
Abstract
Cancer is a critical community health problem. In the future, it is foreseen that there will be more aged people with more diseases, especially diseases like cancer. The critical roles of hypoxia and reactive oxygen species (ROS) on human health have been investigated comprehensively over the years. Numerous scientific evidence from various laboratory and clinical investigations points toward the role of hypoxia in cancer. Besides, ROS also plays a significant role in cancer initiation and progression itself. In living organisms, ROS production is unavoidable because they consume oxygen for their metabolic activity, which generates ROS. Many research outcomes clearly showed that hypoxia and ROS play an essential role in cancer disease, which also is associated with oxygen. This chapter will discuss the crucial role of hypoxia and ROS in cancer initiation and progression. Besides, this chapter also will provide a comprehensive overview, focusing on the contribution of hypoxia and ROS in cancer biology. This chapter also summarizes the updated information on hypoxia and ROS.
Manisekaran Hemagirri and Hong Hui-Jing shared co-first authorship
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Introduction
Cancer is a leading community health problem worldwide, with few efficient treatment options, poor prognosis, and high death rates (Chen et al. 2016). It is foreseen that in the year 2050 there will be more than 20% of aged people with age over 60 years worldwide (Noordin et al. 2020). Meanwhile, the World Health Organization (WHO) estimated in 2019 that cancer will be the first or second important reason of mortality of people with age below 70 years in 112 of 183 countries and ranks third or fourth in the remaining 23 countries (WHO 2020). This crucial statistical data panicked us on the requirement of prompt consideration to increase people’s health internationally. Hypoxia and reactive oxygen species (ROS) play an essential role in cancer biology (Tafani et al. 2016). Hypoxemia is defined as reduced oxygenation of the blood. Numerous scientific evidence from various laboratory and clinical investigations points toward the role of hypoxia in cancer. Furthermore, ROS are incredibly reactive chemicals derived from oxygen, namely superoxide, peroxides, singlet oxygen, alpha-oxygen, and hydroxyl radical. ROS is an unavoidable happening in living organisms that use oxygen for their metabolic activity, followed by ROS production. In addition, an excessive amount of ROS is an introductory course of cancer initiation. Besides, the ROS also plays numerous roles through the normal development of malignant tumors, such as the initial growth of transformed cells, which subsequently help develop the initial tumor mass with or without angiogenesis. When the tumor tissue grows bigger, it might lead to limited diffusion until the tumor tissue becomes hypoxic. These scientific pieces of literature reported the interaction of ROS and hypoxia in cancer origination and development. Although there are many reports on the crucial role of ROS and hypoxia in cancer, there is a lack of literature that combines the subjects on cancer initiation and progression itself. In this chapter, we focus on the relationship between reactive oxygen species (ROS) and hypoxia in cancer in particular on their vital role in cancer initiation and progression. Moreover, this chapter will also summarize the updated information on ROS and hypoxia.
3.2 Cancer
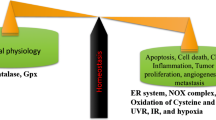
Cancer is an illness in which some of the normal cells grow uncontrollably and spread to other parts of the body. Cancer can be initiated in any part of the human body, which consists of trillions of cells. Usually, human cells grow and multiply in an orderly process of the progression named cell division to create new cells in the body. Sometimes, this orderly process will be disturbed and lead to uncontrollable cell growth and starts to proliferate to form cancerous or noncancerous (benign) tumors, which are lumps of tissue. The mass of cells or a tumor formed of these abnormal cells may stay within the tissue of their origin (in situ cancer) or it may start to invade adjacent tissues (invasive cancer). The invasive tumor is called malignant, and the cells that flow into the bloodstream or lymph from a malignant tumor have the potential to spread to other parts of the body and form new cancers (metastases) (Fig. 3.1). Mortality occurred when the growth of the tumors destroys the vital tissues and organs that are responsible for the individual’s survival (Jena et al. 2012).

Stages of tumor development
3.3 Hypoxia
Hypoxia is a condition where insufficient amounts of oxygen are available in the tissue to preserve satisfactory homeostasis. Hypoxia could result from inadequate oxygen distribution to the tissues either due to low blood supply or low oxygen content in the blood (hypoxemia). Many cellular responses are triggered in the chronic moderate hypoxia stage, such as ion homeostasis, hypoxia-inducible factors (HIFs), erythropoiesis, angiogenesis, cell proliferation, cell differentiation, energy metabolism, ROS generation, and cell death. Hypoxia affects significantly ionic homeostasis, which use much ATP to uphold the ionic gradient (Erecinska and Silver 2001) During hypoxia, a reduction in the intracellular ATP/ADP ratios leads to a series of biological disorders and ROS generation (Bickler and Donohoe 2002; Corbucci et al. 2005; Seta et al. 2004). While activating ion channels at the acute response, at the chronic level, hypoxia implicates differences in the gene expression (Semenza 1999) by activating and stabilizing the hypoxia-inducible factors (HIFs) (Bracken et al. 2003). Chronic hypoxia also upregulates the genes involved in erythropoiesis, which is required for the formation of red blood cells and leads to the increment in the ability of red blood cells to transport oxygen (Farrell and Lee 2004). Furthermore, hypoxia also regulates the iron-metabolizing gene, namely transferrin, transferrin receptor, and ferroxidase, by increasing the expression of these genes to increase the iron resource to the erythroid tissues (Ke and Costa 2006).
Besides, chronic hypoxia also persuades angiogenesis, a multistep process by which new blood vessels grow from present vasculature, responsible for preserving a satisfactory blood flow to areas of inadequate oxygen supply (Liao and Johnson 2007). Chronic hypoxia also encourages the expressions of the numerous growth factors involved in cell proliferation. The promotion of cell proliferation leads typically to cell migration and regeneration after acute or chronic hypoxia injury (Harris 2002). Various scientific evidence also proposes that hypoxia encourages numerous types of cell differentiation, and a strong connection has been established between hypoxia, HIFs, and molecules that are important in cell differentiation, such as Notch, Oct-4, and MYC (Simon and Keith 2008). In normoxic situations, the energy in ATP for cell metabolism is mainly produced via the oxidative metabolism of carbohydrates, fats, and amino acids. However, under the chronic hypoxia stage, the energy production will switch from oxidative phosphorylation to anaerobic glycolysis, which will lead to a noteworthy decrease in the ATP/ADP ratios (Kristian 2004; Hochachka et al. 1996). In normoxic situations, the ROS is produced at numerous cell sites, and the electron transfer chain at the mitochondria produces a significant amount of cellular ROS (Fruehauf and Meyskens 2007). However, although it is uncertain how ROS is formed under the hypoxia stage, researchers have reported both reductions and upsurges of ROS levels (Michiels et al. 2002; Chandel and Budinger 2007; Fandrey and Genius 2000). Nevertheless, it is generally accepted that chronic moderate hypoxia cells induce a relative addition in ROS productions (Chandel et al. 1998), which is toxic to the cells. Unexpectedly, the cells’ reaction to the hypoxia not only leads to the cell proliferation/survival but also the apoptotic cells’ death by regulating the antiapoptotic protein such as BCL-2 proteins to promote apoptosis (Youle and Strasser 2008).
3.4 Role of Hypoxia in Cancer
Hypoxia in cancer can be mainly categorized into two stages: perfusion-restricted (acute) hypoxia and diffusion-restricted (chronic) hypoxia. Acute hypoxia refers to short-term hypoxia (between a few minutes and up to 72 h), with inadequate transport of oxygen as a result of abnormal blood vessels that close and reopen repeatedly to sluggish blood flow and lead to a fluctuation of oxygen supply (Challapalli et al. 2017; Emami Nejad et al. 2021). It usually occurs during the vacillation in tumor perfusion accompanying defective structural and functional vascular network in tumor and is often associated with high-interstitial pressure of extracellular matrix (Challapalli et al. 2017). Chronic hypoxia arises when the diffusion of oxygen is restricted by the aberrant vascular network (Emami Nejad et al. 2021). It usually occurs when the tumor expands and exceeds a diameter of 70 μm, which result in inadequate oxygen consumption for the cells further away from the preexisting nutritive blood vessels (Emami Nejad et al. 2021). Sometimes, anemic hypoxia will occur associated with a reduction in the oxygen transport capacity by the blood. Anemic hypoxia is the consequence of the presence of the tumor or therapy-induced (Emami Nejad et al. 2021).
Folkman was the first to provide seminal evidence regarding the importance of the availability of oxygen and nutrient that can affect the growth of tumors leading to the prediction of the existence of a molecular mechanism allowing tumors to co-opt vessels (Folkman 1995, 1971). This hypothesis was subsequently proved as angiogenesis (a process of development of new blood vessels from preexisting vasculature) is an adaptive pathobiological response that is commonly found in hypoxic tumor cells to promote oxygen delivery (Schito 2019). This is because angiogenesis is required to sustain tumor cells that proliferate rapidly with an adequate supply of oxygen and metabolism. Hence, hypoxia is the key regulator of angiogenesis in cancer. Hypoxia-inducible factor (HIF), a family of transcriptional regulators, had been identified as the primary mediator in response to cellular adaptation to hypoxia (Muz et al. 2015). Intratumoral hypoxia stimulates the expression of several pro-angiogenic and angiogenic factors and their receptors such as vascular endothelial growth factor (VEGF), VEGF receptor-1,-2 (VEGFR-1,-2), platelet-derived growth factor B (PDGF), epidermal growth factors (EFG), stromal-derived growth factor-1 (SDF-1), basic fibroblast growth factor (bFGF), and insulin-like growth factor II (IGF2) in cancer cells via HIFα-dependent transcriptional activity to facilitate tumor angiogenesis (Carmeliet 2005; Emami Nejad et al. 2021). In particular, gene products, including VEGF-A and angiopoïetun-2 (ANG-2), which are mediated by HIF in the hypoxic microenvironment are able to induce the growth of the vascular network and develop new blood supplies (Brahimi-Horn et al. 2007; Seo et al. 2014). Hypoxia also activates the HIFα-independent pro-angiogenic pathways including the unfolded protein response (UPR) and mechanistic target of rapamycin (mTOR) (Schito 2019). Additionally, angiogenesis-related gene products such as endothelin, heme oxygenase 1, inducible nitric oxide synthase (iNOS), and adrenomedullin are also important in modulating local blood flow via regulation of the vascular tone (Emami Nejad et al. 2021).
In tumors, new vessels that develop under hypoxia conditions are often abnormal, immature, and leaky. They are either inadequate or excessive depending on the type of tumor (Muz et al. 2015). These newly developed vessels often displayed substantial abnormalities such as contractile wall components deficiency and physiological or pharmacological receptor deficiency (Emami Nejad et al. 2021). Besides, reported literature showed that these newly developed vessels with insufficient smooth muscle layer, lack of endothelial cell lining and basement membrane, or broken endothelium may increase their dilation and permeability, whereas the newly developed vessels with abnormal elongated, tortuous, or disorganized shape and often containing blind ends may lead to the disruption in blood flow and intermittent stasis due to the formation of geometric resistance (Emami Nejad et al. 2021). Hence, the tumor tissue response to alleviate defective oxygen supply still fails. Consequently, hypoxia-induced tumor angiogenesis will associate with a more invasive tumor phenotype due to more access of metastatic cells to blood vessels. This further allows the cancer cells to escape from the adverse microenvironment and disseminate into secondary sites (Schito 2019). Therefore, hypoxia-induced tumor angiogenesis can also be recognized as one of the important steps in carcinogenesis as it increased tumor progression and impeded the survival rate of cancer patients.
It was demonstrated previously that tumor oxygenation is the primary factor of cancer progression. Cancer stem cells (CSCs) that possess the self-renew ability are considered one of the important agendas for tumor growth and metastasis as they can produce all the heterogeneous cells in tumors (Sun et al. 2020). In a hypoxic environment, the expression of HIF-1α and HIF-α transcriptional regulators can maintain the stemness of cancer stem cells (CSCs) by activating the Notch and Wnt signaling pathway so that the CSCs can continue to proliferate and self-renew (Sun et al. 2020). Besides, hypoxic tumor cells with overexpression of HIF-α transcriptional regulators and enhanced angiogenesis are often associated with metastasis as the newly developed heterogeneous and permeable vessels are able to facilitate the circulation, extravasation, and relocation of tumor cells (Muz et al. 2015). This may cause the tumor cells to metastasize in order to escape from the adverse hypoxic environment and move to new and unaffected tissues (Muz et al. 2015). Furthermore, hypoxic tumor cells also possess a more aggressive and invasive phenotype and have a better ability to metastasis. For instance, the expression of HIFs in hypoxic cancer cells will generate numerous signaling pathways, further resulting in the disruption of the basement membrane and reconstruction of the extracellular matrix (EMC) that causes the tumor tissue to invade and passage the lumen of lymphatic vessels or blood vessels (Schito and Semenza 2016). The expression of HIFs will also inhibit anoikis, which is a programmed cell death occurring upon the detachment of tumor cells from the ECM when transiting in blood or lymph. In addition, the HIF transition factors also induce epithelial-mesenchymal transition (EMT) whereby the tumor epithelial cells lose their polarity and convert into mesenchymal cells, which are more intrusive and invasive and regarded as a critical event in morphogenic changes during cancer metastasis (Jiang et al. 2011; Schito and Semenza 2016). This EMT process can be done by decreasing the expression of epithelial-associated genes including the β-catenin, E-cad, and increasing the expression of mesenchymal-like genes such as SMA, N-cad, CXCR4, and vimentin (Muz et al. 2015).
Moreover, hypoxia is also responsible for modulating the immunosuppressive tumor microenvironment (TME). TME, which consists of immune effector cells and stromal such as CD8+ cytotoxic T cells, CD4+ T helper cells, FoxP3/CD25+ regulatory T cells, myeloid-derived suppressor cells (MDSC), natural killer (NK) cells, NK-like T (NKT) cells, and M1 and M2 macrophages, are sensitive to hypoxia (Multhoff and Vaupel 2020). Several shreds of evidence suggest that a hypoxic microenvironment may inhibit antitumor immune effector cells to protect tumors from the antitumor response and facilitate immune escape (Lee et al. 2010). For example, hypoxia (1) induces cell shedding of immune recognition molecules, (2) inhibits the release of immunostimulatory cytokines, (3) induces the production of immunosuppressive cytokines, (4) induces the expression of the immune checkpoint, and (5) supports the immunosuppressive activity of M2 macrophages (Lee et al. 2010; Multhoff and Vaupel 2020). Besides, hypoxia also increases the production of immunosuppressive Treg cells flowed by decreasing the monocytes and dendritic cells (DC) motility, which further impairs the stimulation of T cells (Ohta 2016).
Otto Warburg was the first to describe the increased conversion of glucose to lactate in cancer cells, leading to the exploration of the enhanced anaerobic glycolysis effect in cancer cells (Warburg 1956). A number of studies have proven that enhanced glucose uptake is one of the hallmarks of cancer. It is also agreed that the “Warburg effect” is part of the essential metabolic changes in cancer cells in order to proliferate uncontrollably and inhibit apoptosis (Matsuura et al. 2016). The alteration of cancer metabolism closely relates to the cancer microenvironment. Under hypoxic conditions, tumor cells will switch their energy-producing metabolism from mitochondria oxidative phosphorylation to glycolysis with the help of HIF-1 transcription factors (Matsuura et al. 2016). This is because the glycolysis pathway provides the precursors for the nucleotides and phospholipids synthesis, which are critical for cells to grow rapidly (Li and Rich 2010). In hypoxic conditions, the von Hippel–Lindau (VHL) protein that is responsible for the degradation of HIF-1α is inhibited. This further results in the accumulation of HIF-1α and lead to the upregulation of HIF-1-related targets such as glucose transporter GLUT1 and GLUT3, hexokinases HK1 and HK2, to promote glycolysis (Matsuura et al. 2016). Besides, HIF-1 transcription regulators also induce the target genes, i.e., (1) lactate dehydrogenase A (LDHA) that is responsible for the conversion of pyruvate to lactate; (2) pyruvate dehydrogenase kinase 1 that is responsible for the inhibition of acetyl-CoA conversion from pyruvate as well as (3) BNIP3 and BNIP3L that is responsible for the mediation of damaged mitochondria clearance in tumor cells (Matsuura et al. 2016).
Additionally, hypoxia also plays an important role in therapy resistance and had been recognized as one of the biggest obstacles in cancer therapy. For example, oxygen deprivation may cause the cancer cells’ resistance to ionizing radiation, multiple forms of chemotherapy, and photodynamic therapy (Emami Nejad et al. 2021). In this case, hypoxia confers treatment resistance to cancer cells by inhibiting apoptosis and senescence of cells, promoting cell cycle arrest, controlling autophagy, and affecting drug delivery and cellular uptake (Muz et al. 2015). Accumulating evidence shows that HIF-1 transcriptional factors play a crucial role in the apoptosis resistance of tumor cells. HIF-1, which is expressed in the hypoxic environment, is able to upregulate the antiapoptotic genes, including MCL-1, BCL-xL, BCL-2, surviving and NF-κB, to protect the cancer cells from hypoxia-induced cell death and confers tumor cells the ability to resist various cancer treatments (Lin et al. 2014). Besides, hypoxia promotes cell cycle arrest and quiescence cellular state that make tumor cells less susceptible to external stress during chemotherapy or radiotherapy, which is commonly targeted at the bulk of rapidly proliferating tumor cells (Emami Nejad et al. 2021; Muz et al. 2015). Under a hypoxic environment, the expression of HIF-2 in tumor cells may result in the promotion of HIF-induced autophagy and inhibition of p53-mediated apoptosis, which increases the ability of cancer cells to resist chemotherapy (Emami Nejad et al. 2021). Hypoxia also leads to the formation of low pH (acidic) tumor microenvironment (TME). The changes in TME will affect the accumulation of chemotherapeutic drugs that are highly pH-dependent in terms of their cellular targets and further reduce the efficacy of drugs, thereby eventually leading to drug resistance (Jing et al. 2019).
The components in the tumor hypoxic microenvironment are associated with the poor prognosis in cancer patients as hypoxia allows the tumor to survive, disseminate, and be invasive. The HIF transcription factors play an important role in the regulation of the hypoxic tumor microenvironment. In hypoxic conditions, the activation of HIF transcription factors will result in the expression of specific target genes encoding proteins to promote angiogenesis, maintain the stemness of cancer cells, switch energy-producing metabolism from mitochondria oxidative phosphorylation to glycolysis, induce metastasis, and resist cancer therapy. Besides, tumor hypoxia may result in an immunosuppression effect in cancer cells as hypoxia promotes the production of cytokines and chemokines that are capable of recruiting pro-tumor immune cells and block tumor immune response. Furthermore, the hypoxia-induced acidic tumor microenvironment is also important for chemoresistance. Since hypoxia significantly increases tumor survival and reduces the survival rate of cancer patients, a better understanding of the hypoxic phenomenon and hypoxia signaling cascade is crucial to develop new strategies to improve treatment outcomes.
3.5 Free Radicals
Oxygen is a vital element of life that cells utilize in the process of generating energy. It is during the production of adenosine triphosphate (ATP) by mitochondria that results in the formation of free radicals. Free radicals can be defined as reactive molecules or molecular fragments containing one or more unpaired electron(s) in their external shell (Valko et al. 2007). Free radicals are said to be unstable and highly reactive because of the odd number of electron(s) in their atomic or molecular shell. The presence of the unpaired electron tends to donate or attract electrons from other compounds to attain its stability. This process results in a chain reaction cascade as the attacked molecule loses its electron and becomes a free radical itself that brings about damage in living cells (Phaniendra et al. 2015). Since these free radicals are produced by losing or accepting a single electron, therefore, they are also known as reductants and oxidants, respectively. The existence of free radicals in biology was discovered less than 50 years ago (Commoner et al. 1954; Droge 2002).
Free radicals were described as Pandora’s Box of evil due to their possibility to account for cellular damage, cancer, as well as the degenerative process of biological aging. In the later years, the discovery of enzyme superoxide dismutase (SOD) brought about the evolution of free radicals in living organisms that convinced most researchers regarding its importance in biology (McCord and Fridovich 1969). Ever since, the mention of free radicals has gained an ever-increasing curiosity due to their pivotal role in various physiological conditions and a diverse range of diseases (Phaniendra et al. 2015). Free radicals typically include both reactive oxygen and nitrogen species referred to as reactive oxygen-nitrogen species (RONS). Both reactive oxygen and nitrogen species are categorized into two compound groups: radicals and nonradicals. Radicals primarily consist of those species that contain at least one unpaired electron in the external shells and are capable of independent existence (Phaniendra et al. 2015). One excellent example of a radical is the oxygen molecule. However, because of the presence of two unpaired electrons in the shell surrounding its atomic nucleus, it is referred to as biradical. Besides oxygen, superoxide, hydroxyl, alkoxyl, peroxyl radical, nitric oxide, and nitrogen dioxide are some other examples of radicals. Nonradical species, on the contrary, are not free radicals but are capable of bringing about their reactions in living organisms. Some examples of nonradical species include hydrogen peroxide (H2O2), hypochlorous acid (HOCl), hypobromous acid (HOBr), ozone (O3), singlet oxygen (O2), nitrous acid (HNO2), nitrosyl cation (NO+), nitroxyl anion (NO−), dinitrogen trioxide (N2O3), dinitrogen tetraoxide (N2O4), and peroxynitrite (ONOOH) (Phaniendra et al. 2015).
ROS and RONS in the human body are produced primarily by employing essential cellular metabolism and also external sources. In other words, these sources of free radicals are conveniently known as endogenous and exogenous sources. Endogenous sources simply refer to internally generated sources of free radicals like mental stress, excessive exercise, infection, inflammation, phagocytosis, reperfusion injury, mitochondria, and peroxisomes (Lobo et al. 2010). Continuous formation of free radicals takes place in the cells as a consequence of both enzymatic and nonenzymatic reactions. Enzymatic reactions contributing to this process include those involved in the respiratory chain, prostaglandin synthesis, phagocytosis, and cytochrome P-450 system. Nonenzymatic reactions include the reaction of oxygen with organic compounds and those initiated by ionization (Lobo et al. 2010). Exogenous free radicals, on the other hand, result from cigarette smoke, air pollutants, industrial solvents, radiation, ozone, heavy and/or transition metals such as mercury, lead, and iron, as well as some drugs like cyclosporine and gentamycin. Upon the absorption/penetration of these external compounds into the body, they undergo decomposition or metabolization, which brings about the formation of free radicals. There are various physiological and pathological conditions in which free radicals play their fundamental part. By targeting and attacking essential macromolecules, reactive oxygen-nitrogen species (RONS) leads to cell damage and disruption of homeostasis (Lobo et al. 2010). Major essential molecules targeted in the body by RONS include nucleic acids (RNA and DNA), lipids, and proteins. Severe damage to these macromolecules occurs abundantly with the accumulation of free radicals as a consequence of antioxidants and oxidants imbalance. This leads to tissue damage in various disease conditions such as diabetes mellitus, neurodegenerative diseases, cancer, cardiovascular diseases, rheumatoid, arthritis, and asthma, therefore speeding the progression and growth of the disease (Phaniendra et al. 2015). Although both reactive oxygen and nitrogen species are frequently studied for their ability to cause extensive damage to some major molecules in the body, these free radicals also are undoubtedly recognized for their dual role as both deleterious and beneficial species (Valko et al. 2007). The harmonious balance between both antagonistic effects plays a significant role in the body. Reactive species, at low or moderate levels, portray beneficial effects in cellular signaling systems and immune function (Pham-Huy et al. 2008). On the contrary, its overproduction leads to the exertion of undesirable effects by inducing oxidative stress that damages the cell.
3.6 Oxidative Stress
The term “stress,” initially used in the literature, was described as the hyperactivity of the hormone system particularly concerning corticosteroids (Breitenbach and Eckl 2015). After about two decades, the understanding of “stress” was brought to considerable attention once again regarding its importance in the study of diseases as well as general physiology. In other words, “stress” was primarily seen as a disease-causing factor. With the evolution of theories over the years, the term “oxidative stress” was proposed based on the profound knowledge of its mechanism and involvement in disease studies. Oxidative stress is the detrimental effect of free radicals that brings about possible biological damage. Oxidative stress occurs because of an imbalance between reactive oxygen species (ROS) formation and cell capacity to eliminate them (Liguori et al. 2018). This arises when the production of ROS overpowers the content level of intrinsic antioxidants, rendering the antioxidant defense mechanism unfavorable. The generation and elimination of ROS are usually very well balanced by extensive regulatory systems to maintain an equilibrium state of ROS level (Lushchak 2014). However, this critical balance can be interrupted by several sources/factors. The factors include depletion of low molecular mass antioxidant reserves, inactivation, and/or decreased production of antioxidant enzymes, as well as the combinations of the stated factors (Lushchak 2014). The degree of outcomes due to the imbalance depends on the location of ROS production, the efficiency of antioxidant defense systems, and the cellular targets with which the free radicals interact (Lushchak 2014).
As mentioned, oxidative stress is a destructive process that negatively affects cellular components like membranes, lipids, proteins, and nucleic acids. This process leads to the oxidation of the cellular components, resulting in their structural and functional changes (Pizzino et al. 2017). Thus, the critical balance between beneficial and deleterious effects of free radicals is vital and is achieved by a mechanism known as redox regulation (Droge 2002). Maintenance and regulation of ROS homeostasis play a crucial role in cellular growth, survival, and metabolism. The presence of ROS at a low level is important in maintaining cellular functions such as viability and apoptosis while the extreme level, on the other hand, causes biological systems to incompletely detoxify the reactive intermediates and prevent the normal functions of biomolecules. Both reactive oxygen species and oxidative stress have been proposed to play significant roles in various illnesses and health conditions. They contribute mainly to the process of aging and diseases such as cancer, inflammatory disorders (arthritis, vasculitis, and systemic lupus erythematosus), ischemic disorders (heart diseases, stroke), acquired immunodeficiency syndrome, hypertension, neurological conditions (Alzheimer’s, Parkinson’s disease, muscular dystrophy), and many more (Bajpai et al. 2014). The metabolism of molecular oxygen by cells brings about the formation of reactive oxygen species that interact with vital macromolecules. This interaction becomes the basis of most diseases and conditions stated. Currently, it is a challenge to mention any illness for which the roles of oxidative stress and ROS have not been postulated (Ghezzi et al. 2017). Many researchers, with the help of strong evidence, suggest that oxidative stress can be associated with numerous diseases.
With virtually three decades passed since the primary definition of oxidative stress was introduced, there still has been no accepted categorization to date. Consequently, in an effort to understand the possible degrees of stress and effects, a basic classification based on intensity was presented. This intensity-based categorization includes basal oxidative stress (BOS), low-intensity oxidative stress (LOS), intermediate intensity oxidative stress (IOS), as well as high-intensity oxidative stress (HOS) (Lushchak 2014). In addition, another classification of potential stress degree too was proposed consisting of three simple terms, namely mild oxidative stress (MOS), temperate oxidative stress (TOS), and severe (strong) oxidative stress (SOS) (Lushchak 2014). Normally, the level of ROS fluctuates to an extent outlined by their generation and elimination. However, because of various endogenous and exogenous factors, its usual level increases beyond the normal range. In circumstances in which the antioxidant defense systems are capable dealing with increased free radical amounts, they are known as “acute oxidative stress” (Lushchak 2012). On the contrary, “chronic oxidative stress” takes place when cells are unable to counteract the enhanced ROS content. At such a state, even an improved expression of antioxidant and related enzymes would be unable to return ROS to its initial range. Stabilization at the new increased ROS level (quasi-stationary level) occurs and modification of cellular components is enhanced, significantly interrupting the redox homeostasis (Lushchak 2012).
3.7 Role of Reactive Oxygen Species in Cancer
Reactive oxygen species (ROS) are highly reactive oxygen species containing a single unpaired electron in their outermost shell of electrons. Among the various types of endogenous and exogenously generated ROS, superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH) are the most well-studied ROS in cancer (Liou and Storz 2010). Today, it is well-known that increased amounts of ROS culminate in oxidative stress, a state of a cell defined by the oxidation of critical biomolecules, that alters cell function, homeostasis, and cell structure. This condition potentially leads to the development of numerous pathologies, including inflammatory, cardiovascular and neurodegenerative disorders, as well as age-related diseases and deadly cancer disease (Snezhkina et al. 2019; Zhang et al. 2016). Though the present antioxidant system, along with the enzymatic and nonenzymatic systems, protects the cells from the deleterious effects of ROS when they are in a state of the homeostatic microenvironment, during an overproduction of ROS, an imbalance between the ROS and antioxidant factors occurs (De Sá Junior et al. 2017). This interruption to the redox balance imbalance is related to the progression of many diseases, especially cancers.
In particular, excessive generation of ROS in cancer cells, both in the various cellular compartments and in the cancer cell microenvironment, are able to disrupt the genetic stability of cells and a variety of other cellular processes. ROS and oxidative stress are important factors in the development of carcinogenesis and have an impact on all the cancer hallmarks (Aggarwal et al. 2019). Indeed, when compared to nearby normal tissues, tumor tissues generation of H2O2 is elevated. Furthermore, in the blood and urine of cancer patients, it is common to detect higher levels of 4-hydroxynonenal (4-HNE) (lipid peroxidation) and 8-oxoguanine (8-oxoG) (oxidative DNA damage) and are linked to a poor prognosis (Assi 2017). Cancer cells have high steady-state ROS levels as a result of both external and internal processes. High metabolic activity, cellular signaling, peroxisomal activity, mitochondrial dysfunction, oncogene activation, and increased enzymatic activity of oxidases, cyclooxygenases, lipoxygenases, and thymidine phosphorylases have all been linked to elevated ROS levels in cancer cells (Kumari et al. 2018). The final impact on cancer biology is dependent on the overall balance of ROS and the combined positive and detrimental effects of ROS. Thus, a better understanding of the intricate roles of ROS in cancer biology could provide insights into the underlying molecular mechanisms and assist in the development of more effective anticancer strategies.
In the course of cancer, the role of ROS is considered a double-edged sword due to their characteristic features in which many studies have defined that the intensity of ROS levels could either play a role of tumor-promoting (carcinogenic role) or tumor-suppressing (antitumorigenic role) depending on several factors (Fig. 3.1). In fact, the intricate roles of ROS in cancer growth are complicated. For normal cell survival, a physiological concentration of ROS must be maintained in balance. Generally, in almost all malignancies, the reprogramming of redox metabolism causes an ectopic ROS accumulation level, therefore functioning as signaling molecules favoring various aspects of tumor growth and progression. Nevertheless, the role of ROS in cancer is not one-sided. ROS functions as anti-tumorigenic at excessive ROS levels triggering oxidative stress-induced cancer cell death due to their detrimental, genotoxic, or even proapoptotic effect on cancer cells (De Sá Junior et al. 2017). The latter is attributable to an increase in antioxidant protein levels in tumor cells in response to the overproduction of ROS, implying that cancer cell function depends on a precise balance of intracellular ROS levels. Taken together, the multifaceted roles of ROS in tumor growth, metastasis, and death are according to the ROS types, various distributions, concentrations, and lifetime in specific subcellular structures (Wang et al. 2021). Besides, recent literature has proposed categorizing the paradoxical roles of ROS in cancer cells depending on two groups: early and late stages of cancer progression. That is to say that intracellular ROS plays a variable role in cancer cell survival depending on the stage of cancer growth (Liao et al. 2019). Moderate ROS levels stimulate carcinogenesis, tumor propagation, angiogenesis, metastasis, and survival in precancerous/early stages of tumor growth. Whereas increased ROS levels beyond the hazardous threshold prompt cell death, apoptosis, and senescence with tumor progressions (Fig. 3.2) (Galadari et al. 2017).

The paradoxical role of reactive oxygen species (ROS) in cancer
3.7.1 ROS as Tumor-Promoting Agent (Carcinogenic Role)
Substantial research over the last two decades has clearly suggested that cancer cells have increased aerobic glycolysis (Warburg effect), which is correlated with augmenting ROS (Hart et al. 2015), and that these elevated ROS levels are thought to play oncogenic roles and act in multiple signaling cascades related to various behaviors in the cancer cell. Increased ROS is usually accompanied by proto-oncogenes activation, which is the initial step of malignant transformation and inactivation of tumor suppressor genes. ROS are thus essential and responsible for the tumorigenesis initiation, development, progression, invasion, as well as metastasis of cancer in several ways (Wang and Yi 2008; Kirtonia et al. 2020). The increase in ROS will boost the mutation rate and promote the transformation of normal cells into tumor cells. ROS can also promote the stability of important signal molecules that drive tumorigenesis and progression (Xie et al. 2021). Overall, these tumor-promoting ROS levels can lead to cell cycle progression, increased proliferation and survival signaling, epithelial-to-mesenchymal transition (EMT), increased motility, genomic instability, and increased angiogenesis and may be negatively regulated by therapeutic antioxidants. ROS plays a vital role at every stage of cancer development in which the prooncogenic role for ROS is usually associated at precancerous and early stages of cancer. ROS has been demonstrated to activate a number of canonical pathways implicated in tumor-promoting inflammation and cell proliferation. The following sections go over some of ROS’s most essential tumor-promoting actions.
3.7.2 ROS Role in Tumorigenesis
ROS can affect cellular proteins, lipids, and DNA, leading to genomic instability and activation of various tumor-related signaling pathways, depending on the concentration and duration of exposure (Galadari et al. 2017). Increased intracellular ROS is widely thought to enhance cancer initiation by triggering oxidative and base-pair substitution mutations in pro-oncogenes that target GC bases or inactivation of tumor suppressor genes, all of which ROS can promote carcinogenesis. One of the crucial phases in carcinogenic mutagenesis and tumor transformation is the accumulation of mutations, which impact genome stability and dynamics of gene expression, resulting in irreversible changes in genetic material. Examples of these mutations can be seen in active mutant Ras, Bcr-Abl, and c-Myc, which are involved in cell proliferation and tumor suppressor genes such as p53 that provokes abnormal mitosis, and promote cancer development (Assi 2017). 8-Hydroxy-2 deoxyguanosine (8-oxo-dG) is a major oxidative DNA damage product that is commonly used to test intracellular oxidative stress levels. It is highly expressed in a variety of malignant tumor tissues compared to matched normal tissues and is well known for inducing adjacent DNA base mutations (Tudek et al. 2010). ROS also functions as signaling molecules that are controlled by oncogene activation or anti-oncogene inactivation apart from the direct carcinogenic consequences of DNA damage and chromosomal instability. For instance, highly expressed proto-oncogene p21RAS in NIH3T3 fibroblast cells produces superoxide through RAC1 activation in NOX complexes in huge amounts, which proceeds to increase the mitogenic activity (Wang et al. 2021). Hence, ROS may contribute to cancer growth and progression by inducing signaling pathways that enhance the rate of mutations (Ramalingam and Rajaram 2021). Apart from triggering mutations, ROS can also affect the side chains of certain amino acids, altering the structure and function of proteins (Kumari et al. 2018), which is usually through oxidizing the disulfide bonds of cysteine residues. Due to the sheer presence of a thiol group, cysteine (Cys) is more susceptible to oxidation by ROS than the other amino acids. Cys appears to be the most important player in redox signaling, working as a reversible regulatory molecular switch.
Furthermore, ROS has the ability to influence the activities of a number of proteins and signaling pathways involved in tumor cell proliferation of several malignancies, including lung, liver, and breast cancers. ROS drives proliferation by activating protein kinase D (PKD), mitogen activated-protein kinase/extracellular-regulated kinase 1/2 (MAPK/ERK 1/2), and phosphoinositide-3-kinase/protein kinase B (PI3K/Akt) signaling pathways (Moloney and Cotter 2018). Increased ROS levels, for example, impede MAPK by oxidizing cysteine residues in the active site, while degradation of MAPK phosphatase 3 (MPK3) significantly lowers ERK 1/2 activity (Chan et al. 2008). Besides, the constitutive activation of cell proliferation-related transcription factors such as nuclear factor-B (NF-B) and activator protein-1, which promote cancer cell proliferation during cancer initiation and development by activating numerous genes, are also activated by the elevated ROS levels (Raza et al. 2017). These findings back up the theory that ROS-induced mutagenesis is a key driver of tumor initiation and progression.
3.7.3 ROS Role in Invasion and Metastasis
Some of the critical requirement events in the metastasis and prognosis of cancer are cell migration and invasion. The crucial characteristics of tumor metastasis processes that are linked to ROS include the loss of cell-to-cell adhesion, survival after matrix dissociation, mitigating capability, and breakthrough into the cell basement membrane (Chiang and Massagué 2008). Metastasis, the primary cause of morbidity and mortality, is a complicated process in which cancer cells migrate from the primary tumor to the surrounding tissues and distant organs. This happens as a result of cancerous cells’ intrinsic mutational burden and bidirectional interaction between nonmalignant and malignant cells (Brooks et al. 2010). The activation of various transcriptional factors, including NF-kB, ETS-1 (ETS proto-oncogene 1, transcription factor), Twist, Snail, AP-1, and Zeb (zinc finger E-box binding homeobox); metalloproteases, viz., MMP-9 and, MMP-2; and chemokines or cytokines like transforming growth factor-beta (TGF-β) results in metastasis (Aggarwal et al. 2019). The epithelial to mesenchymal transition (EMT), in which epithelial cells lose their polarity, cell–cell adhesion, and motility, is the most common cause and beginning of tumor metastasis. Many studies have found that ROS, which is mostly formed as by-products of mitochondrial electron transport in aerobic respiration, plays a crucial role in the migration and invasion of malignant cells, particularly as the primary cause of EMT. ROS activate the Distal-less homeobox-2 (Dlx-2)/Snail axis, triggering the EMT, the glycolytic switch, and mitochondrial suppression, all of which are important in metastasis (Lee et al. 2019). Besides, ROS enhances tumor migration via activating hypoxia-mediated MMPs and cathepsin production, according to another study (Kamiya et al. 2016; Shin et al. 2015). One of the most important players in the EMT induction is TGF-β1, which employs the ROS-dependent pathways to regulate uPA (urokinase type plasminogen activator) and MMP-9 to enhance cell migration and invasion (Tobar et al. 2010). As more information became available, it became clear that ROS plays a diverse role in EMT. The fact that ROS is involved in multiple pathways that are directly linked to many important EMT-inducing pathways emphasizes its significance and key function in EMT.
3.7.4 ROS Role in Angiogenesis
There will be more developments of fledgling blood vessels as highly proliferative tumors outgrow, assisting oxygen and nutrition delivery to the center of the tumor. Angiogenesis is the development of new blood vessels from preexisting capillaries during the early stages of carcinogenesis, which helps tumor proliferation, survival, and metastasis (Liou and Storz 2010). ROS appears to have a role in enhancing angiogenesis for tumor survival and maintenance by modulating important events in tumor angiogenesis such as endothelial cell (EC) proliferation, migration, and tube formation, according to numerous lines of evidence (Potente et al. 2011). Datla et al. (2007) observed that NOX4 mediates EC proliferation by generating H2O2, whereas NOX2 inhibits apoptosis and encourages EC survival. The angiogenesis process is mediated by an angiogenic switch that may be opened and closed by adjusting the balance between angiogenic [vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), and platelet-derived growth factor (PDGFB)] and anti-angiogenic [angiopoietin-1, leptin, endoglin, prominin-1, transforming growth factor beta (TGF-beta), integrins, and matrix metalloproteinase (MMP) enzymes factors] (Varol 2020). The direction of the angiogenic switch is predicted to be regulated in conjunction with the activation of angiogenic or anti-angiogenic factors within cells and microenvironments where ROS and oxidative stress are present through the regulation of transcriptional factors, the release of some growth factors, and the alteration of cellular signaling cascades. ROS, for example, has been linked to numerous pathways that stimulate VEGF-mediated phosphorylation of the cadherin/catenin cell–cell adhesion complex. These ROS-mediated phosphorylation of cadherin/catenin causes EC disassembly, which promotes tumor vascularization and rapid expansion of the tumors (Monaghan-Benson and Burridge 2009). In addition, Redox factor-1 (Ref-1), NF-B, p54, matrix metalloproteinases (MMPs), and cyclooxygenase-2 (COX-2) are all transcription factors and genes involved in angiogenesis that are regulated by ROS (Galadari et al. 2017). The tumor-induced angiogenesis and the re-oxygenation of tumor cells through the development of new capillaries, however, is contradictory to the assumptions as this only exacerbates the problem of cancer cells rather than resolving it (Varol 2017).
3.7.5 ROS as Tumor-Suppressing Agent (Cytotoxic Role)
With the growing plethora of evidence pointing to ROS as a tumor-promoting agent, there is also mounting research demonstrating that in conjunction with the enhanced tumorigenesis, ROS is responsible for tumor degradation by inducing cell death and reversing chemoresistance in tumors (Yang et al. 2018). This apparent contradiction between the positive and negative roles of ROS in tumors stems from the fact that antioxidant therapy, which supposedly eliminates cancer-boosting ROS, bizarrely corresponds with lower survival in clinical trials (Goodman et al. 2011). Although it is rational to treat ROS-triggering tumors with antioxidants, the mechanism underlying the effects of many chemotherapeutic agents and ionizing radiation on tumor cell death is not linked to an upsurge in antioxidants, instead of with irreversible oxidative stress due to an increase in ROS (Wang and Yi 2008). Indeed, numerous therapeutic techniques have been shown to not only rely on ROS but also boost cellular ROS levels could in fact efficiently kill more cancer cells (Ozben 2007). Another explanation for the antioxidant therapy results could be that antioxidants reduce ROS to a level that promotes tumor propagation and migration while diminishing some of ROS’s negative effects in cancer cells (Wang et al. 2016). These events of tumor degeneration occur when ROS levels rise above the tipping threshold, increasing cellular oxidative stress. The increased levels of ROS come as a result of chemotherapy, which affects ROS formation in cells while also inhibiting cellular antioxidant detoxification (Yang et al. 2018). This results in a shift in the role of ROS in cancer from carcinogenic to antitumorigenic by increasing regulated cell death (RCD) programs such as apoptosis, necroptosis, and ferroptosis, as well as triggering cell cycle arrest and senescence, all of which can impede tumor growth (Liao et al. 2019). The development of ROS’ antitumorigenic role is attributed to the phases of cancer, in which ROS suppresses tumor during the late stages of cancer or as cancer develops. This is because the accumulation of excess intracellular ROS might trigger apoptosis as cancer advances (Assi 2017). Apoptosis, however, can be avoided by preserving the delicate balance in ROS level in tumor cells by creating high concentrations of intracellular antioxidants that allows them to proliferate and survive. Meanwhile, metastatic tumors acquire mechanisms that regulate ROS as a stimulant for cancer cell dispersion by lowering the cell’s antioxidant capacity in the late stages of tumor progression. This general impact suggests that therapeutic techniques that either boost ROS production or weaken antioxidant defense may drive cancer cells past their snapping point, activating various cell death pathways and therefore slowing cancer growth. Nonetheless, due to the cell death-inducing action, disproportionately enhanced ROS emerges as a significant strategy for cancer therapeutic techniques.
3.7.6 ROS Role in Cellular Apoptosis
Increases in intracellular ROS that are disproportionally high can cause cancer cell cycle arrest, senescence, and apoptosis. The most prevalent form of cell death is apoptosis, commonly known as type I programmed cell death, and is regulated by extrinsic (death receptor-dependent) and intrinsic (mitochondrial) pathways, which are performed by caspases, specialized cysteine proteases (Hengartner 2000). The ligand–receptor interaction between death-inducing ligands like Fas ligand (FasL) and tumor necrosis factor (TNF) and their respective receptors, Fas receptor (FasR) and TNF receptor, drives the extrinsic apoptotic pathway (TNFR) (Meynier and Rieux-Laucat 2019; Minchenko et al. 2016). Following the ligand–receptor contact, the death-inducing signaling complex (DISC) is formed, which includes an adaptor protein (FADD for FasR and TRADD for TNFR), receptor-interacting protein kinase 1 (RIP1), and procaspase-8 (Wang et al. 2021). On the other hand, the intrinsic apoptotic pathway is activated in a mitochondria-dependent manner by the release of the proapoptotic factors such as cytochrome-c (Cyt-c) and apoptosis-inducing factor (AIF) from mitochondria via the mitochondrial permeability transition pore (MPTP), which then increases the mitochondrial membrane permeability (Burke 2017). Overproduction of ROS by endogenous as well as exogenous sources stimulates both extrinsic and intrinsic apoptosis pathways. ROS activates the extrinsic apoptosis route by speeding up the ubiquitination of the cellular FLICE-inhibitory protein c-FLIP, which then increases the binding between the adaptor protein and pro-caspase-8, facilitating the extrinsic apoptosis process (Wang et al. 2008). ROS, on the other hand, promotes intrinsic apoptosis by accelerating the production of the proapoptotic factor cytochrome-c (Cyt-c). Cyt-c moves from the mitochondria to the cytoplasm, where it forms an apoptosome with caspase-9 and APAF-1, activating the caspase-9 signaling cascade and triggering apoptosis (Dorstyn et al. 2018).
Since the discovery of ROS’s double-edged sword nature, there has been a shift in the understanding of its involvement in cancer. This multifaceted role of ROS in cellular homeostasis and carcinogenesis has been investigated for possible therapeutic effects is critically significant. The perplexing duality of ROS that may either impair antioxidant function or upregulate the apoptosis signaling pathway could be a promising approach in the active search for cancer therapies. The fine-tuning of intracellular ROS signaling to successfully deprive cells of ROS-induced tumor-promoting events in the direction of overturning the balance to ROS-induced apoptotic signaling will be a challenge for emerging therapeutic techniques.
3.8 Conclusions
This chapter provides detailed information on the role of hypoxia and reactive oxygen species in cancer biology. By providing detailed information on the role of hypoxia and reactive oxygen species in cancer biology, we aim to support scientists and young researchers conducting cancer-related studies and serve as resources for researchers working on the development of new strategies to treat cancer by targeting hypoxia and reactive oxygen species.
References
Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, Varol M, Jain A, Khan MA, Sethi G (2019) Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomolecules 9(11). https://doi.org/10.3390/BIOM9110735
Assi M (2017) The differential role of reactive oxygen species in early and late stages of cancer. Am J Physiol Regul Integr Comp Physiol 313:646–653. https://doi.org/10.1152/ajpregu.00247.2017
Bajpai A, Verma AK, Srivastava M, Srivastava R (2014) Oxidative stress and major depression. J Clin Diagn Res 8(12):4–7
Bickler PE, Donohoe PH (2002) Adaptive responses of vertebrate neurons to hypoxia. J Exp Biol 205:3579–3586
Bracken CP, Whitelaw ML, Peet DJ (2003) The hypoxia-inducible factors: key transcriptional regulators of hypoxic responses. Cell Mol Life Sci 60:1376–1393
Brahimi-Horn M, Chiche J, Pouysségur J (2007) Hypoxia and cancer. J Mol Med 85:1301–1307
Breitenbach M, Eckl P (2015) Introduction to oxidative stress in biomedical and biological research. Biomol Ther 5:1169–1177
Brooks SA, Lomax-Browne HJ, Carter TM, Kinch CE, Hall DMS (2010) Molecular interactions in cancer cell metastasis. Acta Histochem 112(1):3–25. https://doi.org/10.1016/J.ACTHIS.2008.11.022
Burke PJ (2017) Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer 3(12):857–870. https://doi.org/10.1016/J.TRECAN.2017.10.006
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936
Challapalli A, Carroll L, Aboagye EO (2017) Molecular mechanisms of hypoxia in cancer. Clin Transl Imaging 5:225–253
Chan DW, Liu VWS, Tsao GSW, Yao KM, Furukawa T, Chan KKL, Ngan HYS (2008) Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 29(9):1742–1750. https://doi.org/10.1093/CARCIN/BGN167
Chandel NS, Budinger GR (2007) The cellular basis for diverse responses to oxygen. Free Radic Biol Med 42:165–174
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 95:11715–11720
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J (2016) Cancer statistics in China, 2015. CA Cancer J Clin 66:115–132
Chiang AC, Massagué J (2008) Molecular basis of metastasis. N Engl J Med 359(26):2814–2823. https://doi.org/10.1056/NEJMRA0805239
Commoner B, Townsend J, Pake GE (1954) Free radicals in biological materials. Nature 174(4432):689–691
Corbucci GG, Marchi A, Lettieri B, Luongo C (2005) Muscle catabolic mechanisms: from disuse atrophy to cachexia. Minerva Anestesiol 71:727–740
Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, Jiang F (2007) Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. https://doi.org/10.1161/ATVBAHA.107.149450
De Sá Junior PL, Câmara DAD, Porcacchia AS, Fonseca PMM, Jorge SD, Araldi RP, Ferreira AK (2017) The roles of ROS in cancer heterogeneity and therapy. Oxidative Med Cell Longev 2017. https://doi.org/10.1155/2017/2467940
Dorstyn L, Akey CW, Kumar S (2018) New insights into apoptosome structure and function. Cell Death Differ 25(7):1194. https://doi.org/10.1038/S41418-017-0025-Z
Droge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82(1):47–95
Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, Nedaeinia R, Haghjooy Javanmard S, Taherian M, Ahmadlou M, Salehi R, Sadeghi B, Manian M (2021) The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment. Cancer Cell Int 21:1–26
Erecinska M, Silver IA (2001) Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol 128:263–276
Fandrey J, Genius J (2000) Reactive oxygen species as regulators of oxygen dependent gene expression. Adv Exp Med Biol 475:153–159
Farrell F, Lee A (2004) The erythropoietin receptor and its expression in tumor cells and other tissues. Oncologist 9(Suppl. 5):18–30
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285:1182–1186
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1:27–30
Fruehauf JP, Meyskens FL Jr (2007) Reactive oxygen species: a breath of life or death? Clin Cancer Res 13:789–794
Galadari S, Rahman A, Pallichankandy S, Thayyullathil F (2017) Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med 104:144–164. https://doi.org/10.1016/J.FREERADBIOMED.2017.01.004
Ghezzi P, Jaquet V, Marcucci F, Schmidt HH (2017) The oxidative stress theory of disease: levels of evidence and epistemological aspects. Br J Pharmacol 174(12):1784–1796
Goodman M, Bostick RM, Kucuk O, Jones DP (2011) Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic Biol Med 51(5):1068–1084. https://doi.org/10.1016/J.FREERADBIOMED.2011.05.018
Harris AL (2002) Hypoxia- a key regulatory factor in tumor growth. Nat Rev Cancer 2:38–47
Hart PC, Mao M, De Abreu ALP, Ansenberger-Fricano K, Ekoue DN, Ganini D, Kajdacsy-Balla A, Diamond AM, Minshall RD, Consolaro MEL, Santos JH, Bonini MG (2015) MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun 6. https://doi.org/10.1038/NCOMMS7053
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407(6805):770–776. https://doi.org/10.1038/35037710
Hochachka PW, Buc LT, Doll CJ, Land SC (1996) Unifying theory of hypoxia tolerance: molecular/ metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 93:9493–9498
Jena J, Ranjan R, Ranjan P, Sarangi MK (2012) A study on natural anticancer plants. Int J Pharmaceut Chem Sci 1(1):365–368
Jiang J, Tang Y, Liang X (2011) EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther 11:714–723
Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, Shu Y (2019) Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer 18:1–15
Kamiya T, Goto A, Kurokawa E, Hara H, Adachi T (2016) Cross talk mechanism among EMT, ROS, and histone acetylation in Phorbol Ester-treated human breast cancer MCF-7 cells. Oxidative Med Cell Longev 2016. https://doi.org/10.1155/2016/1284372
Ke Q, Costa M (2006) Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol 70:1469–1480
Kirtonia A, Sethi G, Garg, · Manoj. (2020) The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci 77:4459–4483. https://doi.org/10.1007/s00018-020-03536-5
Kristian T (2004) Metabolic stages, mitochondria and calcium in hypoxia/ ischemic brain damage. Cell Calcium 36:221–233
Kumari S, Badana AK, Murali Mohan G, Shailender G, Malla RR (2018) Reactive oxygen species: a key constituent in cancer survival. Biomark Insights 13. https://doi.org/10.1177/1177271918755391
Lee C, Mace T, Repasky E (2010) Hypoxia-driven immunosuppression: a new reason to use thermal therapy in the treatment of cancer? Int J Hyperth 26:232–246
Lee SY, Ju MK, Jeun HM, Lee YJ, Kim CH, Park HG, Han SI, Kang HS (2019) Reactive oxygen species induce epithelial-mesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx-2/Snail signaling pathways in MCF-7 cells. Mol Med Rep 20(3):2339–2346. https://doi.org/10.3892/MMR.2019.10466
Li Z, Rich JN (2010) Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Curr Top Microbiol Immunol 345:21–30
Liao D, Johnson RS (2007) Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev 26:281–290
Liao Z, Chua D, Tan NS (2019) Reactive oxygen species: a volatile driver of field cancerization and metastasis. Mol Cancer 18(1):1–10. https://doi.org/10.1186/S12943-019-0961-Y/FIGURES/2
Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D, Abete P (2018) Oxidative stress, aging, and diseases. Clin Interv Aging 13:757–772
Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44(5):479–496. https://doi.org/10.3109/10715761003667554
Lin S, Liao W, Lee J, Tsai S (2014) Hypoxia-regulated gene network in drug resistance and cancer progression. Exp Biol Med 239:779–792
Lobo V, Patil A, Phatak A, Chandra N (2010) Free radicals, antioxidants and functional foods: impact on human health. Pharmacogn Rev 4(8):118–126
Lushchak VI (2012) Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids 2012:1–26
Lushchak VI (2014) Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact 224:1–12
Matsuura K, Canfield K, Feng W, Kurokawaew M (2016) Metabolic regulation of apoptosis in cancer. Int Rev Cell Mol Biol 327:43–87
McCord JM, Fridovich I (1969) Superoxide dismutase an enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244(22):6049–6055
Meynier S, Rieux-Laucat F (2019) FAS and RAS related apoptosis defects: from autoimmunity to leukemia. Immunol Rev 287(1):50–61. https://doi.org/10.1111/IMR.12720
Michiels C, Minet E, Mottet D, Raes M (2002) Regulation of gene expression by oxygen NF-kappaB and HIF-1, two extremes. Free Radic Biol Med 33:1231–1242
Minchenko OH, Tsymbal DO, Minchenko DO, Ratushna OO (2016) The role of the TNF receptors and apoptosis inducing ligands in tumor growth. Ukr Biochem J 88(5) https://doi.org/10.15407/UBJ88.05.018
Moloney JN, Cotter TG (2018) ROS signalling in the biology of cancer. Semin Cell Dev Biol 80:50–64. https://doi.org/10.1016/J.SEMCDB.2017.05.023
Monaghan-Benson E, Burridge K (2009) The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J Biol Chem 284(38):25602–25611. https://doi.org/10.1074/JBC.M109.009894
Multhoff G, Vaupel P (2020) Hypoxia compromises anti-cancer immune responses. Adv Exp Med Biol 1232:131–143
Muz B, de la Puente P, Azab F, Azab A (2015) The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 3:83
Noordin MAM, Noor MM, Aizat WM (2020) The impact of plant bioactive compounds on aging and fertility of diverse organisms: a Review. Mini Rev Med Chem 20(13):1287–1299
Ohta A (2016) A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol 7:109
Ozben T (2007) Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci 96(9):2181–2196. https://doi.org/10.1002/JPS.20874
Pham-Huy LA, He H, Pham-Huy C (2008) Free radicals, antioxidants in disease and health. Int J Biomed Sci 4(2):89–96
Phaniendra A, Jestadi DB, Periyasamy L (2015) Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem 30(1):11–26
Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, Squadrito F, Altavilla D, Bitto A (2017) Oxidative stress: harms and benefits for human health. Oxidative Med Cell Longev 2017:1–13
Potente M, Gerhardt H, Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis. Cell 146(6):873–887. https://doi.org/10.1016/J.CELL.2011.08.039
Ramalingam V, Rajaram R (2021) A paradoxical role of reactive oxygen species in cancer signaling pathway: physiology and pathology. Process Biochem 100:69–81. https://doi.org/10.1016/J.PROCBIO.2020.09.032
Raza MH, Siraj S, Arshad A, Waheed U, Aldakheel F, Alduraywish S, Arshad M (2017) ROS-modulated therapeutic approaches in cancer treatment. J Cancer Res Clin Oncol 143(9):1789–1809. https://doi.org/10.1007/S00432-017-2464-9
Schito L (2019) Hypoxia-dependent angiogenesis and lymphangiogenesis in cancer. Adv Exp Med Biol 1136:71–85
Schito L, Semenza G (2016) Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer 2:758–770
Semenza GL (1999) Perspectives on oxygen sensing. Cell 98:281–284
Seo B, DelNero P, Fischbach C (2014) In vitro models of tumor vessels and matrix: engineering approaches to investigate transport limitations and drug delivery in cancer. Adv Drug Deliv Rev 69:205–216
Seta KA, Yuan Y, Spicer Z, Lu G, Bedard J, Ferguson TK, Pathrose P, Cole-Strauss A, Kaufhold A, Millhorn DE (2004) The role of calcium in hypoxia-induced signal transduction and gene expression. Cell Calcium 36:331–340
Shin DH, Dier U, Melendez JA, Hempel N (2015) Regulation of MMP-1 expression in response to hypoxia is dependent on the intracellular redox status of metastatic bladder cancer cells. Biochim Biophys Acta (BBA) - Mol Basis Dis 1852(12):2593–2602. https://doi.org/10.1016/J.BBADIS.2015.09.001
Simon MC, Keith B (2008) The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol 9:285–296
Snezhkina AV, Kudryavtseva AV, Kardymon OL, Savvateeva MV, Melnikova NV, Krasnov GS, Dmitriev AA (2019) ROS generation and antioxidant defense systems in normal and malignant cells. Oxidative Med Cell Longev 2019. https://doi.org/10.1155/2019/6175804
Sun X, Lv X, Yan Y, Zhao Y, Ma R, He W, Wei M (2020) Hypoxia-mediated cancer stem cell resistance and targeted therapy. Biomed Pharmacother 130:110623
Tafani M, Sansone L, Limana F, Arcangeli T, De Santis E, Polese M, Fini M, Russo MA (2016) The interplay of reactive oxygen species, hypoxia, inflammation, and sirtuins in cancer initiation and progression. Oxidative Med Cell Longev 2016:3907147. https://doi.org/10.1155/2016/3907147
Tobar N, Villar V, Santibanez JF (2010) ROS-NFkappaB mediates TGF-beta1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol Cell Biochem 340(1–2):195–202. https://doi.org/10.1007/S11010-010-0418-5
Tudek B, Winczura A, Janik J, Siomek A, Foksinski M, Oliński R (2010) Involvement of oxidatively damaged DNA and repair in cancer development and aging. Am J Transl Res 2(3):254
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39(1):44–84
Varol M (2017) Angiogenesis as an important target in cancer therapies. In: Researches on science and art in 21st century Turkey. Gece Publishing, Ankara, pp 1971–1981
Varol M (2020) ROS and oxidative stress in cancer: recent advances. In: Drug targets in cellular processes of cancer: from nonclinical to preclinical models. Springer, Cham, pp 109–138. https://doi.org/10.1007/978-981-15-7586-0_6
Wang H, Liu X, Long M, Huang Y, Zhang L, Zhang R, Zheng Y, Liao X, Wang Y, Liao Q, Li W, Tang Z, Tong Q, Wang X, Fang F, De La Vega MR, Ouyang Q, Zhang DD, Yu S, Zheng H (2016) NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med 8(334):334ra51. https://doi.org/10.1126/SCITRANSLMED.AAD6095/SUPPL_FILE/AAD6095_TABLE_S9.XLSX
Wang J, Yi J (2008) Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther 7(12):1875–1884. https://doi.org/10.4161/cbt.7.12.7067
Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang B-H, Rojanasakul Y (2008) The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol 180(5):3072–3080. https://doi.org/10.4049/JIMMUNOL.180.5.3072
Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T, Chen D, Piao HL, Liu HX (2021) The double-edged roles of ROS in cancer prevention and therapy. Theranostics 11(10):4839–4857. https://doi.org/10.7150/THNO.56747
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
World Health Organization (WHO) Global health estimates 2020: deaths by cause, age, sex, by country and by region, 2000–2019. WHO; 2020. Accessed 11 Dec 2020. https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death
Xie A, Li H, Hao Y, Zhang Y (2021) Tuning the toxicity of reactive oxygen species into advanced tumor therapy. Nanoscale Res Lett 16(1):1–10. https://doi.org/10.1186/S11671-021-03599-8/FIGURES/6
Yang H, Villani RM, Wang H, Simpson MJ, Roberts MS, Tang M, Liang X (2018) The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res 37(1):1–10. https://doi.org/10.1186/S13046-018-0909-X/FIGURES/4
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W (2016) ROS and ROS-mediated cellular signaling. Oxidative Med Cell Longev 2016. https://doi.org/10.1155/2016/4350965
Acknowledgments
This work was partly supported by the Fundamental Research Grant Scheme (FRGS) (FRGS/1/2019/STG03/USM/02/9) from the Ministry of Higher Education, Government of Malaysia, Malaysia. Manisekaran Hemagirri and Hong Hui-Jing were supported by GRA-ASSIST scholarship from the University Sains Malaysia, Malaysia.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Hemagirri, M. et al. (2023). Role of Hypoxia and Reactive Oxygen Species in Cancer Biology. In: Mukherjee, S., Kanwar, J.R. (eds) Hypoxia in Cancer: Significance and Impact on Cancer Therapy. Springer, Singapore. https://doi.org/10.1007/978-981-99-0313-9_3
Download citation
DOI: https://doi.org/10.1007/978-981-99-0313-9_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-0312-2
Online ISBN: 978-981-99-0313-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)