
Abstract
Idiopathic pulmonary fibrosis (IPF), the most common form of idiopathic interstitial pneumonia, is a progressive, irreversible, and typically lethal disease characterized by an abnormal fibrotic response involving vast areas of the lungs. Given the poor knowledge of the mechanisms underpinning IPF onset and progression, a better understanding of the cellular processes and molecular pathways involved is essential for the development of effective therapies, currently lacking. Besides a number of established IPF-associated risk factors, such as cigarette smoking, environmental factors, comorbidities, and viral infections, several other processes have been linked with this devastating disease. Apoptosis, senescence, epithelial-mesenchymal transition, endothelial-mesenchymal transition, and epithelial cell migration have been shown to play a key role in IPF-associated tissue remodeling. Moreover, molecules, such as chemokines, cytokines, growth factors, adenosine, glycosaminoglycans, non-coding RNAs, and cellular processes including oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress, hypoxia, and alternative polyadenylation have been linked with IPF development. Importantly, strategies targeting these processes have been investigated to modulate abnormal cellular phenotypes and maintain tissue homeostasis in the lung. This review provides an update regarding the emerging cellular and molecular mechanisms involved in the onset and progression of IPF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary fibrosis (IPF), the most common form of idiopathic interstitial pneumonia, is an irreversibly progressive and usually lethal disease. IPF patients typically succumb to respiratory failure secondary to loss of respiratory function from extensive fibrotic scarring of the lung parenchyma. Following diagnosis, the average life expectancy is 3–5 years. IPF is more common in males and individuals older than 60 years. The histopathological hallmarks include subpleural fibrosis, subepithelial fibroblastic foci, and microscopic honeycombing [1,2,3,4]. The clinical progress is usually complicated by acute episodes of respiratory function deterioration, termed IPF exacerbations. No effective treatments are available in preventing and controlling the acute exacerbations of IPF [5, 6]. The most common complications of IPF include lung cancer, depression, pulmonary hypertension, muscle weakness, heart failure, thrombosis, acute respiratory distress syndrome (ARDS), and respiratory failure. The recent introduction of two anti-fibrotic drugs, pirfenidone and nintedanib, will likely lead to a significant retardation in lung-function decline and a reduction in the incidence and severity of associated complications. However, as these agents are not curative, new therapeutic approaches are needed [7] Given that the exact pathophysiological mechanisms involved in IPF remain elusive. Additional studies on the cellular processes and molecular pathways involved are essential for the development of effective IPF therapies. A number of processes and factors, such as the role of aging and cellular apoptosis, oxidative stress, endoplasmic reticulum stress, cellular plasticity, and non-coding RNAs are the focus of intense research. Their better understanding might lead to the effective modulation of aberrant cellular processes and the maintenance of tissue homeostasis in the lung. This review discusses the key evolving concepts in IPF pathogenesis, the cellular and molecular mechanisms involved in the onset and progression of the disease, and the identification and development of novel targeted therapies.
IPF risk factors
The exact causes of IPF remain unknown. Several studies have identified potential risk factors, including genetic alterations, viral infections, lifestyle habits, environmental influences, occupational hazards. However, current evidence suggests that IPF is the result of complex interactions between genetic and environmental factors, e.g. cigarette smoking, metal and wood dust exposures, and comorbid diseases, such as gastroesophageal reflux, diabetes mellitus, and obstructive sleep apnea (Fig. 1) [8].

Schematic representation of IPF risk factors
Cigarette smoking
As with other lung diseases, cigarette smoking has a strong association with IPF. This association is particularly strong in frequent smokers or in those who have smoked for a long time [9]. For example, a study showed that long-term smoking was an independent factor for IPF development and that IPF patients who were smokers exhibited lower overall cellularity but higher and more severe alveolar space cellularity. This resulted in increased local inflammation, due to the progressive accumulation of macrophages [10]. Subsequently, consistent evidence confirmed that tobacco smoking is associated with an increased rate of lung-function loss over time [11, 12]. Six case–control studies showed a strong association between cigarette smoking and the development of IPF [11, 12]. Further studies reported that current smokers developed the disease at a younger age in comparison to non-smokers and ex-smokers [13], and that IPF patients with a smoking history have a shorter survival when compared to never smokers history [14, 15]. The mechanisms by which tobacco smoking influences IPF onset and progression are not fully understood. It has been reported that cigarette smoke stimulates in vitro overexpression of genes associated with epithelial-to-mesenchymal transition (EMT) and fibroblast-like phenotype [16], in vivo acceleration of telomere shortening [17], endoplasmic reticulum stress [18], repetitive mechanical stretch, and multiple other effects that need to be further investigated [19].
Environmental factors
The main environmental factors contributing to the pathogenesis of IPF include dust, fibers, fumes, and particulate matter. Studies have identified a significant increase in IPF rates in populations exposed to inorganic and animal dusts, as well as to chemical fumes including wood dusts and fumes, metal dusts from brass, lead, and steel, other than bird droppings, and other pollutants [20]. Some of the most common professions involving exposure to such pollutants include, metallurgy, farming, textile work, welding, veterinarians and others [11, 21]. In a multicenter case–control study, Baumgartner et al. identified a dose–response relationship between metal dust exposure and IPF, especially in those with more than five years of exposure [22]. This study was included in a subsequent meta-analysis which showed a significant association between metal dust exposure and the development of IPF (OR 2.44, 95% CI 1.74–3.40) [20]. Hubbard et al. found that there was a significant relationship between the duration of exposure and the risk of IPF-related death in metal-exposed workers in UK [23]. In addition, studies in Sweden and the United States reported a direct relationship between exposure to wood dusts and risk of IPF [24,25,26,27]. Nevertheless, a significant number of IPF patients do not have any history of occupational pollutants inhalation. A study from Sweden failed to identify associations between metal dust exposure and IPF among patients on oxygen therapy [28], suggesting that the impact of professional exposure to pollutants on the development and progress of IPF needs to be further elucidated.
Other occupational and environmental risk factors for IPF have been described, including hairdressing, bird breeding, dental work, and residing in an urban or polluted area [20].
Comorbidities
There is an increased risk of developing IPF in patients affected by other diseases, mainly gastroesophageal reflux disease (GERD), diabetes mellitus (DM), and obstructive sleep apnea (OSA).
The first report linking GERD with IPF was published by Pearson and Wilson in 1971 in 6 patients [29]. Five years later, it was reported that GERD was more common in patients with radiologic evidence of pulmonary fibrosis of unknown origin than in controls [30]. Other studies in GERD patients reported that the reflex of gastric acid contents into the airways, in the presence of impaired esophageal peristalsis, may lead to local irritation and respiratory symptoms [31]. In a retrospective study, Lee et al. observed a lower radiologic fibrosis score in patients with IPF treated with anti-GERD medications, and anti-reflux therapy was identified as an independent predictor of longer survival time [32]. Furthermore, a slower decline in forced vital capacity (FVC) and fewer acute exacerbations were reported in IPF patients receiving anti-acid treatments in three randomized controlled trials [33]. Clinical guidelines for the treatment of IPF that include specific indications for the use of anti-acid medications or anti-GERD interventions in association with anti-fibrotic regimens have been published [34,35,36]. Despite the early identification of the association between IPF and GERD, its pathophysiological mechanisms remain largely unknown. In a post hoc analysis of anti-acid use from the CAPACITY/ASCEND pooled trial data, there was no significant difference in disease progression at 52 weeks between anti-acid users and non-users. Furthermore, use of anti-acids was shown to be potentially associated with an increased risk of infection in advanced disease stages [37].
The lungs are among the target organs affected by DM [38, 39]. In a systematic review, Klein et al. identified seven cross-sectional studies between 1975 and 2009 that reported an association between DM and restrictive patterns based on FVC and diffusion capacity of the lung for carbon monoxide (DLCO) values [23, 40]. The incidence of diabetes in 65 IPF patients was fourfold higher than in 164 controls in a study performed in Japan, [41]. Furthermore, diabetes was found to be an independent risk factor for IPF [14]. The potential mechanisms by which diabetes contributes to the development of IPF are not clear. It has been shown that the accumulation of advanced glycation-end products (AGEs) in alveolar macrophages contributes to IPF, and that inhibition of the formation of AGEs prevents the disease in animal models [42, 43]. In vivo studies in bleomycin mouse models of pulmonary fibrosis have shown that the administration of the hypoglycemic agent metformin attenuates lung fibrosis [44]. However, other studies on the relationship between DM or metformin use in IPF have reported contrasting results [45].
Obstructive sleep apnea (OSA) is common in IPF, with prevalence estimates ranging between 59 and 88% [19]. However, whether OSA precedes IPF or vice versa remains to be determined. It has been hypothesized that OSA causes a recurrent stretch injury of the alveoli in proximity to the pleura. Although this resembles the characteristic distribution of fibrosis in IPF, further studies are warranted to investigate the link with OSA [46].
Viral infections
Viral agents, particularly Hepatitis C Virus (HCV) [47, 48], Adenovirus [49], Torque-Teno (Transfusion-Transmitted) (TT) virus [50], and Herpesviruses may increase the risk of pulmonary fibrosis, the acute exacerbations, and the progression of the disease [51]. Among herpesviruses, the Epstein–Barr Virus (EBV), cytomegalovirus (CMV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) seem to play a particularly important role in this context.
Studies performed in the UK reported that EBV was detected in lung tissues in 40–60% of IPF patients, using immunohistochemistry or protein chain reaction (PCR), but only in 0–4% of control patients [52,53,54]. Tanget al. showed that 97% of IPF lung samples, but only 36% of healthy control samples, harbored at least one of EBV, CMV, KSHV or human herpes virus 7 (HHV-7) [55]. In this study, 57% of IPF cases were infected by two of the aforementioned viruses, in comparison to 8% of the control cases [55]. Additionally, Folik et al. detected Herpesvirus Saimiri DNA in the regenerative epithelium of 21 IPF biopsies, while no virus was identified in healthy control lung epithelial cells [56]. Potential mechanisms linking latent viral infection with the pathogenesis of IPF include activation of EMT and tumoral growth factor (TGF-β) [19]. Animal models of Th2-biased mice lacking IFN-gamma or the IFN-gamma receptor have been used to demonstrate that Herpes viruses can promote pulmonary fibrosis and that antiviral therapy can prevent or limit exacerbations in patients with IPF [57,58,59,60].
Cellular and molecular mechanisms in IPF
Research on the pathogenesis of IPF has focused on chronic inflammation [61], abnormal wound healing [62], and models combining multifactorial and heterogeneous disease processes, such as cellular senescence, oxidative stress, endoplasmic reticulum stress, cellular plasticity, and the role of non-coding RNAs. Exposure to risk factors, such as those previously discussed, leads to phenotypic changes in the structure and function of lung cells, mainly due to EMT and endothelial-mesenchymal transition (EndMT), and ultimately fibrosis. Therefore, IPF can be considered as a pathological and phenotypical result of alterations in the homeostasis and communication between cell populations of the lung and their environment.
Apoptosis and senescence
A seminal study in 1996 showed that patients over 65 years of age had a higher incidence of IPF than younger patients [63]. The higher incidence rates of IPF in advanced age has been subsequently confirmed in numerous epidemiological studies in European and Asian populations [64,65,66,67,68]. Patients under 50 years of age diagnosed with IPF are more likely to be affected by familiar forms [69]. In addition, mortality rates are positively related to older age at diagnosis and to the temporal increase in the number of older people worldwide [70].
Aging is a process characterized by the interplay of multiple complex factors, such as physiological abnormalities, accumulation of the negative effects of risk factors, abnormal cell populations and tissues, with consequent progressive functional impairment of the whole organism [71]. Senescence occurs when the cell cycle is progressively interrupted at abnormal stages, leading to impaired cell replication and tissue regeneration. The impaired tissue homeostasis favors the release of intermediates, such as pro-inflammatory cytokines and metalloproteinases, collectively referred to as the ‘secretory phenotype related to aging’ (SPRA). Factors which further affect premature aging include telomere deficiency, mitochondrial dysfunction, oxidative stress, DNA damage, and altered protein function; most of these factors have been reported to play pivotal roles in the pathophysiology of IPF [72,73,74,75].
The concentrations of a number of SPRA-related mediators including TGF-β and IL-6 in rat lungs have been found to be increased with advancing age. By contrast, the elimination of senescent cells in IPF models reduced SPRA-related factors, such as IL-6, TGF-β and matrix metalloproteinase 12 (MMP12) [76]. An “anti-aging” agent that kills senescent type II alveolar epithelial cells (AECs), ABT-263, has been reported to reverse fibrosis in mice models of radiation-induced lung fibrosis [77]. In other studies, an increase in p16 and p21 concentrations in AEC type II from pulmonary tissues in IPF patients might be associated with higher disease severity, possibly as a result of an impaired transport of carbon monoxide [78].
Bone marrow-derived mesenchymal stem cells (BMSCs), which have the potential to differentiate into lung epithelial cells, have shown slower proliferation, shortening telomeres, and overall low levels of activity in older adults [79]. Because of the gradual instability of this stem cell, any epithelial cells that derive from it are more prone to errors resulting in premature cell-mediated death and genetic instability. This concept was shown in a paper in which the properties of BMSCs were studied in mice of varying ages [80]. Because the BMSCs of older mice failed to differentiate, the B-MSCs of younger mice were administered to the older mice. This restored stem cell potency, further reinforcing the concept that the ability of stem cells to proliferate decreases with age (Fig. 2).

Aging-associated molecular and cellular events linked to IPF the pathogenesis (red lines indicate activation, black lines indicted inhibition)
Chemokines, cytokines, and growth factors
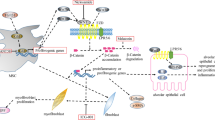
The role of serum chemokines and cytokines in the development of lung fibrosis remains controversial. Studies have shown that some cytokines and chemokines are potential pro-fibrotic agents as their concentrations are increased in the bronchoalveolar lavage fluid (BALF) and/or lung samples of IPF patients. The concentrations of these molecules have been investigated in various cell populations (macrophages, fibroblasts, neutrophils and alveolar epithelial cells) of lungs of IPF patients and healthy controls. Despite the functional roles and pathways of each molecule in the pathogenesis and development of IPF are not extensively known, it is possible that they participate in a complex concert of cellular and extracellular matrix interactions, leading to fibrosis and disease progression (Fig. 3). Their effects tend to be cell-specific, thus causing different effects on different types of lung cells [81, 82]

Inflammation-activated molecular and cellular events associated with IPF pathogenesis (red lines indicate activation)
TGF-β has been identified as a central factor in the development of pulmonary fibrosis. TGF-β has numerous biological effects, such as promoting wound repair through increased extracellular matrix deposition, inflammatory cell recruitment, and fibroblast differentiation. Overexpression of TGF-β via an adenovirus vector caused fibrosis in rat lungs, characterized by proliferation of myofibroblasts and extracellular matrix deposition [83,84,85]. In addition, overexpression of the TGF-β gene in pulmonary fibroblasts, as well as increased concentrations in BALF from IPF patients, has been reported [86]. However, concentrations of TGF-β in BALF increase also in other sclerosing diseases, such as stage IV sarcoidosis and systemic sclerosis [87]. Other than in fibroblasts, TGF-β is located in alveolar epithelial cells and macrophages, as well as in the bronchial epithelium and extracellular matrix [87, 88]. These findings suggest that TGF-β plays a pivotal role in the development of IPF.
Insulin-like growth factor (IGF-1) is another mediator potentially involved in IPF development. IGF-1 modulates cell migration and differentiation, and its concentrations have been found increased in the lungs of IPF patients [83]. Different cell types, particularly macrophages, lymphocytes and epithelial cells produce IGF-1 [89]. IGF-1 contributes to lung re-epithelialization, acting as an anti-apoptotic factor, and stimulates the in vitro synthesis of collagen by fibroblasts, thus leading to the production of extracellular matrix [83, 90, 91]. Fibroblasts from the lungs of IPF patients exhibit increased secretion activity in comparison with healthy tissue fibroblasts, especially in the production of fibronectin [92]. This occurs partly through the interaction between IGF-1 and several binding proteins [93]. In particular, proteins associated with IGF (IGFBP)-3 and -5 were found increased in vivo in IPF lung samples, and in vitro in models of IPF fibroblasts [90,91,92,93,94].
Concentrations of platelet-derived growth factor (PDGF) have also been found increased in BALF and lung samples from patients with IPF [94]. Similarly to IGF-1, in the early stages of IPF, PDGF has been detected in alveolar macrophages, mononuclear phagocytes, fibroblasts, type II pulmonary cells, as well as in endothelial and vascular smooth muscle cells [95]. In later stages, the localization of PDGF, in alveolar macrophages, a type of cells that is consistently increased in this phase, becomes similar to that of healthy controls [96]. PDGF is involved in lung angiogenesis and pulmonary hypertension, as evidenced by its overexpression in vascular endothelial and smooth muscle cells, but the exact mechanisms remain to be elucidated [95,96,97].
The connective tissue growth factor (CTGF) is a pro-fibrotic cytokine that stimulates fibroblast proliferation and increased deposition of extracellular matrix [98]. In lung tissues from IPF patients, CTGF was detected in both type II alveolar cells and interstitial fibroblasts [99]. CTGF is likely to be an important mediator of fibrosis but its role depends on the concentrations of TGF-β; low TGF-β activity is necessary for CTGF-dependent fibrosis [100]. Indeed, in experimental animal models, the overexpression of CTGF alone leads only to mild fibrosis [98,99,100,101,102].
IL-1β is a pro-inflammatory cytokine with prominent roles in wound repair. Adenovirus IL-1β gene transfer in rat lungs resulted in increased pro-inflammatory cytokine IL-6 and tumor necrosis factor-α (TNF-α), as well as increased PDGF and TGF-β concentrations [103]. In addition, in vivo studies showed that fibroblasts treated with IL-1β differentiated to myofibroblasts, which led to increased extracellular matrix (ECM) deposition [103]. Alveolar macrophages increase the production of TNF-α in response to lipopolysaccharide (LPS) treatment [104]. In addition, radiation exposure increases TNF-α concentrations in mice, which increases their susceptibility to fibrosis [105, 106]. In experimental rats treated with bleomycin, there was an increased TNF-α and TGF-β expression [107]. Excessive expression of TNF-α in rats resulted in fibrosis and alveolar inflammation, whereas TNF-α receptor knockout mice were protected from fibrosis caused by asbestos [108,109,110]. In another study, the use of TNF-α receptor knockout mice exposed to bleomycin showed that TNF-α expression was increased, as opposed to that of TGF-β [107]. Taken together, these findings suggest that TNF-α and TGF-β are significantly involved in the development of pulmonary fibrosis.
IL-6 concentrations were shown to be mildly increased in a rat model of experimental fibrosis [111]. While IL-6 is found to have anti-proliferative effects in normal fibroblasts, in IPF fibroblasts a proliferative response has been reported [111,112,113]. IL-8 is also increased both in the BALF and serum from IPF patients [114, 115]. In pulmonary fibrosis, alveolar and neutrophil macrophages are the main cells producing IL-8 [114,115,116]. Furthermore, IL-4 and IL-5, two inflammation mediators of T helper cell type 2 (Th2), have been found expressed, using immuno-histochemical methods, in infiltrating mononuclear cells within the interstitial space of IPF patients [117]. Both colony-stimulating factor (M-CSF) and CC Chemokine Ligand 2 (CCL2)/Monocyte chemoattractant protein-1 (MCP-1) have been found to be increased in the BALF of IPF patients compared with healthy controls [118, 119]. In addition, M-CSF and CCL2 knockout mice exhibit a lesser degree of pulmonary fibrosis following bleomycin treatment [117,118,119,120]. Finally, in vitro studies identified prostaglandin E2 (PGE2) as a regulator of fibroblast proliferation, differentiation and collagen synthesis [121, 122]. Specifically, fibroblasts retrieved from IPF lung tissue showed less PGE2 production in response to IL-1β, LPS, and TGF-β than healthy controls [123, 124]. Overexpression of COX2 in rats increased PGE2 expression in the lungs and subsequently reduced fibroblast proliferation. Although this finding supports the hypothesis that the decrease in PGE2 concentrations contributes to the development of IPF, increased PGE2 concentrations in the BALF of IPF patients have also been reported [121, 122, 124,125,126,127,128].
The above-mentioned mediators are differently produced by the cells involved in the pathological process. A recent in vitro study identified numerous clusters of endothelial, epithelial, and mesothelial cells as well as hematopoietic cells including macrophages, monocytes, neutrophils, dendritic cells, natural killer cells, and lymphocytes with different profiles. In addition, the study described 12 different clusters of collagen-producing cells which are involved in the genesis of fibrosis. These clusters were categorized into two sub-clusters: the first, including clusters 0, 1, 2, 4, 6, 8, 10 with higher collagen a1 expression, and the second, including clusters 3, 5, 7, 9 with higher Acta2 expression. Cluster 11 was characterized by proliferating cells that expressed Mki67 and Cdc20. Clusters 5 and 7 expressed smooth muscle cell markers, such as Acta2 and Myh11. Cluster 9 expressed pericyte markers [129].
Adenosine
It has been recently proposed that adenosine, an endogenous autocoid regulating several body functions, plays a pivotal role in both tissue regeneration and fibrotic processes [130]. Adenosine regulates cell function by binding to specific cell surface receptors [131]. There are four adenosine receptor subtypes. Three of them, subtypes, A1, A2A, and A2B, are highly conserved during evolution, while the A3 receptor subtype differs significantly among species [132]. A2B receptor stimulation has been reported to drive the differentiation of pulmonary fibroblasts [133], and its expression was increased in the lungs of mice deficient in adenosine deaminase, an enzyme responsible for adenosine catabolism [134]. In this context, compared with control animals, Sun et al. reported an increased number of fibroblasts and the accumulation of α-smooth muscle actin in adenosine deaminase-deficient mice, a phenomenon that was prevented by the A2B receptor antagonist CVT-6883 [135]. When compared with adenosine deaminase-deficient mice, CVT-6883-treated animals showed significantly lower levels of lung histological inflammatory markers and reduced fibrosis, collagen deposition and alveolar airspace enlargement. Macrophages-mediated production of pro-fibrotic mediators, such as TGF-β1, was also suppressed by administration of A2B receptor antagonists [135]. A2B receptor-mediated antagonism protection on lung fibrogenesis was also linked with a decreased secretion of chemokines and cytokines including CCL17, chemokine (CXC motif) ligand 1 (CXCL1), chemokine (CC motif) ligand 11 (CCL11) CXCL2, TNF-α and IL-6 [135]. The A2B receptor has also been studied in a model of lung fibrosis caused by bleomycin, where it seems to have different roles during both the acute and the chronic stages of lung injury [136]. Genetic ablation of the A2B receptor in mice systemically exposed to bleomycin (chronic treatment) reduced lung fibrosis exerting modest effects on lung injury, while A2B receptor ablation in mice intra-tracheally treated with bleomycin (acute treatment) increased pulmonary inflammation with little effect on pulmonary fibrosis [136]. Following bleomycin treatment, the effect of the A3 adenosine receptor in lung fibrogenesis has also been investigated in mice [137]. Although this receptor appears to play an important role as anti-inflammatory modulator, it appears unlikely to be primarily involved in the control of lung fibrogenesis [137]. As adenosine appears to exert a key role in the pathophysiological control of pulmonary fibrosis, the development of A2B receptor antagonists may represent a therapeutic option in IPF (Fig. 4).

Schematic representation of the cellular and molecular events linked to the IPF-pathogenesis in the lung environment. NOX NADPH oxidase, ROS reactive oxygen species, cAMP cyclic adenosine monophosphate, DAG diacylglycerol, IP3 inositol trisphosphate, DPI diphenyleneiodonium, CVT-6883A 2B-adenosine receptor antagonist, ER endoplasmic reticulum (Red arrows indicate activation, black lines indicate inhibition)
Oxidative stress
Oxidative stress is defined as an imbalance between oxidant production and antioxidant defense, favoring oxidants. This leads to the production of several reactive oxygen species (ROS), containing one or more unpaired electrons in their atomic or molecular orbitals. These unpaired electrons generate highly reactive radicals, such as the superoxide radical (O2 ˉ), hydrogen peroxide (H2O2), hydroxyl radical (OH), hypochlorous acid (HOCl), and peroxynitrite (ONOO). Excessive ROS production causes oxidative stress, leading to cell damage. Numerous studies have reported that oxidative stress is involved in the pathogenesis of IPF [138,139,140,141]. Recently, Fois et al. performed a systematic review to describe the type of markers of oxidative stress identified in different biological specimens of patients with IPF [142]. In each of the 30 identified studies, the authors critically appraised the type, site (systemic vs. local, e.g. breath, sputum, expired breath condensate, epithelial lining fluid, bronchoalveolar lavage, and lung tissue specimens), and method used for measuring the specific oxidative stress biomarkers. Subsequently, Paliogiannis et al. performed a meta-analysis of 15 studies in patients with IPF, which showed either an increase in the concentrations of oxidative stress markers or a reduction in antioxidant markers, independent of the type of biological sample [143]. These observations strongly support the proposition that oxidative stress is one of the most relevant pathophysiological mechanisms involved in IPF.
The specific mechanisms by which oxidative stress favors the development of IPF are not yet fully understood although they are likely to target both the cell phenotype and life-cycle promoting aging and apoptosis. This, in turn, would lead to alterations in tissue micro-environment that favor fibrosis upon regeneration. It has been shown that increased oxidative stress induces the premature aging of cells [144, 145]. As a result, fibroblasts acquire features of apoptosis resistance [146, 147], and remain metabolically active producing higher levels of ROS [148]. Furthermore, pulmonary myofibroblasts secrete hydrogen peroxide (H2O2), which mediates fibrotic effects in tissues through induction of epithelial apoptosis though paracrine mechanisms [149], or spatial alterations of the extracellular matrix in presence of extracellular cells [150]. In the lungs, various factors contribute to the generation of oxidative stress. Environmental factors, such as cigarette smoking stimulate ROS production through endoplasmic reticulum (ER) stress, disconnection of the mitochondrial enzymatic system [150, 151], and production of NADPH oxidases (NOX), especially NADPH oxidase-4 (NOX4), by both inflammatory and lung cells. ROS stimulate apoptosis in airway epithelial cells, as well as production of cytokines and growth factors (such as TGF-β [152]) which play an important role in myofibroblast differentiation and collagen deposition, resulting in pro-fibrotic events and further reduced antioxidant capacity (Fig. 4).
In a recent study, an increase of reactive oxygen species (ROS) generation induced by sera from IPF patients determined both collagen type I deposition and proliferation of primary human pulmonary artery smooth muscle cells (HPASMCs). IPF sera-induced cellular effects were significantly blunted by the NADPH oxidase inhibitor diphenyleneiodonium (DPI), supporting the causative role of ROS and their cellular source. In contrast to what observed in treatment-naïve IPF patients, serum from IPF patients treated with pirfenidone failed to induce the generation of ROS and collagen synthesis in HPASMC (Fig. 4) [153]. The use of antioxidants in IPF is a focus of current research. N-acetylcysteine has been widely used in IPF as antioxidant and anti-fibrotic agent since it is relatively inexpensive, well-tolerated, and orally available. However, the evidence for its effectiveness is not robust, mainly due to the lack of adequately designed studies using placebo arm comparators [142]
Mitochondrial dysfunction
Mitochondrial-generated increase of ROS has been found to induce lung fibrosis [154, 155]. Although a potentially vicious cycle of TGF-β and ROS interaction exists, where ROS activate TGF-β and TGF-β activates ROS [156,157,158]), TGF-β1 appears to be the most important trigger of mitochondrial (mtROS) production associated with the pro-fibrotic phenotype reprogramming of lung cells [159]. This proposition is further supported by the observation that the deletion of NOX4 abrogates TGF-β1-induced fibrosis in mice [160]. Moreover, TGF-β1 has been reported to activate NOX4-mediated collagen production in myofibroblasts [161], and this signaling pathway is strongly implicated in IPF pathogenesis [162, 163]. Although the role of mitochondrial mtROS in IPF onset and progression is not fully understood approaches to beneficially modulate mtROS levels with antioxidants are currently under investigation (Fig. 4) [164, 165].
Besides ROS homeostasis, healthy mitochondrial function is also maintained by the coordinated activities of mitochondrial biogenesis, dynamics, and mitophagy, also known as Mitochondrial Quality Control (MQC). In this context, emerging evidence suggests that MQC is one of the major contributors to the pathophysiology of IPF [166]. Key proteins involved in controlling MQC include GTPases, those that require oxidative phosphorylation (OXPHOS)-mediated ATP generation, such as mitofusin (MFN1, MFN2), ubiquitinated autophagy-related proteins, and dynamin-related protein [166,167,168]. As mitochondrial OXPHOS provides energy for the above-mentioned processes, the modulation of mitochondrial bio-energetic processes can simultaneously affect mitochondrial function [166]. Alterations in aerobic metabolism have been reported in fibroblasts involved in pulmonary fibrosis [166], particularly increased glycolysis, resistance to apoptosis, and propensity to develop a metabolic-driven fibrotic phenotype, which appears to be linked to decreased ATP synthesis and increased mtROS production [169, 170]. Fatty acid oxidation (FAO) has also been linked with lung fibrosis as pulmonary macrophages with enhanced FAO appear to enhance their pro-fibrotic reprogramming in the lung [171]. This is also facilitated by the tight association between FAO and OXPHOS [172, 173]. This association has been confirmed in a mice model of bleomycin-induced lung fibrosis, where macrophages metabolic reprogramming shifts glycolysis to FAO [171, 174]. Macrophages pro-fibrotic metabolic reprogramming seems to be essential to maintain both regenerative cell activity and apoptotic resistance in the lung [168, 173, 175, 176]. Moreover, while II AECs differentiation into I AECs and subsequent re-epithelialization represents the normal lung response to injury, during pulmonary fibrosis the accelerated biogenesis and mitophagy in type II AECs is associated with increased glycolysis-driven metabolism, leading to apoptosis due to reduced ATP and increased mtROS production [177]. Indeed, while the induction of type II AEC apoptosis is linked with increased fibrotic phenotype [178] its inhibition attenuates pulmonary fibrosis [178,179,180]. In line with these observations, a higher number of cells with compromised mitochondria [167, 181] and elevated mtROS production [182, 183] has been reported in IPF subjects (Fig. 4).
To further support the link between altered cell metabolism and fibrotic cell reprogramming in the lung, an impaired mitochondrial lipid metabolism associated with the fibrotic process in type II AECs has been reported [184]. The impaired synthesis of phospholipids and cholesterol affects the lung surfactant synthesis and alveolar homeostasis by disrupting the epithelial barrier integrity and promoting fibrogenesis [184]. Increased levels of lipids and plasmatic cholesterol have been linked to disease acute exacerbation and high mortality in IPF subjects [185]. In this regard, mitochondrial MFN1 and MFN2 play a crucial role in regulating lipids and cholesterol production and their knockdown affect lipid synthesis in AEC2 cells and exacerbates bleomycin-induced lung fibrosis [184]. Altered lipid profiles have been reported IPF BAL fluid [186], IPF lung [187], and IPF type II AECs [188]. In line with these findings, the cholesterol-lowering agent simvastatin has been shown able to reduce atherogenic diet-elicited pulmonary fibrosis by downregulating Hsp70/Hsp90 protein concentration in the lung [189].
The TGF-β1/NOX4/ROS pathway also appears to be linked with the mitochondrial metabolic pathways implicated in lung fibrogenesis. NOX4 activation has been reported to suppress both mitochondrial biogenesis and bioenergetics in lung fibroblasts, while NOX4′s pharmacological inhibition, or its genetic silencing, has been shown to restore them [190]. Association between ROS and cholesterol in IPF pathogenesis has also been reported in a recent paper indicating the ability of the antioxidant Epigallocatechin Gallate (EGCG) to affect the expression of genes involved in cholesterol synthesis and cholesterol-associated metabolic processes [191]
Mitochondrial DNA (mtDNA) is also emerging as an important player in IPF pathogenesis. As apoptosis is a key step in fibrosis, the lack of appropriate mtDNA repair mechanisms makes the mitochondrion an important trigger of apoptotic-associated fibrogenesis [166, 192]. Exacerbated lung fibrosis has been detected in several animal models with altered mtDNA repair mechanisms, making this phenomenon more than an indicator of excessive oxidative stress [193, 194]. Secreted mtDNA, as well as its putative receptor TLR9, resulted increased in many fibrotic lung diseases and positively correlated with increased mortality [195]. Therapies targeting mitochondria with organ-specific drugs are under development. In this regard, MitoQ, an antioxidant that targets mitochondria, has been shown to decrease the expression of TGF-β1 and NOX4 in IPF pulmonary fibroblasts [154]. Further research in mitochondrial biology might lead to the identification of new druggable cellular and molecular targets (Fig. 4).
Endoplasmic reticulum stress
In physiological conditions, a cell produces up to 4 × 106 proteins per minute, and the role of the ER is to fold and process at least one-third of these proteins [196]. The ER coordinates processing, folding, assembly and protein trading, as well as the degradation of faulty proteins. Its function is regulated by a combination of factors including protein load, cell metabolism, redox balance, and calcium homeostasis. Alterations in any of these factors can lead to ER stress, and activate unopened protein reaction (UPR) [197,198,199,200,201,202,203,204,205,206]. Numerous proteins are related to UPR, including ER proteins, such as Grp78 (BiP), ER kinase-like PKR (PERK), ATP-4, and ATF-6 [207]. UPR tends to limit protein penetration into the ER, facilitating both the folding and the degradation of ER proteins and protecting from cell death [207]. When ER stress occurs due to prolonged or excessive protein mismatch, the process of apoptosis through the Bcl-2 signaling pathway is activated and cell death occurs [207].
The onset and progression of fibrotic diseases, including IPF, have been linked to ER stress (Fig. 4). Gene mutations affecting the surfactant protein A2 coding (SP-A2, SFTPA2) lead to the production of proteins that are unable to be secreted. This leads to their accumulation in the cytoplasm and the activation of ER-association degradation pathways with increased EC stress. This phenomenon has been reported in A549 cells and primary type II alveolar epithelial cells [208]. Mutation in the Surfactant Protein C gene has also been associated with Familial Interstitial Lung Disease [209]. Gene mutations affecting the C-terminal domain of Surfactant Protein C leads to both ER aberrant processing and toxic intracellular accumulation, ultimately promoting alveolar epithelial cell injury [210], a pivotal trigger in the development and progression of IPF [211].
Furthermore, both ER stress and unfolded protein response (UPR) have been reported to be associated with pulmonary fibrosis through AEC apoptosis, EMT, altered myofibroblast differentiation, and M2 macrophage polarization [207, 212, 213]. Studies on IPF lung samples showed increased staining of the ER stress- and UPR activation-associated proteins ER chaperone immunoglobulin heavy-chain-binding protein (BiP), X-box-binding protein 1 (XBP-1), and ER degradation enhancing α-mannosidase-like protein (EDEM) in epithelial cells, in comparison with healthy control lung tissues [214]. In addition, expression of the pro-SP-C mutant protein in A549 cells increased the expression of BiP, demonstrating the presence of ER stress in these cells due to improper fold of the pro- SP-C protein [215, 216]. A relationship between ER stress and aging has also been reported in vitro, and seems to be related to an increase in p16 and p21 in lung epithelial cells in older IPF patients [217,218,219,220].
Cellular plasticity
Cellular "plasticity", the ability of some cells to acquire the characteristics of other cells, is usually manifested under specific conditions, such as tissue regeneration, restoration and fibrosis, and involves particular cell types, including epithelial, endothelial, and mesenchymal cells. Alterations in cellular plasticity, associated with the onset and progression of IPF, can be broadly classified in EMT and EndMT.
Epithelial-mesenchymal transition (EMT)
Since the 1980s, EMT has been recognized as one of the key mechanisms involved in fibrosis in IPF. EMT consists in the loss of a subset of epithelial proteins, such as E-Cadherin, which leads to the conversion of epithelial cells into a mesenchymal phenotype characterized by a different milieu of specific proteins including N-Cadherin [221,222,223,224,225], Vimentin, and Fibronectin, as well as fibroblast transcription factors, such as Snail, Slug and Twist [226].
When tissues are damaged by various insults (i.e. viruses, bacteria, parasites, toxic environmental particles), a series of immunity activating signals are produced, leading to inflammation and promoting EMT. Immune cells, such as macrophages, neutrophils, and eosinophils, are recruited in this process and release cytokines and growth factors to maintain inflammation and tissue repair [227,228,229,230,231,232]. However, when this process is sustained, the development of a chronic inflammatory state enhances and prolongs EMT through increased fibroblast proliferation. In addition, activated fibroblasts stimulate the production of myofibroblasts as well as an excessive amount of mediators that favor extracellular matrix deposition. The accumulation of fibrous tissue progressively replaces functionally normal tissue, resulting in a gradual functional and structural dysfunction of the lung parenchyma [233,234,235].
Numerous cytokines and chemokines involved in the pathogenesis of IPF, included those mentioned before, also play a role in EMT. In particular, TGF-β1 favors EMT by directly activating a series of signaling pathways including Smad, ERK/MAP kinases, and networks involving microRNAs, through the binding with type I and type II TGF receptors (TBR-I and TBR-II). This triggers a series of intracellular signals that ultimately stimulate the transcription of Snail and Twist, which inhibit endothelial markers production and activates mesenchymal markers expression (Fig. 5a–b). As a consequence, there is a loss of the integrity of the epithelium, resulting from alterations of the tight junctions and the intercellular matrix [16, 236,237,238,239,240]. A wide range of additional mediators that trigger EMT through TGF-β1 activation have been described, including hypoxia-mediated ROS, Fibroblast Growth Factor-2 (FGF-2), Epidermal Growth Factor (EGF), CTGF and Transglutaminase 2 (TG2) [241,242,243,244] (Fig. 1). This network of mediators is essential for several cellular processes involved in EMT, including proliferation, cell differentiation and apoptosis (Fig. 5a–b).

a Transforming growth factor-β (TGF-β) signaling. Schematic representation of the cellular and molecular events involved in EMT (b) and EndMT (c). (Red arrows indicate activation, black lines indicate inhibition)
Previous studies have shown that AEC could differentiate into fibroblasts/myofibroblasts increasing fibrotic tissue deposition and that EMT contributes to fibroblast accumulation and consequent development of pulmonary fibrosis in animal models [245,246,247,248]. Studies in models of fibrosis secondary to bleomycin treatment or TGF-β1 overexpression have reported decreased expression of E-cadherin and C-surfactant protein and increased expression of α-smooth muscle actin (α-SMA) and S100A4 [244, 248]. In the study by Tanjore et al., approximately one-third of fibroblasts in mice treated with bleomycin derived from the pulmonary epithelium and stained consistently for S100A4, an EMT-related protein. By contrast, only a few S100A4-positive cells were detected in the lung epithelium of untreated mice [248].
In vitro studies suggest a synergistic effect of TGF-β1 and EGF in stimulating EMT [248]. Immunohistochemical methods have been also used to study the role of the Wnt/β-catenin signaling pathway in IPF [249]. Furthermore, the different expression levels of S100A4 in fibroblasts in several in vivo models suggest that different fibroblast cell subtypes and myofibroblasts coexist during the development of pulmonary fibrosis [248, 250].
Recent studies have investigated the effects of anti-fibrotic drugs, such as tannic and triptolide acid, to better understand the molecular mechanisms underlying IPF. Tannic acid caused TGF-β1 overexpression and a reduction in smad2 and Smad3 phosphorylation. This led to the reversal of the morphological changes observed in epithelial cell cultures. Other medications have been tested, including thalidomide (inhibits Smad pathways), Pirfenidone (inhibits myofibroblast differentiation through inhibition of ROS production, PDGFR-PI3K and Akt) and Tubastatin (inhibits HDAC6 and TGF-β1-PI3K-AKT signaling) [251,252,253,254,255]. Nevertheless, the clinical effectiveness of most of these medications has been shown to be poor.
Endothelial-mesenchymal transition (EndMT)
As previously discussed, both an increase in local fibroblasts and their differentiation in myofibroblasts have been shown to favor the progression of fibrosis. Compared to fibroblasts, myofibroblasts upregulate the expression of α-SMA, and increase the production of extracellular matrix proteins, such as collagen type I, III, V and VI [256, 257]. Furthermore, expression of tissue inhibitors of metalloproteinases (TIMPs) is increased by myofibroblasts, leading to reduced extracellular matrix degradation [257, 258]. Experimental evidence showed that myofibroblasts can originate from cell sources other than fibroblasts. In particular, endothelial cells (ECs) are a potential source acting through EndMT. During this process, ECs acquire a mesenchymal phenotype and present typical markers of myofibroblast differentiation, such as α-SMA, vimentin and collagens, while reducing the expression of vascular endothelial cadherin (VE-cadherin) [259]. It has been reported that 16% of fibroblasts expressing α-SMA and collagen type I were derived from the EC in lungs of mice with bleomycin-induced IPF [259].
The importance of EndMT in experimental fibrosis has been demonstrated both in vitro and in vivo. These studies showed that TGF-β has a central role in promoting EndMT, through a wide network of molecular interactions (Fig. 5a, c). Signals from TGF-β are mediated through Smad transcription modifiers [260, 261]. TGF-β can also activate factors of the mitogen-activated protein kinase (MAPK) pathways, such as the extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK) and c-Jun-N-terminal kinase (JNK) [262, 263]. MAPK-mediated TGF-β responses can be Smad-dependent or independent [262]. TGF-β-induced ERK activation can either potentiate or interfere with Smad signaling [264]. By contrast, p38 MAPK and JNK usually potentiate TGFβ/Smad-induced responses [265, 266]. TGF-β was shown to activate PI3 kinase/Akt and Rho GTPase pathways [263]. Moreover, TGF-β signaling exerts its effects by interacting with other signaling cascades, including Wnt and Notch (Fig. 5a, c) [267,268,269].
The co-activation of the RAS and TGF-β signaling cascades could favor EndMT in pulmonary ECs, with an increase in fibronectin and type I collagen expression [270]. Caveolin-1 (CAV-1) is the main protein component of caveolae, associated with TGF-β receptors localization, trafficking and degradation, and plays an important role in the pathogenesis of different fibrosing diseases. A study by Jimenez et al. investigated the role of CAV-1 in EndMT caused by TGF-β in mice [271]. The authors found that mice lacking CAV-1 exhibited EndMT in pulmonary ECs, which was demonstrated by expression of α-SMA marker, high production level of collagen type I, and high expression of Snail and Slug. Accordingly, spontaneous EndMT and TGF-β1 were reversed by restoring functional CAV-1 [141, 271, 272].
Choi et al. found increased EndMT in blood vessels due to hypoxic injury in radiation-induced pulmonary fibrosis [272]. In this study, EMT was also observed in alveolar epithelial cells, but only after the occurrence of EndMT. In human pulmonary arterial ECs, radiation-induced HIF-1α-dependent EndMT through activation of TGFβ-R1/Smad signals has been also detected (Fig. 5a) [273, 274].
Hypoxia
One feature of many pathological fibrotic disorders is the decreased oxygen availability in the cellular environment, a condition known as hypoxia [275,276,277]. By activating the hypoxia-inducible factors (HIFs), a set of transcription factors that respond to low oxygen tension, the hypoxic environment is able to modulate the expression of several genes [278, 279]. So far, three HIFs isotypes, HIF1, HIF2 and HIF3 have been identified. Each of them is formed by one subunit beta (β) and one alpha (α), with the latter being more sensible to oxygen levels and prominently implicated in disease development [278, 280].
Several studies have shown a correlation between hypoxia and IPF, mediated by signaling mechanisms triggered by the activation of HIFs (Fig. 5).[281,282,283,284]. These include the augmented expression of myofibroblast differentiation markers, such as αSMA and β-actin, in response to increased levels of HIF-1α and HIF-2α in human normal fibroblasts exposed to hypoxic conditions and in lung tissue from IPF patients [284]. Noteworthy, in the same model, HIF-3α expression was decreased, probably as consequence of its hypermethylation, which negatively regulates the hypoxia signaling pathway [284, 285]. In a mice model of bleomycin-induced fibrosis, HIF-1α knockout reduced lung fibrosis and alveolar epithelial cells proliferation [283]. Comparative Expression Profiling experiments performed in the lung of bleomycin-induced mice fibrosis showed significant overexpression of several HIF-1 targets, including VEGF-A, VEGF-C; Siah1, Flt1, Bnip3l and Bhlhb2 [281]. Similarly, high expression levels of HIF-1α and its targets VEGF and p53 have been reported in lung tissue from patients with IPF or cryptogenic organizing pneumonia [281].
The molecular mechanisms involved in HIFs signaling have also been investigated. Activation and nuclear translocation of the transcription factor Nuclear Factor Activated T cell (NAFT) has been reported to be an essential mechanism of the hypoxia signaling pathway in human lung fibroblasts [286]. Hypoxia-induced IPF fibroblast proliferation has been reported to be driven by the miR-210-mediated overexpression of c-Myc [282]. Interestingly, while moderate hypoxia (2% O2) increases human lung fibroblasts proliferation in a p21-dependent and a p53-independent fashion, a more severe hypoxic environment (0.1% O2) leads to cell cycle arrest through both p53 and p21 [287].
The interaction between hypoxia and other factors involved in IPF development has also been reported. For instance, hypoxia and TGF-β act synergistically in regulating a number of mRNAs and lncRNAs in lung fibroblasts, potentially contributing to IPF pathogenesis [288]. Compared to wild-type mice, bleomycin-treated mice deficient of the ER stress-regulated transcription factor C/EBP homologous protein (CHOP) are protected from the exacerbated lung fibrosis elicited by hypoxia [289]. In vitro experiments revealed the involvement of the inositol-requiring enzyme 1α (IRE1α) and the PKR-like ER kinase (PERK) pathways in CHOP-mediated AECs apoptosis [289]. Moreover, both CHOP and hypoxia markers, such as HIF1α, carbonic anhydrase IX, and pyruvate kinase, were found to be increased in the lung tissue of IPF patients when compared to controls [289].
The link between hypoxia and IPF and the underpinning mechanisms controlling this interaction require further investigation. Pending further research addressing this issue, therapeutic strategies ameliorating the hypoxic environment by targeting specific regulating mechanisms might be useful in IPF.
Hyaluronan and other glycosaminoglycans
IPF is characterized by an extensive cellular and molecular restructure of the lung tissue that primarily involves components of the extracellular matrix (ECM), such as collagens, glycoproteins, and proteoglycans, ultimately resulting in the formation of fibrotic tissue and decrease of lung function [290]. ECM plays an essential role in modulating lung structure and function [291, 292], and its alteration can result in lung disease [291, 293, 294]. The latter can also be favored by an abnormal interaction between ECM components, growth factors, such as TGF-β and connective tissue growth factor (CTGF), and lung epithelial, alveolar and fibroblast cells [295, 296].
The role of specific ECM components, particularly the glycosaminoglycans (GAG), in IPF onset and progression is still under investigation. There are two main types of GAGs in the EMC, the non-sulfated GAG hyaluronic acid (HA) and the sulfated GAG (heparan sulphate, heparin, chondroitin sulphate, dermatan sulphate and keratan sulphate) [297]. GAGs play a key role in modulating pulmonary function [297]. Their increased deposition, reported in IPF subjects [298], can aggravate lung inflammation and contribute to disease progression [299].
A number of studies have investigated the association between GAG and IPF. HA synthase 2 (HAS2) deletion affect AECs renewal capacity in vitro, prompting severe fibrosis and mortality in vivo [300]. Furthermore, reduced levels of cell membrane HA and impaired renewal capacity have been observed in AECs from subjects with severe lung fibrosis [300]. Lung fibroblasts appear to be the primary source of HA production. The latter, has been found to be increased in IPF patients when compared to control subjects [301]. Inhibition of HS synthesis ameliorates IPF-associated conditions including pulmonary hypertension [302]. Interestingly, HA-regulated signals in IPF might be mediated by ER stress as the latter appears to be responsible for the regulation of HA secretion in airway epithelial cells [303]. Fibroblasts derived from IPF subjects show an HAS2-dependent invasive phenotype while myofibroblasts overexpressing HAS2 display an aggressive fibrotic phenotype [304]. Further, through the p27-CDK2-SKP2 signaling pathway, HAS2 regulates fibroblast senescence, a phenomenon strongly implicated in the fibrotic process in IPF [305].
The HS-associated effects in IPF appear to be mediated by its primary receptor CD44 as the blockage of this signaling pathway can revert or counteract several HS-induced phenomena, including the development of a cell fibrotic/invasive phenotype (Fig. 6) [304]. In a bleomycin-induced model of fibrosis, CD44 deficient mice, or mice treated with a CD44 blocking antibody, display both reduced lung fibrosis and fibroblast/myofibroblast invasion ability [304]. Similarly, targeting CD44 with monoclonal antibodies has been shown to prevent the migration/invasion of fibroblasts derived from subjects with acute alveolar fibrosis [306]. In vivo, IPF lungs of bleomycin-exposed rat display an up-regulated expression of CD44 receptors [307]. Additionally, as a source of altered fibroblasts in IPF, the CD44/HA signaling pathway appears to play a pivotal role in IPF onset and development [308,309,310].

GAGs-activated molecular and cellular events associated with IPF pathogenesis (red lines indicate activation, blue lines indicted bidirectional interplay)
The increased deposition, or altered structure, of other GAGs, such as heparan, chondroitin, and dermatan sulfate, has also been reported in IPF [298, 311, 312]. In this context, heparan sulphate has been reported to mediate the binding of Coagulation factor XII to IPF fibroblasts, promoting a heparinase-inhibitable cell migration [313].
Therefore, recent studies have highlighted the role of ECM in regulating lung structure and functions, paving the way for the discovery of new therapeutic strategies targeting ECM-driven processes in IPF.
Alternative polyadenylation
Alternative polyadenylation (APA) is a widespread mechanism by which eukaryotic cells regulate gene expression ultimately generating distinct 3′ ends in different transcripts created by the RNA polymerase II [314]. APS is one of the most important post-transcriptional regulatory mechanisms involved in mRNA metabolism modulation, and its dysregulation has been implicated in many disease states [315,316,317]. A number of studies have reported a link between APA and fibrotic conditions including IPF [318,319,320,321]. Recently, Zhou et al. reported that matrix stiffness induces cell fibrotic responses by a signaling mechanism involving the modulation of APA by the mammalian cleavage factor I (CFIm) [318]. These authors demonstrated that a stiff matrix acts as a down-regulator of the CFIm subunits CFIm68, CFIm59 and CFIm25, promoting the APA-dependent up regulation of collagen and fibronectin production in primary human lung fibroblasts [318]. In vivo experiments have also reported a strong association between matrix stiffness and CFIm subunits in a bleomycin-induced mouse model of pulmonary fibrosis [318]. By regulating APA, the CFIm subunit CFIm25 appears to play an essential role in fibroblast-to-myofibroblast differentiation [321]. CFIm25 has been found to be decreased in the lungs of human and mice harboring pulmonary fibrosis and its proteins levels are downregulated in IPF fibroblasts [321]. CFIm25 silencing in normal lung fibroblast induced the expression of fibrotic markers by a mechanism involving APA-mediated 3′-UTR shortening while its overexpression reverted the same phenomena in IPF lung fibroblast [321]. The association between APA and lung fibrosis is also supported by the link between the pro-fibrotic factor TGFβ1 and CFIm25 [320]. Treatment of primary human lung fibroblasts with TGFβ1 downregulates CFIm25 with a mechanism that implicates miR203. In vivo experiments using TGFβ1 transgenic mice confirmed the TGFβ1-mediated inhibition of CFIm25 [320]. APA analysis in TGFβ1-treated human lung fibroblasts revealed a differential mRNA transcript regulation with APA target genes mostly linked to pro-fibrotic-associated pathways.[320].
Further studies are needed to elucidate the role of APA in the IPF pathogenesis and its potential therapeutic applications.
Epithelial cell migration
The presence of mechanisms involving the reduced ability to restore lung tissue chronic micro-injury due to impairment of epithelial cell migration abilities has also been hypothesized in IPF. It has been reported that epithelial-specific deletion of the focal adhesion kinase (FAK), which is necessary for cell migration, leads to epithelial cell apoptosis and enhanced bleomycin injury, while enhancement of the Notch signaling pathway leads to functional impairment of Tp63-expressing stem cells in the bronchiolar epithelium; these cells proliferate and migrate to areas of denuded alveolar epithelium to restore lung damage [322]
Non-coding RNAs in extracellular matrix remodeling and fibrosis
Recently, studies have shown that microRNAs (miRNAs), small non-coding RNAs (21–25 nucleotides) that bind by base pairing to the 3′ untranslated region of their target mRNAs, play important roles in the evolution of IPF. Several miRNAs showed altered concentrations in blood and lung samples from IPF patients both during early and advanced disease stages. Those miRNAs were generally associated with one or more pathophysiological mechanisms of IPF, including EMT, fibroblast activation, myofibroblast differentiation, pulmonary epithelium repair, macrophage polarization, AEC senescence, and collagen production [323]. Milosevic et al. identified 43 significantly up-regulated microRNAs in IPF lungs [324]. Twenty four of these 43 microRNAs were localized in the chromosome 14q32 microRNA cluster. The authors validated the increased expression of miR-154, miR-134, miR-299-5p, miR-410, miR-382, miR-409-3p, miR-487b, miR-31, and miR-127 by quantitative RT-PCR. Furthermore, they showed that transfection with miR-154 caused activation of the WNT pathway in normal human lung fibroblasts. In another study, miR-29 was downregulated in mice lungs during remission of bleomycin-induced fibrosis [325]. mir-29b is a key suppressor of many downstream target genes involved in fibrogenesis, including COL1A1, COL3A1, and FBN1, which are regulated by the TGF-b/smad3 pathway, and transfection with miR-29 mimics and miR-29 inhibitors suggests that miR-29 regulates the expression of mortality receptor Fas, leading to apoptosis [326, 327]. Other miRNAs, such as let-7, mir199, mir21, mir-31, mir200, and mir17-92 cluster [328], have been found altered in IPF studies, and active research testing a large number of further molecules is still ongoing. Several miRNAs also appear to contribute to the pathogenesis of IP by inducing the trans-differentiation of lung cells, such us fibroblast, epithelial and endothelial cells (Fig. 7) [329].

miRNAs-activated molecular and cellular events associated with IPF pathogenesis (red lines indicate activation)
Another class of non-coding RNAs currently under investigation in the pathogenesis of IPF is the long non-coding RNAs (lncRNAs). LncRNAs are greater than 200 nucleotide long molecules which attracted increasing attention in recent years as studies reported their crucial roles in regulating embryogenesis, stem cell biology, development and cancer [330]. Only a few studies have been published to date investigating lncRNAs in IPF. A microarray study by Cao et al. using the bleomycin-induced lung fibrosis rat model was one of the first to identify differential expression levels of multiple lncRNAs and mRNAs, most significantly lncRNAs AJ005396 and S69206 [331]; in situ hybridization confirmed the expression of these lncRNAs and located their expression in the cytoplasm of interstitial lung cells. Hadjicharalambous et al. used next-generation sequencing to identify 14 lncRNAs that are differentially expressed in human lung fibroblasts following the induction of inflammation using interleukin-1β [332]. Knockdown of the two most highly expressed lncRNAs, IL7AS, and MIR3142HG, showed that IL7AS negatively regulated IL-6 release whilst MIR3142HG was a positive regulator of IL-8 and CCL2 release, supporting their potential role in regulating the inflammatory response in IPF.
Lu et al. showed that the expression of lnc RNA H19 was significantly increased in transforming growth factor-β (TGF-β)-induced fibroblast proliferation and bleomycin (BLM)-induced lung fibrosis (p < 0.05) [333]. The authors also showed that H19 was a direct target of miR-196a and was associated with COL1A1 expression by sponging miR-196a. Moreover, downregulation of H19 alleviated fibroblast activation and lung fibrosis, and this effect was blocked by a miR-196a inhibitor. The expression of lncRNA NONMMUT065582, also known as pulmonary fibrosis-associated RNA (PFAR), was found to be elevated in the fibrotic lungs of mice as well as mouse fibroblasts [334]. The LncRNA, PFAR, was shown to promote the development of fibrosis by acting as ceRNA for miR-138 and regulating the expression of yes-associated protein 1 (YAP1). Other lnc RNAs, MRAK088388, MRAK081523, CD99P1, n341773, CHRF, IL7AS and MIR3142GHG, have also been associated with IPF [335]
Conclusion and future perspectives
IPF is a lethal disease of unknown etiology. Numerous pathophysiological factors are implied in the genesis and progression of the disease, and most of them are not yet fully elucidated. Medications currently employed in clinical practice display therapeutic effects that target most of the pathophysiological processes involved in IPF, especially, aging, oxidative stress and EMT. Senescence and apoptosis, stimulated by a large number of mediators, including several cytokines and growth factors, such as interleukines 1β, 6 and 8 and TGF-β, play a central role. Several new drugs under evaluation, e.g. bosentan, macitentan, ambrisentan, interferon, sildenafil, TNF-α inhibitors and imatinib mesylate, target cytokines or chemokines. Similarly, oxidative stress is essential for the development of IPF, and has been the target, albeit with poor results to date, of therapeutic agents, such as N-acetylcysteine. Further research is likely to identify novel therapeutic agents, to use singly or in combination with anti-fibrotic agents, to improve clinical outcomes. Promising results have been reported with non-coding RNAs, however, translation into clinical use is premature. Interestingly, targeted therapies used in lung cancer patients have shown promising results in IPF; this is a noteworthy observation, given the epidemiological association between lung cancer and IPF patients, which is likely to stimulate additional studies to identify the cellular and molecular mechanisms underpinning these conditions.
References
Jones MG, Fabre A, Schneider P, Cinetto F, Sgalla G, Mavrogordato M, Jogai S, Alzetani A, Marshall BG, O’Reilly KM, Warner JA, Lackie PM, Davies DE, Hansell DM, Nicholson AG, Sinclair I, Brown KK, Richeldi L (2016) Three-dimensional characterization of fibroblast foci in idiopathic pulmonary fibrosis. JCI Insight. https://doi.org/10.1172/jci.insight.86375
King TE Jr, Pardo A, Selman M (2011) Idiopathic pulmonary fibrosis. Lancet (London, England) 378(9807):1949–1961. https://doi.org/10.1016/s0140-6736(11)60052-4
Wolters PJ, Collard HR, Jones KD (2014) Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol 9:157–179. https://doi.org/10.1146/annurev-pathol-012513-104706
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G (2006) Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 174(7):810–816. https://doi.org/10.1164/rccm.200602-163OC
Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr, Lasky JA, Loyd JE, Noth I, Olman MA, Raghu G, Roman J, Ryu JH, Zisman DA, Hunninghake GW, Colby TV, Egan JJ, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kondoh Y, Lynch DA, Muller-Quernheim J, Myers JL, Nicholson AG, Selman M, Toews GB, Wells AU, Martinez FJ (2007) Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 176(7):636–643. https://doi.org/10.1164/rccm.200703-463PP
Kottmann RM, Hogan CM, Phipps RP, Sime PJ (2009) Determinants of initiation and progression of idiopathic pulmonary fibrosis. Respirology 14(7):917–933. https://doi.org/10.1111/j.1440-1843.2009.01624.x
Somogyi V, Chaudhuri N, Torrisi SE, Kahn N, Müller V, Kreuter M (2019) The therapy of idiopathic pulmonary fibrosis: what is next? Eur Resp Rev 28(153):190021
Garcia-Sancho C, Buendia-Roldan I, Fernandez-Plata MR, Navarro C, Perez-Padilla R, Vargas MH, Loyd JE, Selman M (2011) Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir Med 105(12):1902–1907. https://doi.org/10.1016/j.rmed.2011.08.022
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183(6):788–824. https://doi.org/10.1164/rccm.2009-040GL
Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA (1997) Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 155(1):242–248. https://doi.org/10.1164/ajrccm.155.1.9001319
Baumgartner KB, Samet JM, Coultas DB, Stidley CA, Hunt WC, Colby TV, Waldron JA (2000) Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am J Epidemiol 152(4):307–315. https://doi.org/10.1093/aje/152.4.307
Miyake Y, Sasaki S, Yokoyama T, Chida K, Azuma A, Suda T, Kudoh S, Sakamoto N, Okamoto K, Kobashi G, Washio M, Inaba Y, Tanaka H (2005) Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Ann Occup Hyg 49(3):259–265. https://doi.org/10.1093/annhyg/meh090
Kärkkäinen M, Kettunen H-P, Nurmi H, Selander T, Purokivi M, Kaarteenaho R (2017) Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir Res 18(1):160. https://doi.org/10.1186/s12931-017-0642-6
Garcia-Sancho Figueroa MC, Carrillo G, Perez-Padilla R, Fernandez-Plata MR, Buendia-Roldan I, Vargas MH, Selman M (2010) Risk factors for idiopathic pulmonary fibrosis in a Mexican population. A case-control study. Resp Med 104(2):305–309. https://doi.org/10.1016/j.rmed.2009.08.013
Antoniou KM, Hansell DM, Rubens MB, Marten K, Desai SR, Siafakas NM, Nicholson AG, du Bois RM, Wells AU (2008) Idiopathic pulmonary fibrosis: outcome in relation to smoking status. Am J Respir Crit Care Med 177(2):190–194. https://doi.org/10.1164/rccm.200612-1759OC
Milara J, Peiró T, Serrano A, Guijarro R, Zaragozá C, Tenor H, Cortijo J (2014) Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther 28(2):138–148. https://doi.org/10.1016/j.pupt.2014.02.001
Huzen J, Wong L, Van Veldhuisen D, Samani N, Zwinderman A, Codd V, Cawthon R, Benus G, Van Der Horst I, Navis G (2014) Telomere length loss due to smoking and metabolic traits. J Intern Med 275(2):155–163
Tanjore H, Blackwell TS, Lawson WE (2012) Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol-Lung Cell Mol Physiol 302(8):L721–L729
Zaman T, Lee JS (2018) Risk factors for the development of idiopathic pulmonary fibrosis: a review. Curr Pulmonol Rep 7(4):118–125. https://doi.org/10.1007/s13665-018-0210-7
Mohning MP, Swigris JJ, Olson AL (2019) Idiopathic pulmonary fibrosis: the epidemiology and natural history of disease. In: Idiopathic pulmonary fibrosis. Springer, pp 11–35
American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001 (2002) Am J Resp Crit Care Med 165 (2):277–304. https://doi.org/10.1164/ajrccm.165.2.ats01
Baumgartner KB, Samet JM, Coultas DB, Stidley CA, Hunt WC, Colby TV, Waldron JA, Centers C (2000) Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Am J Epidemiol 152(4):307–315
Hubbard R, Cooper M, Antoniak M, Venn A, Khan S, Johnston I, Lewis S, Britton J (2000) Risk of cryptogenic fibrosing alveolitis in metal workers. Lancet (London, England) 355(9202):466–467. https://doi.org/10.1016/S0140-6736(00)82017-6
Scott J, Johnston I, Britton J (1990) What causes cryptogenic fibrosing alveolitis? A case-control study of environmental exposure to dust. BMJ (Clinical research ed) 301(6759):1015–1017. https://doi.org/10.1136/bmj.301.6759.1015
Hubbard R, Lewis S, Richards K, Johnston I, Britton J (1996) Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet (London, England) 347(8997):284–289. https://doi.org/10.1016/s0140-6736(96)90465-1
Iwai K, Mori T, Yamada N, Yamaguchi M, Hosoda Y (1994) Idiopathic pulmonary fibrosis Epidemiologic approaches to occupational exposure. Am J Resp Crit Care Med 150(3):670–675. https://doi.org/10.1164/ajrccm.150.3.8087336
Pinheiro GA, Antao VC, Wood JM, Wassell JT (2008) Occupational risks for idiopathic pulmonary fibrosis mortality in the United States. Int J Occup Environ Health 14(2):117–123. https://doi.org/10.1179/oeh.2008.14.2.117
Gustafson T, Dahlman-Höglund A, Nilsson K, Ström K, Tornling G, Torén K (2007) Occupational exposure and severe pulmonary fibrosis. Respir Med 101(10):2207–2212
Pearson JE, Wilson RS (1971) Diffuse pulmonary fibrosis and hiatus hernia. Thorax 26(3):300–305. https://doi.org/10.1136/thx.26.3.300
Mays EE, Dubois JJ, Hamilton GB (1976) Pulmonary fibrosis associated with tracheobronchial aspiration. A study of the frequency of hiatal hernia and gastroesophageal reflux in interstitial pulmonary fibrosis of obscure etiology. Chest 69(4):512–515. https://doi.org/10.1378/chest.69.4.512
Pellegrini CA, DeMeester TR, Johnson LF, Skinner DB (1979) Gastroesophageal reflux and pulmonary aspiration: incidence, functional abnormality, and results of surgical therapy. Surgery 86(1):110–119
Lee JS, Ryu JH, Elicker BM, Lydell CP, Jones KD, Wolters PJ, King TE Jr, Collard HR (2011) Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 184(12):1390–1394
Lee JS, Collard HR, Anstrom KJ, Martinez FJ, Noth I, Roberts RS, Yow E, Raghu G, Investigators I (2013) Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Resp Med 1(5):369–376
Savarino E, Bazzica M, Zentilin P, Pohl D, Parodi A, Cittadini G, Negrini S, Indiveri F, Tutuian R, Savarino V, Ghio M (2009) Gastroesophageal reflux and pulmonary fibrosis in scleroderma: a study using pH-impedance monitoring. Am J Respir Crit Care Med 179(5):408–413. https://doi.org/10.1164/rccm.200808-1359OC
Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, Brozek JL, Collard HR, Cunningham W, Homma S, Johkoh T, Martinez FJ, Myers J, Protzko SL, Richeldi L, Rind D, Selman M, Theodore A, Wells AU, Hoogsteden H, Schunemann HJ (2015) An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the (2011) Clinical practice guideline. Am J Respir Crit Care Med 192(2):e3-19. https://doi.org/10.1164/rccm.201506-1063ST
Allaix ME, Rebecchi F, Morino M, Schlottmann F, Patti MG (2017) Gastroesophageal reflux and idiopathic pulmonary fibrosis. World J Surg 41(7):1691–1697. https://doi.org/10.1007/s00268-017-3956-0
Kreuter M, Wuyts W, Renzoni E, Koschel D, Maher TM, Kolb M, Weycker D, Spagnolo P, Kirchgaessler K-U, Herth FJ (2016) Antacid therapy and disease outcomes in idiopathic pulmonary fibrosis: a pooled analysis. Lancet Resp Med 4(5):381–389
Klein OL, Kalhan R, Williams MV, Tipping M, Lee J, Peng J, Smith LJ (2012) Lung spirometry parameters and diffusion capacity are decreased in patients with Type 2 diabetes. Diabet Med 29(2):212–219. https://doi.org/10.1111/j.1464-5491.2011.03394.x
Irfan M, Jabbar A, Haque A, Awan S, Hussain S (2011) Pulmonary functions in patients with diabetes mellitus. Lung India 28(2):89–92. https://doi.org/10.4103/0970-2113.80314
Klein O, Krishnan J, Glick S, Smith LJ (2010) Systematic review of the association between lung function and Type 2 diabetes mellitus. Diabet Med 27(9):977–987
Enomoto T, Usuki J, Azuma A, Nakagawa T, Kudoh S (2003) Diabetes mellitus may increase risk for idiopathic pulmonary fibrosis. Chest 123(6):2007–2011. https://doi.org/10.1378/chest.123.6.2007
Matsuse T, Ohga E, Teramoto S, Fukayama M, Nagai R, Horiuchi S, Ouchi Y (1998) Immunohistochemical localisation of advanced glycation end products in pulmonary fibrosis. J Clin Pathol 51(7):515–519. https://doi.org/10.1136/jcp.51.7.515
Chen L, Wang T, Wang X, Sun BB, Li JQ, Liu DS, Zhang SF, Liu L, Xu D, Chen YJ, Wen FQ (2009) Blockade of advanced glycation end product formation attenuates bleomycin-induced pulmonary fibrosis in rats. Respir Res 10(1):55. https://doi.org/10.1186/1465-9921-10-55
Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J, Saito N, Fujita Y, Kurita Y, Kobayashi K, Ito S, Hara H, Kadota T, Yanagisawa H, Hashimoto M, Utsumi H, Wakui H, Kojima J, Numata T, Kaneko Y, Odaka M, Morikawa T, Nakayama K, Kohrogi H, Kuwano K (2016) Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res 17(1):107. https://doi.org/10.1186/s12931-016-0420-x
Spagnolo P, Kreuter M, Maher TM, Wuyts W, Bonella F, Corte TJ, Kopf S, Weycker D, Kirchgaessler K-U, Ryerson CJ (2018) Metformin does not affect clinically relevant outcomes in patients with idiopathic pulmonary fibrosis. Respiration 96(4):314–322
Leslie KO (2012) Idiopathic pulmonary fibrosis may be a disease of recurrent, tractional injury to the periphery of the aging lung: a unifying hypothesis regarding etiology and pathogenesis. Arch Pathol Lab Med 136(6):591–600
Ueda T, Ohta K, Suzuki N, Yamaguchi M, Hirai K, Horiuchi T, Watanabe J, Miyamoto T, Ito K (1992) Idiopathic pulmonary fibrosis and high prevalence of serum antibodies to hepatitis C virus. Am Rev Respir Dis 146(1):266–268. https://doi.org/10.1164/ajrccm/146.1.266
Arase Y, Suzuki F, Suzuki Y, Akuta N, Kobayashi M, Kawamura Y, Yatsuji H, Sezaki H, Hosaka T, Hirakawa M, Saito S, Ikeda K, Kumada H (2008) Hepatitis C virus enhances incidence of idiopathic pulmonary fibrosis. World J Gastroenterol 14(38):5880–5886. https://doi.org/10.3748/wjg.14.5880
Hayashi S, Hogg JC (2007) Adenovirus infections and lung disease. Curr Opin Pharmacol 7(3):237–243. https://doi.org/10.1016/j.coph.2006.11.014
Bando M, Ohno S, Oshikawa K, Takahashi M, Okamoto H, Sugiyama Y (2001) Infection of TT virus in patients with idiopathic pulmonary fibrosis. Respir Med 95(12):935–942. https://doi.org/10.1053/rmed.2001.1151
Wootton SC, Kim DS, Kondoh Y, Chen E, Lee JS, Song JW, Huh JW, Taniguchi H, Chiu C, Boushey H, Lancaster LH, Wolters PJ, DeRisi J, Ganem D, Collard HR (2011) Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 183(12):1698–1702. https://doi.org/10.1164/rccm.201010-1752OC
Kelly BG, Lok SS, Hasleton PS, Egan JJ, Stewart JP (2002) A rearranged form of Epstein-Barr virus DNA is associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 166(4):510–513
Lok SS, Stewart JP, Kelly BG, Hasleton PS, Egan JJ (2001) Epstein-Barr virus and wild p53 in idiopathic pulmonary fibrosis. Respir Med 95(10):787–791. https://doi.org/10.1053/rmed.2001.1152
Stewart JP, Egan JJ, Ross AJ, Kelly BG, Lok SS, Hasleton PS, Woodcock AA (1999) The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 159(4 Pt 1):1336–1341. https://doi.org/10.1164/ajrccm.159.4.9807077
Tang YW, Johnson JE, Browning PJ, Cruz-Gervis RA, Davis A, Graham BS, Brigham KL, Oates JA Jr, Loyd JE, Stecenko AA (2003) Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol 41(6):2633–2640. https://doi.org/10.1128/jcm.41.6.2633-2640.2003
Folcik VA, Garofalo M, Coleman J, Donegan JJ, Rabbani E, Suster S, Nuovo A, Magro CM, Di Leva G, Nuovo GJ (2013) Idiopathic pulmonary fibrosis is strongly associated with productive infection by herpesvirus saimiri. Mod Pathol 27:851. https://doi.org/10.1038/modpathol.2013.198
Dutia BM, Clarke CJ, Allen DJ, Nash AA (1997) Pathological changes in the spleens of gamma interferon receptor-deficient mice infected with murine gammaherpesvirus: a role for CD8 T cells. J Virol 71(6):4278–4283
Ebrahimi B, Dutia BM, Brownstein DG, Nash AA (2001) Murine gammaherpesvirus-68 infection causes multi-organ fibrosis and alters leukocyte trafficking in interferon-gamma receptor knockout mice. Am J Pathol 158(6):2117–2125. https://doi.org/10.1016/s0002-9440(10)64683-4
Bartholdy C, Hogh-Petersen M, Storm P, Holst PJ, Orskov C, Christensen JP, Thomsen AR (2014) IFNgamma and perforin cooperate to control infection and prevent fatal pathology during persistent gammaherpesvirus infection in mice. Scand J Immunol 79(6):395–403. https://doi.org/10.1111/sji.12176
Mora AL, Torres-Gonzalez E, Rojas M, Xu J, Ritzenthaler J, Speck SH, Roman J, Brigham K, Stecenko A (2007) Control of virus reactivation arrests pulmonary herpesvirus-induced fibrosis in IFN-gamma receptor-deficient mice. Am J Respir Crit Care Med 175(11):1139–1150. https://doi.org/10.1164/rccm.200610-1426OC
Bitterman PB, Adelberg S, Crystal RG (1983) Mechanisms of pulmonary fibrosis. Spontaneous release of the alveolar macrophage-derived growth factor in the interstitial lung disorders. J Clin Investig 72(5):1801–1813. https://doi.org/10.1172/jci111140
Selman M, King TE, Pardo A (2001) Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 134(2):136–151. https://doi.org/10.7326/0003-4819-134-2-200101160-00015
Coultas DB, Zumwalt RE, Black WC, Sobonya RE (1994) The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med 150(4):967–972. https://doi.org/10.1164/ajrccm.150.4.7921471
Fernandez Perez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, Yi ES, Ryu JH (2010) Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest 137(1):129–137. https://doi.org/10.1378/chest.09-1002
Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ (2006) Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 61(11):980–985. https://doi.org/10.1136/thx.2006.062836
Natsuizaka M, Chiba H, Kuronuma K, Otsuka M, Kudo K, Mori M, Bando M, Sugiyama Y, Takahashi H (2014) Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med 190(7):773–779. https://doi.org/10.1164/rccm.201403-0566OC
Navaratnam V, Fleming KM, West J, Smith CJ, Jenkins RG, Fogarty A, Hubbard RB (2011) The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax 66(6):462–467. https://doi.org/10.1136/thx.2010.148031
Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR (2016) Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Resp J 48(1):179–186. https://doi.org/10.1183/13993003.01653-2015
Ley B, Collard HR (2013) Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol 5:483–492. https://doi.org/10.2147/clep.s54815
Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB (2014) Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Ann Am Thorac Soc 11(8):1176–1185. https://doi.org/10.1513/AnnalsATS.201404-145OC
Harman D (2003) The free radical theory of aging. Antioxid Redox Signal 5(5):557–561. https://doi.org/10.1089/152308603770310202
López-Ramírez C, Suarez Valdivia L, Rodríguez Portal JA (2018) Causes of Pulmonary Fibrosis in the Elderly. Medical sciences (Basel, Switzerland) 6 (3). doi:https://doi.org/10.3390/medsci6030058
Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK (2007) Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA 104(18):7552–7557. https://doi.org/10.1073/pnas.0701009104
Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, Phillips JA 3rd, Lansdorp PM, Loyd JE, Armanios MY (2008) Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA 105(35):13051–13056. https://doi.org/10.1073/pnas.0804280105
Cronkhite JT, Xing C, Raghu G, Chin KM, Torres F, Rosenblatt RL, Garcia CK (2008) Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med 178(7):729–737. https://doi.org/10.1164/rccm.200804-550OC
Calhoun C, Shivshankar P, Saker M, Sloane LB, Livi CB, Sharp ZD, Orihuela CJ, Adnot S, White ES, Richardson A, Le Saux CJ (2016) Senescent cells contribute to the physiological remodeling of aged lungs. J Gerontol A 71(2):153–160. https://doi.org/10.1093/gerona/glu241
Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, Lin S, Huang L, Chung EJ, Citrin DE, Wang Y, Hauer-Jensen M, Zhou D, Meng A (2017) Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys 99(2):353–361. https://doi.org/10.1016/j.ijrobp.2017.02.216
Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Gunther A, Konigshoff M (2017) Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Resp J. https://doi.org/10.1183/13993003.02367-2016
Leung J, Cho Y, Lockey RF, Kolliputi N (2015) The role of aging in idiopathic pulmonary fibrosis. Lung 193(4):605–610. https://doi.org/10.1007/s00408-015-9729-3
Phadwal K, Watson AS, Simon AK (2013) Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol Life Sci 70(1):89–103. https://doi.org/10.1007/s00018-012-1032-3
Agostini C, Gurrieri C (2006) Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc Am Thorac Soc 3(4):357–363. https://doi.org/10.1513/pats.200601-010TK
Scotton CJ, Chambers RC (2007) Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest 132(4):1311–1321. https://doi.org/10.1378/chest.06-2568
Krein PM, Winston BW (2002) Roles for insulin-like growth factor I and transforming growth factor-beta in fibrotic lung disease. Chest 122(6 Suppl):289s–293s. https://doi.org/10.1378/chest.122.6_suppl.289s
Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J (1997) Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Investig 100(4):768–776. https://doi.org/10.1172/jci119590
Sporn MB, Roberts AB (1992) Transforming growth factor-beta: recent progress and new challenges. J Cell Biol 119(5):1017–1021. https://doi.org/10.1083/jcb.119.5.1017
Ramos C, Montano M, Garcia-Alvarez J, Ruiz V, Uhal BD, Selman M, Pardo A (2001) Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Resp Cell Mol Biol 24(5):591–598
Meloni F, Caporali R, Marone Bianco A, Paschetto E, Morosini M, Fietta AM, Patrizio V, Bobbio-Pallavicini F, Pozzi E, Montecucco C (2004) BAL cytokine profile in different interstitial lung diseases: a focus on systemic sclerosis. Sarcoidosis Vasc Diffuse Lung Dis 21(2):111–118
Khalil N, O’Connor RN, Flanders KC, Unruh H (1996) TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am J Respir Cell Mol Biol 14(2):131–138. https://doi.org/10.1165/ajrcmb.14.2.8630262
Homma S, Nagaoka I, Abe H, Takahashi K, Seyama K, Nukiwa T, Kira S (1995a) Localization of platelet-derived growth factor and insulin-like growth factor I in the fibrotic lung. Am J Respir Crit Care Med 152(6):2084–2089
Gillery P, Leperre A, Maquart FX, Borel JP (1992) Insulin-like growth factor-I (IGF-I) stimulates protein synthesis and collagen gene expression in monolayer and lattice cultures of fibroblasts. J Cell Physiol 152(2):389–396. https://doi.org/10.1002/jcp.1041520221
Goldstein RH, Poliks CF, Pilch PF, Smith BD, Fine A (1989) Stimulation of collagen formation by insulin and insulin-like growth factor I in cultures of human lung fibroblasts. Endocrinology 124(2):964–970. https://doi.org/10.1210/endo-124-2-964
Hetzel M, Bachem M, Anders D, Trischler G, Faehling M (2005) Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung 183(4):225–237. https://doi.org/10.1007/s00408-004-2534-z
Pilewski JM, Liu L, Henry AC, Knauer AV, Feghali-Bostwick CA (2005) Insulin-like growth factor binding proteins 3 and 5 are overexpressed in idiopathic pulmonary fibrosis and contribute to extracellular matrix deposition. Am J Pathol 166(2):399–407. https://doi.org/10.1016/s0002-9440(10)62263-8
Homma S, Nagaoka I, Abe H, Takahashi K, Seyama K, Nukiwa T, Kira S (1995b) Localization of platelet-derived growth factor and insulin-like growth factor I in the fibrotic lung. Am J Respir Crit Care Med 152(6 Pt 1):2084–2089. https://doi.org/10.1164/ajrccm.152.6.8520779
Nagaoka I, Trapnell BC, Crystal RG (1990) Upregulation of platelet-derived growth factor-A and -B gene expression in alveolar macrophages of individuals with idiopathic pulmonary fibrosis. J Clin Investig 85(6):2023–2027. https://doi.org/10.1172/jci114669
Vignaud JM, Allam M, Martinet N, Pech M, Plenat F, Martinet Y (1991) Presence of platelet-derived growth factor in normal and fibrotic lung is specifically associated with interstitial macrophages, while both interstitial macrophages and alveolar epithelial cells express the c-sis proto-oncogene. Am J Respir Cell Mol Biol 5(6):531–538. https://doi.org/10.1165/ajrcmb/5.6.531
Cao B, Guo Z, Xu W, Zhu Y (1999) The potential role of cytokine expression and its relation to angiogenesis in idiopathic pulmonary fibrosis. Zhonghua nei ke za zhi 38(12):814–816
Lasky JA, Ortiz LA, Tonthat B, Hoyle GW, Corti M, Athas G, Lungarella G, Brody A, Friedman M (1998) Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am J Physiol 275(2):L365-371. https://doi.org/10.1152/ajplung.1998.275.2.L365
Pan LH, Yamauchi K, Uzuki M, Nakanishi T, Takigawa M, Inoue H, Sawai T (2001) Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur Resp J 17(6):1220–1227
Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, Grotendorst GR (1999) Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J 13(13):1774–1786
Grotendorst GR (1997) Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev 8(3):171–179
Kelly M, Kolb M, Bonniaud P, Gauldie J (2003) Re-evaluation of fibrogenic cytokines in lung fibrosis. Curr Pharm Des 9(1):39–49. https://doi.org/10.2174/1381612033392341
Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J (2001) Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Investig 107(12):1529–1536. https://doi.org/10.1172/jci12568
Freeburn RW, Armstrong L, Millar AB (2005) Cultured alveolar macrophages from patients with idiopathic pulmonary fibrosis (IPF) show dysregulation of lipopolysaccharide-induced tumor necrosis factor-alpha (TNF-alpha) and interleukin-10 (IL-10) inductions. Eur Cytokine Netw 16(1):5–16
Johnston CJ, Piedboeuf B, Rubin P, Williams JP, Baggs R, Finkelstein JN (1996) Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumor necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res 145(6):762–767
Thrall RS, Vogel SN, Evans R, Shultz LD (1997) Role of tumor necrosis factor-alpha in the spontaneous development of pulmonary fibrosis in viable motheaten mutant mice. Am J Pathol 151(5):1303–1310
Ortiz LA, Lasky J, Hamilton RF Jr, Holian A, Hoyle GW, Banks W, Peschon JJ, Brody AR, Lungarella G, Friedman M (1998) Expression of TNF and the necessity of TNF receptors in bleomycin-induced lung injury in mice. Exp Lung Res 24(6):721–743
Miyazaki Y, Araki K, Vesin C, Garcia I, Kapanci Y, Whitsett JA, Piguet PF, Vassalli P (1995) Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Investig 96(1):250–259. https://doi.org/10.1172/jci118029
Sime PJ, Marr RA, Gauldie D, Xing Z, Hewlett BR, Graham FL, Gauldie J (1998) Transfer of tumor necrosis factor-alpha to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor-beta1 and myofibroblasts. Am J Pathol 153(3):825–832. https://doi.org/10.1016/s0002-9440(10)65624-6
Liu JY, Brass DM, Hoyle GW, Brody AR (1998) TNF-alpha receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am J Pathol 153(6):1839–1847. https://doi.org/10.1016/s0002-9440(10)65698-2
DiCosmo BF, Geba GP, Picarella D, Elias JA, Rankin JA, Stripp BR, Whitsett JA, Flavell RA (1994) Airway epithelial cell expression of interleukin-6 in transgenic mice. Uncoupling of airway inflammation and bronchial hyperreactivity. J Clin Investig 94(5):2028–2035. https://doi.org/10.1172/jci117556
Moodley YP, Misso NL, Scaffidi AK, Fogel-Petrovic M, McAnulty RJ, Laurent GJ, Thompson PJ, Knight DA (2003) Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol 29(4):490–498. https://doi.org/10.1165/rcmb.2002-0262OC
Eickelberg O, Pansky A, Mussmann R, Bihl M, Tamm M, Hildebrand P, Perruchoud AP, Roth M (1999) Transforming growth factor-beta1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J Biol Chem 274(18):12933–12938. https://doi.org/10.1074/jbc.274.18.12933
Ziegenhagen MW, Zabel P, Zissel G, Schlaak M, Muller-Quernheim J (1998) Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am J Respir Crit Care Med 157(3 Pt 1):762–768. https://doi.org/10.1164/ajrccm.157.3.9705014
Car BD, Meloni F, Luisetti M, Semenzato G, Gialdroni-Grassi G, Walz A (1994) Elevated IL-8 and MCP-1 in the bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am J Respir Crit Care Med 149(3 Pt 1):655–659. https://doi.org/10.1164/ajrccm.149.3.8118632
Carre PC, Mortenson RL, King TE Jr, Noble PW, Sable CL, Riches DW (1991) Increased expression of the interleukin-8 gene by alveolar macrophages in idiopathic pulmonary fibrosis. A potential mechanism for the recruitment and activation of neutrophils in lung fibrosis. J Clin Investig 88(6):1802–1810. https://doi.org/10.1172/jci115501
Furuie H, Yamasaki H, Suga M, Ando M (1997) Altered accessory cell function of alveolar macrophages: a possible mechanism for induction of Th2 secretory profile in idiopathic pulmonary fibrosis. Eur Resp J 10(4):787–794
Baran CP, Opalek JM, McMaken S, Newland CA, O’Brien JM Jr, Hunter MG, Bringardner BD, Monick MM, Brigstock DR, Stromberg PC, Hunninghake GW, Marsh CB (2007) Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 176(1):78–89. https://doi.org/10.1164/rccm.200609-1279OC
Capelli A, Di Stefano A, Gnemmi I, Donner CF (2005) CCR5 expression and CC chemokine levels in idiopathic pulmonary fibrosis. Eur Resp J 25(4):701–707. https://doi.org/10.1183/09031936.05.00082604
Mosmann TR, Sad S (1996) The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today 17(3):138–146
Elias JA, Rossman MD, Zurier RB, Daniele RP (1985) Human alveolar macrophage inhibition of lung fibroblast growth. A prostaglandin-dependent process. Am Rev Resp Dis 131(1):94–99. https://doi.org/10.1164/arrd.1985.131.1.94
Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB (2003) Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol 29(5):537–544. https://doi.org/10.1165/rcmb.2002-0243OC
Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M (1995) Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Investig 95(4):1861–1868
Keerthisingam CB, Jenkins RG, Harrison NK, Hernandez-Rodriguez NA, Booth H, Laurent GJ, Hart SL, Foster ML, McAnulty RJ (2001) Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol 158(4):1411–1422. https://doi.org/10.1016/s0002-9440(10)64092-8
Jenkins G, Hart SL, Hodges RJ, Meng QH, Kinnon C, Laurent GJ, McAnulty RJ (2002) Cyclooxygenase-2 overexpression, using an integrin-targeted gene delivery system (the LID vector), inhibits fibroblast proliferation in vitro and leads to increased prostaglandin E(2) in the lung. Chest 121(3 Suppl):102s–104s. https://doi.org/10.1378/chest.121.3_suppl.102s
Li JZ, Li ZH, Kang J, Hou XM, Yu RJ (2004) [Change of prostaglandin E2 and interleukin-12, interleukin-13 in the bronchoalveolar lavage fluid and the serum of the patients with idiopathic pulmonary fibrosis]. Zhonghua jie he he hu xi za zhi = Zhonghua jiehe he huxi zazhi = Chinese journal of tuberculosis and respiratory diseases 27 (6):378–380
Saltzman LE, Moss J, Berg RA, Hom B, Crystal RG (1982) Modulation of collagen production by fibroblasts. Effects of chronic exposure to agonists that increase intracellular cyclic AMP. Biochem J 204(1):25–30. https://doi.org/10.1042/bj2040025
Hodges RJ, Jenkins RG, Wheeler-Jones CP, Copeman DM, Bottoms SE, Bellingan GJ, Nanthakumar CB, Laurent GJ, Hart SL, Foster ML, McAnulty RJ (2004) Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E(2) production. Am J Pathol 165(5):1663–1676. https://doi.org/10.1016/s0002-9440(10)63423-2
Tsukui T, Sun K-H, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, Adams TS, Schupp JC, Poli SD, Rosas IO (2020) Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun 11(1):1–16
Chan ES, Cronstein BN (2010) Adenosine in fibrosis. Mod Rheumatol 20(2):114–122
Sattin A, Rall T (1970) The effect of adenosine and adenine nucleotides on the cyclic adenosine 3’, 5’-phosphate content of guinea pig cerebral cortex slices. Mol Pharmacol 6(1):13–23
Olah ME, Stiles GL (1995) Adenosine receptor subtypes: characterization and therapeutic regulation. Annu Rev Pharmacol Toxicol 35(1):581–606
Zhong H, Belardinelli L, Maa T, Zeng D (2005) Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol 32(1):2–8
Chunn JL, Young HW, Banerjee SK, Colasurdo GN, Blackburn MR (2001) Adenosine-dependent airway inflammation and hyperresponsiveness in partially adenosine deaminase-deficient mice. J Immunol 167(8):4676–4685
Sun C-X, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, Belardinelli L, Zeng D, Blackburn MR (2006) Role of A 2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Investig 116(8):2173–2182
Zhou Y, Schneider DJ, Morschl E, Song L, Pedroza M, Karmouty-Quintana H, Le T, Sun C-X, Blackburn MR (2011) Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J Immunol 186(2):1097–1106
Morschl E, Molina JG, Volmer JB, Mohsenin A, Pero RS, Hong J-S, Kheradmand F, Lee JJ, Blackburn MR (2008) A3 adenosine receptor signaling influences pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol 39(6):697–705
Kinnula VL, Fattman CL, Tan RJ, Oury TD (2005) Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med 172(4):417–422. https://doi.org/10.1164/rccm.200501-017PP
Brenner DA (2009) Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc 120:361–368
Cheresh P, Kim SJ, Tulasiram S (1832) Kamp DW (2013) Oxidative stress and pulmonary fibrosis. Biochem Biophys Acta 7:1028–1040. https://doi.org/10.1016/j.bbadis.2012.11.021
Kliment CR, Oury TD (2010) Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radical Biol Med 49(5):707–717. https://doi.org/10.1016/j.freeradbiomed.2010.04.036
Fois AG, Paliogiannis P, Sotgia S, Mangoni AA, Zinellu E, Pirina P, Carru C, Zinellu A (2018) Evaluation of oxidative stress biomarkers in idiopathic pulmonary fibrosis and therapeutic applications: a systematic review. Respir Res 19(1):51. https://doi.org/10.1186/s12931-018-0754-7
Paliogiannis P, Fois AG, Collu C, Bandinu A, Zinellu E, Carru C, Pirina P, Mangoni AA, Zinellu A (2018) Oxidative stress-linked biomarkers in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Biomark Med 12(10):1175–1184. https://doi.org/10.2217/bmm-2018-0108
Dasari A, Bartholomew JN, Volonte D, Galbiati F (2006) Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Can Res 66(22):10805–10814. https://doi.org/10.1158/0008-5472.can-06-1236
MacNee W (2009) Accelerated lung aging: a novel pathogenic mechanism of chronic obstructive pulmonary disease (COPD). Biochem Soc Trans 37(Pt 4):819–823. https://doi.org/10.1042/bst0370819
Wang E (1995) Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Can Res 55(11):2284–2292
Campisi J, d’Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8(9):729–740. https://doi.org/10.1038/nrm2233
Toussaint O, Medrano EE, von Zglinicki T (2000) Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol 35(8):927–945
Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ (2005) Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J 19(7):854–856. https://doi.org/10.1096/fj.04-2882fje
Larios JM, Budhiraja R, Fanburg BL, Thannickal VJ (2001) Oxidative protein cross-linking reactions involving L-tyrosine in transforming growth factor-beta1-stimulated fibroblasts. J Biol Chem 276(20):17437–17441. https://doi.org/10.1074/jbc.M100426200
Liu RM, Gaston Pravia KA (2010) Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radical Biol Med 48(1):1–15. https://doi.org/10.1016/j.freeradbiomed.2009.09.026
Ricci A, Cherubini E, Ulivieri A, Lavra L, Sciacchitano S, Scozzi D, Mancini R, Ciliberto G, Bartolazzi A, Bruno P, Graziano P, Mariotta S (2013) Homeodomain-interacting protein kinase2 in human idiopathic pulmonary fibrosis. J Cell Physiol 228(1):235–241. https://doi.org/10.1002/jcp.24129
Fois AG, Posadino AM, Giordo R, Cossu A, Agouni A, Rizk NM, Pirina P, Carru C, Zinellu A, Pintus G (2018) Antioxidant activity mediates pirfenidone antifibrotic effects in human pulmonary vascular smooth muscle cells exposed to sera of idiopathic pulmonary fibrosis patients. Oxid Med Cell Longev 2018:2639081. https://doi.org/10.1155/2018/2639081
Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GS, Chandel NS (2013) Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J Biol Chem 288(2):770–777
Gonzalez-Gonzalez FJ, Chandel NS, Jain M, Budinger GS (2017) Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl Res 190:61–68
Liu R-M, Pravia KG (2010) Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Radical Biol Med 48(1):1–15
Thuan DTB, Zayed H, Eid AH, Abou-Saleh H, Nasrallah GK, Mangoni AA, Pintus G (2018) A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front Immunol 9:1985
Rangarajan S, Bernard K, Thannickal VJ (2017) Mitochondrial dysfunction in pulmonary fibrosis. Ann Am Thorac Soc 14(Supplement 5):S383–S388
Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15(9):1077–1081
Wermuth PJ, Mendoza FA, Jimenez SA (2019) Abrogation of transforming growth factor-β-induced tissue fibrosis in mice with a global genetic deletion of Nox4. Lab Invest 99(4):470–482
Hotta Y, Uchiyama K, Takagi T, Kashiwagi S, Nakano T, Mukai R, Toyokawa Y, Yasuda T, Ueda T, Suyama Y (2018) Transforming growth factor β1-induced collagen production in myofibroblasts is mediated by reactive oxygen species derived from NADPH oxidase 4. Biochem Biophys Res Commun 506(3):557–562
Ghatak S, Hascall VC, Markwald RR, Feghali-Bostwick C, Artlett CM, Gooz M, Bogatkevich GS, Atanelishvili I, Silver RM, Wood J (2017) Transforming growth factor β1 (TGFβ1)-induced CD44V6-NOX4 signaling in pathogenesis of idiopathic pulmonary fibrosis. J Biol Chem 292(25):10490–10519
Sanders YY, Lyv X, Zhou QJ, Xiang Z, Stanford D, Bodduluri S, Rowe SM, Thannickal VJ (2020) Brd4-p300 inhibition downregulates Nox4 and accelerates lung fibrosis resolution in aged mice. JCI Insight. https://doi.org/10.1172/jci.insight.137127
Otoupalova E, Smith S, Cheng G, Thannickal VJ (2020) Oxidative stress in pulmonary fibrosis. Compr Physiol 10(2):509–547
Posadino AM, Cossu A, Giordo R, Zinellu A, Sotgia S, Vardeu A, Hoa PT, Carru C, Pintus G (2015) Resveratrol alters human endothelial cells redox state and causes mitochondrial-dependent cell death. Food Chem Toxicol 78:10–16
Larson-Casey JL, He C, Carter AB (2020) Mitochondrial quality control in pulmonary fibrosis. Redox Biol 33:101426
Bueno M, Lai Y-C, Romero Y, Brands J, Croix CMS, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS (2015) PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Investig 125(2):521–538
Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB (2016) Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 44(3):582–596
Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ (2012) Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med 186(8):740–751
Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu R-M, Bernard K, Thannickal VJ, Liu G (2015) Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med 192(12):1462–1474
Gu L, Casey JLL, Andrabi SA, Lee JH, Meza-Perez S, Randall TD, Carter AB (2019) Mitochondrial calcium uniporter regulates PGC-1α expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol 26:101307
Huang SC-C, Everts B, Ivanova Y, O’sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’neill CM (2014) Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15(9):846–855
Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, Cascante M, Boscá L (2010) Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185(1):605–614
Ryu C, Sun H, Gulati M, Herazo-Maya JD, Chen Y, Osafo-Addo A, Brandsdorfer C, Winkler J, Blaul C, Faunce J (2017) Extracellular mitochondrial DNA is generated by fibroblasts and predicts death in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 196(12):1571–1581
van der Windt GJ, Everts B, Chang C-H, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL (2012) Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36(1):68–78
Larson-Casey JL, Murthy S, Ryan AJ, Carter AB (2014) Modulation of the mevalonate pathway by akt regulates macrophage survival and development of pulmonary fibrosis. J Biol Chem 289(52):36204–36219
Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D (2005) Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest 127(1):266–274
Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES (2010) Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med 181(3):254–263
Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J (2004) Early growth response gene 1–mediated apoptosis is essential for transforming growth factor β1–induced pulmonary fibrosis. J Exp Med 200(3):377–389
Kang H-R, Cho SJ, Lee CG, Homer RJ, Elias JA (2007) Transforming growth factor (TGF)-β1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, Bid-activated pathway that involves matrix metalloproteinase-12. J Biol Chem 282(10):7723–7732
Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, El-Chemaly S, Lee CG, Rosas IO, Elias JA (2015) Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 10(3):e0121246
Panduri V, Weitzman SA, Chandel NS, Kamp DW (2004) Mitochondrial-derived free radicals mediate asbestos-induced alveolar epithelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol 286(6):L1220–L1227
Kim S-J, Cheresh P, Jablonski RP, Morales-Nebreda L, Cheng Y, Hogan E, Yeldandi A, Chi M, Piseaux R, Ridge K (2016) Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radical Biol Med 101:482–490
Chung K-P, Hsu C-L, Fan L-C, Huang Z, Bhatia D, Chen Y-J, Hisata S, Cho SJ, Nakahira K, Imamura M (2019) Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat Commun 10(1):1–17
Hachisu Y, Murata K, Takei K, Tsuchiya T, Tsurumaki H, Koga Y, Horie T, Takise A, Hisada T (2019) Possible serological markers to predict mortality in acute exacerbation of idiopathic pulmonary fibrosis. Medicina 55(5):132
McCormack FX, King TE Jr, Voelker DR, Robinson PC, Mason RJ (1991) Idiopathic pulmonary fibrosis: abnormalities in the bronchoalveolar lavage content of surfactant protein A. Am Rev Respir Dis 144(1):160–166
Zhao YD, Yin L, Archer S, Lu C, Zhao G, Yao Y, Wu L, Hsin M, Waddell TK, Keshavjee S (2017) Metabolic heterogeneity of idiopathic pulmonary fibrosis: a metabolomic study. BMJ Open Respir Res 4(1):e000183
Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl A-KT, Funari VA, Gokey JJ (2016) Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. https://doi.org/10.1172/jci.insight.90558
Kruzliak P, Hare DL, Zvonicek V, Klimas J, Zulli A (2015) Simvastatin impairs the induction of pulmonary fibrosis caused by a western style diet: a preliminary study. J Cell Mol Med 19(11):2647–2654
Bernard K, Logsdon NJ, Miguel V, Benavides GA, Zhang J, Carter AB, Darley-Usmar VM, Thannickal VJ (2017) NADPH oxidase 4 (Nox4) suppresses mitochondrial biogenesis and bioenergetics in lung fibroblasts via a nuclear factor erythroid-derived 2-like 2 (Nrf2)-dependent pathway. J Biol Chem 292(7):3029–3038
Tsai M-J, Chang W-A, Liao S-H, Chang K-F, Sheu C-C, Kuo P-L (2019) The effects of epigallocatechin gallate (EGCG) on pulmonary fibroblasts of idiopathic pulmonary fibrosis (IPF)—a next-generation sequencing and bioinformatic approach. Int J Mol Sci 20(8):1958
Yakes FM, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci 94(2):514–519
Fang F, Marangoni RG, Zhou X, Yang Y, Ye B, Shangguang A, Qin W, Wang W, Bhattacharyya S, Wei J (2016) Toll-like receptor 9 signaling is augmented in systemic sclerosis and elicits transforming growth factor β-dependent fibroblast activation. Arthritis Rheumatol 68(8):1989–2002
Meneghin A, Choi E, Evanoff H, Kunkel S, Martinez F, Flaherty K, Toews G, Hogaboam C (2008) TLR9 is expressed in idiopathic interstitial pneumonia and its activation promotes in vitro myofibroblast differentiation. Histochem Cell Biol 130(5):979–992
Trujillo G, Meneghin A, Flaherty KR, Sholl LM, Myers JL, Kazerooni EA, Gross BH, Oak SR, Coelho AL, Evanoff H (2010) TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med 2(57):57ra82
Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, Bennink JR, Yewdell JW (2003) Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 18(3):343–354
Guerriero CJ, Brodsky JL (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev 92(2):537–576. https://doi.org/10.1152/physrev.00027.2011
Frakes AE, Dillin A (2017) The UPRER: sensor and coordinator of organismal homeostasis. Mol Cell 66(6):761–771. https://doi.org/10.1016/j.molcel.2017.05.031
Hetz C, Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13(8):477–491. https://doi.org/10.1038/nrneurol.2017.99
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11(4):381–389. https://doi.org/10.1038/sj.cdd.4401373
Wouters BG, Koritzinsky M (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8(11):851–864. https://doi.org/10.1038/nrc2501
Oslowski CM, Urano F (2011) Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 490:71–92. https://doi.org/10.1016/b978-0-12-385114-7.00004-0
Mandl J, Meszaros T, Banhegyi G, Csala M (2013) Minireview: endoplasmic reticulum stress: control in protein, lipid, and signal homeostasis. Mol Endocrinol (Baltimore, Md) 27(3):384–393. https://doi.org/10.1210/me.2012-1317
Fu S, Watkins SM, Hotamisligil GS (2012) The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab 15(5):623–634. https://doi.org/10.1016/j.cmet.2012.03.007
Ma Y, Hendershot LM (2004) ER chaperone functions during normal and stress conditions. J Chem Neuroanat 28(1–2):51–65. https://doi.org/10.1016/j.jchemneu.2003.08.007
Kabore AF, Wang WJ, Russo SJ, Beers MF (2001) Biosynthesis of surfactant protein C: characterization of aggresome formation by EGFP chimeras containing propeptide mutants lacking conserved cysteine residues. J Cell Sci 114(Pt 2):293–302
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Investig 115(10):2656–2664. https://doi.org/10.1172/jci26373
Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK (2010) Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem 285(29):22103–22113. https://doi.org/10.1074/jbc.M110.121467
Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA (2001) A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 344(8):573–579. https://doi.org/10.1056/nejm200102223440805
Thomas AQ, Lane K, Phillips J 3rd, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, Roberts R, Haines J, Stahlman M, Loyd JE (2002) Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 165(9):1322–1328. https://doi.org/10.1164/rccm.200112-123OC
Selman M, Pardo A (2006) Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc 3(4):364–372. https://doi.org/10.1513/pats.200601-003TK
Byrne AJ, Maher TM, Lloyd CM (2016) Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med 22(4):303–316. https://doi.org/10.1016/j.molmed.2016.02.004
Byrne AJ, Mathie SA, Gregory LG, Lloyd CM (2015) Pulmonary macrophages: key players in the innate defence of the airways. Thorax 70(12):1189–1196. https://doi.org/10.1136/thoraxjnl-2015-207020
Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng D-S, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB (2008) Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol 294(6):L1119–L1126
Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, Weaver TE, Guenther A (2008) Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 178(8):838–846. https://doi.org/10.1164/rccm.200802-313OC
Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, Hara N (1996) P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 154(2 Pt 1):477–483. https://doi.org/10.1164/ajrccm.154.2.8756825
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8:14532. https://doi.org/10.1038/ncomms14532
Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, Kaufman RJ, Bastian BC, Soengas MS (2006) Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol 8(10):1053–1063. https://doi.org/10.1038/ncb1471
Matos L, Gouveia AM, Almeida H (2015) ER stress response in human cellular models of senescence. J Gerontol A 70(8):924–935. https://doi.org/10.1093/gerona/glu129
Greenburg G, Hay ED (1986) Cytodifferentiation and tissue phenotype change during transformation of embryonic lens epithelium to mesenchyme-like cells in vitro. Dev Biol 115(2):363–379. https://doi.org/10.1016/0012-1606(86)90256-3
Karicheva O, Rodriguez-Vargas JM, Wadier N, Martin-Hernandez K, Vauchelles R, Magroun N, Tissier A, Schreiber V, Dantzer F (2016) PARP3 controls TGFbeta and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget 7(39):64109–64123. https://doi.org/10.18632/oncotarget.11627
Yang ZC, Yi MJ, Ran N, Wang C, Fu P, Feng XY, Xu L, Qu ZH (2013) Transforming growth factor-beta1 induces bronchial epithelial cells to mesenchymal transition by activating the Snail pathway and promotes airway remodeling in asthma. Mol Med Rep 8(6):1663–1668. https://doi.org/10.3892/mmr.2013.1728
Korpal M, Lee ES, Hu G, Kang Y (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283(22):14910–14914. https://doi.org/10.1074/jbc.C800074200
Johnson JR, Nishioka M, Chakir J, Risse PA, Almaghlouth I, Bazarbashi AN, Plante S, Martin JG, Eidelman D, Hamid Q (2013) IL-22 contributes to TGF-beta1-mediated epithelial-mesenchymal transition in asthmatic bronchial epithelial cells. Respir Res 14:118. https://doi.org/10.1186/1465-9921-14-118
Baulida J (2017) Epithelial-to-mesenchymal transition transcription factors in cancer-associated fibroblasts. Mol Oncol 11(7):847–859. https://doi.org/10.1002/1878-0261.12080
Sun Q, Fan J, Billiar TR, Scott MJ (2017) Inflammasome and autophagy regulation—a two-way street. Mol Med (Cambridge, Mass) 23:188–195. https://doi.org/10.2119/molmed.2017.00077
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Investig 119(6):1420–1428. https://doi.org/10.1172/jci39104
Yang Z, Xie H, He D, Li L (2016) Infiltrating macrophages increase RCC epithelial mesenchymal transition (EMT) and stem cell-like populations via AKT and mTOR signaling. Oncotarget 7(28):44478–44491. https://doi.org/10.18632/oncotarget.9873
Elizur A, Cannon CL, Ferkol TW (2008) Airway inflammation in cystic fibrosis. Chest 133(2):489–495. https://doi.org/10.1378/chest.07-1631
Wang Y, Jia M, Yan X, Cao L, Barnes PJ, Adcock IM, Huang M, Yao X (2017) Increased neutrophil gelatinase-associated lipocalin (NGAL) promotes airway remodelling in chronic obstructive pulmonary disease. Clin Sci (London, England: 1979) 131(11):1147–1159. https://doi.org/10.1042/cs20170096
Yasukawa A, Hosoki K, Toda M, Miyake Y, Matsushima Y, Matsumoto T, Boveda-Ruiz D, Gil-Bernabe P, Nagao M, Sugimoto M, Hiraguchi Y, Tokuda R, Naito M, Takagi T, D’Alessandro-Gabazza CN, Suga S, Kobayashi T, Fujisawa T, Taguchi O, Gabazza EC (2013) Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS ONE 8(5):e64281. https://doi.org/10.1371/journal.pone.0064281
Zhao YL, Zhu RT, Sun YL (2016) Epithelial-mesenchymal transition in liver fibrosis. Biomed Rep 4(3):269–274. https://doi.org/10.3892/br.2016.578
Ganesan S, Sajjan US (2013) Repair and remodeling of airway epithelium after injury in chronic obstructive pulmonary disease. Curr Resp Care Rep. https://doi.org/10.1007/s13665-013-0052-2
Shaykhiev R, Zuo WL, Chao I, Fukui T, Witover B, Brekman A, Crystal RG (2013) EGF shifts human airway basal cell fate toward a smoking-associated airway epithelial phenotype. Proc Natl Acad Sci USA 110(29):12102–12107. https://doi.org/10.1073/pnas.1303058110
Heldin CH, Miyazono K, ten Dijke P (1997) TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390(6659):465–471. https://doi.org/10.1038/37284
Mahmood MQ, Reid D, Ward C, Muller HK, Knight DA, Sohal SS, Walters EH (2017) Transforming growth factor (TGF) beta1 and Smad signalling pathways: a likely key to EMT-associated COPD pathogenesis. Respirology 22(1):133–140. https://doi.org/10.1111/resp.12882
Banerjee P, Surendran H, Chowdhury DR, Prabhakar K, Pal R (2016) Metformin mediated reversal of epithelial to mesenchymal transition is triggered by epigenetic changes in E-cadherin promoter. J Mol Med (Berlin, Germany) 94(12):1397–1409. https://doi.org/10.1007/s00109-016-1455-7
Lutful Kabir F, Ambalavanan N, Liu G, Li P, Solomon GM, Lal CV, Mazur M, Halloran B, Szul T, Gerthoffer WT, Rowe SM, Harris WT (2018) MicroRNA-145 antagonism reverses TGF-beta inhibition of F508del CFTR correction in airway epithelia. Am J Respir Crit Care Med 197(5):632–643. https://doi.org/10.1164/rccm.201704-0732OC
Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, Haitchi HM, Vernon-Wilson E, Sammut D, Bedke N, Cremin C, Sones J, Djukanovic R, Howarth PH, Collins JE, Holgate ST, Monk P, Davies DE (2011) Defective epithelial barrier function in asthma. J Allergy Clin Immunol 128(3):549–556. https://doi.org/10.1016/j.jaci.2011.05.038
Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI (2009) Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol 297(6):L1120-1130. https://doi.org/10.1152/ajplung.00007.2009
Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB (2005) Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol 16(3):667–675. https://doi.org/10.1681/asn.2004050425
Zavadil J, Bottinger EP (2005) TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24(37):5764–5774. https://doi.org/10.1038/sj.onc.1208927
Nyabam S, Wang Z, Thibault T, Oluseyi A, Basar R, Marshall L, Griffinb M (2016) A novel regulatory role for tissue transglutaminase in epithelial-mesenchymal transition in cystic fibrosis. Biochem Biophys Acta 1863 9:2234–2244. https://doi.org/10.1016/j.bbamcr.2016.05.012
Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA (2006a) Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA 103(35):13180–13185. https://doi.org/10.1073/pnas.0605669103
Adamson IY, Young L, Bowden DH (1988) Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol 130(2):377–383
Selman M, Pardo A (2002) Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res 3:3. https://doi.org/10.1186/rr175
Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA (2006b) Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci 103(35):13180–13185
Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, Lawson WE (2009) Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 180(7):657–665. https://doi.org/10.1164/rccm.200903-0322OC
Chen H, Chen Q, Jiang CM, Shi GY, Sui BW, Zhang W, Yang LZ, Li ZY, Liu L, Su YM, Zhao WC, Sun HQ, Li ZZ, Fu Z (2018) Triptolide suppresses paraquat induced idiopathic pulmonary fibrosis by inhibiting TGFB1-dependent epithelial mesenchymal transition. Toxicol Lett 284:1–9. https://doi.org/10.1016/j.toxlet.2017.11.030
Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z (2005) Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol 166(5):1321–1332. https://doi.org/10.1016/s0002-9440(10)62351-6
Pattarayan D, Sivanantham A, Krishnaswami V, Loganathan L, Palanichamy R, Natesan S, Muthusamy K, Rajasekaran S (2018) Tannic acid attenuates TGF-beta1-induced epithelial-to-mesenchymal transition by effectively intervening TGF-beta signaling in lung epithelial cells. J Cell Physiol 233(3):2513–2525. https://doi.org/10.1002/jcp.26127
Kurita Y, Araya J, Minagawa S, Hara H, Ichikawa A, Saito N, Kadota T, Tsubouchi K, Sato N, Yoshida M, Kobayashi K, Ito S, Fujita Y, Utsumi H, Yanagisawa H, Hashimoto M, Wakui H, Yoshii Y, Ishikawa T, Numata T, Kaneko Y, Asano H, Yamashita M, Odaka M, Morikawa T, Nakayama K, Kuwano K (2017) Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir Res 18(1):114. https://doi.org/10.1186/s12931-017-0600-3
Saito S, Zhuang Y, Shan B, Danchuk S, Luo F, Korfei M, Guenther A, Lasky JA (2017) Tubastatin ameliorates pulmonary fibrosis by targeting the TGFbeta-PI3K-Akt pathway. PLoS ONE 12(10):e0186615. https://doi.org/10.1371/journal.pone.0186615
Zhou XL, Xu P, Chen HH, Zhao Y, Shen J, Jiang C, Jiang S, Ni SZ, Xu B, Li L (2017) Thalidomide inhibits TGF-beta1-induced epithelial to mesenchymal transition in alveolar epithelial cells via smad-dependent and smad-independent signaling pathways. Sci Rep 7(1):14727. https://doi.org/10.1038/s41598-017-15239-2
Karsdal MA, Manon-Jensen T, Genovese F, Kristensen JH, Nielsen MJ, Sand JM, Hansen NU, Bay-Jensen AC, Bager CL, Krag A, Blanchard A, Krarup H, Leeming DJ, Schuppan D (2015) Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am J Physiol Gastrointest Liver Physiol 308(10):G807-830. https://doi.org/10.1152/ajpgi.00447.2014
Ho YY, Lagares D, Tager AM, Kapoor M (2014) Fibrosis–a lethal component of systemic sclerosis. Nat Rev Rheumatol 10(7):390–402. https://doi.org/10.1038/nrrheum.2014.53
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180(4):1340–1355. https://doi.org/10.1016/j.ajpath.2012.02.004
Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13(8):952–961. https://doi.org/10.1038/nm1613
Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, Shimokata K, Hasegawa Y (2010) Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 43(2):161–172. https://doi.org/10.1165/rcmb.2009-0031OC
Pardali E, Goumans MJ, ten Dijke P (2010) Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol 20(9):556–567. https://doi.org/10.1016/j.tcb.2010.06.006
Ten Dijke P, Arthur HM (2007) Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol 8(11):857–869
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425(6958):577–584. https://doi.org/10.1038/nature02006
Guo X, Wang X-F (2009) Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res 19(1):71–88
Funaba M, Zimmerman CM, Mathews LS (2002) Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J Biol Chem 277(44):41361–41368. https://doi.org/10.1074/jbc.M204597200
Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K (2005) Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol 166(4):1029–1039. https://doi.org/10.1016/s0002-9440(10)62324-3
Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y (2003) p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 38(4):879–889
Sanchez-Duffhues G, Garcia de Vinuesa A, Ten Dijke P (2018) Endothelial-to-mesenchymal transition in cardiovascular diseases: developmental signaling pathways gone awry. Dev Dyn 247(3):492–508. https://doi.org/10.1002/dvdy.24589
van Meeteren LA, ten Dijke P (2012) Regulation of endothelial cell plasticity by TGF-beta. Cell Tissue Res 347(1):177–186. https://doi.org/10.1007/s00441-011-1222-6
Yang R, Yang F, Hu Y, Chen M, Liu Y, Li J, Zhong W (2019) Hepatocyte growth factor attenuates the development of TGF-beta1-induced EndMT through down-regulating the Notch signaling. Endocr Metab Immune Disord Drug Targets. https://doi.org/10.2174/1871530319666191023141638
Miyasato SK, Loeffler J, Shohet R, Zhang J, Lindsey M, Le Saux CJ (2011) Caveolin-1 modulates TGF-beta1 signaling in cardiac remodeling. Matrix Biol 30(5–6):318–329. https://doi.org/10.1016/j.matbio.2011.05.003
Li Z, Wermuth PJ, Benn BS, Lisanti MP, Jimenez SA (2013) Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am J Pathol 182(2):325–331. https://doi.org/10.1016/j.ajpath.2012.10.022
Choi SH, Hong ZY, Nam JK, Lee HJ, Jang J, Yoo RJ, Lee YJ, Lee CY, Kim KH, Park S, Ji YH, Lee YS, Cho J, Lee YJ (2015) A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res 21(16):3716–3726. https://doi.org/10.1158/1078-0432.ccr-14-3193
Walton KL, Johnson KE, Harrison CA (2017) Targeting TGF-beta mediated SMAD signaling for the prevention of fibrosis. Front Pharmacol 8:461. https://doi.org/10.3389/fphar.2017.00461
Bonniaud P, Margetts PJ, Ask K, Flanders K, Gauldie J, Kolb M (2005) TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. J Immunol (Baltimore, Md : 1950) 175(8):5390–5395. https://doi.org/10.4049/jimmunol.175.8.5390
Kawakami T, Mimura I, Shoji K, Tanaka T, Nangaku M (2014) Hypoxia and fibrosis in chronic kidney disease: crossing at pericytes. Kidney Int Suppl 4(1):107–112
Czubryt MP (2012) Common threads in cardiac fibrosis, infarct scar formation, and wound healing. Fibrogenesis & tissue repair 5(1):1–11
Troy L, Corte T (2015) Interstitial lung disease in 2015: where are we now? Aust Fam Phys 44(8):546
Dengler VL, Galbraith MD, Espinosa JM (2014) Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol 49(1):1–15
Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5(5):343–354
Semenza G (2011) Mechanisms of disease oxygen sensing, homeostasis, and disease (vol 365, pg 537, 2011). N Engl J Med 365(10):968–968
Tzouvelekis A, Harokopos V, Paparountas T, Oikonomou N, Chatziioannou A, Vilaras G, Tsiambas E, Karameris A, Bouros D, Aidinis V (2007) Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1α in disease pathogenesis. Am J Respir Crit Care Med 176(11):1108–1119
Bodempudi V, Hergert P, Smith K, Xia H, Herrera J, Peterson M, Khalil W, Kahm J, Bitterman PB, Henke CA (2014) miR-210 promotes IPF fibroblast proliferation in response to hypoxia. Am J Physiol Lung Cell Mol Physiol 307(4):L283–L294
Weng T, Poth JM, Karmouty-Quintana H, Garcia-Morales LJ, Melicoff E, Luo F, Chen N-y, Evans CM, Bunge RR, Bruckner BA (2014) Hypoxia-induced deoxycytidine kinase contributes to epithelial proliferation in pulmonary fibrosis. Am J Respir Crit Care Med 190(12):1402–1412
Aquino-Gálvez A, González-Ávila G, Jiménez-Sánchez LL, Maldonado-Martínez HA, Cisneros J, Toscano-Marquez F, Castillejos-López M, Torres-Espíndola LM, Velázquez-Cruz R, Rodríguez VHO (2019) Dysregulated expression of hypoxia-inducible factors augments myofibroblasts differentiation in idiopathic pulmonary fibrosis. Respir Res 20(1):130
Ravenna L, Salvatori L, Russo MA (2016) HIF 3α: the little we know. FEBS J 283(6):993–1003
Senavirathna LK, Huang C, Yang X, Munteanu MC, Sathiaseelan R, Xu D, Henke CA, Liu L (2018) Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci Rep 8(1):1–16
Mizuno S, Bogaard HJ, Voelkel NF, Umeda Y, Kadowaki M, Ameshima S, Miyamori I, Ishizaki T (2009) Hypoxia regulates human lung fibroblast proliferation via p53-dependent and-independent pathways. Respir Res 10(1):1–12
Senavirathna LK, Huang C, Pushparaj S, Xu D, Liu L (2020) Hypoxia and transforming growth factor β1 regulation of long non-coding RNA transcriptomes in human pulmonary fibroblasts. Physiol Rep 8(1):e14343
Burman A, Kropski JA, Calvi CL, Serezani AP, Pascoalino BD, Han W, Sherrill T, Gleaves L, Lawson WE, Young LR (2018) Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight. https://doi.org/10.1172/jci.insight.99543
Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP (1998) Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 157(1):199–203
Noble PW, Jiang D (2006) Matrix regulation of lung injury, inflammation, and repair: the role of innate immunity. Proc Am Thorac Soc 3(5):401–404
Souza-Fernandes AB, Pelosi P, Rocco PR (2006) Bench-to-bedside review: the role of glycosaminoglycans in respiratory disease. Crit Care 10(6):237
Laurent G, Chambers R, Hill M, McAnulty R (2007) Regulation of matrix turnover: fibroblasts, forces, factors and fibrosis. Portland Press Ltd, London
Negrini D, Passi A, de Luca G, Miserocchi G (2002) Matrix proteoglycans in development of pulmonary edema. In: Proteoglycans in lung disease. CRC Press, Boca Raton, pp 172–197
Hewlett JC, Kropski JA, Blackwell TS (2018) Idiopathic pulmonary fibrosis: epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol 71:112–127
Wigén J, Elowsson-Rendin L, Karlsson L, Tykesson E, Westergren-Thorsson G (2019) Glycosaminoglycans: a link between development and regeneration in the lung. Stem Cells Dev 28(13):823–832
Pelosi P, Rocco PR (2008) Effects of mechanical ventilation on the extracellular matrix. Intensive Care Med 34(4):631–639
Westergren-Thorsson G, Hedström U, Nybom A, Tykesson E, Åhrman E, Hornfelt M, Maccarana M, van Kuppevelt TH, Dellgren G, Wildt M (2017) Increased deposition of glycosaminoglycans and altered structure of heparan sulfate in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol 83:27–38
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA (2005) Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11(11):1173–1179
Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, Kurkciyan A, Mena JM, Stripp BR, Jiang D (2016) Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med 22(11):1285–1293
Bjermer L, Lundgren R, Hällgren R (1989) Hyaluronan and type III procollagen peptide concentrations in bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. Thorax 44(2):126–131
Collum SD, Chen NY, Hernandez AM, Hanmandlu A, Sweeney H, Mertens TC, Weng T, Luo F, Molina JG, Davies J (2017) Inhibition of hyaluronan synthesis attenuates pulmonary hypertension associated with lung fibrosis. Br J Pharmacol 174(19):3284–3301
Lauer ME, Erzurum SC, Mukhopadhyay D, Vasanji A, Drazba J, Wang A, Fulop C, Hascall VC (2008) Differentiated murine airway epithelial cells synthesize a leukocyte-adhesive hyaluronan matrix in response to endoplasmic reticulum stress. J Biol Chem 283(38):26283–26296
Li Y, Jiang D, Liang J, Meltzer EB, Gray A, Miura R, Wogensen L, Yamaguchi Y, Noble PW (2011) Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med 208(7):1459–1471
Li Y, Liang J, Yang T, Mena JM, Huan C, Xie T, Kurkciyan A, Liu N, Jiang D, Noble PW (2016) Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix Biol 55:35–48
Svee K, White J, Vaillant P, Jessurun J, Roongta U, Krumwiede M, Johnson D, Henke C (1996) Acute lung injury fibroblast migration and invasion of a fibrin matrix is mediated by CD44. J Clin Investig 98(8):1713–1727
Teder P, Heldin P (1997) Mechanism of impaired local hyaluronan turnover in bleomycin-induced lung injury in rat. Am J Respir Cell Mol Biol 17(3):376–385
Mubarak KK, Montes-Worboys A, Regev D, Nasreen N, Mohammed KA, Faruqi I, Hensel E, Baz MA, Akindipe OA, Fernandez-Bussy S (2012) Parenchymal trafficking of pleural mesothelial cells in idiopathic pulmonary fibrosis. Eur Respir J 39(1):133–140
Zolak JS, Jagirdar R, Surolia R, Karki S, Oliva O, Hock T, Guroji P, Ding Q, Liu R-M, Bolisetty S (2013) Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am J Pathol 182(4):1239–1247
Koistinen V, Härkönen K, Kärnä R, Arasu UT, Oikari S, Rilla K (2017) EMT induced by EGF and wounding activates hyaluronan synthesis machinery and EV shedding in rat primary mesothelial cells. Matrix Biol 63:38–54
Venkatesan N, Ouzzine M, Kolb M, Netter P, Ludwig MS (2011) Increased deposition of chondroitin/dermatan sulfate glycosaminoglycan and upregulation of β1, 3-glucuronosyltransferase I in pulmonary fibrosis. American Journal of Physiology-Lung Cellular and Molecular Physiology 300(2):L191–L203
Lu J, Auduong L, White ES, Yue X (2014) Up-regulation of heparan sulfate 6-O-sulfation in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 50(1):106–114
Wujak L, Didiasova M, Zakrzewicz D, Frey H, Schaefer L, Wygrecka M (2015) Heparan sulfate proteoglycans mediate factor XIIa binding to the cell surface. J Biol Chem 290(11):7027–7039
Tian B, Manley JL (2017) Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol 18(1):18–30
Chang J-W, Yeh H-S, Yong J (2017) Alternative polyadenylation in human diseases. Endocrinol Metab 32(4):413–421
Liaw H-H, Lin C-C, Juan H-F, Huang H-C (2013) Differential microRNA regulation correlates with alternative polyadenylation pattern between breast cancer and normal cells. PLoS ONE 8(2):e56958
Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu A-B, Li W, Wagner EJ (2014) CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510(7505):412–416
Zhou Z, Qu J, He L, Zhu Y, Yang S-Z, Zhang F, Guo T, Peng H, Chen P, Zhou Y (2020) Stiff matrix instigates type I collagen biogenesis by mammalian cleavage factor I complex-mediated alternative polyadenylation. JCI Insight. https://doi.org/10.1172/jci.insight.133972
Weng T, Huang J, Wagner EJ, Ko J, Wu M, Wareing NE, Xiang Y, Chen N-Y, Ji P, Molina JG (2020) Downregulation of CFIm25 amplifies dermal fibrosis through alternative polyadenylation. J Exp Med 217(2):e20181384
Ko J, Mills T, Huang J, Chen N-y, Mertens TC, Collum SD, Lee G, Xiang Y, Han L, Zhou Y (2019) Transforming growth factor β1 alters the 3′-UTR of mRNA to promote lung fibrosis. J Biol Chem 294(43):15781–15794
Weng T, Ko J, Masamha CP, Xia Z, Xiang Y, Chen N-y, Molina JG, Collum S, Mertens TC, Luo F (2019) Cleavage factor 25 deregulation contributes to pulmonary fibrosis through alternative polyadenylation. J Clin Investig 129(5):1984–1999
Wheaton AK, Agarwal M, Jia S, Kim KK (2017) Lung epithelial cell focal adhesion kinase signaling inhibits lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol 312(5):L722–L730. https://doi.org/10.1152/ajplung.00478.2016
Li H, Zhao X, Shan H, Liang H (2016) MicroRNAs in idiopathic pulmonary fibrosis: involvement in pathogenesis and potential use in diagnosis and therapeutics. Acta Pharm Sin B 6(6):531–539
Milosevic J, Pandit K, Magister M, Rabinovich E, Ellwanger DC, Yu G, Vuga LJ, Weksler B, Benos PV, Gibson KF, McMillan M, Kahn M, Kaminski N (2012) Profibrotic role of miR-154 in pulmonary fibrosis. Am J Respir Cell Mol Biol 47(6):879–887. https://doi.org/10.1165/rcmb.2011-0377OC
Matsushima S, Ishiyama J (2016) MicroRNA-29c regulates apoptosis sensitivity via modulation of the cell-surface death receptor, Fas, in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 311(6):L1050-l1061. https://doi.org/10.1152/ajplung.00252.2016
Kurowska-Stolarska M, Hasoo MK, Welsh DJ, Stewart L, McIntyre D, Morton BE, Johnstone S, Miller AM, Asquith DL, Millar NL, Millar AB, Feghali-Bostwick CA, Hirani N, Crick PJ, Wang Y, Griffiths WJ, McInnes IB, McSharry C (2017) The role of microRNA-155/liver X receptor pathway in experimental and idiopathic pulmonary fibrosis. J Allergy Clin Immunol 139(6):1946–1956. https://doi.org/10.1016/j.jaci.2016.09.021
Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, Abraham E (2010) miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med 207(8):1589–1597. https://doi.org/10.1084/jem.20100035
Meyer KC, Nathan SD (2018) Idiopathic pulmonary fibrosis: a comprehensive clinical guide. Springer, New York
Takagi K, Yamakuchi M, Hashiguchi T, Inoue H (2019) The role of miRNAs in idiopathic pulmonary fibrosis. In: Interstitial lung diseases. IntechOpen
Paliogiannis P, Putzu C, Fois AG, Pirina P, Vidili G, Madonia M, Calvisi DF, Sotgia S, Carru C, Zinellu A. Pathophysiological roles, molecular interactions and clinical implications of long non-coding RNA CCAT2 in human cancer
Cao G, Zhang J, Wang M, Song X, Liu W, Mao C, Lv C (2013) Differential expression of long non-coding RNAs in bleomycin-induced lung fibrosis. Int J Mol Med 32(2):355–364. https://doi.org/10.3892/ijmm.2013.1404
Hadjicharalambous MR, Roux BT, Feghali-Bostwick CA, Murray LA, Clarke DL, Lindsay MA (2018) Long non-coding RNAs are central regulators of the IL-1beta-induced inflammatory response in normal and idiopathic pulmonary lung fibroblasts. Front Immunol 9:2906. https://doi.org/10.3389/fimmu.2018.02906
Lu Q, Guo Z, Xie W, Jin W, Zhu D, Chen S, Ren T (2018) The lncRNA H19 mediates pulmonary fibrosis by regulating the miR-196a/COL1A1 axis. Inflammation 41(3):896–903. https://doi.org/10.1007/s10753-018-0744-4
Zhao X, Sun J, Chen Y, Su W, Shan H, Li Y, Wang Y, Zheng N, Shan H, Liang H (2018) lncRNA PFAR promotes lung fibroblast activation and fibrosis by targeting miR-138 to regulate the YAP1-twist axis. Mol Ther 26(9):2206–2217. https://doi.org/10.1016/j.ymthe.2018.06.020
Hadjicharalambous MR, Lindsay MA (2020) Idiopathic pulmonary fibrosis: pathogenesis and the emerging role of long non-coding RNAs. Int J Mol Sci. https://doi.org/10.3390/ijms21020524
Acknowledgements
This work has been made possible thanks to grants from Sardinian Region (Fondo di Sviluppo e Coesione 2014–2020, Patto per lo Sviluppo della Regione Sardegna, L.R.7-2017 #RASSR82005), to GP, AZ, AGF; Qatar University (IRCC-2019-007) to GKN,GP; (fondo UNISS di Ateneo per la Ricerca 2020) to GP, AZ, AGF; and University of Sharjah (Seed 2001050151) to GP
Author information
Authors and Affiliations
Contributions
Manuscript conceptualization GP and PTHG; Literature search and articles acquisition AHE, AGF, AZ, RG; Funding acquisition, GKN, GP, AZ, AGF; Writing original manuscript draft PTHG, GP, PP, AAM; Figures drawing, PTHG, GP, PP, RG, AAM.; Final manuscript review and editing, AAM, GKN, PP, AHE, AF, AZ RG and GP.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Phan, T.H.G., Paliogiannis, P., Nasrallah, G.K. et al. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 78, 2031–2057 (2021). https://doi.org/10.1007/s00018-020-03693-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03693-7