetic, environmental, and underlying pulmonary problems could set off a number of molecular pathways which is involved in the development of IPF. However, several genetic loci and genetic polymorphisms linked to IPF have been examined by genome-wide association studies and whole-genome sequencing. The newly found gene may clarify key elements in the identification, prognosis, and treatments of IPF. Additionally IPF can result from a variety of epigenetic alternations, including modification of histone, methylation of DNA, and non-coding RNA. This book chapter summarises the pathogenesis of pulmonary fibrosis, available treatment and the pathways that involved in IPF progression and may develop into lung cancer. Furthermore, this highlights epigenetic and molecular mechanism of IPF progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
9.1 Introduction
Idiopathic pulmonary fibrosis (IPF) is a long-term, persistent illness associated with lungs and marked by scarred lungs and the typical interstitial pneumonia histology. Further it is characterised by intensifying cough, dyspnoea, and worsening of life quality [1]. Extra-cellular matrix deposition causes fibrosis in which functional tissue is replaced by non-functional scarred tissue, affect lung function and often causes death [2]. Among the various type of lung diseases IPF is becoming the centre of attraction for many researchers, because its development is slow and it is resistant to traditional treatments as well. IPF has a 3-year median life expectancy and leads to dyspnoea and final respiratory collapse [3]. There is a considerable risk of death in many IPF patients who encounter acute episodes of respiratory deterioration [4]. Acute exacerbations are idiopathic acute worsening when there is no known reason for more than 50% of these acute episodes [5]. It is believed that acute IPF exacerbations signify an inherent speeding up of the underlying disease process, possibly brought on by a hidden stressor like a viral infection, micro aspiration, or ambient pollution [6]. The most prevalent form of idiopathic interstitial pneumonia is IPF. Although the condition has been regarded as rare, its frequency of occurrence is less than testicular, brain, and stomach cancers. Over time, the prevalence of IPF has increased with estimates for Europe and North America ranging from 2 to 18 cases per 100,000 persons per year. Little information about global variance is available, however, Asia and South America, where incidence is thought to be between 0 and 42 instances per 100,000 people annually, may have lower rates. People over the age of 50 are more susceptible to IPF, and men are more affected than women by the disease. The typical time to survive after diagnosis is between 2 and 4 years [2]. IPF occurs between 0.09 and 1.30 and 0.33 and 4.51 out of every 10,000 people, respectively [7]. In the US, it is reported that the occurrence of IPF is higher and increased rates of IPF-related hospital admissions and fatalities also point to a rising disease burden [3]. Identification of the underlying cause is necessary for the diagnosis: interstitial pneumonia histology pattern, typically with high-resolution computerised tomography scanning; lung biopsy perhaps necessary in some patients. The diagnostic process also requires ruling out any coexisting illnesses or other interstitial lung disorders [1]. Comprehensive management of the patient with IPF entails making a precise diagnosis using an interdisciplinary review that is meticulous; managing common comorbidities like depression, gastroesophageal reflux disease, obstructive sleep apnea; immunising against influenza and pneumococcal infection; providing education and structured exercise and through formal pulmonary rehabilitation classes; and, in cases of severe or worsening disease, determining whether or not lung transplantation is appropriate. All IPF patients should be examined for clinical trials of innovative therapeutic agents in the absence of effective treatments. Patients who do not meet the requirements for a clinical trial should be given the option of empiric therapy with acetylcysteine or proton pump inhibition [6]. Although pirfenidone, a new antifibrotic medication, has received approval for usage in some countries, there are no widely accepted treatments for IPF [6]. Pirfenidone and nintedanib are approved for the treatment of IPF because they can halt the spread of the illness, and functional decline, but they don’t provide a cure and have tolerability problems [4]. Notably, prednisone and immunomodulatory drugs like azathioprine are ineffective in treating interstitial lung disease (Fig. 9.1). The active therapy arm of a recent randomised controlled study of prednisone, azathioprine, and the antioxidant acetylcysteine was terminated early due to safety and efficacy concerns. Development of drugs has focused on fibrosis and its proliferation as a result of this lack of efficacy, and a growing number of targeted treatments are currently undergoing early-phase clinical studies. In the coming 10 years, it appears likely that the therapeutic environment for IPF will undergo significant change [6]. There are more therapy options available for IPF. However, a combination of treatment approaches with various mechanisms of action may be necessary to target the large number of profibrotic cytokines and growth factors implicated in disease development [4]. Building on the advances in our understanding of IPF pathobiology, it is hoped that future research into the role of gene variants, epigenetic changes, and other molecular biomarkers reflecting disease activity and behaviour will help the development of innovative medications for individualised therapy of IPF, enable earlier and more definite diagnosis, and better disease phenotyping [5].

Pathogenesis of idiopathic pulmonary fibrosis and various risk factors for pulmonary alterations that result in idiopathic pulmonary fibrosis, which develops into malignancy, respiratory failure, and death. Abbreviations: SFTPC surfactant protein C (SP-C) gene, TERT telomerase reverse transcriptase, TERC telomerase RNA component, MUC5B mucin 5B, ABCA3 ATP-binding cassette sub-family A member 3, ELMOD 2 ELMO domain containing 2, AEC 1 alveolar epithelial type-1 cells, AEC 2 alveolar epithelial type-2 cells
9.2 Involvement of Environmental Factor in the Pathophysiology of IPF
Earlier, it was considered that the primary cause of IPF is inflammation, which later develops into fibrosis. However, both anti-inflammatory therapy and immunosuppressive agent were unable to mitigate the IPF. Recent studies suggested that various environmental factors, genetic factors, and microbial infection lead to fibrosis development. It has been determined that progressive injury of the alveolar epithelium is the primary cause of an altered healing process in which numerous lung cells exhibit abnormal behaviours, resulting in the onset and maintenance of the fibrotic process [5]. Fibrotic foci are the principal histological characteristics of IPF which is triggered by alveolar epithelial damage, which also appears to stimulate fibroblast proliferation, differentiation of myofibroblast, and collagen accumulation. This extracellular matrix and collagen accumulation stiffens the lungs and obliterates the delicate lace-like structure of the alveolar gaps, leading to impairment in gaseous exchange, and ultimately, respiratory failure and death [8]. Type 2 alveolar epithelial cells (AEC2) are the stem cells in the pulmonary cells; persistent dysregulation of AEC2 has been identified as the main mechanism behind the development of fibrosis. AEC2 also involve significantly in the regeneration of AEC2 after lungs injury. The dysregulated AEC2 and loss of AEC1 are observed in IPF tissue [2] (Fig. 9.2).

Progression of idiopathic pulmonary fibrosis to pulmonary cancer: Idiopathic pulmonary fibrosis can proceed to lung cancer as a result of the activation of several molecular pathways that result in genetic alterations. Abbreviations: TGF transforming growth factor, CD 90/Thy-1 cluster of differentiation 90, SMAD 2 suppressor of mothers against decapentaplegic 2, SMAD 3 suppressor of mothers against decapentaplegic 3, PI3K phosphoinositide 3-kinase, MEK mitogen-activated protein kinase, mTOR mammalian target of rapamycin, ECM extracellular matrix, CAF cancer-associated fibroblast, EMT epithelial-mesenchymal transition
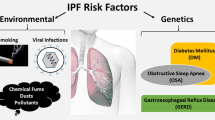
IPF susceptibility is likely influenced by several genetic characteristics, including gene variants and transcriptional changes that lead to epithelial integrity loss. Currently genome-wide association studies (GWAS) found that common genetic variations that are essential for the integrity of the epithelium have been defined as IPF risk factors. These studies suggested that telomere biology such as Telomerase reverse transcriptase (TERT), Telomerase RNA component (TERC), OB Fold-containing Protein 1 (OBFC1), host defence (mucin 5B (MUC5B), ATPase phospholipid transporting 11A, and toll-interacting protein (TOLLIP)), and cellular barrier function (desmoplakin, dipeptidyl peptidase 9) could play a role in the development of disease. Both the GWAS identified additional common variants linked to illness and established the promoter of the MUC5B gene as a risk factor. The functional gain of the promoter region of MUC5B, i.e., rs35705950, has been established as a major risk factor for the development of sporadic IPF [5, 9]. Apart from genetic factors, environmental exposures and microbes play significant roles in the progression of IPF. Cigarette smoking, wood dust, metal dust, coal dust, elements such as silicon and beryllium, asbestos, and radiation are some of the substances which are potential risk of IPF [2, 9] (Fig.9.1). Bacterial, fungal, and viral (hepatitis C, Epstein-Barr, Cytomegalovirus, herpesvirus) infections also lead to the development of IPF [9, 10].
9.3 IPF Association with Lung Cancer
There are various molecular and cellular mechanisms which are common in both such as endoplasmic reticulum stress, and fibroblast transition proliferation, activation, and oxidative stress, along with several epigenetic markers and genetics which make IPF patients more vulnerable to developing Lung cancer (LC) (Fig. 9.2) [11, 12]. According to earlier research, IPF patients had more sensitivity in developing primary lung cancer (22% of patients) in comparison to the normal public [13,14,15,16,17]. Similarly to this, individuals who receive lung transplants for IPF have also become vulnerable for primary lung cancer of more than 20 times than that of the general population [18, 19]. LC prevalence among patients with IPF reportedly ranges between 2.7 to 48% [13]. The study also shows the overall lung cancer occurrence in IPF. IPF cumulative incidence was reported by Ozawa et al. to be 3.3, 15.4, and 54.7% after one, five, and ten years of follow-up [14]. In a second experiment, individuals with IPF had cumulative lung cancer occurrence rates of 41% at 1 year and 82% at 3 years [20]. Additionally identified as complicating variables in emergence of LC in individuals having IPF are their age and smoking history. Finger clubbing is observed in nearly all individuals with IPF and lung cancer (95%) against around 60% of IPF patients who are alone.
Numerous research contrast IPF with pulmonary cancer to shed light on the aetiology of these two conditions, which have poor prognoses. Homogeneity, metastasis, and laterality of cancer are used to refute the parallels between IPF and cancer. Furthermore, IPF exists when both lungs are affected at once. However, it is predicated on the generally held notion that cancers usually typically grow in a single lung before colonising and metastasising various body part. Fibrosis in IPF patients initiate with the lower lobes and lung periphery, that are frequently the sites of lung malignancies from an anatomical perspective [21]. Additionally, altered cell-cell interactions, unchecked multiplication, and aberrant actuation of particular signal transduction pathways are characteristics of these two disorders [22, 23] (Fig. 9.3).

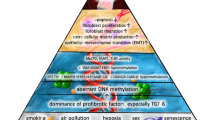
A diagrammatic representation of various risk factors and epigenetic changes that includes DNA methylation, histone modification and non-coding RNA which lead to the development of idiopathic pulmonary fibrosis and outlining ongoing and potential therapeutic treatments for idiopathic pulmonary fibrosis. Abbreviations: α-SMA alpha smooth muscle actin, PTGER2 prostaglandin E receptor 2, H3K4 histone H3 lysine K4, H3K27 histone H3 lysine K27, Cav-1 caveolin 1, CXC10 C-X-C chemokine ligand 10, COX-2 cyclooxygenase-2, miR MicroRNA, let-7d lethal-7d microRNA, hTERT human telomerase reverse transcriptase
9.3.1 Pathways Involved in the Progression of IPF into LC
9.3.1.1 Transforming Growth Factor-β1
Profibrotic mediators that support the onset of IPF include platelet-derived growth factor (PDGF), transforming growth factor β-1(TGFβ-1), tumour necrosis factor (TNF), endothelin-1, chemokine connective tissue growth factor, and osteopontin. Since it controls the activity of myofibroblasts and the ensuing remodelling of extracellular matrix deposition (ECM), TGFβ-1 stands out among them as the chief regulator of fibrotic development [24]. Chemotactic factors include TGF β-1 which attracts macrophages and monocytes, causing the release of PDGF from these sorts of cells, interleukin-1 (IL-1), basic Fibroblast growth factors, and TNF. Because of this, expression of TGFβ-1 has been increased in macrophages and alveolar epithelial cells from the lung tissues of IPF patients [25]. Furthermore, in the bleomycin mice, an in vivo IPF model [26] was demonstrated in the research [27], which found that 9 out of 12 standardised lung tissue samples from patients with IPF showed high membrane PD-L1 expression in alveolar macrophages. These macrophages play a crucial part in the development of lung cancer and IPF. Additionally, they discovered that IPF patients had considerably greater serum levels of soluble PD-L1 (sPD-L1) than a healthy control group. Checkpoint inhibitors that target PD-1/PD-L1 unleash antitumor T lymphocytes in cancer biology, enabling their activation, proliferation, and tumour cell eradication. Activating the TGF β-1 receptor complex also activates downstream canonical (Suppressor of mothers against decapentaplegic (SMAD2 and 3)) and non-canonical (PI3K, Mitogen-activated protein kinase (MEK), mammalian target of rapamycin (mTOR), etc.) signalling cascades, that ultimately impact the transcription of ECM proteins, profibrotic mediators, growth factors, and microRNAs [28, 29]. Particularly, phosphorylated SMAD2 and SMAD3 are translocated into the nucleus to modulate transcriptional responses as a result of activated TGFβ-1 receptors. For this reason, Zhao et al. showed that SMAD3 deletion mice reduced bleomycin-induced pulmonary fibrosis in mice [30]. The Wnt/catenin system and TGFβ-1 signalling have interactions that may promote the epithelial-mesenchymal transition and myofibroblast activation [31]. The stimulation of Wnt/ß-catenin signalling in IPF patients’ alveolar epithelium has recently been shown to hinder lung healing and accelerate AEC2 ageing [32]. Indeed, it has been discovered that lung fibrosis causes the Hippo pathway’s yes-associated protein and transcriptional coactivator with PDZ-binding motif to become activated. For this reason, YAP/TAZ, TGF-ß, Wnt, and PI3K axis activation was discovered by Xu et al. using single-cell RNA sequencing [33, 34]. They hypothesised that mTOR/PI3K/AKT signalling aided in the aetiology of IPF by promoting the growth of lung fibroblasts and epithelial cells. TGF-ß1 may consequently be in charge of the hedgehog pathway (Shh), whose activation in IPF lungs accelerates the disease’s progression with fibroblast proliferation and aberrant extracellular matrix deposition [35]. The activation of v6 integrin, one of the additional pathways implicated in TGF-ß1 synthesis by the AECs of IPF lungs, triggers the unfolded protein response, a defence biological process attributed to the build-up of misfolded proteins [36].
9.3.1.2 Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs) function the same as myofibroblasts in IPF and they are significant elements in the tumour microenvironment and are largely attracted to and activated by cytokines produced by cancer cells and immune cells that have infiltrated the tumour [37, 38]. Due to their significance in the tumour microenvironment’s pathways of proliferation, invasion, inflammation, angiogenesis, and metastasis, CAFs, which make up the majority of the cells in tumour stroma, significantly contribute to the biology of tumours. Additionally, stromal cells, which are both malignant and non-cancerous, produce different growth factors, such as FGF, TGF-β1, TNF, EGF, IL-1, vascular endothelial growth factor, and IL-6 [39, 40], which promote CAF trans-differentiation and activation, which also has the effect of boosting inflammation and carcinogenesis. After accounting for everything, it is possible to draw comparisons between fibrosis and LC, and among the various growth factors, TGF-β1 represents the key driving signalling for both fibroblasts and CAFs’ trans-differentiation, that is important for tumour development and therapeutic resistance [41, 42]. Numerous in vitro studies conducted in recent years have demonstrated that TGF-β triggered epithelial-mesenchymal transition into non-cancerous epithelial cells by activating the mTOR and PI3K pathways resulting in cancer cell’s resistance to apoptosis, encouraging CAFs to transdifferentiate [43]. Furthermore, CAFs exhibit great heterogeneity, which is most likely resulting from their liability to various tumour-secreted components, which prompt them to communicate various molecular markers. Although they are diverse, the molecular processes that activate CAFs and involved in the progression of cancer may be shared by different malignancies. In addition to TGF-β, epidermal growth factor receptor, the Wnt/ß-catenin, Hippo pathways, and JAK/STAT are other important molecular pathways for this function [44].
By interacting with or activating a number of additional downstream pathways, the profibrotic stimuli enhance the degree of TGFß expression that fosters activation of myofibroblast further which leads to the aberrant ECM accumulation and progression of IPF (such as the canonical SMAD3 pathway or non-canonical pathway and growth factors). The LC counterpart to the myofibroblasts, the CAFs, can come from different sources, such as cancer-associated adipocytes, and resident fibroblasts, which can originate from circulating progenitors in the bone marrow and are prevalent in tumour stroma [35] (Fig. 9.4).

Idiopathic pulmonary fibrosis management
9.3.1.3 Abnormal Cell-Cell Communication
Cxs (connexions) family made intracellular channels that connect cells metabolically and electrically. The coordination of cell growth and tissue repair depends on Cxs [45]. Cx43, one of them, is engaged in wound healing and tissue repair healing and is the one that is most prevalent on fibroblast membranes. Repression of Cx43 at wound sites aids in the healing of damaged skin tissues by promoting keratinocyte and fibroblast migration and proliferation. Therefore, Cx43 downregulation is linked with greater levels of TGF-β expression, collagen formation, and accelerated myofibroblast differentiation, all of which may aid in the promotion of healing. These modifications help explain how aberrant healing and fibrosis are characterised by a loss of control over fibroblast growth, a decline in control of fibroblast proliferation that distinguishes fibrosis and aberrant repair. The discovery that keloids and hypertrophic scars exhibit lower levels of Cx43 expression than normal skin tissues does corroborate this claim [46]. Despite the fact that human lung carcinoma cell lines expressing enhanced level of Cx43 demonstrated lower proliferation, decrease expression of Cxs are usually linked to the onset of cancer and the breakdown of intercellular communication [47]. Primary lung fibroblasts from IPF patients were found to have decreased intercellular communication as well as decreased Cx43 expression [48]. Limited cell-cell communication is frequently observed in cancer cells and fibroblasts from IPF patients, suggesting shared abnormalities of contact inhibition and unchecked growth [12].
9.3.1.4 Wnt/β-catenin Signalling Pathway
Matrilysin, laminin, and cyclin-D1 are a few examples of molecules that the Wnt/ß-catenin signalling pathway controls. But it might be argued that mediating interaction with TGF-β is the Wnt/ß-catenin pathway’s most significant role. Researchers reported that this pathway is inappropriately active in some cancers [49]. Recent research has shown that fibroproliferative disorders of the liver and kidneys activate the Wnt/ß-catenin pathway [50]. Patients with IPF have highly active Wnt/ß-catenin pathways in their lung tissues, which may be a reflection of TGF-β activity [51]. Particularly, TGFβ may activate a protein controlled by extracellular signals. Apoptosis and proliferation are regulated by the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, which is activated by the kinases 1 and 2 and target genes of this pathway. The functions of PI3K in myofibroblast development and proliferation have been demonstrated after TGF-β stimulation [52]. When the PI3K pathway is activated in cancer cells, the regulatory mechanisms that regulate cell proliferation break down. Blockers of the PI3K pathway have been utilised as therapeutic targets, and researchers are examining how they affect the growth and survival of tumours in different types of cancer [53].
On the other hand, unnatural actions of these kinases have been linked to a variety of cancers’ growth, development, and spread [54]. Common carcinogenic and fibrogenic mediators include TGF-β, Platelet-derived growth factor(PDGF), Vascular endothelial growth factor(VEGF), and Fibroblast growth factors (FGF). VEGF, in particular, may stimulate ERK1/2 and PI3K in a manner that directly or indirectly promotes cell survival and proliferation. Accordingly, it was discovered that endothelial progenitor cells (EPCs) from patients with IPF had increased VEGF mRNA expression levels [55].
9.3.1.5 Hepatocyte Growth Factor
Mesenchymal cells produce hepatocyte growth factor (HGF), which has been found to be an effective mitogen for mature hepatocytes. The c-Met proto-oncogene product known as the HGF receptor is mostly expressed in different kinds of epithelial cells. IPF patients’ bronchoalveolar lavage fluid and serum have greater amounts of HGF than serum from healthy individuals [56, 57]. In the IPF patients lung tissue, hyperplastic AECs and macrophages also show elevated levels of HGF [58]. In addition, Trkb treatment stopped mice from developing lung fibrosis in the bleomycin model [59]. In light of these findings, treatment with HGF could offer a new way for the inhibition of lung injury with in pulmonary fibrosis patients [60].
9.4 Several Aberrant Genes Expression Are Involved in the Pathogenesis of IPF
Both sporadic and familial forms of pulmonary fibrosis are correlated with genetic variations. Rare and frequent genetic variations are linked to five genes associated with telomere biology—TERT, TERC, DKC1 (Dyskerin pseudouridine synthase 1), TINF2 (TERF1-interacting nuclear factor 2), RTEL1 (Regulator of telomere elongation helicase 1), and PARN (Poly(A)-specific ribonuclease) as well as FIP (feline infectious peritonitis)-related mutations connected to alveolar stability SFTPA1, SFTPC, SFTPA2, NAF1, and ATP-binding cassette-type 3 have also been discovered through familial studies. Common variations, which are minor allele frequencies of >5%, also seem to affect the risk of FIP [61,62,63,64,65].
The promoter region of MUC5B has the most frequently repeated risk mutation (rs35705950), which was first discovered and has been tightly linked with IPF and familial IPF in a combined linkage and association analysis. In pulmonary fibrosis, there have been two substantial GWAS of IPF individuals (both familial and sporadic) with controls. Along with confirming TERT’s function at 5p15, MUC5B at 11p15, and the 3q26 area close to TERC, the seven novel linked loci were discovered by GWAS, including DSP (6q24), FAM13A(4q22), ATP11A (13q34), OBFC1 (10q24), DPP9(19q13), and the chromosomal regions 7q22 and 15q14-15, respectively, those who have nominally coupled with others. In contrast, common variations typically have a smaller effect magnitude, but they occur more frequently, particularly together, and may increase the percentage of disease risk [66].
9.4.1 Surfactant Protein-C (SFTPC)
According to numerous reports, SFTPC is linked with IPF. The gene SFTPC, which has gene ID 6440, is found on the short arm of chromosome 8 and codes for a 197 amino acid precursor protein through six exons. The first example of newborn ILD having a heterozygous change from A to G in the intron 4 splice donor site was described by Nogee et al. in 2001 [67]. This nucleotide mutation resulted in the protein precursor’s C-terminal region losing 37 amino acids and bypassing exon 4. As a result, the lung was deficient in mature surfactant protein C (SP-C) tissue and fluid from bronchoalveolar lavage. Afterwards, two more Leu188 to Gln (L188Q), a heterozygous mutation, and Ile73 were found in the SFTPC gene and correspond to Thr (I73T) [68, 69]. Prosurfactant protein C (pro-SP-C) builds up within alveoli and altered protein intracellular trafficking are potential effects of these two mutations. The mature 35-residue hydrophobic protein (SP-C), which is encoded by the SFTPC, is created by many rounds of proteolytic cleavage of the pro-SP-C. SP-C is subsequently secreted into the alveolar space. The SP-C is crucial for preserving alveolar stability, along with surfactant phospholipids and other surfactant proteins [70]. The aberrant SP-C proteins produced by SFTPC mutations can activate apoptotic pathways, instigate ER stress, and block the ubiquitin/proteasome system [71]. When SFTPCL188Q was transfected into cultured type II alveolar epithelial cells (AECII), aberrant lamellar structures, ER stress, and an unfolded protein response appeared [72]. Pro-SP-BRICHOS C’s domain, a unique domain identified in 12 protein families with a variety of functions and illness correlations, has been revealed to include certain fatal mutations [73]. When SP-C is made from the pro-SP-C, the BRICHOS domain acts as a chaperone. It is essential for the maturation of pro-SP-C, the precise folding procedure, and the correct packaging before it joins other surfactant components in the lamellar bodies for exocytosis. This mechanism may explain why infant/adult respiratory distress is not the same as pulmonary fibrosis, which is a condition caused by unfolded pro-SP-C but not a deficit. It has also been demonstrated that pulmonary fibrosis and the process of collagen deposition in the alveolus wall both include epithelial-to-mesenchymal transition (EMT). In A549 cells, targeted expression of SFTPCL188Q led to reduced zonula occludin-I and E-cadherin expression enhanced smooth mesenchymal marker expression muscle actin, demonstrating the connection between the variations of EMT and SFTPC [74]. In humans, SP-C makes up 10% of the protein components of pulmonary surfactants together with SP-A, SP-B, and SP-D, with lipids making up the remaining 90%. The main function of surfactants is to reduce surface tension at the air-water interface and prevent alveolar collapse. The surfactant-related genetic and acquired disorders attracted attention in the aetiology study of IPF since they had been linked in the pathophysiology of IPF together with surfactant modifications and alveolar type II cell death [75].
9.4.2 Telomerase Reverse Transcriptase and Telomerase RNA Component
Chromosome ends have telomeres, which are repeating nucleotide sequences that prevent the chromosomes from gradually shrinking when cells replicate normally. Telomere length is restored by telomerases’ two primary components, telomerase RNA (encoded by TERC) and telomerase reverse transcriptase (encoded by TERT). Oral leukoplakia, unusual skin darkening, and dystrophic nails are the hallmarks of the rare inherited telomere shortening condition known as dyskeratosis congenita (DKC). About 20% of individuals have lung fibrosis, and DKC complications including bone marrow failure can also occur. DKC served as the setting for the initial discovery of telomerase component mutations. More recent studies have found associations between FIP and a number of telomerase maintenance pathway genes, including those implicated in telomere stabilisation (DKC1, PARN, and RTELI) and catalytic activity (TERT and TERC) [76].
In peripheral blood and the lung, these pathogenic mutations promote telomere shortening by impairing telomerase activity. TERT variations have so far been found in 15% of FIP cases and 1–3% of sporadic cases, making them the uncommon variants associated with pulmonary fibrosis. TERT, RTEL1, and PARN variations were previously identified by a study associated with occasional IPF being linked to FIP, further supporting the pathophysiology of IPF in which telomere dysfunction plays a role as well as emphasising the genetic similarities between FIP and IPF sporadic. Further research has linked IPF to telomere dysfunction as there is evidence that telomere length is not the only connected to rare variant mutations in telomerase. A study discovered that without known mutations for TERT or TERC, short telomeres were present in 25% of sporadic IPF participants and 24% of familial IPF participants. Additionally, all participants in this study who had pulmonary fibrosis and those with TERC or TERT mutations also showed short telomeres. Uncertainty exists regarding the exact processes through which lung disease is brought on by telomere abnormalities. Epithelial cell senescence and a reduced ability to respond to epithelial damage have both been linked to defects in telomere maintenance. Telomere shortening happens throughout subsequent cycles of cell division and eventually triggers DNA damage pathways that result in apoptosis or senescence. Although early senescence can disrupt lung epithelial homeostasis and cause a remodelling response that leads to fibrotic lesions, cellular senescence is sometimes acceptable [66].
9.4.3 Pulmonary-Surfactant Associated Proteins (SFTPA2) Mutations
Researchers thought that mutations in the other surfactant proteins (A, B, and D) might also be discovered after discovering SFTPC mutations in FIP. The discovery of two families with FIP caused by mutations in SFTPA2 was reported by Wang et al. in 2009. Neighbouring genes encode two SPA isoforms (SFTPA1 and SFTPA2) and Wang et al. used whole genome linkage on a family of 15 individuals who had FIP, bronchoalveolar cell carcinoma, or unspecified lung disease and were connected to an area around SFTPA1 and SFTPA2 [63].
9.4.4 Mucin 5B (MUC5B)
A genome-wide linkage study was conducted in 2011 and discovered a locus on chromosome 11 that was strongly connected to the possibility of IPF [77]. After resequencing this region, a frequent single nucleotide polymorphism (rs35705950) in the promoter of the mucin 5B (Muc5B) gene was discovered, and it was connected to a six- to eightfold increased risk for IPF. Additional independent cohorts have now verified the link between this MUC5B promoter polymorphism and IPF, mostly made up of Caucasians [78]. It’s interesting to note that the MUC5B SNP appears to be frequent in cases of FIP and sporadic IPF [77]. The results reveal that rs35705950 did not raise the risk of scleroderma-related interstitial lung diseases or sarcoidosis. However, rs35705950 was discovered to be uncommon in the Korean population. This connection between rs35705950 and IPF was verified in a cohort of Mexican patients [79]. Similar to this, rs35705950 was uncommon in IPF patients in a Chinese community, while various MUC5B polymorphisms were linked to the condition [80].
MUC5B expression was uniformly higher in patients of IPF lungs compared with controls, despite the MUC5B SNP being present, despite the rs35705950 MUC5B SNP being linked with increased MUC5B mRNA expression in control people’s lungs. This result is in line with individuals with IPF having more MUC5B-expressing cells in the distal airways [81]. In the Framingham cohort, MUC5B rs35705950 has also been identified as a risk factor for asymptomatic interstitial lung abnormalities detected on CT scans in persons over 50. Uncommonly, those with the minor allele of rs35705950 are more likely to experience the syndrome, despite the fact that IPF patients who have the risk allele appear to have increased mortality in comparison to non-carriers. According to past animal studies, MUC5B may alter airway host defence [82], despite the fact that it is not yet known how MUC5B influences fibrotic remodelling [83].
9.4.5 Toll-Like Receptor
TLR signalling disruption has been identified in patients with IPF as a bridge between innate and adaptive immune responses [84]. Yet, the precise role of these signalling cascades in the fibroproliferative response is still largely unknown. There are ten functioning TLRs in humans, each of which has a unique receptor-ligand relationship. To recognise distinct external and intracellular signals, the [85] TLRs are either localised to the cell membrane (TLR1, 2, 4, 5, 6) or endosomal compartments (TLR3, 7, 8, 9), correspondingly. The majority of TLRs signal via a MyD88-dependent pathway, which ultimately leads to NF-қB activation and transcription of proinflammatory cytokine genes. A MyD88-independent mechanism underlies TLR3 and alternative TLR4 signalling, with TRIF recruitment ultimately leading to IRF3 or IRF7 transcription of type I interferon (IFN) genes [85].
Below is a description of the genetic risk mutations influencing TLR family signalling that are linked to IPF. GWAS results revealed three typical variations (rs111521887, rs5743894, rs574389) which were found in the toll-interacting protein gene connected with IPF. There has been conflicting research regarding whether these variations are providing separate associations with IPF or are in linkage disequilibrium. However, the IL-1 receptor-associated kinase inhibition (IRAK) phosphorylation of TLRs, notably TLR2 and TLR4, have been shown to limit TLR activity [86]. It is reported that IPF epithelia have increased levels of TLR2 and TLR4 activation, possibly as a result of continuous exposure to pathogenic microorganisms [86]. It has been demonstrated that decreased toll-interacting protein (TOLLIP) expression causes macrophages to secrete more pro-inflammatory cytokines as a result of TLR activation [87]. Through TLR4-dependent signalling, TOLLIP stimulates the production of IL-10, which protects mice from a fibrosis model caused by bleomycin. Additionally, TOLLIP inhibits TGF-β signalling by destroying TGF-ß1 receptors via interactions with SMAD7. These findings collectively imply that TOLLIP expression may protect against IPF by reducing pro-inflammatory and profibrotic pathways [88]. The rs1278769 variation in ATP11A discovered in the GWAS, however, less functionally characterised, might affect TLR4 signalling. It has been demonstrated that the gene 13 ATP11A, which codes for a phospholipid flippase, increases MyD88-dependent NF-kB activation and the generation of proinflammatory cytokines by inhibiting TLR4 endocytosis [89].
Dysregulated TLR9 activation encourages fibroblast-to-myofibroblast differentiation and has been associated with a more aggressive IPF phenotype. In most cases, microbial genetic material contains hypomethylated CpG nucleotides [90]. Since TLR9 also recognises mitochondrial DNA released from wounded cells, it’s conceivable that the cell deterioration and non-apoptotic cell death observed in the IPF distal airway may likewise play a substantial role in TLR9-mediated fibrosis. IL1RN gene (rs408392 and rs419598) was considerably connected to the IPF condition [91]. These risk alleles cause a decrease in IL-1Ra expression, which results in unrestricted IL-1R activation in innate immune cells and airway/alveolar epithelial cells. In contrast to the conventional bleomycin model, adenoviral-mediated IL-1 overexpression is employed to study the role of IL-1R signalling in animal models of pulmonary fibrosis [92, 93].
IPF patients’ alveolar macrophages exhibit an increased IL-1:IL-1Ra ratio indicating this pro-inflammatory condition. It is reported that the IL-1R/MyD88 axis reduces fibrosis in bleomycin and silica-induced fibrosis through genetic and pharmaceutical targeting. Both non-pathogenic and pathogenic bacterial signals are directly responsive to many of these hyperactive TLR signalling pathways, including TLR2 and TLR4 [94].
9.4.6 Adenosine Triphosphate-Binding Cassette Transporter A3(ABCA3)
The protein ABCA3 has been discovered in children with ILD or newborns with respiratory distress syndrome. The transfer of lipids across plasma membranes is facilitated by this transporter protein, which is mostly expressed in the AECII. The ABCA3 gene results in the production of a 1704 amino acid protein which can be found on the surfactant secretory vesicle on the limiting membrane of lamellar bodies, indicating that it may be involved in the metabolism and transport of surfactant. The ABCA3 gene has a large range of variations, and the genetic defects in surfactant metabolism that result in neonatal respiratory distress and paediatric ILD have been linked to 150 different mutations [95,96,97].
9.5 Epigenetic and Genetic Abnormalities
For the majority of cancers, documented pathogenic mechanisms include hypo-methylation of oncogenes and methylation of tumour suppressor genes. Patients with IPF have recently been found to exhibit epigenetic responses to environmental exposures, such as smoking and nutritional variables, as well as ageing. Additionally, current research has shown that individuals with IPF experience global methylation pattern changes that are similar to those in lung cancer patients [98]. Hypermethylation of the CD90/Thy-1 promoter region reduces the expression of the glycoprotein Thy-1, which is generally produced by fibroblasts, in the context of IPF [99, 100]. In addition, the conversion of fibroblasts into myofibroblasts and the invasive nature of malignancies are linked to the absence of this molecule in IPF patients. This suggests a unique therapeutic approach for this condition: pharmacological inhibition of Thy-1 gene methylation may restore Thy-1 production. In addition, particular gene changes have been implicated in the occurrence and progression of cancer [101]. Similarly, the oncogene p53 was expressed in almost half of the cases of IPF, along with fragile histidine triads, microsatellite instability, and loss of heterozygosity, commonly in the peripheral honeycombed lung areas that are unique to idiopathic pulmonary fibrosis [101,102,103,104]. Also, familial IPF has been linked to mutations that are typically linked to the occurrence and progression of cancer, such as those that influence telomere shortening and telomerase production [105, 106]. Recent studies have looked at circulating and cell-free DNA as a cancer biomarker for diagnosis and prognosis. In these studies, individuals with cancer and IPF had higher free circulating DNA concentrations than those with other fibrotic lung illnesses. The pathophysiology of both disorders was linked to aberrant expression levels of mRNA in addition to circulating DNA. According to these findings, short non-protein-coding RNAs control genes relevant to carcinogenesis, which are implicated in the growth, invasion, and metastasis of cancer cells [98, 107, 108]. According to recent studies, 10% of mRNAs are expressed abnormally in IPF patients. Among them, miR-21 and miR-155 were upregulated, whereas lethal-7 (let-7), miR-29, miR-30, and miR-200 were downregulated. These alterations correlated with gene families involved in apoptosis, ECM modulation, fibrosis, and the formation of epithelial-to-mesenchymal transition (EMT). Some of these mRNAs might affect TGF-β expression and be affected by it, hastening the functional loss in IPF patients [109,110,111].
9.6 Epigenetic Regulation of Gene Expression
The chromatin regulation, expression of genes, and particular cell type activity which participates in disease pathophysiology are mediated by various environmental exposures which have been translated by epigenetic processes that is linked to the incidence of disease. Environmental factors lead to change in non-inherent epigenetic marks, which are more dynamic than inheritable epigenetic marks [112]. Methylation of DNA, modification of histones, and long-chain non-coding RNAs are the processes which are part of the epigenome (Table 9.1).
Epigenetic factors are influenced by environmental exposure, ageing, and the diet of an individual. The progression of IPF depends upon a complex interaction between various environmental and genetic factors [113]. Inhalation of several environmental factors like metal dust, wood dust, industries, and dust from textile, and cigarette smoking affects the genetic and epigenetic marks. Several studies show that modulation of epigenetic factors regulates the expressions of genes which are involved in the occurrence of IPF.
The link between DNA methylation and histone alterations is an emerging model for epigenetic control of gene expression. One illustration of these relationships is the association of the regulatory component DNMT (DNA methyltransferases) 3L with the N-terminal of the histone H3 tail, which is linked in sequence to the mammalian new genetic methyltransferases like DNMT3A and DNMT3B. These data support the notion that the histone H3 tail’s unmethylated lysine 4 serves as a chromatin regulator of DNA methylation. DNMT3L identifies unmethylated histone H3 tails at lysine 4 and triggers de novo methylation of DNA by recruiting or activating DNA methyltransferase3A2. DNA methyltransferases similarly favour targeting nucleosome-bound DNA [114].
It is already evident that epigenetic marks which are related to the progression of IPF are also being regulated by environmental exposure [115] such as wood dust, textile dust, metal dust, silica, cigarette smoking, and many more [112, 115]. Out of all the environmental exposures, cigarette smoking is one of the major causes which has been linked to the development of IPF. Some studies related with case control have shown a link between cigarette smoking [116], occupational exposure, and the progression of IPF [115]. Various epigenome-wide association studies have confirmed that cigarette smoke can alter the DNA methylation at multiple CpG sites, and it can also regulate the miRNA expression in epithelial cells of bronchioles [117]. Cigarette smoking also involves in the methylation of Wnt7a, which is a specific promoter of a gene that is involved in progression of IPF [112]. Diet and ageing are also an equal contributors in the alteration of epigenomes. The pathobiology of ageing includes abnormal telomere shortening and dysfunction which is also linked to the development of IPF [10]. Other ageing-associated alterations, like the dysfunctioning of mitochondria and changed proteostasis enhance cell senescence. Persistent deposition of senescent cells is found in IPF lungs and it is responsible for stem cell dysfunctioning and produces pro-inflammatory cytokines such as IL-6, TGF-β, and metalloproteases which may cause fibrosis [118]. Exposure to fine particulate matter 2.5 (PM2.5), pesticides, and fungicides can also lead to the methylation of DNA [115]. The initiation and progression of IPF can also be a result of viral infection. A meta-analysis including 634 IPF patients, 653 control and 19 virus species (such as Epstein-Barr Virus, CHV, Human herpesvirus), suggested that viral infection is strongly associated with higher risk of IPF development [118]. Many case control studies have suggested that the exposure of cigarette smoke can lead to the development and progression of IPF, however, there is still need of evident result to confirm the linkage and exposure of smoking and epigenomic of IPF (Fig. 9.3).
9.6.1 DNA Methylation
In numerous targeted studies, it is already shown that modulation of epigenetics controls the expression of genes which are involved in the development and progression of IPF. Likewise, Thy-1(CD90) is a glycosylphosphatidylinositol-linked membranous (outer) glycoprotein and is a major regulator of cell-matrix and cell-cell interactions [119] but in IPF patients, its expression is absent in fibroblastic foci and myofibroblasts [112]. Thy-1 suppresses myofibroblast differentiation and its expression is downregulated by hypermethylation of promoter gene DNA in rat lung fibroblast [119] and also by histone modification [115]. The expression of the α-smooth muscle actin (α-SMA) gene varies depending on the degree of methylation of three CpG islands in the promoter of the α-SMA gene in fibroblasts, myofibroblasts, and alveolar epithelial type II cells. Inhibition of DNMT by siRNA induces the expression of α-SMA while its overexpression downregulates the α-SMA expression [120]. p14 (ARF) and prostaglandin E receptor 2 gene (PTGER2) are two examples of genes involved in fibroblast apoptosis whose expression was reduced in IPF lung fibroblasts due to hypermethylation of their promoters. It has been seen that prostaglandin E2 increases the DNMT3a activity, which leads to global hypermethylation and also results in the upregulation of those genes which are responsible for the suppression of cell proliferation in pulmonary fibroblast [121]. In IPF patients, TP53INP1 protein, which is a cell stress response protein induced by p53, is the most hypomethylated and upregulated gene [121].
9.6.2 Histone Modification
Histone deacetylases are the enzymes that deacetylase specific non-histone and histone proteins at particular locations [122]. In the process of deacetylation, histone deacetylases remove the acetyl group from lysine and retain the positive charge of histone, which results in the development of dense chromatin, hence inhibiting gene expression. On the other hand, histone acetyltransferases, i.e., HATs are another enzymes that acetylate the amino acids having conserved lysine by transferring the acetyl group from acetyl coenzyme A to form ε-N-acetyl-lysine, that attach with positive charged histone lysine and increases hydrophobicity. This binding leads to the emergence of a loose chromatin structure that promotes the transcription of uncoiled DNA [123]. Defects in the acetylation of histone can lead to the repression of expression of two genes which are antifibrotic, COX2 (cyclooxygenase-2), and chemokine IP-10 [115, 124]. C-X-C motif chemokine 10 is an antifibrotic molecule, produced by lung fibroblast, as a counterregulatory factor. It has been shown that the methylation of histone H3 lysine 27 by histone lysine methyltransferases (EZH2, G9a, G9a-like protein) represses the CXCL10 gene [124].
Recent studies demonstrated the suppression of the caveolin (Cav-1) gene by TGF-β leads to fibroblast proliferation and attains anti-apoptotic tendency. Cav-1 is constitutively repressed in IPF and the repression of the Cav-1 gene is regulated by histone modification (trimethylation of H3 lysine 4) [125]. It has been also reported that class III histone deacetylase, sirtuin, encourages proteasomal degradation of p21 to lessen the senescence brought on by TGF-β in human bronchial epithelial cells [121].
9.6.3 Non-Coding RNA Regulation
After exploring global miRNA profiles in lung tissue of IPF patients, epigenetic regulators of fibroproliferation, miR-29, and let-7d have been recognised while miR-155, miR-154, and miR-21 were found to be upregulated [114, 121]. The analysis of the lung fibrosis model of murine shows that let-7d has a protective role through the restriction of SMAD-3-dependent EMT. According to a subsequent investigation, in IPF lungs, miR-154 was one of the miRs that was most highly elevated. miR-154 appears to be a viable therapeutic target since transfection of primary fibroblasts with it led to substantial increases in cell migration and proliferation by activating a Wnt pathway [121]. A study reported that in IPF patients’ lung tissue, the miR17~92 clusters were reduced, while DNA methylation of its promoters’ region by DNMT1 was increased [112, 126]. miR17~92 is crucial in the development of lung epithelial cells and targets essential fibrotic genes [126]. A short non-coding RNA, miR-29 has also shown antifibrotic action in lung fibrosis, and it is majorly expressed during the fibrosis of various tissues. miR-21 has been demonstrated to be increased in the lungs of individuals with IPF and an experimental model of pulmonary fibrosis, primarily in fibroblastic foci [121].
9.7 Treatment of Idiopathic Pulmonary Fibrosis
Since delayed access to care and treatment is independently related to a higher risk of mortality, it is indicated that patients with known or suspected IPF be sent as soon as possible to a facility with experience in providing care for the condition [127]. The step-by-step treatment approach should be followed in the management of patients with IPF (Fig. 9.4). There is currently no cure for IPF, although nintedanib and pirfenidone are two drugs that work to delay the disease’s development and reduce mortality. The patient’s tolerance for side effects will determine which medicine is preferred because, based on the available evidence, none is more effective than the other [128]. Pirfenidone, a pyridone, is an orally available, small molecule having multiple effects such as anti-inflammatory, antioxidant and antifibrotic [8, 123]. Pirfenidone inhibits ECM production and fibrogenesis by suppressing the transcription of profibrotic and procollagen factors, such as TGF-β and PGDF, and it also suppresses the production of reactive oxygen species [123]. Pirfenidone also inhibits the proliferation of fibroblasts [128].
Nintedanib, a tyrosine kinase inhibitor, was developed for use in cancer treatment because it inhibits the proangiogenic factors [8]. Nintedanib targets platelet-derived growth factor receptors, receptor, and vascular endothelial growth factor receptor, that are highly upregulated in the lung tissue of IPF patients [123]. In a phase 2 study of patients with IPF, nintedanib showed a dose-dependent tendency to slow the loss of lung function and lower the frequency of acute exacerbations. In two additional phase 3 studies, nintedanib was compared to a placebo. For inclusion, the forced vital capacity had to be larger than or equal to 50% of the expected value, and the predicted lung carbon monoxide diffusion capacity had to be between 30 and 79%. In both studies, the relative fall in forced vital capacity after 52 weeks was 47.9 and 55.1% lower in the treatment group compared to the control group [2]. Both the above drugs have the same effect on the rate of decrement in forced vital capacity. Till date, none of the drugs, whether it is nintedanib or pirfenidone, has shown a survival benefit in clinical trials. However, these drugs have proven their effect on decreasing the mortality of IPF patients [2]. Various studies had been done using different drugs to treat IPF. However, none of them proved to be an effective and a reliable treatment therapy. Drugs, such as ambrisentan, everolimus, prednisolone, azathioprine, acetylcysteine, and warfarin had been identified as potentially harmful therapies in clinical trials. However, some therapies were potentially ineffective such as bosentan, imatinib, macitentan, and sildenafil [2]. Further, several drugs were tried and under clinical trials unfortunately no success has been achieved (Table 9.2).
9.7.1 Symptom-Focused Therapy
Due to the significant symptom load of IPF, including dyspnoea and cough, adjunctive symptom-based therapy is crucial. Corticosteroids and opiates might help in reducing chronic cough, anxiety, dyspnoea respectively [2]. Further, studies have demonstrated that pulmonary rehabilitation can reduce these symptoms, enhancing the quality of life. Moreover, LTOT (long-term oxygen therapy) is a necessary treatment in patients with IPF suffering from disease progression [128]. To treat low oxygen levels in IPF patients, supplemental oxygen therapy should be considered [2].
9.8 Future Perspective of IPF Treatments
There are several approaches that should be considered in the treatment of IPF such as alveolar epithelial injury prevention, targeting the coagulation cascade, boosting epithelial proliferation and collagen synthesis, preventing fibroblast proliferation and extracellular matrix direct targeting, inhibiting TGF-β and other profibrotic cytokines, and increasing epithelial resistance to injury [8]. The drugs which are in advanced stages in clinical trials are Simtuzumab (monoclonal antibody against lysyl oxidase-like 2), Lebrikizumab (monoclonal antibody against IL-13), and STX-100 [8]. An approach to combating telomere shortening, a known contributor to epithelial senescence is an appealing therapeutic approach for the treatment of IPF. hTERT administered by using an adenoviral vector has shown the potential treatment to express enzyme telomerase in other human diseases. Additionally, by inhibiting the development of senescent cells as a result of mitochondrial malfunction, treating lung fibrosis may be possible by enhancing mitochondrial biogenesis or mitophagy by utilising activators of PINK1 or SIRT3 [118]. Brd4 inhibitor JQ1 and HDAC inhibitors such as Spiruchostatin A (SpA), and Suberoylanilide hydroxamic acid (SAHA) have shown the reversal of profibrotic phenotype in primary fibroblasts and bleomycin mouse model [137].
9.9 Conclusion
In the last 15 years, the perspective of IPF has been changed enormously. Earlier IPF was considered idiopathic, but series of findings such as genetic polymorphisms, ageing, epigenetic alterations put the light on various pathogenesis through which IPF is characterised, that are ECM deposition, inflammation, and fibrosis. Several studies have also shown that alteration in epigenetics leads to the progression of IPF. Some animal studies have shown effectiveness in mitigating the IPF by targeting these epigenetic alterations. So far, the management of IPF relies on two newly approved drugs, that are pirfenidone and nintedanib as they have shown effect in reducing the IPF severity, yet these two are unable to prove efficiency in reducing mortality of IPF patients. Some drugs, like Simtuzumab, which is a monoclonal antibody, Lebrikizumab, monoclonal antibody against IL-13, and STX-100 are in advanced stage of clinical trials. Apart from these drugs, various other drugs were proved ineffective and hazardous during clinical trials. Other approaches should also be considered in the therapy of IPF, such as alveolar epithelial injury prevention, preventing fibroblast proliferation, ECM accumulation, and inhibiting profibrotic and TGF-β. Targeting telomere shortening by administration of hTERT has potential to combat IPF. Due to the significant symptom load of idiopathic pulmonary fibrosis, including dyspnoea and cough, adjunctive symptom-based therapy is crucial. Consider probable contributory comorbidities in individuals with persistent cough, such as gastroesophageal reflux disease. Corticosteroids and opiates might help in reducing chronic cough, anxiety, dyspnoea, respectively. Supplemental Oxygen for long-term is also a necessary treatment in patients with IPF suffering disease progression. To treat low oxygen level in IPF patients, supplemental oxygen therapy should be considered. Altogether, IPF is a disease of concern as till date no effective treatment is available and targeting root cause of disease such as epigenetic alterations is a crucial approach to mitigate the disease.
References
Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074. https://doi.org/10.1038/nrdp.2017.74.
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–52. https://doi.org/10.1016/s0140-6736(17)30866-8.
Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378(19):1811–23. https://doi.org/10.1056/NEJMra1705751.
Spagnolo P, Kropski JA, Jones MG, Lee JS, Rossi G, Karampitsakos T, et al. Idiopathic pulmonary fibrosis: disease mechanisms and drug development. Pharmacol Ther. 2021;222:107798. https://doi.org/10.1016/j.pharmthera.2020.107798.
Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res. 2018;19(1):32. https://doi.org/10.1186/s12931-018-0730-2.
Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–79. https://doi.org/10.1146/annurev-pathol-012513-104706.
Maher TM, Bendstrup E, Dron L, Langley J, Smith G, Khalid JM, et al. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res. 2021;22(1):197. https://doi.org/10.1186/s12931-021-01791-z.
Woodcock HV, Maher TM. The treatment of idiopathic pulmonary fibrosis. F1000Prime Rep. 2014;6:16. https://doi.org/10.12703/P6-16.
Mustafin RN. Molecular genetics of idiopathic pulmonary fibrosis. Vavilovskii Zhurnal Genet Selektsii. 2022;26(3):308–18. https://doi.org/10.18699/VJGB-22-37.
Leung J, Cho Y, Lockey RF, Kolliputi N. The role of aging in idiopathic pulmonary fibrosis. Lung. 2015;193(4):605–10. https://doi.org/10.1007/s00408-015-9729-3.
Samarelli AV, Masciale V, Aramini B, Coló GP, Tonelli R, Marchioni A, et al. Molecular mechanisms and cellular contribution from lung fibrosis to lung cancer development. Int J Mol Sci. 2021;22(22) https://doi.org/10.3390/ijms222212179.
Kinoshita T, Goto T. Molecular mechanisms of pulmonary fibrogenesis and its progression to lung cancer: a review. Int J Mol Sci. 2019;20(6) https://doi.org/10.3390/ijms20061461.
Karampitsakos T, Tzilas V, Tringidou R, Steiropoulos P, Aidinis V, Papiris SA, et al. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2017;45:1–10. https://doi.org/10.1016/j.pupt.2017.03.016.
Ozawa Y, Suda T, Naito T, Enomoto N, Hashimoto D, Fujisawa T, et al. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology. 2009;14(5):723–8. https://doi.org/10.1111/j.1440-1843.2009.01547.x.
Goto T, Maeshima A, Akanabe K, Oyamada Y, Kato R. Acute exacerbation of idiopathic pulmonary fibrosis of microscopic usual interstitial pneumonia pattern after lung cancer surgery<en-aut-mei> </en-aut-mei>. Ann Thorac Cardiovasc Surg. 2011;17(6):573–6. https://doi.org/10.5761/atcs.cr.10.01619.
Goto T, Maeshima A, Oyamada Y, Kato R. Idiopathic pulmonary fibrosis as a prognostic factor in non-small cell lung cancer. Int J Clin Oncol. 2014;19(2):266–73. https://doi.org/10.1007/s10147-013-0566-1.
Goto T. Measuring surgery outcomes of lung cancer patients with concomitant pulmonary fibrosis: a review of the literature. Cancers. 2018;10(7):223.
Hendriks LE, Drent M, van Haren EH, Verschakelen JA, Verleden GM. Lung cancer in idiopathic pulmonary fibrosis patients diagnosed during or after lung transplantation. Respir Med Case Rep. 2012;5:37–9. https://doi.org/10.1016/j.rmedc.2011.10.003.
Daniels CE, Jett JR. Does interstitial lung disease predispose to lung cancer? Curr Opin Pulm Med. 2005;11(5):431–7. https://doi.org/10.1097/01.mcp.0000170521.71497.ba.
Tomassetti S, Gurioli C, Ryu JH, Decker PA, Ravaglia C, Tantalocco P, et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest. 2015;147(1):157–64. https://doi.org/10.1378/chest.14-0359.
Antoniou KM, Tomassetti S, Tsitoura E, Vancheri C. Idiopathic pulmonary fibrosis and lung cancer: a clinical and pathogenesis update. Curr Opin Pulm Med. 2015;21(6):626–33. https://doi.org/10.1097/mcp.0000000000000217.
Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35(3):496–504. https://doi.org/10.1183/09031936.00077309.
Vancheri C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur Respir Rev. 2013;22(129):265–72. https://doi.org/10.1183/09059180.00003613.
El Agha E, Moiseenko A, Kheirollahi V, De Langhe S, Crnkovic S, Kwapiszewska G, et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell. 2017;20(2):261–73.e3. https://doi.org/10.1016/j.stem.2016.10.004.
Luo YH, Wang C, Xu WT, Zhang Y, Zhang T, Xue H, et al. 18β-glycyrrhetinic acid has anti-cancer effects via inducing apoptosis and G2/M cell cycle arrest, and inhibiting migration of A549 lung cancer cells. Onco Targets Ther. 2021;14:5131–44. https://doi.org/10.2147/ott.S322852.
Pallante P, Malapelle U, Nacchio M, Sgariglia R, Galati D, Capitelli L, et al. Liquid biopsy is a promising tool for genetic testing in idiopathic pulmonary fibrosis. Diagnostics. 2021;11(7):1202.
Jovanovic D, Roksandic Milenkovic M, Kotur Stevuljevic J, Markovic J, Ceriman V, Kontic M, et al. Membrane PD-L1 expression and soluble PD-L1 plasma levels in idiopathic pulmonary fibrosis-a pilot study. J Thorac Dis. 2018;10(12):6660–9. https://doi.org/10.21037/jtd.2018.11.16.
Marriott S, Baskir RS, Gaskill C, Menon S, Carrier EJ, Williams J, et al. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am J Physiol Cell Physiol. 2014;307(8):C684–98. https://doi.org/10.1152/ajpcell.00114.2014.
Karki S, Surolia R, Hock TD, Guroji P, Zolak JS, Duggal R, et al. Wilms’ tumor 1 (Wt1) regulates pleural mesothelial cell plasticity and transition into myofibroblasts in idiopathic pulmonary fibrosis. FASEB J. 2014;28(3):1122–31. https://doi.org/10.1096/fj.13-236828.
Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108(52):E1475–83. https://doi.org/10.1073/pnas.1117988108.
Strieter RM, Keeley EC, Hughes MA, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J Leuk Biol. 2009;86(5):1111–8. https://doi.org/10.1189/jlb.0309132.
Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel). 2015;7(4):2443–58. https://doi.org/10.3390/cancers7040902.
Chen Q, Chen S, Zhao J, Zhou Y, Xu L. MicroRNA-126: a new and promising player in lung cancer. Oncol Lett. 2021;21(1):35. https://doi.org/10.3892/ol.2020.12296.
Domen A, Quatannens D, Zanivan S, Deben C, Van Audenaerde J, Smits E, et al. Cancer-associated fibroblasts as a common orchestrator of therapy resistance in lung and pancreatic cancer. Cancers (Basel). 2021;13(5) https://doi.org/10.3390/cancers13050987.
Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73(18):5657–68. https://doi.org/10.1158/0008-5472.Can-13-0530.
De P, Aske J, Dey N. Cancer-associated fibroblast functions as a road-block in cancer therapy. Cancers (Basel). 2021;13(20) https://doi.org/10.3390/cancers13205246.
Monteran L, Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol. 2019;1835
De Jaeghere EA, Denys HG, De Wever O. Fibroblasts fuel immune escape in the tumor microenvironment. Trends Cancer. 2019;5(11):704–23. https://doi.org/10.1016/j.trecan.2019.09.009.
Yazdani S, Bansal R, Prakash J. Drug targeting to myofibroblasts: implications for fibrosis and cancer. Adv Drug Deliv Rev. 2017;121:101–16. https://doi.org/10.1016/j.addr.2017.07.010.
Caja L, Dituri F, Mancarella S, Caballero-Diaz D, Moustakas A, Giannelli G, et al. TGF-β and the tissue microenvironment: relevance in fibrosis and cancer. Int J Mol Sci. 2018;19(5):1294.
Chen P-Y, Wei W-F, Wu H-Z, Fan L-S, Wang W. Cancer-associated fibroblast heterogeneity: a factor that cannot be ignored in immune microenvironment remodeling. Front Immunol. 2021;12:2760.
Ji X, Ji J, Shan F, Zhang Y, Chen Y, Lu X. Cancer-associated fibroblasts from NSCLC promote the radioresistance in lung cancer cell lines. Int J Clin Exp Med. 2015;8(5):7002–8.
Hua W, ten Dijke P, Kostidis S, Giera M, Hornsveld M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci. 2020;77(11):2103–23. https://doi.org/10.1007/s00018-019-03398-6.
Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP, et al. CAF subpopulations: a new reservoir of stromal targets in pancreatic cancer. Trends Cancer. 2019;5(11):724–41. https://doi.org/10.1016/j.trecan.2019.09.010.
Losa D, Chanson M, Crespin S. Connexins as therapeutic targets in lung disease. Expert Opin Ther Targets. 2011;15(8):989–1002. https://doi.org/10.1517/14728222.2011.584875.
Mori R, Power KT, Wang CM, Martin P, Becker DL. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J Cell Sci. 2006;119(24):5193–203. https://doi.org/10.1242/jcs.03320.
Cesen-Cummings K, Fernstrom MJ, Malkinson AM, Ruch RJ. Frequent reduction of gap junctional intercellular communication and connexin43 expression in human and mouse lung carcinoma cells. Carcinogenesis. 1998;19(1):61–7. https://doi.org/10.1093/carcin/19.1.61.
Trovato-Salinaro A, Trovato-Salinaro E, Failla M, Mastruzzo C, Tomaselli V, Gili E, et al. Altered intercellular communication in lung fibroblast cultures from patients with idiopathic pulmonary fibrosis. Respir Res. 2006;7(1):122. https://doi.org/10.1186/1465-9921-7-122.
Mazieres J, He B, You L, Xu Z, Jablons DM. Wnt signaling in lung cancer. Cancer Lett. 2005;222(1):1–10. https://doi.org/10.1016/j.canlet.2004.08.040.
Hoguin A, Rastogi A, Bowler C, Tirichine L. Genome-wide analysis of allele-specific expression of genes in the model diatom Phaeodactylum tricornutum. Sci Rep. 2021;11(1):2954. https://doi.org/10.1038/s41598-021-82529-1.
Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N, et al. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res. 2008;57(4):274–82. https://doi.org/10.1016/j.phrs.2008.02.001.
Conte E, Fruciano M, Fagone E, Gili E, Caraci F, Iemmolo M, et al. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: the role of class I P110 isoforms. PLoS One. 2011;6(10):e24663.
Guerreiro AS, Fattet S, Kulesza DW, Atamer A, Elsing AN, Shalaby T, et al. A Sensitized RNA interference screen identifies a novel role for the PI3K p110γ isoform in medulloblastoma cell proliferation and chemoresistance. Mol Cancer Res. 2011;9(7):925–35. https://doi.org/10.1158/1541-7786.Mcr-10-0200.
Grimminger F, Schermuly RT, Ghofrani HA. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat Rev Drug Discov. 2010;9(12):956–70. https://doi.org/10.1038/nrd3297.
Malli F, Koutsokera A, Paraskeva E, Zakynthinos E, Papagianni M, Makris D, et al. Endothelial progenitor cells in the pathogenesis of idiopathic pulmonary fibrosis: an evolving concept. PLoS One. 2013;8(1):e53658. https://doi.org/10.1371/journal.pone.0053658.
Sakai T, Satoh K, Matsushima K, Shindo S, Abe S, Abe T, et al. Hepatocyte growth factor in bronchoalveolar lavage fluids and cells in patients with inflammatory chest diseases of the lower respiratory tract: detection by RIA and in situ hybridization. Am J Respir Cell Mol Biol. 1997;16(4):388–97. https://doi.org/10.1165/ajrcmb.16.4.9115749.
Maeda J, Ueki N, Hada T, Higashino K. Elevated serum hepatocyte growth factor/scatter factor levels in inflammatory lung disease. Am J Respir Crit Care Med. 1995;152(5):1587–91. https://doi.org/10.1164/ajrccm.152.5.7582299.
Shiratori M, Michalopoulos G, Shinozuka H, Singh G, Ogasawara H, Katyal SL. Hepatocyte growth factor stimulates DNA synthesis in alveolar epithelial type II cells in vitro. Am J Respir Cell Mol Biol. 1995;12(2):171–80. https://doi.org/10.1165/ajrcmb.12.2.7532419.
Yaekashiwa M, Nakayama S, Ohnuma K, Sakai T, Abe T, Satoh K, et al. Simultaneous or delayed administration of hepatocyte growth factor equally represses the fibrotic changes in murine lung injury induced by bleomycin. Am J Respir Crit Care Med. 1997;156(6):1937–44. https://doi.org/10.1164/ajrccm.156.6.9611057.
Panganiban RA, Day RM. Hepatocyte growth factor in lung repair and pulmonary fibrosis. Acta Pharmacol Sin. 2011;32(1):12–20. https://doi.org/10.1038/aps.2010.90.
Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1(4):309–17. https://doi.org/10.1016/s2213-2600(13)70045-6.
Nathan N, Giraud V, Picard C, Nunes H, Dastot-Le Moal F, Copin B, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet. 2016;25(8):1457–67. https://doi.org/10.1093/hmg/ddw014.
Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52–9. https://doi.org/10.1016/j.ajhg.2008.11.010.
Crossno PF, Polosukhin VV, Blackwell TS, Johnson JE, Markin C, Moore PE, et al. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest. 2010;137(4):969–73. https://doi.org/10.1378/chest.09-0790.
Stanley SE, Gable DL, Wagner CL, Carlile TM, Hanumanthu VS, Podlevsky JD, et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med. 2016;8(351):351ra107. https://doi.org/10.1126/scitranslmed.aaf7837.
Kaur A, Mathai SK, Schwartz DA. Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front Med. 2017;4 https://doi.org/10.3389/fmed.2017.00154.
Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344(8):573–9. https://doi.org/10.1056/nejm200102223440805.
Thomas AQ, Lane KF, Phillips JA, Prince MA, Markin CR, Speer MC, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165(9):1322–8.
Brasch F, Griese M, Tredano M, Johnen G, Ochs M, Rieger C, et al. Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene. Eur Respir J. 2004;24(1):30–9. https://doi.org/10.1183/09031936.04.00000104.
Li J, Liepinsh E, Almlén A, Thyberg J, Curstedt T, Jörnvall H, et al. Structure and influence on stability and activity of the N-terminal propeptide part of lung surfactant protein C. FEBS J. 2006;273(5):926–35. https://doi.org/10.1111/j.1742-4658.2006.05124.x.
Maguire JA, Mulugeta S, Beers MF. Multiple ways to die: delineation of the unfolded protein response and apoptosis induced by Surfactant Protein C BRICHOS mutants. Int J Biochem Cell Biol. 2012;44(1):101–12. https://doi.org/10.1016/j.biocel.2011.10.003.
Lawson WE, Cheng D-S, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A. 2011;108(26):10562–7. https://doi.org/10.1073/pnas.1107559108.
Willander H, Hermansson E, Johansson J, Presto J. BRICHOS domain associated with lung fibrosis, dementia and cancer--a chaperone that prevents amyloid fibril formation? FEBS J. 2011;278(20):3893–904. https://doi.org/10.1111/j.1742-4658.2011.08209.x.
Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2015;290(6):3277. https://doi.org/10.1074/jbc.A110.181164.
Zhou W, Wang Y. Candidate genes of idiopathic pulmonary fibrosis: current evidence and research. Appl Clin Genet. 2016;9:5–13. https://doi.org/10.2147/tacg.S61999.
Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res. 2012;730(1–2):52–8. https://doi.org/10.1016/j.mrfmmm.2011.10.013.
Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–12. https://doi.org/10.1056/NEJMoa1013660.
Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45(6):613–20. https://doi.org/10.1038/ng.2609.
Peljto AL, Selman M, Kim DS, Murphy E, Tucker L, Pardo A, et al. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a Mexican cohort but is rare among Asian ancestries. Chest. 2015;147(2):460–4.
Wei R, Li C, Zhang M, Jones-Hall YL, Myers JL, Noth I, et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res. 2014;163(5):494–502.
Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8(3):e58658. https://doi.org/10.1371/journal.pone.0058658.
Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature. 2014;505(7483):412–6. https://doi.org/10.1038/nature12807.
Yang IV, Fingerlin TE, Evans CM, Schwarz MI, Schwartz DA. MUC5B and idiopathic pulmonary fibrosis. Ann Am Thoracic Soc. 2015;12 Suppl 2(Suppl 2):S193–9. https://doi.org/10.1513/AnnalsATS.201503-110AW.
Karampitsakos T, Woolard T, Bouros D, Tzouvelekis A. Toll-like receptors in the pathogenesis of pulmonary fibrosis. Eur J Pharmacol. 2017;808:35–43. https://doi.org/10.1016/j.ejphar.2016.06.045.
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5 https://doi.org/10.3389/fimmu.2014.00461.
Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by tollip*. J Biol Chem. 2002;277(9):7059–65. https://doi.org/10.1074/jbc.M109537200.
Didierlaurent A, Brissoni B, Velin D, Aebi N, Tardivel A, Käslin E, et al. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol Cell Biol. 2006;26(3):735–42. https://doi.org/10.1128/MCB.26.3.735-742.2006.
Kowalski EJA, Li L. Toll-interacting protein in resolving and non-resolving inflammation. (1664–3224 (Print)).
van der Mark VA, Ghiboub M, Marsman C, Zhao J, van Dijk R, Hiralall JK, et al. Phospholipid flippases attenuate LPS-induced TLR4 signaling by mediating endocytic retrieval of Toll-like receptor 4. Cell Mol Life Sci. 2017;74(4):715–30. https://doi.org/10.1007/s00018-016-2360-5.
Trujillo G, Meneghin A, Flaherty KR, Sholl LM, Myers JL, Kazerooni EA, et al. TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med. 2010;2(57):57ra82. https://doi.org/10.1126/scitranslmed.3001510.
Korthagen NM, van Moorsel CHM, Kazemier KM, Ruven HJT, Grutters JC. IL1RN genetic variations and risk of IPF: a meta-analysis and mRNA expression study. Immunogenetics. 2012;64(5):371–7. https://doi.org/10.1007/s00251-012-0604-6.
Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117(12):3786–99. https://doi.org/10.1172/jci32285.
Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol. 2016;38(4):517–34. https://doi.org/10.1007/s00281-016-0559-z.
Michalski JE, Schwartz DA. Genetic risk factors for idiopathic pulmonary fibrosis: insights into immunopathogenesis. J Inflamm Res. 2020;13:1305–18. https://doi.org/10.2147/jir.S280958.
Bruder E, Hofmeister J, Aslanidis C, Hammer J, Bubendorf L, Schmitz G, et al. Ultrastructural and molecular analysis in fatal neonatal interstitial pneumonia caused by a novel ABCA3 mutation. Mod Pathol. 2007;20(10):1009–18. https://doi.org/10.1038/modpathol.3800928.
Campo I, Zorzetto M, Mariani F, Kadija Z, Morbini P, Dore R, et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir Res. 2014;15(1):43. https://doi.org/10.1186/1465-9921-15-43.
Ciantelli M, Ghirri P, Presi S, Sigali E, Vuerich M, Somaschini M, et al. Fatal respiratory failure in a full-term newborn with two ABCA3 gene mutations: a case report. J Perinatol. 2011;31(1):70–2. https://doi.org/10.1038/jp.2010.122.
Pandit KV, Milosevic J, Kaminski N. MicroRNAs in idiopathic pulmonary fibrosis. Transl Res. 2011;157(4):191–9. https://doi.org/10.1016/j.trsl.2011.01.012.
Sanders YY, Kumbla P, Hagood JS. Enhanced myofibroblastic differentiation and survival in Thy-1 (−) lung fibroblasts. Am J Respir Cell Mol Biol. 2007;36(2):226–35.
Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39(5):610–8.
Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, et al. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1996;154(2 Pt 1):477–83. https://doi.org/10.1164/ajrccm.154.2.8756825.
Hojo S, Fujita J, Yamadori I, Kamei T, Yoshinouchi T, Ohtsuki Y, et al. Heterogeneous point mutations of the p53 gene in pulmonary fibrosis. Eur Respir J. 1998;12(6):1404–8. https://doi.org/10.1183/09031936.98.12061404.
Uematsu K, Yoshimura A, Gemma A, Mochimaru H, Hosoya Y, Kunugi S, et al. Aberrations in the fragile histidine triad (FHIT) gene in idiopathic pulmonary fibrosis. Cancer Res. 2001;61(23):8527–33.
Demopoulos K, Arvanitis DA, Vassilakis DA, Siafakas NM, Spandidos DA. MYCL1, FHIT, SPARC, p16(INK4) and TP53 genes associated to lung cancer in idiopathic pulmonary fibrosis. J Cell Mol Med. 2002;6(2):215–22. https://doi.org/10.1111/j.1582-4934.2002.tb00188.x.
Rosales W, Carulla J, García J, Vargas D, Lizcano F. Role of histone demethylases in cardiomyocytes induced to hypertrophy. Biomed Res Int. 2016;2016:2634976. https://doi.org/10.1155/2016/2634976.
Correll KA, Edeen KE, Redente EF, Zemans RL, Edelman BL, Danhorn T, et al. TGF beta inhibits HGF, FGF7, and FGF10 expression in normal and IPF lung fibroblasts. Physiol Rep. 2018;6(16):e13794. https://doi.org/10.14814/phy2.13794.
Lovat F, Valeri N, Croce CM. MicroRNAs in the pathogenesis of Cancer. Semin Oncol. 2011;38(6):724–33. https://doi.org/10.1053/j.seminoncol.2011.08.006.
Oak SR, Murray L, Herath A, Sleeman M, Anderson I, Joshi AD, et al. A micro RNA processing defect in rapidly progressing idiopathic pulmonary fibrosis. PLoS One. 2011;6(6):e21253. https://doi.org/10.1371/journal.pone.0021253.
Corella D, Ordovas JM. Basic concepts in molecular biology related to genetics and epigenetics. Rev Española Cardiol (English Edition). 2017;70(9):744–53. https://doi.org/10.1016/j.rec.2017.05.011.
Roach KM, Feghali-Bostwick CA, Amrani Y, Bradding P. Lipoxin A4 attenuates constitutive and TGF-β1–dependent profibrotic activity in human lung myofibroblasts. J Immunol. 2015;195(6):2852–60.
Samara KD, Trachalaki A, Tsitoura E, Koutsopoulos AV, Lagoudaki ED, Lasithiotaki I, et al. Upregulation of citrullination pathway: from autoimmune to idiopathic lung fibrosis. Respir Res. 2017;18(1):218. https://doi.org/10.1186/s12931-017-0692-9.
Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res. 2015;165(1):48–60. https://doi.org/10.1016/j.trsl.2014.03.011.
Yang IV. Epigenomics of idiopathic pulmonary fibrosis. (1750-192X (Electronic)).
Yang IV, Schwartz DA. Epigenetic control of gene expression in the lung. Am J Respir Crit Care Med. 2011;183(10):1295–301. https://doi.org/10.1164/rccm.201010-1579PP.
Penz-Österreicher M, Österreicher CH, Trauner M. Fibrosis in autoimmune and cholestatic liver disease. Best Pract Res Clin Gastroenterol. 2011;25(2):245–58. https://doi.org/10.1016/j.bpg.2011.02.001.
Ding Q, Luckhardt T, Hecker L, Zhou Y, Liu G, Antony VB, deAndrade J, et al. New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis. (1179–1950 (Electronic)).
Zong D, Liu X, Li J, Ouyang R, Chen P. The role of cigarette smoke-induced epigenetic alterations in inflammation. Epigenetics Chromatin. 2019;12(1):65. https://doi.org/10.1186/s13072-019-0311-8.
Pardo A, Selman M. The interplay of the genetic architecture, aging, and environmental factors in the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2021;64(2):163–72. https://doi.org/10.1165/rcmb.2020-0373PS.
Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39(5):610–8. https://doi.org/10.1165/rcmb.2007-0322OC.
Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Epigenetic regulation of myofibroblast differentiation by DNA methylation. (1525–2191 (Electronic)).
Tzouvelekis A, Kaminski N. Epigenetics in idiopathic pulmonary fibrosis. Biochem Cell Biol. 2015;93(2):159–70. https://doi.org/10.1139/bcb-2014-0126.
Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. (1943–0264 (Electronic)).
Sehgal M, Jakhete SM, Manekar AG, Sasikumar S. Specific epigenetic regulators serve as potential therapeutic targets in idiopathic pulmonary fibrosis. Heliyon. 2022;8(8):e09773. https://doi.org/10.1016/j.heliyon.2022.e09773.
Zhang M, Serna-Salas S, Damba T, Borghesan M, Demaria M, Moshage H. Hepatic stellate cell senescence in liver fibrosis: characteristics, mechanisms and perspectives. Mech Ageing Dev. 2021;199:111572. https://doi.org/10.1016/j.mad.2021.111572.
Sanders YY, Liu H, Scruggs AM, Duncan SR, Huang SK, Thannickal VJ. Epigenetic regulation of caveolin-1 gene expression in lung fibroblasts. Am J Respir Cell Mol Biol. 2017;56(1):50–61. https://doi.org/10.1165/rcmb.2016-0034OC.
Dakhlallah D, Batte K, Wang Y, Cantemir-Stone CZ, Yan P, Nuovo G, et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187(4):397–405. https://doi.org/10.1164/rccm.201205-0888OC.
Lamas DJ, Kawut Smu, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. (1535–4970 (Electronic)).
Abuserewa ST, Duff R, Becker G. Treatment of idiopathic pulmonary fibrosis. Cureus. 2021;13(5):e15360. https://doi.org/10.7759/cureus.15360.
Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, Kaner RJ, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. (1535–4970 (Electronic)).
Malouf MA, Hopkins P, Snell G, Glanville AR. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. (1440–1843 (Electronic)).
King TE, Jr., Behr J, Brown KK, du Bois RM, Lancaster L, de Andrade JA, Stähler G, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. (1535–4970 (Electronic)).
Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. (1539–3704 (Electronic)).
Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr J. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. (1399–3003 (Electronic)).
Zisman Da, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. (1533–4406 (Electronic)).
Raghu G, Brown Kk, Costabel U, Cottin V, du Bois RM, Lasky JA, Thomeer M, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. (1535–4970 (Electronic)).
Daniels CE, Lasky Ja, Limper AH, Mieras K, Gabor E, Schroeder DR. Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results. (1535–4970 (Electronic)).
Helling BA, Yang IV. Epigenetics in lung fibrosis: from pathobiology to treatment perspective. Curr Opin Pulm Med. 2015;21(5):454–62. https://doi.org/10.1097/MCP.0000000000000191.
Acknowledgements
Author would like to acknowledge infrastructure facility and Research Seed Money provided by the Central University of Punjab, Bathinda, and UGC Start-Up grant from UGC-BSR and PECFAR fellowship by IGSTC. PY would like to acknowledge the SRF fellowship received from ICMR.
Conflict of Interest
None.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Singh, S.K. et al. (2023). Epigenetics of Idiopathic Pulmonary Fibrosis. In: Gupta, G., Oliver, B.G., Dua, K., Ali, M.K., Dave, P. (eds) Targeting Epigenetics in Inflammatory Lung Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-99-4780-5_9
Download citation
DOI: https://doi.org/10.1007/978-981-99-4780-5_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-4779-9
Online ISBN: 978-981-99-4780-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)