
Abstract
Mitochondria are the main provider of adenosine triphosphate (ATP) and help in maintaining optimum calcium homeostasis while also participating in cell death cascades. Hence, the fate of cells depends on the optimum functioning and positioning of the mitochondria. Perturbations to the normal functioning of these organelles play a central role in a wide range of mitochondrial diseases, which affect multiple organs with varying severity. Due to this heterogeneity, multiple diagnostic modalities including combinations of clinical, biochemical, and structural criteria have been developed. Imaging techniques such as computed tomography (CT), magnetic resonance imaging (MRI), and magnetic resonance spectroscopy (MRS) have been particularly useful in the diagnosis of mitochondrial diseases at the level of the organ as the central nervous system (CNS) is the second most frequently affected organ, while imaging via microscopy is crucial to detect changes in mitochondria at the cellular level. This review provides a detailed overview of the application of imaging modality in the diagnosis of mitochondrial disorders, from the organ to the cellular level.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Magnetic Resonance Spectroscopy
- Mitochondrial Disease
- Mitochondrial Disorder
- Mitochondrial Dynamic
- Mitochondrial Fusion
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction
5.1.1 The Mitochondrion
Mitochondria are ubiquitous in eukaryotic cells and are the site where oxidative phosphorylation (OXPHOS) for production of energy in the form of adenosine triphosphate (ATP) occurs (Saks et al. 2006; Stanley et al. 2005; Zeviani and Di Donato 2004). Mitochondria are known to have a double membrane and are found in the cytosol. Compartmentalization of the mitochondria into (1) the outer mitochondrial membrane (OMM), (2) the inter-membrane space (IMS), (3) the inner mitochondrial membrane (IMM) with the folding of cristae, and (4) the matrix enables the mitochondria to perform specialized functions such as OXPHOS, signaling, mitophagy, induction of apoptosis, and programmed cell division (McBride et al. 2006). A total of 3,000 genes have been documented as fundamental to produce a mitochondrion (DiMauro and Schon 2003; Schapira 2006). Thirty-seven of the genes are encoded by mitochondrial DNA (mtDNA) where the remaining ones are encoded in the nucleus (DiMauro and Schon 2003; Schapira 2006). The proteins produced are transported to the mitochondria (Anderson et al. 1981; Chan 2006a; Ingman et al. 2000). Only 3 % of these genes are responsible for production of ATP, while the remaining genes are involved in other physiological processes such as urea detoxification, cholesterol metabolism, and heme synthesis (Anderson et al. 1981; Chan 2006a; Galluzzi et al. 2012; Ingman et al. 2000; McBride et al. 2006).
5.1.2 What is Mitochondrial Disorder?
The presence of mitochondrial disease, a clinically heterogeneous group of disorders, signifies a failure of the mitochondrion, a key organelle responsible for producing energy in every cell of the body except red blood cells. The failure of this organelle appears to mostly impact cells of the brain, heart, liver, skeletal muscles, kidney, and the endocrine and respiratory systems, albeit in varying levels of severity (DiMauro et al. 1990, 1985; Petty et al. 1986). The criteria for diagnosis of mitochondrial disease should include (1) clinically complete respiratory chain (RC) encephalomyopathy and (2) molecular identification of an mtDNA mutation of undisputed pathogenicity (Bernier et al. 2002). Mitochondrial disease can be caused by either inherited or spontaneous mutations in mtDNA or nuclear DNA (nDNA) (DiMauro and Hirano 2005). These mutations can alter the functions of proteins produced by the mitochondria or even mitochondrial proteins themselves. Identical mtDNA mutations may not produce identical diseases due to the complexity of the interplay between genes and proteins in the cells. Conversely, different mutations can lead to the same diseases (Friedman et al. 2010; McFarland and Turnbull 2009). The typical mitochondrial disorder starts with an isolated organ but often evolves into a multisystem disease. Mitochondrial disease exists in many forms with onset occurring during the congenital stage although adult onset is becoming common (McFarland and Turnbull 2009). Symptoms of the disease include muscle weakness and pain, general fatigue, growth defects, loss of motor control and coordination, gastrointestinal disorders, and susceptibility to infection (Mattman et al. 2011; Morava et al. 2006; Schapira 2006). Proper diagnosis may be masked by pre-conceived assumptions that mitochondrial diseases may be other diseases. Most of the mitochondrial diseases in patients are not detected at an early stage due to the complexity of the disease.
5.1.3 Mitochondrial Disease Clinical Manifestations: An Overview
Mitochondrial diseases produce a plethora of manifestations and can be present at any age. The manifestations of mitochondrial diseases range from acute metabolic derangement to intermittent episodes of dysfunction to gradual progressive neurodevelopmental decline or regression. Understanding the clinical manifestations of pediatric and adult-onset mitochondrial disease will help family physicians to properly diagnose and treat the patients (Mattman et al. 2011).
5.1.3.1 General Characteristics of Pediatric and Adult-Onset Disease
The pediatric mitochondrial disease ranges from lethargy, hypotonia, failure to thrive, seizures, cardiomyopathy, deafness, blindness, movement disorder, and lactic acidosis to progressive neurological, cardiac, and liver dysfunction (DiMauro et al. 1985, 1990; DiMauro 2004). The patients suspected of harboring mitochondrial diseases based on the presence of these symptoms should be referred to a tertiary care center for proper evaluation and diagnosis. Manifestations of the symptoms may vary between family members with a maternally derived history of illness (Mattman et al. 2011).
Adult-onset mitochondrial diseases vary in the presentation times and can be triggered off during adulthood or discovered in adulthood following childhood symptoms (DiMauro et al. 1985; Mattman et al. 2011; Zeviani and Di Donato 2004). Adult-onset mitochondrial disease is typically a progressive multisystem disorder and is often discovered following physical examination and laboratory evaluation, even though the initial onset may have happened in a specific organ. The presence of a mitochondrial disease should be suspected when specific clinical manifestations are present and accompanied by one or more of the following: (a) involvement of different organ systems and/or (b) abnormal severity (i.e., early onset with progression over time), and/or (c) maternal inheritance pattern (Mattman et al. 2011).
5.2 Imaging and Diagnosis of Mitochondrial Diseases
Diagnosis of mitochondrial diseases is complicated by the complex interaction between the nuclear genome and mitochondrial genome (Zeviani and Di Donato 2004). Solely relying on enzymological studies is not sufficient to differentiate between mitochondrial and nuclear origins of diseases (Rötig et al. 2004). An alternative would be to study the phenotype of the resulting fused cell between the patient’s fibroblast and mtDNA-null (p0) fibroblast (Rötig et al. 2004). Nevertheless, this approach is not feasible as it can only be performed in patients expressing the RC deficiency in the fibroblasts, in addition to the high-cost and time-consuming protocol. The central nervous system (CNS) is the second most frequently affected organ in mitochondrial disorders, alone or in combination (Finsterer et al. 2001). The abnormalities detected during brain imaging can indicate metabolic disease, albeit non-specific. The varying stages of mitochondrial diseases further add to the complexity of proper diagnosis (Bernier et al. 2002; Morava et al. 2006). A combination of modalities for diagnostic confirmation is required due to the presence of non-specific symptoms and mixed etiologies (Haas et al. 2007; Thorburn et al. 2004). The most commonly used and clinically relevant imaging techniques for visualization of CNS abnormalities in mitochondrial disorders include computed tomography (CT), magnetic resonance imaging (MRI), and magnetic resonance spectroscopy (MRS) (Finsterer 2009).
5.2.1 Computed Tomography (CT) Scans
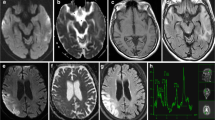
Computed tomography (CT) was the primary neuroimaging method prior to the advent of MRI and was mainly used for the detection of calcification, a pathophysiological process of mitochondrial disease, focal or diffuse atrophy, ischemic lesions, white matter lesions, and demyelination (Finsterer 2009). Intracranial calcification has been detected in a variety of disorders including idiopathic basal ganglia calcifications and spinocerebellar ataxia 20 (Knight et al. 2004). Extensive calcification on CT scans has also been detected in a patient with a novel polymerase gamma 1 (POLG1) mutation (Sidiropoulos et al. 2013). Nevertheless, CT scans face limitations when used to visualize certain abnormalities, such as strokelike lesions, symmetric necrosis of the thalami, basal ganglia, diencephalon, or brain stem in patients with Leigh’s syndrome (Finsterer 2009; Iizuka et al. 2002; McFarland et al. 2002). Current diagnostic protocols rely on the detection of changes in the neurological system at the initial stage, followed by confirmation via the usage of MRI in the CNS. The confirmation via MRI relies on the detection of key characteristics feature of the disease and non-specific abnormalities (Barragán-Campos et al. 2005; Muñoz et al. 1999). Detailed measurements of the changes in chemicals in the brain as indices for disorder characterization can be performed using MRS. The usage of MRI and MRS is extremely safe and non-invasive, hence rendering them suitable for monitoring of disease progression and changes in metabolic markers.
5.2.2 Magnetic Resonance Imaging (MRI)
Structural abnormalities in mitochondrial disorders are detected by MRI. The most common manifestation of mitochondrial disease is a global delay in myelination pattern at the initial stage of the disease—an indication of abnormal metabolic process, which is only resolved at a later stage following development (Dinopoulos et al. 2005; Muñoz et al. 1999). Hypointensity on T1 images as well as a symmetric signal abnormality of deep gray matter presenting with hyperintensity on T2 and fluid attenuation inversion recovery (FLAIR) images constitute the most common specific MRI findings indicative of mitochondrial disease (Gire et al. 2002; Haas and Dietrich 2004). Strokelike lesions affecting the white matter and gray matter are regarded as manifestations of a vasogenic edema and show dynamic changes in intensity and extensiveness over varying periods of time (Iizuka et al. 2007). Diffusion-weighted imaging (DWI) is considered to be more sensitive than T2-weighted images to demonstrate strokelike lesions (Abe 2004). The lesions can be either patchy or homogeneous with the presence of varying degrees of cerebral and cerebellar atrophy (Saneto et al. 2008). Cerebral atrophy may be categorized as focal or diffuse, cortical or subcortical, supratentorial or infratentorial, and primary or secondary after strokelike lesions (Finsterer 2009). Some patients may develop cerebellar atrophy with or without cerebral atrophy (Scaglia et al. 2005; Van Goethem et al. 2004). These MRI findings are mostly associated with syndromic phenotypes as discussed in Sect. 2.4. MRI, by itself, however, still lacks adequate sensitivity or specificity for an accurate diagnosis of certain mitochondrial diseases and is more commonly paired with other imaging modalities such as MRS for determining mitochondrial diseases (Barkovich et al. 1993; Friedman et al. 2010; Valanne et al. 1998).
5.2.3 Magnetic Resonance Spectroscopy
Magnetic resonance spectroscopy (MRS), capable of measuring biochemical changes in the form of metabolites possessing resonating nuclei (hydrogen-1, 1H; phosphorous-31, 31P; carbon-13, 13C) in the mM range, provides valuable metabolic information as a complement to conventional MRI (Saneto et al. 2008). The 1H-MRS is advantageous in that it shares a similar radiofrequency range compared with conventional MRI and thus can be performed simultaneously in the same examination (Saneto et al. 2008). 1H-MRS aids in predicting and classifying childhood white matter diseases, brain creatine deficiency syndromes, and phenotypic mitochondrial diseases (Chi et al. 2011a). The other nuclei, such as phosphorous-31 (31P) and carbon-13 (13C), although used to measure phosphocreatine and metabolite production degradation rates, respectively, are less utilized due to (1) the need of specific hardware for measurement, (2) lower signal to noise per unit time, and (3) the requirement of costly labeled substrate (13C glucose or acetate) as in the case of 13C MRS (Saneto et al. 2008).
Approximately 30 or more brain metabolites can be distinguished using specialized 1H acquisition approaches compared with only about 5–10 brain metabolites using routine clinical 1H-MRS (Ross 2000). Using specific spatial acquisition techniques such as point-resolved spectroscopy (PRESS), stimulated echo acquisition mode (STEAM), or image selected in vivo spectroscopy (ISIS) (Keevil 2006), grid (multivoxel also referred to as chemical shift imaging: [CSI or MRSI]) acquisitions can be obtained, although longer acquisition times will be needed to obtain sufficient signal-to-noise ratio (Saneto et al. 2008).
MRS signals are shown as spectra with different peaks corresponding to the various specific chemical entities. The magnetic field strength produced by nearby electrons for the nucleus determines the position of each resonance peak in the spectrum. The signal shifts to a lower frequency when a higher density of electrons is present. Conversely, a signal of higher frequency is present when the density of electrons is lowered (Saneto et al. 2008). J-coupling refers to the interaction between electrons on close neighboring protons within a molecule. Depending on the acquisition delay (echo times or TE) which can be modified to 20–30, 135–144, and 288 ms, the peak of a particular metabolite can be altered by j-coupling, which is advantageous to detection in clinical 1H-MRS (Saneto et al. 2008). As an example, the interaction between two protons (OH–H) with the magnetic field creates a characteristic doublet for the lactate peak at 1.33 parts per million (ppm) (Rand et al. 1999; Saneto et al. 2008). Depending on the echo time, the doublet can be either pointing up (TE = 288 ms) or pointing down (TE = 144 ms) and this specific feature can be utilized for unequivocal measurement of lactate, compared with the overlapping peaks from lipid/macromolecule signal in brain (Rand et al. 1999; Saneto et al. 2008; Yablonskiy et al. 1998). Short TE (20–30 ms) is more useful for the detection of glutamine, glutamate, and myo-inositol although the baseline value may include lipid signals between 1 and 2 ppm (Yablonskiy et al. 1998). Using a higher TE, the lipid peaks can be ruled out while providing a lower signal-to-noise ratio (S/N) (Yablonskiy et al. 1998). The relationship between TE and magnetic field strength, tesla (T), is also crucial for sensitivity of MRS. An improved sensitivity for the spectra at 3.0 T was detected compared with 1.5 T at short TE (Barker et al. 2001). Conversely, the 3.0 T MRS has a reduced or absent signal intensity using an intermediate echo time (Lange et al. 2006). No differences were noted using a long TE, irrespective of magnetic field strength (Barker et al. 2001).
Single-voxel (one region, typically 2–10 cc’s) technique can be advantageous for specific localization in areas near the air/brain interfaces (e.g., cerebellum) and optimal signal to noise per unit time, but it has the downside of limited spatial coverage (Saneto et al. 2008) as well as the possibility of underestimating the lactate concentration (José da Rocha et al. 2008). Multiple voxel acquisition (in a single plane) is more commonly performed using chemical shift imaging techniques. The grids of data in matrices of (1) 16 × 16, (2) 24 × 24, or (3) 32 × 32 having individual voxels of approximately 1–2 cc after reconstruction filtering confer the benefit of being able to study many different brain regions at the same time, particularly in regions with inconsistent patterns of lactate elevations (Saneto et al. 2008). The downside is that individual voxels have to be summed to facilitate measurement of lactate with sufficient signal-to-noise ratio. The measurement in voxels may be further complicated with the difference in visibility of lactate in different compartments, particularly when only a partial volume is sampled (Saneto et al. 2008).
5.2.3.1 MRS in Mitochondrial Diseases: Lactate and NAA Changes
MRS is used to diagnose mitochondrial disease based on the changes in different markers following impaired OXPHOS or disruption of electron transport chain (ETC). The shift of catabolic metabolism from the TCA cycle to anaerobic glycolysis produces cerebral lactate in the range of 3–11 mM (Wilichowski et al. 1999) or an average of ∼6 mM (Isobe et al. 2007; Saitoh et al. 1998), which can be measured along with other markers of cellular integrity and energetic such as myo-inositol, choline, creatine, and N-acetylaspartate (NAA). Cellular compromise is almost always denoted by increased lactate and decreased NAA (Jeppesen et al. 2003; Moroni et al. 2002). Due to disease variability and regional sampling, lactate elevations may not be detected in some of the patients, including patients with a normal brain MRI (Bianchi et al. 2003; Chi et al. 2011b; Lin et al. 2003). Cady et al. showcased the possibility of confusion in identification of lactate with propan-1,2-diol, localized at 1.1 ppm (Cady et al. 1994). Most of the increased lactates are detected in lesions, and the intensities of the lactate peaks change according to severity of the disease and the developmental course of lesions (Bianchi et al. 2003; Lin et al. 2003). Saneto et al. (2008) recommend using multivoxel acquisition for MRS, followed by reformatting to identify regions of interest so as to study multiple brain sites and compare contralateral regions. Monovoxel acquisition is useful to detect the lactate spikes in the putamen and the cerebellar dentate nucleus (Delonlay et al. 2013). As opposed to the elevation of lactate levels, a reduction in lactate levels can also be detected depending on the type of mitochondrial disease, time course of disease development, or location of the brain sampled (Saneto et al. 2008).
The presence of elevated lactate is almost universally indicative of neuronal/axonal compromise and decreases in NAA. In a survey of mitochondrial patients, 93 % showed NAA/creatine ratio reductions in cerebellum, 87 % in cortical gray matter regions, though few changes were observed in white matter (Bianchi et al. 2003). Dinopoulos et al. (2005) showed that 11 patients with definitive mitochondrial disease had decreased NAA/creatine ratio in both gray matter and white matter.
Voxel placement is crucial for the detection of abnormal lactate and can be missed if the voxel is placed over the incorrect brain region. This is evident in the case study of Boddaert et al. (2008) where lactate peaks were detected in 9 out of 11 patients with cerebellar involvement when voxels were placed over the cerebellum, while only 3 out of 11 showed lactate voxels over the putamen. In addition, elevated levels of alanine, glucose, or pyruvate can also be detected due to monoxidative glycolysis or pyruvate dehydrogenase complex deficiency (Medina et al. 1990; Wilichowski et al. 1999).
To summarize, although MRS can complement conventional MRI in studying the underlying biochemical changes, the optimal protocol for determining the abnormalities in the brain has yet to be clearly defined.
5.2.4 Mitochondrial Disorders
5.2.4.1 Leigh’s Syndrome
Leigh’s syndrome or subacute necrotizing encephalomyelopathy is a progressive degenerative disorder caused by mtDNA or nDNA mutations that affect infants, children, and sometimes adults (DiMauro 2004). The most common biochemical abnormality of Leigh’s syndrome lies in the defective complex IV (cytochrome C oxidase) (Rahman et al. 1996). Clinical presentation includes global developmental delay, feeding and swallowing difficulties, vomiting, spasticity, brainstem dysfunction, dystonia, abnormal eye movements, and multiple organ involvement (Barkovich et al. 1993; Rahman et al. 1996).
The disease is named after Denis Leigh who first described the neuropathological features of the disease, which include focal, bilateral, and symmetric necrotic lesions associated with demyelination, vascular proliferation, and gliosis in the brain stem, diencephalon, basal ganglia, and cerebellum (Leigh 1951). Progressive signal abnormalities with the highest frequency in the lentiform nuclei and caudate nuclei are detected with MRI. Besides that, abnormalities involving the thalamus, periaqueductal gray, tegmentum, red nuclei, and dentate nuclei can also be commonly seen (Saneto et al. 2008). The spongiform changes and vacuolation in the deep gray structures are reflected by the high T2 signal of MRI. Extensive gliosis and cystic degeneration may occur in the white matter. Compared to the cerebral organic acidurias that lead to severe global degeneration, the volumes of the lesion(s) are actually preserved on follow-up studies. In some cases of Leigh’s syndrome, a marked global atrophy over the course of time can be detected even though lesions in the basal ganglia are preserved. As the disease progresses, the basal ganglia can also show volume loss (Saneto et al. 2008).
The etiologies of Leigh’s syndrome can be differentiated by using MRI. MRI scans of patients with SURF-1 (a major gene associated with complex IV deficiency) mutations reveal symmetric lesions in the brain stem, subthalamic nuclei, and possibly cerebellum, while some of the patients show basal ganglia abnormalities as well as signal hyperintensities in bilateral optic radiation on T2-weighted imaging and DWI (Farina et al. 2002; Tanigawa et al. 2012; Zhu et al. 1998). Conversely, patients with Leigh’s syndrome from other etiologies exhibit T2 hyperintensities in the putamina with involvement of the caudate nuclei, globus pallidi, thalami, and brain stem (Farina et al. 2002; Quinonez et al. 2013; Valanne et al. 1998). Furthermore, there were less symmetric areas on increased T2 signal in the cortex and the subcortical white matter of the right and left occipital lobes and in the perirolandic region (Quinonez et al. 2013).
A lactate doublet peak on MRS and hyperperfusion in the patients at baseline with persistent hyperintensities on DWI at follow-ups were also reported (Chen et al. 2012b). In the absence of hypoxia, ischemia, or infection, the presence of symmetric deep gray structures or lactate peaks in the area of MRI abnormality on 1H-MRS warrants further investigation for mitochondrial defects. High choline levels were also detected in the white matter by MRS (Sijens et al. 2008). A diffused supratentorial leukodystrophy involving the deep lobar white matter can also be detected by the MRI in a small cohort of patients with Leigh’s syndrome. Initial swelling followed by cystic degeneration from the posterior to anterior has been reported (Lerman-Sagie et al. 2005; Moroni et al. 2002).
5.2.4.2 Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Strokelike Episodes (MELAS)
Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS) are maternally inherited progressive neurodegenerative disorders with various symptoms such as headache with nausea preceding strokelike events, treatment-resistant partial seizures, short stature, muscle weakness, exercise intolerance, deafness, diabetes, and slow progressive dementia (Hirano and Pavlakis 1994; Pavlakis et al. 1984). In addition to different mitochondrial and nDNA mutations (Emmanuele et al. 2013; Lamperti et al. 2012; Rossmanith et al. 2008; Sproule and Kaufmann 2008; Tam et al. 2008), an estimated 80 % of MELAS patients have an adenine-to-guanine transition at the tRNA for leucine at position 3,243 in the mtDNA (Goto et al. 1990; Janssen et al. 2006; Lamperti et al. 2012; Malfatti et al. 2007). This results in impaired ATP production, following failed synthesis of mitochondrial proteins.
Hallmark MRI features for MELAS patients include transient strokelike lesions predominantly affecting gray matter with the occasional diffused white matter lesions involving periventricular white matter, centrum semiovale as well as the corpus callosum (Apostolova et al. 2005; Barkovich et al. 1993; Conway et al. 2011; Hirano and Pavlakis 1994; Matthews et al. 1991). Areas of hypoattenuation in the left occipital and partially parietal lobe (Pauli et al. 2013) as well as bipallidal microcalcifications (Renard et al. 2012) have been detected using CT scans. Large bilateral hypointensities in the pallidum and the substantia nigra were detected using 3-T T2-weighted MRI (Renard et al. 2012). Strokelike lesions (accompanying strokelike symptoms) can be seen on MRI at different stages of the disease (Renard et al. 2012). Similarly, brain-MRS-based detection of lactate, increased alanine and glucose, decreased N-acetylaspartate, choline, and creatine can be used to resolve the different stages of MELAS progression and treatment (Chen et al. 2012a; José da Rocha et al. 2008).
The difference between chronic lesions and acute ischemic stroke episodes lies in the diffusion coefficient. Intense T2 and FLAIR signals are common features of both events. However, chronic lesion displays a normal to slightly increased apparent diffusion coefficient (ADC) due to vasogenic edema (Oppenheim et al. 2000; Pauli et al. 2013; Yonemura et al. 2001), while acute ischemic stroke episode has a significant reduction in ADC due to cytotoxic edema (Pauli et al. 2013). Therefore, the DWI sequences during the acute event are fundamental to differentiating between the chronic nature and acute nature of the lesions.
Mutations in the POLG1 gene encoding for the DNA polymerase γ, pol γ which replicates the human mitochondrial genome, produce MELAS-like symptoms (Cheldi et al. 2013; Deschauer et al. 2007). Nevertheless, it is difficult to directly compare between MELAS- and POLG1-associated encephalopathy, which has a heterogeneous clinical presentation (Cheldi et al. 2013). In addition, there have also been documented cases of sole involvement of the basal ganglia as well as ETC defects with strokelike brain lesions that do not fulfill the criteria for full MELAS (Kim et al. 2001), hence warranting the need for multiple diagnostic modalities beyond imaging alone to ensure proper and correct diagnosis of MELAS. MELAS syndrome has also been documented to mimic the clinical and radiological signs of herpes simplex encephalitis (Gieraerts et al. 2013). Effective differentiation was achieved by observing diffusion restriction in some parts of the lesions but not throughout the entire lesions following DWI and biochemical investigations on cerebrospinal fluid, electromyogram, muscle biopsy, and genetic analysis (Gieraerts et al. 2013).
5.2.4.3 Pearson’s Syndrome/Kearns–Sayre’s Syndrome
Pearson’s syndrome, first described in 1979, is an uncommon, multisystem mitochondrial disorder caused by single mtDNA deletions (Lee et al. 2007; Pearson et al. 1979). Pearson’s syndrome was first characterized in 4 patients with refractory sideroblastic anemia and vacuolization of marrow precursors and exocrine pancreatic dysfunction (Pearson et al. 1979). Following this characterization, small deletions of 4,799 and 5,500 base pairs in the mtDNA have been shown to be responsible for Pearson’s syndrome (McShane et al. 1991; Rotig et al. 1989).
Clinical manifestations of Pearson’s syndrome include hypotonia, developmental delay, ataxia, refractory sideroblastic anemia, pancytopenia, exocrine pancreatic dysfunction, and sometimes hepatic or renal failure (Lee et al. 2007). Premature death during infancy or childhood following infections or metabolic failure often occurs, whereas those that do survive into adulthood develop Kearns–Sayre’s syndrome (KSS), even with the initial spontaneous remission of infantile sideroblastic anemia (Larsson et al. 1990; Lee et al. 2007; Rotig et al. 1989).
There is relatively little information on CNS imaging of patients with Pearson’s syndrome. In 2007, Lee et al. documented a review study of 55 cases of patients with Pearson’s syndrome. Neuroimaging findings for Pearson’s syndrome were variable, with a myriad of normal, non-specific finding or prominent abnormal signal intensity over the white matter, basal ganglion, or brain stem (Lee et al. 2007). Cortical atrophy has also been reported (Lee et al. 2007).
Kearns–Sayre syndrome (KSS) is a progressive external ophthalmoplegia present before 20 years of age and is commonly associated with atypical retinal pigmentary degeneration and heart cardiac conduction defects (Kearns and Sayre 1958). Cerebellar ataxia, deafness, diabetes, short stature, hypoparathyroidism, and other endocrinopathies may also occur (Mayer et al. 2011). The presence of heteroplasmic rearrangements of multiple DNA deletions in mtDNA denotes the presence of this syndrome (Zeviani et al. 1988).
Typical histopathological findings of this syndrome include status spongiosus involving both gray and white matters, specifically the brainstem tegmentum, basal ganglia, and white matter of the cerebrum and cerebellum (Chi et al. 2011; McKelvie et al. 1991; Tanji et al. 1999). Calcifications are commonly detected in the basal ganglia (José da Rocha et al. 2008). Commonly detected MRI characteristics include cerebral and cerebellar atrophy with early T2/FLAIR hyperintense bilateral lesions in subcortical white matter, thalamus, basal ganglia (substantia nigra and globus pallidus), and brain stem (Chu et al. 1999; Duning et al. 2009; Hourani et al. 2006; Kamata et al. 1998; Leutner et al. 1994; Wray et al. 1995). Cardiovascular MRI has also revealed a potentially typical pattern of diffuse intramural late-gadolinium-enhancement in the left ventricular inferolateral segments of patients suffering from KSS (Yilmaz et al. 2012). Lactate peak on the right putaminal lesion can be detected by MRS (José da Rocha et al. 2008).
5.2.4.4 Alpers’ Syndrome
Alpers’ syndrome, first described in 1931, is a severe hepatocerebral disease caused by different types of mtDNA depletion that is present at various ages depending on the type of mutation within the polymerase gamma 1 gene (POLG) (Alpers 1931; Huttenlocher et al. 1976; Naviaux and Nguyen 2004). Compound heterozygote mutations, usually one of which within the linker region, cause a more severe disease phenotype with onset before 2 years of age, whereas patients with homozygous mutations within the linker region usually have a later onset and milder form of Alpers’ syndrome (Ashley et al. 2008; Saneto et al. 2008; Uusimaa et al. 2013). Mutations in the mitochondrial replicative helicase Twinkle (Hakonen et al. 2007) or the heterozygous presence of a spacer region mutation in trans with another recessive POLG mutation (Kurt et al. 2010) has also been shown to demonstrate phenotypes reminiscent of Alpers’ syndrome, thus suggesting that there may be other etiologies as well. Nevertheless, the current definitive diagnosis for Alpers’ syndrome is the identification of mutations in POLG (Hunter et al. 2011).
Patients with Alpers’ syndrome usually suffer from refractory seizures, psychomotor regression, and a characteristic liver disease (Naviaux and Nguyen 2004). Depending on the site of the POLG gene mutation, the onset and severity vary from infancy to adulthood, while infantile Alpers’ syndrome can slowly progress to external ophthalmoplegia and ataxia (Horvath et al. 2006).
The syndrome is denoted by extensive gliosis and neuronal loss in the occipital cortices and cerebellar cortex (Purkinje cells). In addition, there is necrosis of subcortical deep nuclei, hippocampi, lateral geniculate body of the thalamus, and amygdala (Harding 1990). There are also T2/FLAIR hyperintensities in the MRI scans of patients within the occipital regions, deep cerebellar nuclei, thalamus, and basal ganglia (Tzoulis et al. 2006), albeit these rely on the mutations present. Compound heterozygotes are more involved, while homozygotes have lesions that can reverse during quiescent periods.
In the study of Saneto et al. (2008), they found that the MRI lesions evolved initially from the cerebellum to the occipital regions and pre-motor cortex in one of the patients with compound heterozygous mutations in polymerase gamma 1 (p.Q67X and p.A467T). The patient exhibits clinical symptoms such as focal seizures, nystagmoid eye movement and visual hallucinations (Saneto et al. 2008). Following a rapid course of decline, the patient became blind and developed liver failure after valproic acid exposure. In the same study, a patient with a homozygous mutation in the linker region of polymerase gamma 1 (p.A467T) developed mild sensorineural hearing loss, ataxia, and clumsiness at 5 years of years (Saneto et al. 2008). This patient then developed partial status epilepticus at the age of 15 years. A stable quiescent state for 6–7 years ensued. The patient passed away at the age of 23 years from liver failure, following valproic acid exposure (Saneto et al. 2008). Although the patient’s MRI findings were initially normal, occipital T2/FLAIR hyperintensities were detected at the time of partial status epilepticus which also resolved over time, rendering a virtually normal MRI study just before her death (Saneto et al. 2008). Based on this study, it can be concluded that although MRI may be useful in differentiating the severity of Alpers’ syndrome, one should always be wary of the possible changes in MRI scans and further studies are needed to correlate MRI findings with corresponding disease phenotype and genotype.
The review of the case records of 12 patients with Alpers’ syndrome by Hunter et al. (2011) showed CT scans of focal areas of low attenuation or diffuses cerebral atrophy in four out of eight children scanned. Ten MRI scans of four children dating back to 1992 showed focal high signal changes on T2-weighted images, albeit the positioning of the abnormalities varies. MRS scans were not performed for those patients (Hunter et al. 2011). According to the findings of McCoy et al. (2011), diffusion abnormalities correlated with the T2 abnormalities were acute rather than chronic, while MRS demonstrated the presence of lactate, suggesting a RC defect. Most recently, Uusimaa et al. (2013) also elaborated the importance of increased lactate on MRS and suggestive brain MRI changes (with thalamic predominance) as the impetus for screening of the common POLG mutations and POLG sequencing for the detection of Alpers’ disease.
5.2.4.5 Mitochondrial Neuro-Gastrointestinal Encephalomyopathy
Mitochondrial neuro-gastrointestinal encephalomyopathy (MNGIE), first described in 1983, is derived from mutations within the thymidine phosphorylase gene (TYMP) located on chromosome 22q13.32 and causes loss of mtDNA via DNA deletions and point mutations within mtDNA (Ionasescu 1983; Nishino 1999; Nishino et al. 2000) although novel pathogenic mutations have also been reported (Libernini et al. 2012). Intestinal pseudoobstruction, recurrent diarrhea, nausea, and vomiting are the most dominant symptoms. Other symptoms such as ophthalmoparesis, ptosis, and peripheral neuropathy may also be present. The symptoms generally appear before 20 years of age, and younger patients typically have more severe symptoms with death occurring by 35 years of age for approximately 50 % of patients. There have been cases of patients remaining undiagnosed for many years who later undergo extensive workup to Crohn’s disease (Perez-Atayde 2013).
Prominent leukoencephalopathy can be detected via MRI in almost all patients. The changes in white matter are diffused with increased T2 signal abnormalities, with a case of widespread supratentorial cortical atrophy described in one study (Barragán-Campos et al. 2005). Mild white matter edema or mild demyelination has also been shown in postmortem studies (Bardosi et al. 1987). The presence of megamitochondria in gastrointestinal ganglion cells and in smooth muscle cells of muscularis mucosae and muscularis propria also aids in the diagnosis of MNGIE (Perez-Atayde 2013).
There have been reported cases of observance of MNGIE-like phenotypes in patients with mutations in the ribonucleoside diphosphate reductase subunit M2 B gene (RRM2B) (Shaibani et al. 2009) as well as the POLG1 gene (Tang et al. 2011, 2012; Goethem et al. 2003). Brain MRI of the RRM2B patient showed patchy leukoencephalopathy and also bilateral basal ganglia signal abnormality compared with the commonly observed diffuse leukoencephalopathy in MNGIE due to TYMP mutations (Shaibani et al. 2009). Depending on the mutations in the POLG1 gene, findings from brain MRI range from normal with no leukoencephalopathy (Goethem et al. 2003) to global cerebral atrophy or increased T2 signal in the basal ganglia (Tang et al. 2012).
5.2.4.6 Isolated Electron Transport Chain Disorders
The mitochondrial and nuclear genomes encode for subunits of all RC complexes, except complex II (Rötig et al. 2004). Nuclear genetic defects in OXPHOS and non-OXPHOS proteins involved in the assembly or maintenance of the ETC are the main cause of pediatric cases of mitochondrial disease (Salvatore DiMauro 2004; Shoubridge 2001). Diagnosis is hampered by the clinically heterogeneous nature of these disorders, mainly caused by the interplay of mtDNA and nDNA expression.
Non-specific diffused white matter changes have been found in some patients expressing ETC defects, involving individual and combinations of complexes I–IV deficiencies (Lerman-Sagie et al. 2005). Progressive macrocystic leukodystrophy or diffuse white matter disease including cerebellar white matter loss has been described in four patients with defects in complex I (Schuelke et al. 1999). In the study of Lebre et al. (2011) consisting of 30 patients with complex I deficiency, bilateral brainstem lesions were observed in all cases with 23 patients exhibiting anomalies of the putamen. Patients harboring mtDNA mutations showed supratentorial strokelike lesions, whereas necrotizing leukoencephalopathy was observed in patients with nDNA mutations (Lebre et al. 2011). Baertling et al. (2013) showcased the cerebral MRI with bilateral cystic lesions in the centrum semiovale from an 8-month-old infant girl with rapid developmental regression, who was found to harbor an isolated mitochondrial complex I deficiency due to an NDUFS1 mutation (encoding NADH-dehydrogenase-ubiquinone Fe–S protein 1). Demyelination of the supratentorial white matter ensued in the follow-up imaging after 3 months (Baertling et al. 2013).
Complex II, also known as succinate: ubiquinone oxidoreductase or succinate dehydrogenase, is exclusively nuclear-encoded and participates in the citric acid cycle by oxidizing succinate to fumarate(Rutter et al. 2010; Sun et al. 2005). In the mitochondrial ETC, complex II functions to shuttle electrons to ubiquinone (Rutter et al. 2010; Sun et al. 2005). Various studies also reported neurological disorder with leukoencephalopathy, Leigh’s syndrome, or cerebellar atrophy in isolated complex II defect (Brockmann et al. 2002; Burgeois et al. 1992; Jain-Ghai et al. 2013; Moroni et al. 2002). In these studies reported, MRI shows abnormal intensities in various sections of the brain as well as multiple foci of diffusion restriction with symmetric distribution (Brockmann et al. 2002; Burgeois et al. 1992; Jain-Ghai et al. 2013; Moroni et al. 2002).
Among two patients with defects in complex III, one had a mutation in cytochrome b subunit of complex III and the other was without a recognized mutation but a defect in enzyme activity (Majoie et al. 2002). White matter changes have been described in at least 5 patients with defects in complex IV (Jaksch et al. 2001; Rahman et al. 2001). Similarly, rapid progression of cerebral/cerebellum atrophy on axial T2-weighted and FLAIR are well visualized in complex IV disorders (Castro-Gago et al. 1999; Scaglia et al. 2005; Valanne et al. 1998). A patient having multiple ETC defects coupled with leukodystrophy has also been reported (Moroni et al. 2002). Leukodystrophy in patients is usually detected at infancy or at a very young age. Multisystem disorders with a neurodegenerative course can be present as well. MRI detection of a diffuse leukodystrophy within the clinical context of multisystem involvement compels the need for further investigation for a mitochondrial disorder. In terms of MRS, lactate elevation is a consistent feature of patients with various ETC defects (Dinopoulos et al. 2005).
5.3 Imaging at the Cellular Level
The existence of healthy mitochondria in the cell relies on maintenance of proper morphology, membrane potential, and calcium signaling, while dysfunctional or senescent mitochondria are ‘pruned’ by mitophagy. Any deviation will result in mitochondrial disorder which will ultimately lead to a disease state. Hence, an understanding of the mitochondrial function in normal and pathophysiological states is critical for the monitoring and treatment for mitochondria-related diseases. Methods for imaging the changes in mitochondria at the cellular level have been relatively well established using epifluorescence, confocal laser, and electron microscopy (Chan 2006b; Chen and Chan 2004; Gieraerts et al. 2013; Karbowski et al. 2004; Ong and Gustafsson 2012; Ong and Hausenloy 2010; Ong et al. 2010; Rötig et al. 2004). More recently, multiphoton microscopy has also been explored for imaging of mitochondrial function (Davidson and Duchen 2012; Hall et al. 2009, 2013). In the following sections, I will review some of the latest innovations in mitochondrial imaging at the cellular level.
5.3.1 Imaging Mitochondrial Dynamics and Its Disorders
Research for the past decade has revealed a unique feature of the mitochondria, in that the mitochondria exist in a dynamic equilibrium between a fused (elongated) (Figs. 5.1a, 5.2) and fragmented (Fig. 5.1b) state (Chan 2006b; Chen and Chan 2004; Liesa et al. 2009; Ong et al. 2012; Ong and Hausenloy 2010).

Representative confocal microscopy images showing an endothelial cell with a fused mitochondria and b fragmented mitochondria

Representative electron micrograph depicting an elongated mitochondrion (in oval) in the adult heart tissue
The movement of the mitochondria and subsequent changes in shapes (morphology) of the mitochondria constitute the term of ‘mitochondrial dynamics’ and have since led to an impetus of research in this particular area. The number and shapes of mitochondria vary according to cell type and energy requirement. The balance between fused and fragmented forms of the mitochondria is governed by specific mitochondrial-shaping proteins, mitofusin 1 (Mfn1) (Santel et al. 2003), mitofusin 2 (Mfn2) (Bach et al. 2003), and optic atrophy 1 (OPA1) (Cipolat et al. 2004) for mitochondrial fusion and dynamin-related protein 1 (Drp1) (Smirnova et al. 1998) and human fission 1 (hFis1) (James et al. 2003) for mitochondrial fission. Mitochondrial fusion serves to facilitate the exchange of contents, DNA, and metabolites between neighboring mitochondria and to compensate for mutations in mtDNA (Chen et al. 2010; Nakada et al. 2001) and prevents healthy mitochondria from being removed via autophagy, a process of recycling dysfunctional mitochondria (Gomes et al. 2011). Mitochondrial fusion by inhibition of Drp1 or upregulation of Mfn1 and Mfn2 has been shown to protect the heart from ischamia–reperfusion injury via inhibition of the mitochondrial permeability transition pore (mPTP), a channel in the IMM that opens upon calcium overloading and accumulation of oxidative stress (Ong et al. 2010). Conversely, fission is crucial for enhancing optimum mitochondrial transport, to subcellular regions of specific energy demand. Fragmentation of the mitochondria by upregulation of Drp1 predisposes the cardiac cells to autophagy and cell death upon stress insults such as oxidative stress buildup and calcium overloading (Chen et al. 2005; Gomes et al. 2011; Jheng et al. 2012; Lee et al. 2011).
A variety of methods have been employed to monitor the changes in mitochondrial morphology. Traditional studies of mitochondrial morphology employ electron microscopy. However, this only provides a snapshot in time and space. Mitochondrial dynamics, which encompass the movement, change in shapes, and positioning, can be monitored using photoactivatable fluorescent proteins (mtPA-GFP). The photoactivated GFP diffuses rapidly within the entire mitochondrial matrix and can therefore be used to tag individual mitochondria (Lovy et al. 2012; Twig et al. 2006, 2008). In addition, monitoring mitochondrial fusion using the Renilla luciferase complementation assay has also been described (Huang et al. 2010; Schauss et al. 2010).
Contact between two mitochondria does not necessarily lead to fusion. Fission, conversely, can occur without movement and connection of two juxtaposed mitochondria. Fluorophores are targeted to the mitochondria via the differences in mitochondrial membrane potential to visualize the intermixing and segregation of the inner mitochondrial components. The study of mitochondrial dynamics using this tool enables a better understanding of physiological and pathophysiological processes affecting mitochondrial activity, positioning, and the number of mitochondria (Molina and Shirihai 2009). Certain dyes such as MitoTracker® Green FM, Orange CMTMRos, Red CMXRos, and Deep Red FM probes are not affected by the energetic state of the mitochondria, compared with conventional dyes such as rhodamine 123 and tetramethylrhodamine methyl ester (TMRM). Novel, non-cytotoxic dyes, e.g., AcQCy7, have also been formulated to specifically target mitochondria (Han et al. 2013). Different model organisms, most of which express fluorescent mitochondria in vivo, have also been developed or modified to study mitochondrial dynamics in disease systems (Chandrasekaran et al. 2006; Misgeld et al. 2007; Plucińska et al. 2012). The presence of giant mitochondria can also be detected by electron microscopy (Haas et al. 2008).
The change in mitochondrial morphology has been demonstrated to affect various physiological processes. Mutations in OPA1 lead to type 1 dominant optic atrophy, a common cause of inherited visual failure starting in early childhood characterized by irreversible loss of retinal ganglion cells (RGCs) (Alavi and Fuhrmann 2013; Amati-Bonneau et al. 2009; Eiberg et al. 1994; Galvez-Ruiz et al. 2013; Kjer 1959; Lenaers et al. 2012; Lunkes et al. 1995) as well as impairment of neuronal maturation (Bertholet et al. 2013). Disruption or loss of OPA1 has been demonstrated to be associated with complex I deficiency (Ramonet et al. 2013) and Parkinson’s disease (Anglade et al. 1997; Fernandes and Rao 2011; Sekiya et al. 1982; Trimmer et al. 2000) as well as affecting vertebrate development (Rahn et al. 2013). Mfn2 mutations lead to primary axonal Charcot–Marie–Tooth disease type 2A, an autosomal dominant neuropathy that impairs motor and sensory neurons with the longest axons, resulting in earliest symptoms in distal extremities (Züchner et al. 2004). Similar to OPA1, a missense mutation in Mfn2 can lead to instability of mtDNA and optic atrophy ‘plus’ phenotypes such as syndromic forms of autosomal dominant optic atrophy associated with sensorineural deafness, axonal sensory motor polyneuropathy, ataxia, chronic progressive external ophthalmoplegia, and mitochondrial myopathy with cytochrome c oxidase (COX)-negative and ragged-red fibers (Amati-Bonneau et al. 2008; Hudson et al. 2008; Rouzier et al. 2012). Brain MRI from the patient revealed multiple periventricular white matter lesions and severe diffuse cerebral atrophy (Rouzier et al. 2012). Fragmentation of the mitochondrial network was also observed in the fibroblasts of the patient (Rouzier et al. 2012). Concurrent with reduced levels of Mfn1, Mfn2, and OPA1 and high levels of Fis1 and Drp1, increased mitochondrial fragmentation has also been observed in cells and tissues from patients with Alzheimer’s disease (Manczak et al. 2011; Wang et al. 2009). Mfn2 ablation promotes axon degeneration and disrupts axonal mitochondrial positioning in parkinsonism (Lee et al. 2012; Misko et al. 2012).
5.3.2 Determining Mitochondrial Membrane Potential
Adenosine triphosphate (ATP), the source of cellular energy from the mitochondria, is mainly produced via OXPHOS (Kaim and Dimroth 1999). The OXPHOS system is made up of five multiprotein complexes embedded within the IMM: complex I (reduced nicotinamide adenine dinucleotide: ubiquinone oxidoreductase), complex II (succinate: ubiquinone oxidoreductase), complex III (ubiquinol: cytochrome c oxidoreductase), complex IV (cytochrome c oxidase), and complex V (ATP synthase). Protons, donated by reduced nicotinamide adenine dinucleotide, ubiquinone, and cytochrome c, are channeled by complexes I, III, and IV, respectively, into the mitochondrial IMS. The channeling of protons through the complexes creates a proton gradient between the mitochondrial matrix and the IMS. The proton gradient is crucial for generating and maintaining the electrochemical membrane potential (ΔΨ), which is the driving force for the conversion of ADP and inorganic phosphate into ATP (Kaim and Dimroth 1999).
Mitochondrial membrane potential (ΔΨm) can be estimated using fluorescent voltage-sensitive dyes that are membrane-permeant lipophilic cationic compounds that distribute across the IMM (Lemasters and Ramshesh 2007) based on the Nernst equation. Changes in ΔΨm have been usually expressed in percentage over the basal level (Dedkova and Blatter 2012). Rhodamine 123, tetramethylrhodamine methyl, and ethyl ester (TMRM and TMRE) are specifically used to monitor changes in ΔΨm, which are useful for monitoring pathophysiological states or pharmacological treatment that induces changes in mitochondrial energetic states. Although it is least toxic to cells, TMRM should be used at a low concentration. An alternative fluorescent probe is the carbocyanine compound, JC-1, which exists as a green fluorescent monomer at low membrane potential but switches to form red fluorescent aggregates at higher potentials (Di Lisa et al. 1995). This change in fluorescence is solely based on membrane potential and independent of size and density of the mitochondria. Various factors such as loading times and dye concentration affect the formation of aggregates (Diaz et al. 2001; Duchen et al. 2003). Defects in complexes of the RC, which concurs with defects in OXPHOS, were detected using JC-1, as the buildup of mitochondrial membrane potential is disrupted by impairment of OXPHOS (De Paepe et al. 2012). Simultaneous detection of both ΔΨm and morphology can also be achieved via a combination of potential-insensitive mitochondrial GFP and a potential-sensitive probe such as TMRM (Distelmaier et al. 2008). This will also help in correcting for movement artefacts by applying a ratiometric approach (Twig et al. 2006). Nevertheless, it is important to ascertain that certain considerations should be taken into account when using these dyes, e.g., the optimum dye concentration to be used, the presence of the dye in superfusion solution throughout the experiment, timing for addition of dye, and other factors affecting the localization of the dye such as movement and changes in volume of the mitochondria (Dedkova and Blatter 2012; Duchen et al. 2003; O’Reilly et al. 2003). The potential occurrence of FRET phenomena (Förster resonance energy transfer or fluorescence resonance energy transfer) between two different dyes that can lead to erroneous interpretations of the results should also be evaluated (Aon et al. 2007; Dedkova and Blatter 2012; Honda et al. 2005; Hüser et al. 1998; Slodzinski et al. 2008). The plasma membrane potential, together with the ΔΨm, dictates the accumulation of the dye (Davidson et al. 2007). Upon opening of the mPTP, dyes may leak out of the mitochondria via simple diffusion (O’Reilly et al. 2003; Twig et al. 2006). Quantitative calibration of experimental data with potentiometric probes is usually expressed as a percentage change from basal levels (Dedkova and Blatter 2012). The standard deviation (SD) of the measured fluorescence signal is important as the SD may vary from high (in polarized mitochondria) to low (in depolarized mitochondria) (Duchen et al. 2003). Calibration of ΔΨm can be achieved by obtaining the ratio of mean fluorescence to SD (mean/SD), which gives a quantitative measure of dye localization (Dedkova and Blatter 2012; Duchen et al. 2003).
5.3.3 Detecting Mitochondrial Calcium Flux
Calcium (Ca2+) is an important signaling molecule regulating the process of ATP synthesis and hydrolysis. In response to physiological stimuli, the concentration of mitochondrial Ca2+ is increased in line with the activity of mitochondrial RC to cope with enhanced energy demand and ATP synthesis (Jouaville et al. 1999; Rizzuto and Pozzan 2006). Mitochondria are crucial in maintaining intracellular calcium homeostasis. The mitochondria act as a Ca2+ sink to prevent propagation of large calcium waves by taking up Ca2+ released from the endoplasmic reticulum (ER) into the cytosol (Baughman et al. 2011; Boitier et al. 1999; Jouaville et al. 1995; Mallilankaraman et al. 2012; Plovanich et al. 2013; Tinel et al. 1999; Williams et al. 2013). The Ca2+ will then be released back into the cytosol by release mechanisms (Ca2+/Na exchanger, Ca2+/H+ exchanger, and transient opening of the mPTP) (Ichas et al. 1997; Pinton and Rizzuto 2006; Putney and Thomas 2006; Rizzuto et al. 1993; Tian et al. 2005). Perturbations in the normal functions of cells may result in pathophysiological Ca2+ overloading in mitochondria, followed by the initiation of a cascade of events that lead to cell death (Hajnóczky et al. 2006). Permeabilization of the mitochondrial membrane by prolonged opening of the mPTP leads to the release of apoptogenic proteins from the IMS and subsequent cell death (Hausenloy et al. 2009; Martinou et al. 2000). Mitochondrial calcium can be measured using fluorescent Ca2+ indicators such as indo-1 (Miyata et al. 1991; Schreur et al. 1996), fura-2 (Abdallah et al. 2011), fluo-3 (Sedova et al. 2006), and rhod-2 (Jou et al. 1996) as well as luminescent and fluorescent targeted proteins such as aequorin, pericam, and camaleons (Bonora et al. 2013; De la Fuente et al. 2012; Fonteriz et al. 2010). The membrane-permeant fluorescent Ca2+ indicators [typically acetoxymethyl (AM) esters] are entrapped in the mitochondrial matrix via a de-esterification process by intracellular (cytosolic and intramitochondrial) esterases (Dedkova and Blatter 2012). Combining the use of rhod-2 with mitochondrial-targeted GFP, the mitochondrial positioning, morphology, and relation to sequestration of calcium can be concurrently studied. Potential downsides to using these dyes include the non-ratiometric nature of all rhodamine-derived indicators which may cause errors from changes in dye concentration and motion artifacts (Dedkova and Blatter 2012). Following repetitive simulation with certain agonists, Rhod-2 has been reported to induce Ca2+-dependent inhibition of mitochondrial Ca2+ uptake (Fonteriz et al. 2010). Besides that, the optimum dye concentration has to be determined as this affects mitochondrial membrane potential, morphology, and toxin production (Fonteriz et al. 2010). Classical ratiometric indicators such as fura-2 and indo-1, conversely, have low dissociation constant (Kd) and may accumulate in the cytosol (Dedkova and Blatter 2012). Recent innovations in mitochondrial calcium measurement include the development of Mt-pericam, a mitochondrial-matrix-targeted, circularly permuted green fluorescent protein fused to calmodulin and its target peptide M13 (Nagai et al. 2001), Mitycam which is targeted to mitochondria with a standard mitochondrial-targeting sequence (subunit VIII of human cytochrome c oxidase) (Kettlewell et al. 2009; Nagai et al. 2001), a low-Ca2+ affinity aequorin probe which is able to measure [Ca2+] in the millimolar range for long period of time without problems derived from aequorin consumption (De la Fuente et al. 2012), and, most recently, GCaMP2-mt, consisting of a mitochondrial-targeting sequence attached to a high signal-to-noise Ca2+ sensor protein GCaMP2 to measure oxidant-induced responses of [Ca2+]m in cultured neonatal myocytes (Iguchi et al. 2012).
5.3.4 Monitoring Mitochondrial Autophagy
Autophagy refers to the evolutionarily conserved process of segregation and recycling of damaged or unused cellular components (Klionsky 2007; Mizushima 2007). Autophagy is defined by the formation of autophagosomes—double-membrane structures that sequester defective organelles and cytotoxic protein aggregates, or even viruses or bacteria (Fig. 5.3) (Hayashi-Nishino et al. 2009; Klionsky 2007; Mizushima et al. 2011; Mizushima 2007). The autophagosomes subsequently fuse with lysosomes leading to bulk degradation of their content, and the produced nutrients will then be recycled back to the cytoplasm (Hayashi-Nishino et al. 2009; Klionsky 2007, 2008; Mizushima et al. 2011; Mizushima 2007; Ylä-Anttila et al. 2009a). Specific removal of mitochondria is termed mitophagy (Kissová et al. 2004; Rodriguez-Enriquez et al. 2004). Through mitophagy, dysfunctional mitochondria are recycled to produce useful amino acids and other nutrients.

Representative electron micrograph depicting an autophagosome with an engulfed mitochondrion in the heart
Pathologic dysregulation of autophagy results in various disorders such as synaptic dysfunction, stroke, brain trauma, Parkinson’s, Alzheimer’s, Huntington’s, and other neurodegenerative diseases (Chu et al. 2007; Lai et al. 2008; Liu et al. 2008; Nixon et al. 2005; Ong and Gustafsson 2012; Rudnicki et al. 2008).
Mitophagy is generally visualized via the protein LC3B, a general marker for autophagic membranes (Dagda et al. 2008; Du et al. 2009; Klionsky et al. 2012) or using electron microscopy (Klionsky et al. 2012). Autophagosomes are visualized on TEM as two parallel membrane bilayers separated by an electron-lucent cleft (Eskelinen and Kovács 2011), containing organelles that look morphologically intact, i.e., similar to the cytosol and organelles elsewhere in the cell (Ylä-Anttila et al. 2009b). Autolysosomes usually have only one limiting membrane, while the engulfed cytoplasmic material and/or organelles are at various stages of degradation (Ylä-Anttila et al. 2009b). The recommended quantification of TEM results is to obtain the volume occupied by autophagic structures and express it as a percentage of cytoplasmic or cellular volume in a predetermined number of cells (by power analysis) per sample (Klionsky et al. 2012; Sigmond et al. 2008). Autophagosomes in the brain have been evaluated using confocal immunohistochemistry and antibodies against LC3 (Lai et al. 2008). Caution should be exercised when interpreting the increase in autophagosomes as this can reflect induction of autophagy, impairment of autophagosome turnover (Kovács et al. 1986, 1987, 1988), or the inability of turnover to keep pace with increased autophagosome formation (Chu 2006).
Similar to the cases described before, LC3B-GFP or LC3B-RFP can be used in conjunction with mitochondrial-targeted fluorescent proteins to concurrently study mitochondrial dynamics and mitophagy. High-quality images at a high magnification from epifluorescence or confocal laser microscope are used to quantify LC3B puncta colocalized with mitochondria, average number of LC3 puncta per cell or percentage of cells displaying punctate LC3 that exceeds a particular pre-defined threshold (Klionsky et al. 2012). Anti-LC3 antibodies for immunocytochemistry or immunohistochemistry can be used to detect the endogenous protein without the need for transfection as the overexpression of GFP-LC3 may cause the nuclear localization of the marker (Elsässer et al. 2004; Klionsky et al. 2012; Ost et al. 2010). The number of puncta corresponding to GFP-LC3 on a per cell basis rather than simply the total number of cells displaying puncta can be quantified to monitor autophagy (Klionsky et al. 2012). Sizes of autophagosomes may vary, but additional assays will be required to correlate autophagosome size with autophagic activity (Klionsky et al. 2012; Wu and Pollard 2005). When using fluorescence microscopy to monitor autophagy, a ratio for establishing differences in the degree of autophagy between cells should be obtained by calculating the SD of pixel intensities within the fluorescence image and dividing this by the mean intensity of the pixels within the area of analysis (Klionsky et al. 2012). This is crucial as the expression of GFP-LC3 may not be the same in all cells. Multispectral imaging cytometry has been proposed to assist in quantifying GFP-LC3 puncta in a large number of cells (Dolloff et al. 2011). An additional advantage of this method is the ability of quantification of endogenous LC3 in non-transfected primary cells (Phadwal et al. 2012). A caveat to using GFP-LC3 is the potential association of this chimera with protein aggregates (Kuma et al. 2007). Furthermore, using antibodies will obviate the need for generation of a transgenic organism. The late stage of mitophagy where mitochondria undergo lysosomal degradation is monitored by labeling lysosomes with lysotracker and mitochondria with MitoTracker or mitochondrial-targeted fluorescent proteins (Rodriguez-Enriquez et al. 2006). Autophagic flux can also be measured by a tandem monomeric RFP-GFP-tagged LC3 (Kimura et al. 2007) with a more sensitive and accurate reporter described in 2012 (Zhou et al. 2012). The signal elicited from mRFP is not affected by environmental conditions. The GFP signal, however, is sensitive to the acidic and/or proteolytic conditions of the lysosome lumen. Colocalization of both GFP and mRFP fluorescence indicates a phagophore or an autophagosome that has not fuse with a lysosome. In contrast, an mRFP signal without GFP may correspond to an amphisome or autolysosome. The Rosella biosensor, a dual fluorescence assay, complements the tandem mRFP/mCherry-GFP reporter by monitoring the uptake of specific cellular components to the lysosome/vacuole and the internal environment of the biosensor during autophagy (Rosado et al. 2008). The biosensor Rosella consists of a relatively pH-stable fast-maturing RFP variant and a pH-sensitive GFP variant (Rosado et al. 2008). Novel tools for studying mitochondrial turnover and biogenesis include MitoTimer, a fluorescent reporter protein that changes fluorescence from green to red concurrent with protein maturation in whole cells and isolated mitochondria (Hernandez et al. 2013), the use of Keima, a coral-derived acid-stable fluorescent protein that emits different-colored signals at acidic and neutral pHs to quantify autophagy at a single time point (Katayama et al. 2011). The correlation between component proteins and organelles involved in the timing of the autophagic process has also been studied (Karanasios et al. 2013; Katayama et al. 2011).
5.4 Conclusion
The onset of mitochondrial diseases can occur at any age with multifaceted symptoms in various organs, such as the CNS, visual system, and neuromuscular system. Diagnosis of mitochondrial diseases is not easily achievable, due to the underlying factors such as double genetic origin of RC proteins, heterogenous clinical presentations of patients, and variables between genotype and phenotype. Coupled with other investigation tools such as genetic or biochemical analysis, imaging methods are crucial to visualize and differentiate CNS abnormalities in mitochondrial disorders. Findings from the different imaging methods, however, have to be corroborated to ensure that a proper and accurate diagnosis is achieved. Advances in biomedical imaging enhance a better understanding of mitochondrial integrity and function which will help develop future therapeutics against mitochondrial diseases.
References
Abdallah Y, Kasseckert SA, Iraqi W, Said M, Shahzad T, Erdogan A, Neuhof C et al (2011) Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J Cell Mol Med 15(11):2478–2485. doi:10.1111/j.1582-4934.2010.01249.x
Abe K (2004) Cerebral lactic acidosis correlates with neurological impairment in MELAS. Neurology 63(12):2458; author reply 2458. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15623741
Alavi MV, Fuhrmann N (2013) Dominant optic atrophy, OPA1, and mitochondrial quality control: understanding mitochondrial network dynamics. Mol Neurodegeneration 8(1):32. doi:10.1186/1750-1326-8-32
Alpers BJ (1931) Diffuse progressive degeneration of the gray matter of the cerebrum. Arch Neurol Psychiatry 25(3):469. doi:10.1001/archneurpsyc.1931.02230030027002
Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissière A, Campos Y et al (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain: J Neurol 131(Pt 2):338–351. doi:10.1093/brain/awm298
Amati-Bonneau P, Milea D, Bonneau D, Chevrollier A, Ferré M, Guillet V, Gueguen N et al (2009) OPA1-associated disorders: phenotypes and pathophysiology. Int J Biochem Cell Biol 41(10):1855–1865. doi:10.1016/j.biocel.2009.04.012
Anderson S, Bankier AT, Barrell BG, De Bruijn MH, Coulson AR, Drouin J, Eperon IC (1981) Sequence and organization of the human mitochondrial genome. Nature 290(5806):457–465. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7219534
Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A et al (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol 12(1):25–31. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9046040
Aon MA, Cortassa S, Maack C, O’Rourke B (2007) Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem 282(30):21889–21900. doi:10.1074/jbc.M702841200
Apostolova LG, White M, Moore SA, Davis PH (2005) Deep white matter pathologic features in watershed regions: a novel pattern of central nervous system involvement in MELAS. Arch Neurol 62(7):1154–1156. doi:10.1001/archneur.62.7.1154
Ashley N, O’Rourke A, Smith C, Adams S, Gowda V, Zeviani M, Brown GK et al (2008) Depletion of mitochondrial DNA in fibroblast cultures from patients with POLG1 mutations is a consequence of catalytic mutations. Hum Mol Genet 17(16):2496–2506. doi:10.1093/hmg/ddn150
Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR et al (2003) Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem 278(19):17190–17197. doi:10.1074/jbc.M212754200
Baertling F, Schaper J, Mayatepek E, Distelmaier F (2013) Teaching NeuroImages: rapidly progressive leukoencephalopathy in mitochondrial complex I deficiency. Neurology 81(2):e10–e11. doi:10.1212/WNL.0b013e31829a339b
Bardosi A, Creutzfeldt W, DiMauro S, Felgenhauer K, Friede RL, Goebel H, Kohlschütter A et al (1987) Myo-, neuro-, gastrointestinal encephalopathy (MNGIE syndrome) due to partial deficiency of cytochrome-c-oxidase. A new mitochondrial multisystem disorder. Acta Neuropathol 74(3):248–258. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2823522
Barker PB, Hearshen DO, Boska MD (2001) Single-voxel proton MRS of the human brain at 1.5T and 3.0T. Magn Reson Med: Official J Soc Magn Reson Med/Soc Magn Reson Med 45(5):765–769. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11323802
Barkovich AJ, Good WV, Koch TK, Berg BO (1993) Mitochondrial disorders: analysis of their clinical and imaging characteristics. AJNR Am J Neuroradiol 14(5):1119–1137. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8237691
Barragán-Campos HM, Vallée J-N, Lô D, Barrera-Ramírez CF, Argote-Greene M, Sánchez-Guerrero J, Estañol B et al (2005) Brain magnetic resonance imaging findings in patients with mitochondrial cytopathies. Arch Neurol 62(5):737–742. doi:10.1001/archneur.62.5.737
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR et al (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476(7360):341–345. doi:10.1038/nature10234
Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR (2002) Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 59(9):1406–1411. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12427892
Bertholet AM, Millet AME, Guillermin O, Daloyau M, Davezac N, Miquel M-C, Belenguer P (2013) OPA1 loss of function affects in vitro neuronal maturation. Brain: J Neurol 136(Pt 5):1518–1533. doi:10.1093/brain/awt060
Bianchi MC, Tosetti M, Battini R, Manca ML, Mancuso M, Cioni G, Canapicchi R et al (2003) Proton MR spectroscopy of mitochondrial diseases: analysis of brain metabolic abnormalities and their possible diagnostic relevance. AJNR Am J Neuroradiol 24(10):1958–1966. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/14625217
Boddaert N, Romano S, Funalot B, Rio M, Sarzi E, Lebre AS, Bahi-Buisson N et al (2008) 1H MRS spectroscopy evidence of cerebellar high lactate in mitochondrial respiratory chain deficiency. Mol Genet Metab 93(1):85–88. doi:10.1016/j.ymgme.2007.09.003
Boitier E, Rea R, Duchen MR (1999) Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. J Cell Biol 145(4):795–808. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2133193&tool=pmcentrez&rendertype=abstract
Bonora M, Giorgi C, Bononi A, Marchi S, Patergnani S, Rimessi A, Rizzuto R et al (2013) Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat Protoc 8(11):2105–2118. doi:10.1038/nprot.2013.127
Brockmann K, Bjornstad A, Dechent P, Korenke CG, Smeitink J, Trijbels JMF, Athanassopoulos S et al (2002) Succinate in dystrophic white matter: a proton magnetic resonance spectroscopy finding characteristic for complex II deficiency. Ann Neurol 52(1):38–46. doi:10.1002/ana.10232
Burgeois M, Goutieres F, Chretien D, Rustin P, Munnich A, Aicardi J (1992) Deficiency in complex II of the respiratory chain, presenting as a leukodystrophy in two sisters with Leigh syndrome. Brain Dev 14(6):404–408. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1492653
Cady EB, Lorek A, Penrice J, Reynolds EO, Iles RA, Burns SP, Coutts GA et al (1994) Detection of propan-1,2-diol in neonatal brain by in vivo proton magnetic resonance spectroscopy. Mag Reson Med: Official J Soc Mag Reson Med/Soc Mag Reson Med 32(6):764–767. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7869898
Castro-Gago M, González-Conde V, Fernández-Seara MJ, Rodrigo-Sáez E, Fernández-Cebrián S, Alonso-Martín A, Campos Y et al (1999) Early mitochondrial encephalomyopathy due to complex IV deficiency consistent with Alpers-Huttenlocher syndrome: report of two cases. Rev Neurol 29(10):912–917. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10637838
Chan DC (2006a) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125(7):1241–1252. doi:10.1016/j.cell.2006.06.010
Chan DC (2006b) Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22:79–99. doi:10.1146/annurev.cellbio.22.010305.104638
Chandrasekaran K, Hazelton JL, Wang Y, Fiskum G, Kristian T (2006) Neuron-specific conditional expression of a mitochondrially targeted fluorescent protein in mice. J Neurosci: Official J Soc Neurosci 26(51):13123–13127. doi:10.1523/JNEUROSCI.4191-06.2006
Cheldi A, Ronchi D, Bordoni A, Bordo B, Lanfranconi S, Bellotti MG, Corti S et al (2013) POLG1 mutations and stroke like episodes: a distinct clinical entity rather than an atypical MELAS syndrome. BMC Neurol 13:8. doi:10.1186/1471-2377-13-8
Chen H, Chan DC (2004) Mitochondrial dynamics in mammals. Curr Top Dev Biol 59:119–144. doi:10.1016/S0070-2153(04)59005-1
Chen H, Chomyn A, Chan DC (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280(28):26185–26192. doi:10.1074/jbc.M503062200
Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141(2):280–289. doi:10.1016/j.cell.2010.02.026
Chen C, Xiong N, Wang Y, Xiong J, Huang J, Zhang Z, Wang T (2012a) A study of familial MELAS: evaluation of A3243G mutation, clinical phenotype, and magnetic resonance spectroscopy-monitored progression. Neurol India 60(1):86–89. doi:10.4103/0028-3886.93609
Chen Z, Li J, Lou X, Ma L (2012b) Sequential evaluation of brain lesions using functional magnetic resonance imaging in patients with Leigh syndrome. Nan fang yi ke da xue xue bao = J South Med Univ 32(10):1474–1477. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/23076188
Chi C-S, Lee H-F, Tsai C-R, Chen CC-C, Tung J-N (2011a) Cranial magnetic resonance imaging findings in children with nonsyndromic mitochondrial diseases. Pediatr Neurol 44(3):171–176. doi:10.1016/j.pediatrneurol.2010.09.009
Chi C-S, Lee H-F, Tsai C-R, Chen W-S, Tung J-N, Hung H-C (2011b) Lactate peak on brain MRS in children with syndromic mitochondrial diseases. JCMA J Chin Med Assoc 74(7):305–309. doi:10.1016/j.jcma.2011.05.006
Chu CT (2006) Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol 65(5):423–432. doi:10.1097/01.jnen.0000229233.75253.be
Chu BC, Terae S, Takahashi C, Kikuchi Y, Miyasaka K, Abe S, Minowa K et al (1999) MRI of the brain in the Kearns-Sayre syndrome: report of four cases and a review. Neuroradiology 41(10):759–764. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10552027
Chu CT, Zhu J, Dagda R (2007) Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 3(6):663–666. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2779565&tool=pmcentrez&rendertype=abstract
Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101(45):15927–15932. doi:10.1073/pnas.0407043101
Conway LJ, Robertson TE, McGill JJ, Hanson JP (2011) MELAS syndrome in an Indigenous Australian woman. Med J Aust 195(10):581–582. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/22107001
Dagda RK, Zhu J, Kulich SM, Chu CT (2008) Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy 4(6):770–782. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2574804&tool=pmcentrez&rendertype=abstract
Davidson SM, Duchen MR (2012) Imaging mitochondrial calcium signalling with fluorescent probes and single or two photon confocal microscopy. Methods Mol Biol (Clifton, NJ) 810:219–234. doi:10.1007/978-1-61779-382-0_14
Davidson SM, Yellon D, Duchen MR (2007) Assessing mitochondrial potential, calcium, and redox state in isolated mammalian cells using confocal microscopy. Methods Mol Biol (Clifton, NJ) 372:421–430. doi:10.1007/978-1-59745-365-3_30
De la Fuente S, Fonteriz RI, De la Cruz PJ, Montero M, Alvarez J (2012) Mitochondrial free [Ca(2+)] dynamics measured with a novel low-Ca(2+) affinity aequorin probe. Biochem J 445(3):371–376. doi:10.1042/BJ20120423
De Paepe B, Smet J, Vanlander A, Seneca S, Lissens W, De Meirleir L, Vandewoestyne M et al (2012) Fluorescence imaging of mitochondria in cultured skin fibroblasts: a useful method for the detection of oxidative phosphorylation defects. Pediatr Res 72(3):232–240. doi:10.1038/pr.2012.84
Dedkova EN, Blatter LA (2012) Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol 52(1):48–61. doi:10.1016/j.yjmcc.2011.08.030
Delonlay P, Rötig A, Sarnat HB (2013) Respiratory chain deficiencies. Handb Clin Neurol 113:1651–1666. doi:10.1016/B978-0-444-59565-2.00033-2
Deschauer M, Tennant S, Rokicka A, He L, Kraya T, Turnbull DM, Zierz S et al (2007) MELAS associated with mutations in the POLG1 gene. Neurology 68(20):1741–1742. doi:10.1212/01.wnl.0000261929.92478.3e
Di Lisa F, Blank PS, Colonna R, Gambassi G, Silverman HS, Stern MD, Hansford RG (1995) Mitochondrial membrane potential in single living adult rat cardiac myocytes exposed to anoxia or metabolic inhibition. J Physiol 486 (Pt 1):1–13. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1156492&tool=pmcentrez&rendertype=abstract
Diaz G, Diana A, Falchi AM, Gremo F, Pani A, Batetta B, Dessì S et al (2001) Intra- and intercellular distribution of mitochondrial probes and changes after treatment with MDR modulators. IUBMB Life 51(2):121–126. doi:10.1080/15216540119470
DiMauro S (2004) Mitochondrial diseases. Biochim Biophys Acta 1658(1–2):80–88. doi:10.1016/j.bbabio.2004.03.014
DiMauro S, Hirano M (2005) Mitochondrial encephalomyopathies: an update. Neuromuscul Disord: NMD 15(4):276–286. doi:10.1016/j.nmd.2004.12.008
DiMauro S, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348(26):2656–2668. doi:10.1056/NEJMra022567
DiMauro S, Bonilla E, Zeviani M, Nakagawa M, DeVivo DC (1985) Mitochondrial myopathies. Ann Neurol 17(6):521–538. doi:10.1002/ana.410170602
DiMauro S, Bonilla E, Lombes A, Shanske S, Minetti C, Moraes CT (1990) Mitochondrial encephalomyopathies. Neurologic clinics 8(3):483–506. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2170831
Dinopoulos A, Cecil KM, Schapiro MB, Papadimitriou A, Hadjigeorgiou GM, Wong B, de Grauw T et al (2005) Brain MRI and proton MRS findings in infants and children with respiratory chain defects. Neuropediatrics 36(5):290–301. doi:10.1055/s-2005-872807
Distelmaier F, Koopman WJH, Testa ER, De Jong AS, Swarts HG, Mayatepek E, Smeitink JAM et al (2008) Life cell quantification of mitochondrial membrane potential at the single organelle level. Cytometry Part A: J Int Soc Anal Cytol 73(2):129–138. doi:10.1002/cyto.a.20503
Dolloff NG, Ma X, Dicker DT, Humphreys RC, Li LZ, El-Deiry WS (2011) Spectral imaging-based methods for quantifying autophagy and apoptosis. Cancer Biol Ther 12(4):349–356. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3230317&tool=pmcentrez&rendertype=abstract
Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM et al (2009) Starving neurons show sex difference in autophagy. J Biol Chem 284(4):2383–2396. doi:10.1074/jbc.M804396200
Duchen MR, Surin A, Jacobson J (2003) Imaging mitochondrial function in intact cells. Methods Enzymol 361:353–389. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12624920
Duning T, Deppe M, Keller S, Mohammadi S, Schiffbauer H, Marziniak M (2009) Diffusion tensor imaging in a case of Kearns-Sayre syndrome: striking brainstem involvement as a possible cause of oculomotor symptoms. J Neurol Sci 281(1–2):110–112. doi:10.1016/j.jns.2009.03.007
Eiberg H, Kjer B, Kjer P, Rosenberg T (1994) Dominant optic atrophy (OPA1) mapped to chromosome 3q region. I. Linkage analysis. Hum Mol Genet 3(6):977–980. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7951248
Elsässer A, Vogt AM, Nef H, Kostin S, Möllmann H, Skwara W, Bode C et al (2004) Human hibernating myocardium is jeopardized by apoptotic and autophagic cell death. J Am Coll Cardiol 43(12):2191–2199. doi:10.1016/j.jacc.2004.02.053
Emmanuele V, Sotiriou E, Rios PG, Ganesh J, Ichord R, Foley AR, Akman HO et al (2013) A novel mutation in the mitochondrial DNA cytochrome b gene (MTCYB) in a patient with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes syndrome. J Child Neurol 28(2):236–242. doi:10.1177/0883073812445787
Eskelinen E-L, Kovács AL (2011) Double membranes vs. lipid bilayers, and their significance for correct identification of macroautophagic structures. Autophagy 7(9):931–932. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/21642767
Farina L, Chiapparini L, Uziel G, Bugiani M, Zeviani M, Savoiardo M (2002) MR findings in Leigh syndrome with COX deficiency and SURF-1 mutations. AJNR Am J Neuroradiol 23(7):1095–1100. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12169463
Fernandes C, Rao Y (2011) Genome-wide screen for modifiers of Parkinson’s disease genes in Drosophila. Mol brain 4:17. doi:10.1186/1756-6606-4-17
Finsterer J (2009) Central nervous system imaging in mitochondrial disorders. Can J Neurol Sci. Le journal canadien des sciences neurologiques 36(2):143–153. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19378706
Finsterer J, Jarius C, Eichberger H (2001) Phenotype variability in 130 adult patients with respiratory chain disorders. J Inherit Metab Dis 24(5):560–576. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11757584
Fonteriz RI, De la Fuente S, Moreno A, Lobatón CD, Montero M, Alvarez J (2010) Monitoring mitochondrial [Ca(2+)] dynamics with rhod-2, ratiometric pericam and aequorin. Cell Calcium 48(1):61–69. doi:10.1016/j.ceca.2010.07.001
Friedman SD, Shaw DWW, Ishak G, Gropman AL, Saneto RP (2010) The use of neuroimaging in the diagnosis of mitochondrial disease. Dev Disabil Res Rev 16(2):129–135. doi:10.1002/ddrr.103
Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G (2012) Mitochondrial control of cellular life, stress, and death. Circ Res 111(9):1198–1207. doi:10.1161/CIRCRESAHA.112.268946
Galvez-Ruiz A, Neuhaus C, Bergmann C, Bolz H (2013) First cases of dominant optic atrophy in Saudi Arabia: report of two novel OPA1 mutations. J Neuroophthalmol: Official J N Am Neuroophthalmol Soc. doi:10.1097/WNO.0b013e31829ffb9a
Gieraerts C, Demaerel P, Van Damme P, Wilms G (2013) Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome mimicking herpes simplex encephalitis on imaging studies. J Comput Assist Tomogr 37(2):279–281. doi:10.1097/RCT.0b013e3182811170
Gire C, Girard N, Nicaise C, Einaudi MA, Montfort MF, Dejode JM (2002) Clinical features and neuroradiological findings of mitochondrial pathology in six neonates. Child’s Nerv Syst: ChNS: Official J Int Soc Pediatr Neurosurg 18(11):621–628. doi:10.1007/s00381-002-0621-0
Goethem Van, Gert Schwartz M, Löfgren A, Dermaut B, Van Broeckhoven C, Vissing J (2003) Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. EJHG Eur J Hum Genet 11(7):547–549. doi:10.1038/sj.ejhg.5201002
Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13(5):589–598. doi:10.1038/ncb2220
Goto Y, Nonaka I, Horai S (1990) A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348(6302):651–653. doi:10.1038/348651a0
Haas R, Dietrich R (2004) Neuroimaging of mitochondrial disorders. Mitochondrion 4(5–6):471–490. doi:10.1016/j.mito.2004.07.008
Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Cohen BH (2007) Mitochondrial disease: a practical approach for primary care physicians. Pediatrics 120(6):1326–1333. doi:10.1542/peds.2007-0391
Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Wong L-J et al (2008) The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab 94(1):16–37. doi:10.1016/j.ymgme.2007.11.018
Hajnóczky G, Csordás G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M (2006) Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40(5–6):553–560. doi:10.1016/j.ceca.2006.08.016
Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lönnqvist T (2007) Recessive twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain: J Neurol 130(Pt 11):3032–3040. doi:10.1093/brain/awm242
Hall AM, Unwin RJ, Parker N, Duchen MR (2009) Multiphoton imaging reveals differences in mitochondrial function between nephron segments. JASN J Am Soc Nephrol 20(6):1293–1302. doi:10.1681/ASN.2008070759
Hall AM, Rhodes GJ, Sandoval RM, Corridon PR, Molitoris BA (2013) In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int 83(1):72–83. doi:10.1038/ki.2012.328
Han J, Han MS, Tung C-H (2013) A non-toxic fluorogenic dye for mitochondria labeling. Biochim Biophys Acta 1830(11):5130–5135. doi:10.1016/j.bbagen.2013.07.001
Harding BN (1990) Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): a personal review. J Child Neurol 5(4):273–287. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2246481
Hausenloy DJ, Ong S-B, Yellon DM (2009) The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol 104(2):189–202. doi:10.1007/s00395-009-0010-x
Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A (2009) A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11(12):1433–1437. doi:10.1038/ncb1991
Hernandez G, Thornton C, Stotland A, Lui D, Sin J, Ramil J, Magee N et al (2013) MitoTimer: a novel tool for monitoring mitochondrial turnover. Autophagy 9(11):1852–1861. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/24128932
Hirano M, Pavlakis SG (1994) Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol 9(1):4–13. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8151079
Honda HM, Korge P, Weiss JN (2005) Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci 1047:248–258. doi:10.1196/annals.1341.022
Horvath R, Hudson G, Ferrari G, Fütterer N, Ahola S, Lamantea E, Prokisch H et al (2006) Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain: J Neurol 129(Pt 7):1674–1684. doi:10.1093/brain/awl088
Hourani RG, Barada WM, Al-Kutoubi AM, Hourani MH (2006) Atypical MRI findings in Kearns-Sayre syndrome: T2 radial stripes. Neuropediatrics 37(2):110–113. doi:10.1055/s-2006-924226
Huang H, Choi S-Y, Frohman MA (2010) A quantitative assay for mitochondrial fusion using Renilla luciferase complementation. Mitochondrion 10(5):559–566. doi:10.1016/j.mito.2010.05.003
Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, Griffiths PG et al (2008) Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain: J Neurol 131(Pt 2):329–337. doi:10.1093/brain/awm272
Hunter MF, Peters H, Salemi R, Thorburn D, Mackay MT (2011) Alpers syndrome with mutations in POLG: clinical and investigative features. Pediatr Neurol 45(5):311–318. doi:10.1016/j.pediatrneurol.2011.07.008
Hüser J, Rechenmacher CE, Blatter LA (1998) Imaging the permeability pore transition in single mitochondria. Biophys J 74(4):2129–2137. doi:10.1016/S0006-3495(98)77920-2
Huttenlocher PR, Solitare GB, Adams G (1976) Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol 33(3):186–192. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1252162
Ichas F, Jouaville LS, Mazat JP (1997) Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89(7):1145–1153. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9215636
Iguchi M, Kato M, Nakai J, Takeda T, Matsumoto-Ida M, Kita T, Kimura T et al (2012) Direct monitoring of mitochondrial calcium levels in cultured cardiac myocytes using a novel fluorescent indicator protein, GCaMP2-mt. Int J Cardiol 158(2):225–234. doi:10.1016/j.ijcard.2011.01.034
Iizuka T, Sakai F, Suzuki N, Hata T, Tsukahara S, Fukuda M, Takiyama Y (2002) Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology 59(6):816–824. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12297560
Iizuka T, Sakai F, Ide T, Miyakawa S, Sato M, Yoshii S (2007) Regional cerebral blood flow and cerebrovascular reactivity during chronic stage of stroke-like episodes in MELAS—implication of neurovascular cellular mechanism. J Neurol Sci 257(1–2):126–138. doi:10.1016/j.jns.2007.01.040
Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408(6813):708–713. doi:10.1038/35047064
Ionasescu V (1983) Oculogastrointestinal muscular dystrophy. Am J Med Genet 15(1):103–112. doi:10.1002/ajmg.1320150114
Isobe T, Matsumura A, Anno I, Kawamura H, Shibata Y, Muraishi H, Minami M (2007) Lactate quantification by proton magnetic resonance spectroscopy using a clinical MRI machine: a basic study. Australas Radiol 51(4):330–333. doi:10.1111/j.1440-1673.2007.01745.x
Jain-Ghai S, Cameron JM, Al Maawali A, Blaser S, MacKay N, Robinson B, Raiman J (2013) Complex II deficiency–a case report and review of the literature. Am J Med Genet. Part A 161A(2):285–294. doi:10.1002/ajmg.a.35714
Jaksch M, Horvath R, Horn N, Auer DP, Macmillan C, Peters J, Gerbitz KD et al (2001) Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy. Neurology 57(8):1440–1146. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11673586
James DI, Parone PA, Mattenberger Y, Martinou J-C (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278(38):36373–36379. doi:10.1074/jbc.M303758200
Janssen RJRJ, Nijtmans LG, Van den Heuvel LP, Smeitink JAM (2006) Mitochondrial complex I: structure, function and pathology. J Inherit Metab Dis 29(4):499–515. doi:10.1007/s10545-006-0362-4
Jeppesen TD, Schwartz M, Hansen K, Danielsen ER, Wibrand F, Vissing J (2003) Late onset of stroke-like episode associated with a 3256C–> T point mutation of mitochondrial DNA. J Neurol Sci 214(1–2):17–20. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12972383
Jheng H-F, Tsai P-J, Guo S-M, Kuo L-H, Chang C-S, Su I-J, Chang C-R et al (2012) Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 32(2):309–319. doi:10.1128/MCB.05603-11
José da Rocha A, Túlio Braga F, Carlos Martins Maia A, Jorge da Silva C, Toyama C, Pereira Pinto Gama H, Kok F et al (2008) Lactate detection by MRS in mitochondrial encephalopathy: optimization of technical parameters. J Neuroimaging: Official J Am Soc Neuroimaging 18(1):1–8. doi:10.1111/j.1552-6569.2007.00205.x
Jou MJ, Peng TI, Sheu SS (1996) Histamine induces oscillations of mitochondrial free Ca2+ concentration in single cultured rat brain astrocytes. J Physiol 497(Pt 2):299–308. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1160985&tool=pmcentrez&rendertype=abstract
Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD (1995) Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature 377(6548):438–441. doi:10.1038/377438a0
Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R (1999) Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Nat Acad Sci USA 96(24):13807–13812. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=24146&tool=pmcentrez&rendertype=abstract
Kaim G, Dimroth P (1999) ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. EMBO J 18(15):4118–4127. doi:10.1093/emboj/18.15.4118
Kamata Y, Mashima Y, Yokoyama M, Tanaka K, Goto Y, Oguchi Y (1998) Patient with Kearns-Sayre syndrome exhibiting abnormal magnetic resonance image of the brain. J Neuroophthalmol: Official J North Am Neuroophthalmol Soc 18(4):284–288. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9858014
Karanasios E, Stapleton E, Walker SA, Manifava M, Ktistakis NT (2013) Live cell imaging of early autophagy events: omegasomes and beyond. JoVE J Visualized Exp (77). doi:10.3791/50484
Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ (2004) Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol 164(4):493–499. doi:10.1083/jcb.200309082
Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18(8):1042–1052. doi:10.1016/j.chembiol.2011.05.013
Kearns TP, Sayre GP (1958) Retinitis pigmentosa, external ophthalmoplegia, and complete heart block: unusual syndrome with histologic study in one of two cases. AMA Arch Ophthalmol 60(2):280–289. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/13558799
Keevil SF (2006) Spatial localization in nuclear magnetic resonance spectroscopy. Phys Med Biol 51(16):R579–R636. doi:10.1088/0031-9155/51/16/R01
Kettlewell S, Cabrero P, Nicklin SA, Dow JAT, Davies S, Smith GL (2009) Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. J Mol Cell Cardiol 46(6):891–901. doi:10.1016/j.yjmcc.2009.02.016
Kim HS, Kim DI, Lee BI, Jeong EK, Choi C, Lee JD, Yoon PH et al (2001) Diffusion-weighted image and MR spectroscopic analysis of a case of MELAS with repeated attacks. Yonsei Med J 42(1):128–133. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11293491
Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3(5):452–460. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17534139
Kissová I, Deffieu M, Manon S, Camougrand N (2004) Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem 279(37):39068–39074. doi:10.1074/jbc.M406960200
Kjer P (1959) Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Suppl 164(Supp 54):1–147. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/13660776
Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8(11):931–937. doi:10.1038/nrm2245
Klionsky DJ (2008) Autophagy revisited: a conversation with Christian de Duve. Autophagy 4(6):740–743. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18567941
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8(4):445–544
Knight MA, Gardner RJM, Bahlo M, Matsuura T, Dixon JA, Forrest SM, Storey E (2004) Dominantly inherited ataxia and dysphonia with dentate calcification: spinocerebellar ataxia type 20. Brain: J Neurol 127(Pt 5):1172–1181. doi:10.1093/brain/awh139
Kovács J, Fellinger E, Kárpáti PA, Kovács AL, László L (1986) The turnover of autophagic vacuoles: evaluation by quantitative electron microscopy. Biomed Biochim Acta 45(11–12):1543–1547. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3579875
Kovács J, Fellinger E, Kárpáti AP, Kovács AL, László L, Réz G (1987) Morphometric evaluation of the turnover of autophagic vacuoles after treatment with Triton X-100 and vinblastine in murine pancreatic acinar and seminal vesicle epithelial cells. Virchows Archiv B, Cell Pathol Incl Mol Pathol 53(3):183–190. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2888237
Kovács J, László L, Kovács AL (1988) Regression of autophagic vacuoles in pancreatic acinar, seminal vesicle epithelial, and liver parenchymal cells: a comparative morphometric study of the effect of vinblastine and leupeptin followed by cycloheximide treatment. Exp Cell Res 174(1):244–251. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3335225
Kuma A, Matsui M, Mizushima N (2007) LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 3(4):323–328. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17387262
Kurt B, Jaeken J, Van Hove J, Lagae L, Löfgren A, Everman DB, Jayakar P et al (2010) A novel POLG gene mutation in 4 children with Alpers-like hepatocerebral syndromes. Arch Neurol 67(2):239–244. doi:10.1001/archneurol.2009.332
Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, Chu CT, Kochanek PM et al (2008) Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab: Official J Int Soc Cereb Blood Flow Metab 28(3):540–550. doi:10.1038/sj.jcbfm.9600551
Lamperti C, Diodato D, Lamantea E, Carrara F, Ghezzi D, Mereghetti P, Rizzi R et al (2012) MELAS-like encephalomyopathy caused by a new pathogenic mutation in the mitochondrial DNA encoded cytochrome c oxidase subunit I. Neuromuscul Disord. Retrieved from http://www.sciencedirect.com/science/article/pii/S096089661200185X
Lange T, Dydak U, Roberts TPL, Rowley HA, Bjeljac M, Boesiger P (2006) Pitfalls in lactate measurements at 3T. AJNR Am J Neuroradiol 27(4):895–901. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/16611787
Larsson NG, Holme E, Kristiansson B, Oldfors A, Tulinius M (1990) Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr Res 28(2):131–6. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2395603
Lebre AS, Rio M, Faivre d’Arcier L, Vernerey D, Landrieu P, Slama A, Jardel C et al (2011) A common pattern of brain MRI imaging in mitochondrial diseases with complex I deficiency. J Med Genet 48(1):16–23. doi:10.1136/jmg.2010.079624
Lee H-F, Lee H-J, Chi C-S, Tsai C-R, Chang T-K, Wang C-J (2007) The neurological evolution of Pearson syndrome: case report and literature review. Eur J Paediatr Neurol: EJPN: Official J Eur Paediatr Neurol Soc 11(4):208–214. doi:10.1016/j.ejpn.2006.12.008
Lee Y, Lee H-Y, Hanna RA, Gustafsson ÅB (2011) Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301(5):H1924–H1931. doi:10.1152/ajpheart.0 0368.2011
Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson N-G (2012) Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet 21(22):4827–4835. doi:10.1093/hmg/dds352
Leigh D (1951) Subacute necrotizing encephalomyelopathy in an infant. J Neurol, Neurosurg, Psychiatry 14(3):216–221. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=499520&tool=pmcentrez&rendertype=abstract
Lemasters JJ, Ramshesh VK (2007) Imaging of mitochondrial polarization and depolarization with cationic fluorophores. Methods Cell Biol 80:283–295. doi:10.1016/S0091-679X(06)80014-2
Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, Bonneau D, Reynier P et al (2012) Dominant optic atrophy. Orphanet J Rare Dis 7:46. doi:10.1186/1750-1172-7-46
Lerman-Sagie T, Leshinsky-Silver E, Watemberg N, Luckman Y, Lev D (2005) White matter involvement in mitochondrial diseases. Mol Genet Metab 84(2):127–136. doi:10.1016/j.ymgme.2004.09.008
Leutner C, Layer G, Zierz S, Solymosi L, Dewes W, Reiser M (1994) Cerebral MR in ophthalmoplegia plus. AJNR Am J Neuroradiol 15(4):681–687. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8010270
Libernini L, Lupis C, Mastrangelo M, Carrozzo R, Santorelli FM, Inghilleri M, Leuzzi V (2012) Mitochondrial neurogastrointestinal encephalomyopathy: novel pathogenic mutations in thymidine phosphorylase gene in two Italian brothers. Neuropediatrics 43(4):201–208. doi:10.1055/s-0032-1315431
Liesa M, Palacín M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89(3):799–845. doi:10.1152/physrev.0 0030.2008
Lin DDM, Crawford TO, Barker PB (2003) Proton MR spectroscopy in the diagnostic evaluation of suspected mitochondrial disease. AJNR Am J Neuroradiol 24(1):33–41. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12533324
Liu CL, Chen S, Dietrich D, Hu BR (2008) Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab: Official J Int Soc Cereb Blood Flow Metab 28(4):674–683. doi:10.1038/sj.jcbfm.9600587
Lovy A, Molina AJA, Cerqueira FM, Trudeau K, Shirihai OS (2012) A faster, high resolution, mtPA-GFP-based mitochondrial fusion assay acquiring kinetic data of multiple cells in parallel using confocal microscopy. JoVE J Visualized Exp 65:e3991. doi:10.3791/3991
Lunkes A, Hartung U, Magariño C, Rodríguez M, Palmero A, Rodríguez L, Heredero L et al (1995) Refinement of the OPA1 gene locus on chromosome 3q28-q29 to a region of 2–8 cM, in one Cuban pedigree with autosomal dominant optic atrophy type Kjer. Am J Hum Genet 57(4):968–970. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1801490&tool=pmcentrez&rendertype=abstract
Majoie CB, Akkerman EM, Blank C, Barth PG, Poll-The BT, Den Heeten GJ (2002) Mitochondrial encephalomyopathy: comparison of conventional MR imaging with diffusion-weighted and diffusion tensor imaging: case report. AJNR Am J Neuroradiol 23(5):813–816. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12006283
Malfatti E, Bugiani M, Invernizzi F, De Souza CF-M, Farina L, Carrara F, Lamantea E et al (2007) Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain: J Neurol 130(Pt 7):1894–1904. doi:10.1093/brain/awm114
Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE et al (2012) MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151(3):630–644. doi:10.1016/j.cell.2012.10.011
Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20(13):2495–2509. doi:10.1093/hmg/ddr139
Martinou JC, Desagher S, Antonsson B (2000) Cytochrome c release from mitochondria: all or nothing. Nat Cell Biol 2(3):E41–E43. doi:10.1038/35004069
Matthews PM, Tampieri D, Berkovic SF, Andermann F, Silver K, Chityat D, Arnold DL (1991) Magnetic resonance imaging shows specific abnormalities in the MELAS syndrome. Neurology 41(7):1043–1046. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2067632
Mattman A, Sirrs S, Mezei M, Salvarinova-Zivkovic R, Alfadhel M, Lillquist Y (2011) Mitochondrial disease clinical manifestations: an overview. BCMJ 53(4):183–187. Retrieved from http://www.bcmj.org/articles/mitochondrial-disease-clinical-manifestations-overview
Mayer WJ, Remy M, Rudolph G (2011) Kearns-Sayre syndrome: a mitochondrial disease (OMIM #530000). Der Ophthalmologe: Zeitschrift der Deutschen Ophthalmologischen Gesellschaft 108(5):459–462. doi:10.1007/s00347-010-2296-3
McBride HM, Neuspiel M, Wasiak S (2006) Mitochondria: more than just a powerhouse. CB Curr Biol 16(14):R551–R560. doi:10.1016/j.cub.2006.06.054
McCoy B, Owens C, Howley R, Ryan S, King M, Farrell MA, Lynch BJ (2011) Partial status epilepticus—rapid genetic diagnosis of Alpers’ disease. Eur J Paediatr Neurol: EJPN: Official J Eur Paediatr Neurol Soc 15(6):558–562. doi:10.1016/j.ejpn.2011.05.012
McFarland R, Turnbull DM (2009) Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med 265(2):210–228. doi:10.1111/j.1365-2796.2008.02066.x
McFarland, Robert, Taylor RW, Turnbull DM (2002) The neurology of mitochondrial DNA disease. Lancet Neurol 1(6):343–351. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12849395
McKelvie PA, Morley JB, Byrne E, Marzuki S (1991) Mitochondrial encephalomyopathies: a correlation between neuropathological findings and defects in mitochondrial DNA. J Neurol Sci 102(1):51–60. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1906931
McShane MA, Hammans SR, Sweeney M, Holt IJ, Beattie TJ, Brett EM, Harding AE (1991) Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am J Hum Genet 48(1):39–42. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1682744&tool=pmcentrez&rendertype=abstract
Medina L, Chi TL, DeVivo DC, Hilal SK (1990) MR findings in patients with subacute necrotizing encephalomyelopathy (Leigh syndrome): correlation with biochemical defect. AJNR Am J Neuroradiol 11(2):379–384. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2156413
Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW (2007) Imaging axonal transport of mitochondria in vivo. Nat Methods 4(7):559–561. doi:10.1038/nmeth1055
Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH (2012) Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci: Official J Soc Neurosci 32(12):4145–4155. doi:10.1523/JNEUROSCI.6338-11.2012
Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG (1991) Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol 261(4 Pt 2):H1123–H1134. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1928394
Mizushima N (2007) Autophagy: process and function. Genes Dev 21(22):2861–2873. doi:10.1101/gad.1599207
Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132. doi:10.1146/annurev-cellbio-092910-154005
Molina AJ, Shirihai OS (2009) Monitoring mitochondrial dynamics with photoactivatable [corrected] green fluorescent protein. Methods Enzymol 457:289–304. doi:10.1016/S0076-6879(09)05016-2
Morava E, Van den Heuvel L, Hol F, De Vries MC, Hogeveen M, Rodenburg RJ, Smeitink JAM (2006) Mitochondrial disease criteria: diagnostic applications in children. Neurology 67(10):1823–1826. doi:10.1212/01.wnl.0000244435.27645.54
Moroni I, Bugiani M, Bizzi A, Castelli G, Lamantea E, Uziel G (2002) Cerebral white matter involvement in children with mitochondrial encephalopathies. Neuropediatrics 33(2):79–85. doi:10.1055/s-2002-32372
Muñoz A, Mateos F, Simón R, García-Silva MT, Cabello S, Arenas J (1999) Mitochondrial diseases in children: neuroradiological and clinical features in 17 patients. Neuroradiology 41(12):920–928. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10639669
Nagai T, Sawano A, Park ES, Miyawaki A (2001) Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc Natl Acad Sci USA 98(6):3197–3202. doi:10.1073/pnas.051636098
Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I et al (2001) Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med 7(8):934–940. doi:10.1038/90976
Naviaux RK, Nguyen KV (2004) POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol 55(5):706–712. doi:10.1002/ana.20079
Nishino I (1999) Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283(5402):689–692. doi:10.1126/science.283.5402.689
Nishino I, Spinazzola A, Papadimitriou A, Hammans S, Steiner I, Hahn CD, Connolly AM et al (2000) Mitochondrial neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol 47(6):792–800. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10852545
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64(2):113–122. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15751225
O’Reilly CM, Fogarty KE, Drummond RM, Tuft RA, Walsh JV (2003) Quantitative analysis of spontaneous mitochondrial depolarizations. Biophys J 85(5):3350–3357. doi:10.1016/S0006-3495(03)74754-7
Ong S-B, Gustafsson AB (2012) New roles for mitochondria in cell death in the reperfused myocardium. Cardiovasc Res 94(2):190–196. doi:10.1093/cvr/cvr312
Ong S-B, Hausenloy DJ (2010) Mitochondrial morphology and cardiovascular disease. Cardiovasc Res 88(1):16–29. doi:10.1093/cvr/cvq237
Ong S-B, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121(18):2012–2022. doi:10.1161/CIRCULATIONAHA.109.906610
Ong S-B, Hall AR, Hausenloy DJ (2012) Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Signal. doi:10.1089/ars.2012.4777
Oppenheim C, Galanaud D, Samson Y, Sahel M, Dormont D, Wechsler B, Marsault C (2000) Can diffusion weighted magnetic resonance imaging help differentiate stroke from stroke-like events in MELAS? J Neurol, Neurosurg, Psychiatry 69(2):248–50. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1737057&tool=pmcentrez&rendertype=abstract
Ost A, Svensson K, Ruishalme I, Brännmark C, Franck N, Krook H, Sandström P et al (2010) Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med (Cambridge, MA) 16(7–8):235–246. doi:10.2119/molmed.2010.00023
Pauli W, Zarzycki A, Krzyształowski A, Walecka A (2013) CT and MRI imaging of the brain in MELAS syndrome. Pol J Radiol/Pol Med Soc Radiol 78(3):61–65. doi:10.12659/PJR.884010
Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP (1984) Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol 16(4):481–488. doi:10.1002/ana.410160409
Pearson HA, Lobel JS, Kocoshis SA, Naiman JL, Windmiller J, Lammi AT, Hoffman R et al (1979) A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr 95(6):976–984. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/501502
Perez-Atayde AR (2013) Diagnosis of mitochondrial neurogastrointestinal encephalopathy disease in gastrointestinal biopsies. Hum Pathol. Retrieved from http://www.sciencedirect.com/science/article/pii/S0046817713000026
Petty RK, Harding AE, Morgan-Hughes JA (1986) The clinical features of mitochondrial myopathy. Brain: J Neurol 109(Pt 5):915–938. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3779373
Phadwal K, Alegre-Abarrategui J, Watson AS, Pike L, Anbalagan S, Hammond EM, Wade-Martins R et al (2012) A novel method for autophagy detection in primary cells: impaired levels of macroautophagy in immunosenescent T cells. Autophagy 8(4):677–689. doi:10.4161/auto.18935
Pinton P, Rizzuto R (2006) Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ 13(8):1409–1418. doi:10.1038/sj.cdd.4401960
Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS et al (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 8(2):e55785. doi:10.1371/journal.pone.0055785
Plucińska G, Paquet D, Hruscha A, Godinho L, Haass C, Schmid B, Misgeld T (2012) In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J Neurosci: Official J Soc Neurosci 32(46):16203–16212. doi:10.1523/JNEUROSCI.1327-12.2012
Putney JW, Thomas AP (2006) Calcium signaling: double duty for calcium at the mitochondrial uniporter. CB Curr Biol 16(18):R812–R815. doi:10.1016/j.cub.2006.08.040
Quinonez SC, Leber SM, Martin DM, Thoene JG, Bedoyan JK (2013) Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr Neurol. Retrieved from http://www.sciencedirect.com/science/article/pii/S088789941200450X
Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J et al (1996) Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 39(3):343–351. doi:10.1002/ana.410390311
Rahman S, Brown RM, Chong WK, Wilson CJ, Brown GK (2001) A SURF1 gene mutation presenting as isolated leukodystrophy. Ann Neurol 49(6):797–800. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11409433
Rahn JJ, Stackley KD, Chan SSL (2013) Opa1 is required for proper mitochondrial metabolism in early development. PLoS ONE 8(3):e59218. doi:10.1371/journal.pone.0059218
Ramonet D, Perier C, Recasens A, Dehay B, Bové J, Costa V, Scorrano L et al (2013) Optic atrophy 1 mediates mitochondria remodeling and dopaminergic neurodegeneration linked to complex I deficiency. Cell Death Differ 20(1):77–85. doi:10.1038/cdd.2012.95
Rand SD, Prost R, Li SJ (1999) Proton MR spectroscopy of the brain. Neuroimaging Clin North Am 9(2):379–395. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10318721
Renard D, Campello C, Le Floch A, Castelnovo G, Taieb G (2012) Globus pallidus and substantia nigra hypointensities on T2-weighted imaging in MELAS. J Neurol 259(12):2720–2722. doi:10.1007/s00415-012-6633-0
Rizzuto Rosario, Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86(1):369–408. doi:10.1152/physrev.0 0004.2005
Rizzuto R, Brini M, Murgia M, Pozzan T (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science (New York, NY) 262(5134):744–747. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8235595
Rodriguez-Enriquez S, He L, Lemasters JJ (2004) Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int J Biochem Cell Biol 36(12):2463–2472. doi:10.1016/j.biocel.2004.04.009
Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ (2006) Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2(1):39–46. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/16874071
Rosado CJ, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ (2008) Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy 4(2):205–213. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18094608
Ross BD (2000) Real or imaginary? Human metabolism through nuclear magnetism. IUBMB Life 50(3):177–187. doi:10.1080/152165400300001499
Rossmanith W, Freilinger M, Roka J, Raffelsberger T, Moser-Thier K, Prayer D, Bernert G et al (2008) Isolated cytochrome c oxidase deficiency as a cause of MELAS. J Med Genet 45(2):117–121. doi:10.1136/jmg.2007.052076
Rotig A, Colonna M, Bonnefont JP, Blanche S, Fischer A, Saudubray JM, Munnich A (1989) Mitochondrial DNA deletion in Pearson’s marrow/pancreas syndrome. Lancet 1(8643):902–903. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2564980
Rötig A, Lebon S, Zinovieva E, Mollet J, Sarzi E, Bonnefont J-P, Munnich A (2004) Molecular diagnostics of mitochondrial disorders. Biochim Biophys Acta 1659(2–3):129–135. doi:10.1016/j.bbabio.2004.07.007
Rouzier C, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, Bonello-Palot N, Fragaki K et al (2012) The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy “plus” phenotype. Brain: J Neurol 135(Pt 1):23–34. doi:10.1093/brain/awr323
Rudnicki DD, Pletnikova O, Vonsattel J-PG, Ross CA, Margolis RL (2008) A comparison of huntington disease and huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol 67(4):366–374. doi:10.1097/NEN.0b013e31816b4aee
Rutter J, Winge DR, Schiffman JD (2010) Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 10(4):393–401. Retrieved from http://www.sciencedirect.com/science/article/pii/S1567724910000358
Saitoh S, Momoi MY, Yamagata T, Mori Y, Imai M (1998) Effects of dichloroacetate in three patients with MELAS. Neurology 50(2):531–534. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9484392
Saks V, Favier R, Guzun R, Schlattner U, Wallimann T (2006) Molecular system bioenergetics: regulation of substrate supply in response to heart energy demands. J Physiol 577(Pt 3):769–777. doi:10.1113/jphysiol.2006.120584
Saneto RP, Friedman SD, Shaw DWW (2008) Neuroimaging of mitochondrial disease. Mitochondrion 8(5–6):396–413. doi:10.1016/j.mito.2008.05.003
Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT (2003) Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci 116(Pt 13):2763–2774. doi:10.1242/jcs.00479
Scaglia F, Wong L-JC, Vladutiu GD, Hunter JV (2005) Predominant cerebellar volume loss as a neuroradiologic feature of pediatric respiratory chain defects. AJNR Am J Neuroradiol 26(7):1675–1680. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/16091512
Schapira AHV (2006) Mitochondrial disease. Lancet 368(9529):70–82. doi:10.1016/S0140-6736(06)68970-8
Schauss AC, Huang H, Choi S-Y, Xu L, Soubeyrand S, Bilodeau P, Zunino R et al (2010) A novel cell-free mitochondrial fusion assay amenable for high-throughput screenings of fusion modulators. BMC Biol 8:100. doi:10.1186/1741-7007-8-100
Schreur JH, Figueredo VM, Miyamae M, Shames DM, Baker AJ, Camacho SA (1996) Cytosolic and mitochondrial [Ca2+] in whole hearts using indo-1 acetoxymethyl ester: effects of high extracellular Ca2+. Biophys J 70(6):2571–2580. doi:10.1016/S0006-3495(96)79828-4
Schuelke M, Smeitink J, Mariman E, Loeffen J, Plecko B, Trijbels F, Stöckler-Ipsiroglu S et al (1999) Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat Genet 21(3):260–261. doi:10.1038/6772
Sedova M, Dedkova EN, Blatter LA (2006) Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am J Physiol Cell Physiol 291(5):C840–C850. doi:10.1152/ajpcell.0 0619.2005
Sekiya S, Tanaka M, Hayashi S, Oyanagi S (1982) Light- and electron-microscopic studies of intracytoplasmic acidophilic granules in the human locus coeruleus and substantia nigra. Acta Neuropathol 56(1):78–80. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/6278812
Shaibani A, Shchelochkov OA, Zhang S, Katsonis P, Lichtarge O, Wong L-J, Shinawi M (2009) Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol 66(8):1028–1032. doi:10.1001/archneurol.2009.139
Shoubridge EA (2001) Nuclear genetic defects of oxidative phosphorylation. Hum Mol Genet 10(20):2277–2284. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11673411
Sidiropoulos C, Moro E, Lang AE (2013) Extensive intracranial calcifications in a patient with a novel polymerase γ-1 mutation. Neurology 81(2):197–198. doi:10.1212/WNL.0b013e31829a3438
Sigmond T, Fehér J, Baksa A, Pásti G, Pálfia Z, Takács-Vellai K, Kovács J et al (2008) Qualitative and quantitative characterization of autophagy in Caenorhabditis elegans by electron microscopy. Methods Enzymol 451:467–491. doi:10.1016/S0076-6879(08)03228-X
Sijens PE, Smit GPA, Rödiger LA, Van Spronsen FJ, Oudkerk M, Rodenburg RJ, Lunsing RJ (2008) MR spectroscopy of the brain in Leigh syndrome. Brain Dev 30(9):579–583. doi:10.1016/j.braindev.2008.01.011
Slodzinski MK, Aon MA, O’Rourke B (2008) Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol 45(5):650–660. doi:10.1016/j.yjmcc.2008.07.017
Smirnova E, Shurland DL, Ryazantsev SN, Van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143(2):351–358. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2132828&tool=pmcentrez&rendertype=abstract
Sproule DM, Kaufmann P (2008) Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci 1142:133–158. doi:10.1196/annals.1444.011
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85(3):1093–1129. doi:10.1152/physrev.00006.2004
Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M et al (2005) Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121(7):1043–1057. doi:10.1016/j.cell.2005.05.025
Tam EWY, Feigenbaum A, Addis JBL, Blaser S, Mackay N, Al-Dosary M, Taylor RW et al (2008) A novel mitochondrial DNA mutation in COX1 leads to strokes, seizures, and lactic acidosis. Neuropediatrics 39(6):328–334. doi:10.1055/s-0029-1202287
Tang S, Wang J, Lee N-C, Milone M, Halberg MC, Schmitt ES, Craigen WJ et al (2011) Mitochondrial DNA polymerase gamma mutations: an ever expanding molecular and clinical spectrum. J Med Genet 48(10):669–681. doi:10.1136/jmedgenet-2011-100222
Tang S, Dimberg EL, Milone M, Wong L-JC (2012) Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol 259(5):862–868. doi:10.1007/s00415-011-6268-6
Tanigawa J, Kaneko K, Honda M, Harashima H, Murayama K, Wada T, Takano K et al (2012) Two Japanese patients with Leigh syndrome caused by novel SURF1 mutations. Brain Dev. Retrieved from http://www.sciencedirect.com/science/article/pii/S0387760412000393
Tanji K, DiMauro S, Bonilla E (1999) Disconnection of cerebellar Purkinje cells in Kearns-Sayre syndrome. J Neurol Sci 166(1):64–70. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10465502
Thorburn DR, Sugiana C, Salemi R, Kirby DM, Worgan L, Ohtake A, Ryan MT (2004) Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim Biophys Acta 1659(2–3):121–128. doi:10.1016/j.bbabio.2004.08.006
Tian X, Ma X, Qiao D, Ma A, Yan F, Huang X (2005) mCICR is required for As2O3-induced permeability transition pore opening and cytochrome c release from mitochondria. Mol Cell Biochem 277(1–2):33–42. doi:10.1007/s11010-005-4818-x
Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, Petersen OH (1999) Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca(2+) signals. EMBO J 18(18):4999–5008. doi:10.1093/emboj/18.18.4999
Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Miller SW, Davis RE et al (2000) Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol 162(1):37–50. doi:10.1006/exnr 2000.7333
Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A, Deutsch M et al (2006) Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am J Physiol Cell Physiol 291(1):C176–C184. doi:10.1152/ajpcell.00348.2005
Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27(2):433–446. doi:10.1038/sj.emboj.7601963
Tzoulis C, Engelsen BA, Telstad W, Aasly J, Zeviani M, Winterthun S, Ferrari G et al (2006) The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain: J Neurol 129(Pt 7):1685–1692. doi:10.1093/brain/awl097
Uusimaa J, Gowda V, McShane A, Smith C, Evans J, Shrier A, Narasimhan M et al (2013) Prospective study of POLG mutations presenting in children with intractable epilepsy: prevalence and clinical features. Epilepsia 54(6):1002–1011. doi:10.1111/epi.12115
Valanne L, Ketonen L, Majander A, Suomalainen A, Pihko H (1998) Neuroradiologic findings in children with mitochondrial disorders. AJNR Am J Neuroradiol 19(2):369–377. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9504497
Van Goethem G, Luoma P, Rantamäki M, Al Memar A, Kaakkola S, Hackman P, Krahe R et al (2004) POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 63(7):1251–1257. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15477547
Wang X, Su B, Lee H, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci: Official J Soc Neurosci 29(28):9090–9103. doi:10.1523/JNEUROSCI.1357-09.2009
Wilichowski E, Pouwels PJ, Frahm J, Hanefeld F (1999) Quantitative proton magnetic resonance spectroscopy of cerebral metabolic disturbances in patients with MELAS. Neuropediatrics 30(5):256–263. doi:10.1055/s-2007-973500
Williams GSB, Boyman L, Chikando AC, Khairallah RJ, Lederer WJ (2013) Mitochondrial calcium uptake. Proc Natl Acad Sci USA 110(26):10479–10486. doi:10.1073/pnas.1300410110
Wray SH, Provenzale JM, Johns DR, Thulborn KR (1995) MR of the brain in mitochondrial myopathy. AJNR Am J Neuroradiol 16(5):1167–1173. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7639148
Wu J-Q, Pollard TD (2005) Counting cytokinesis proteins globally and locally in fission yeast. Science (New York, NY) 310(5746):310–314. doi:10.1126/science.1113230
Yablonskiy DA, Neil JJ, Raichle ME, Ackerman JJH (1998) Homonuclear J coupling effects in volume localized NMR spectroscopy: pitfalls and solutions. Magn Reson Med 39(2):169–178. doi:10.1002/mrm.1910390202
Yilmaz A, Gdynia H-J, Ponfick M, Rösch S, Lindner A, Ludolph AC, Sechtem U (2012) Cardiovascular magnetic resonance imaging (CMR) reveals characteristic pattern of myocardial damage in patients with mitochondrial myopathy. Clinical Res Cardiol: Official J German Cardiac Soc 101(4):255–261. doi:10.1007/s00392-011-0387-z
Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen E-L (2009a) 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 5(8):1180–1185. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19855179
Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen E-L (2009b) Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol 452:143–164. doi:10.1016/S0076-6879(08)03610-0
Yonemura K, Hasegawa Y, Kimura K, Minematsu K, Yamaguchi T (2001) Diffusion-weighted MR imaging in a case of mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. AJNR Am J Neuroradiol 22(2):269–272. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11156767
Zeviani Massimo, Di Donato S (2004) Mitochondrial disorders. Brain: J Neurol 127(Pt 10):2153–2172. doi:10.1093/brain/awh259
Zeviani M, Moraes CT, DiMauro S, Nakase H, Bonilla E, Schon EA, Rowland LP (1988) Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 38(9):1339–1346. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3412580
Zhou C, Zhong W, Zhou J, Sheng F, Fang Z, Wei Y, Chen Y et al (2012) Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 8(8):1215–1226. doi:10.4161/auto.20284
Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, Cuthbert AP et al (1998) SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet 20(4):337–343. doi:10.1038/3804
Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M et al (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36(5):449–451. doi:10.1038/ng1341
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Ong, SB. (2014). Imaging of Mitochondrial Disorders: A Review. In: Advances in Medical Diagnostic Technology. Lecture Notes in Bioengineering. Springer, Singapore. https://doi.org/10.1007/978-981-4585-72-9_5
Download citation
DOI: https://doi.org/10.1007/978-981-4585-72-9_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-4585-71-2
Online ISBN: 978-981-4585-72-9
eBook Packages: EngineeringEngineering (R0)