
Abstract
Neuroblastoma is the most common extracranial solid tumor in children. Although low-risk neuroblastoma is mostly curable, the outcome of high-risk neuroblastoma, which comprises ~50% of neuroblastoma, is dismal. Abundant expression of disialoganglioside GD2 antigens is a feature of neuroblastoma. The success of immunotherapy targeting the GD2 antigen with dinutuximab has fueled the interest in developing improved immunotherapeutics including humanized anti-GD2 antibodies, GD2 mimotopes, cytokine-fused GD2-specific antibodies, GD2-specific chimeric antigen receptor T/NKT cells, GD2 vaccine, anti-GD2 idiotype monoclonal antibody, and anti-O-acetyl GD2 antibody. Advance in these active or passive cancer immunotherapies hold promises to further improve the outcome of neuroblastoma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Neuroblastoma is the most common malignancy in children aged under 1 year. It is a neuroendocrine tumor that originates from embryonal neural crest tissue. The common lesions of primary tumors are the adrenal medulla, neck, chest, and pelvis. About 8–10% of all childhood cancer cases are neuroblastoma. The incidence of neuroblastoma is 10.54 cases per million young children (<15 years old) per year. According to the International Neuroblastoma Risk Group classification system based on clinical and molecular features, the high-risk group, which represents about 50% of neuroblastoma, has a poor prognosis (Cohn et al. 2009). Genetic alterations are frequent in neuroblastoma. MYCN amplification, ranging from 100 kb to >1 Mb (Amler and Schwab 1989), is associated with the rapid progression of neuroblastoma (Seeger et al. 1985). ALK with gain-of-function mutations (Mosse et al. 2008) and paired mesoderm homeobox protein 2B (PHOX2B) with loss-of-function mutations (Trochet et al. 2004) are two predisposing factors for familial neuroblastoma. Lin-28 homologue B (LIN28B), which can suppress let-7 to increase MYCN expression, is an independent risk factor for high-risk neuroblastoma (Molenaar et al. 2012). On the other hand, altered glycosylation is a hallmark during tumorigenesis (Reis et al. 2010). GD2 is a tumor-associated carbohydrate antigen abundantly expressed on the surface of neuroblastoma cells with ~107 GD2 molecules per neuroblastoma cell (Wu et al. 1986), but limited expression in normal tissues, making it an attractive target for the development of immunotherapies against neuroblastoma.
Disialoganglioside GD2 is a b-series ganglioside consisting of GalNAcβ1-4(NeuAcα2-8NeuAcα2-3)Galβ1-4Glcβ1-1ceramide. Several enzymes are involved in the synthesis of GD2, including galactosyltransferase I, GM3 synthase, GD3 synthase, and GM2/GD2 synthase (Furukawa et al. 2002; Kolter et al. 2002). The biological functions of GD2 have not been fully explored. GD2 ceramide inhibited proliferation and induced apoptosis of T cells (Ladisch et al. 1992; Biswas et al. 2009). Recently, Siglect-7 has been identified to be a ligand for GD2 (Theruvath et al. 2022). Upon binding to GD2, Siglect-7 might suppress immune cell activity through its cytoplasmic immunoreceptor-tyrosine-based inhibitory motif domain. In normal tissues, GD2 expression is weak and restricted to the central nervous system, peripheral pain fibers, and skin melanocytes (Svennerholm et al. 1994; Yuki et al. 1997). In contrast, GD2 is overexpressed in a variety of cancers that include neuroblastoma, small cell lung cancer, melanoma, glioma, osteosarcoma, rhabdomyosarcoma, and Ewing sarcoma (Schulz et al. 1984; Heiner et al. 1987; Tsuchida et al. 1987; Kailayangiri et al. 2012). In addition, expression of GD2 has been reported on cancer stem cells of breast cancer (Battula et al. 2012; Liang et al. 2013) and malignant phyllodes tumors of the breast (Lin et al. 2014). Thus, GD2 is an ideal target for cancer immunotherapy.
Prior to 2015, the standard therapy for high-risk neuroblastoma is chemotherapy, surgical resection, myeloablative therapy, autologous hematopoietic stem cell transplantation, and isotretinoin. Addition of immunotherapy with anti-GD2 (dinutuximab), granulocyte-macrophage colony-stimulating factor (GM-CSF), and interleukin-2 (IL-2) to maintenance phase of treatment significantly improved the 2-year event-free survival from 46% to 66% (Yu et al. 2010), leading to the regulatory approval of dinutuximab in 2015. Thus, immunotherapy with dinutuximab has since been considered as the standard of care for high-risk neuroblastoma. In this chapter, we will focus on the immunotherapeutic strategies including immune checkpoint blockade, adoptive cellular therapies, antibody therapies, and cancer vaccines for further improvement of the outcome of neuroblastoma.
2 Antibody Therapies
Several GD2-specific antibodies have been investigated for the treatment of neuroblastoma, such as murine monoclonal antibody, monoclonal antibody fused with cytokine, chimeric monoclonal antibody, humanized monoclonal antibody, and bispecific antibody.
2.1 Murine Anti-GD2 Antibodies
Monoclonal antibody (mAb) 14G2a (Fig. 1a) is an IgG isotype switch variant of a GD2-specific murine IgG3 14.18 (Mujoo et al. 1989). The mAb 14G2a could change morphology and increase apoptosis of GD2-positive mouse lymphoma EL4 cells (Doronin et al. 2014). Similarly, mAb 14G2a activated caspase 3 to induce apoptosis of human neuroblastoma IMR-32 cells (Kowalczyk et al. 2009). Moreover, mAb 14G2a decreased metalloproteinase-2 to inhibit invasion of SJSA-1, MG-62, and Saos-2 cells (Liu et al. 2014). Besides, many studies showed that mAb 14G2a could inhibit p70S6, Akt, 4E-BP1, mTOR, MYCN, Aurora kinases, an inhibitor of DNA binding 1, and T cell leukemia homeobox 2, and increase p53, PHLDA1, JUN, Ras association RalGDS/AF-6 domain family member 6, and supervillain (Wierzbicki et al. 2008; Horwacik et al. 2013, 2015; Durbas et al. 2015).

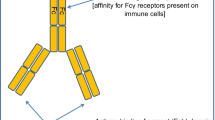
Various GD2-specific antibodies and CAR constructs. (a) Various GD2-specific antibodies are shown: 3F8, 14G2a, chimeric 14.18 (ch14.18), ch14.18-IL-2, hu14.18K322A, hu14.18-IL-2, Surek (anti-GD2 x anti-mouse-CD3 trifunctional bispecific antibody), 5F11 BsAb (anti-GD2 x anti-CD3 bispecific antibody), and 5F11-HDD BsAb. (HDD: dimerization domain of human hepatocyte nuclear factor 1α). (b) Various GD2-specific CAR constructs are shown: GD2 CAR, GD2-BBz CAR, and 4SCAR-GD2 for T cells and G28BBz CAR and GD2.28z.IL-15 CAR for NKT cells. (BB: 4-1BB, z: CD3 zeta signaling domain, 28: CD28, TM: transmembrane domain, ED: endodomain)
Another murine anti-GD2 IgG3 mAb 3F8 (Fig. 1a) has been extensively investigated by Cheng’s group. It altered morphology of SH-SY5Y-TrkB cells by Src-dependent activation of N-methyl-D-aspartate receptor signaling (Tong et al. 2015). The mAb 3F8 also activated caspases 3, 7, and 8 and inhibited survivin and AKT, leading to apoptosis of human melanoma HTB63 cells (Tsao et al. 2015).
The mAb 14G2a not only induced apoptosis of cancer cells but also killed cancer cells by complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC) (Mujoo et al. 1989). Moreover, mAb 14G2a suppressed the growth of M21 xenografts in athymic nude mice (Mueller et al. 1990). Phase I clinical trials of mAb 14G2a was conducted in patients with neuroblastoma or osteosarcoma. The dosage of mAb 14G2a was escalated up to 500 mg/m2/course. Infusion of mAb 14G2a caused pain, hypotension, fever, urticarial, hypertension, tachycardia, and hyponatremia (Saleh et al. 1992; Murray et al. 1994; Uttenreuther-Fischer et al. 1995a). The pain was attributable to the binding of mAb 14G2a to peripheral nerve fibers, which express GD2 (Svennerholm et al. 1994). Some patients with partial or mixed responses were observed in these early phase trials (Saleh et al. 1992; Murray et al. 1994).
In a phase I study, mAb 3F8 had similar side effects as mAb 14G2a (Cheung et al. 1987). A phase II clinical trial of mAb 3F8 showed long-term survival (79–130+ months) of three patients (3/16) with stage 4 neuroblastoma (Cheung et al. 1998). Other phase II studies showed 62% 5-year EFS for stage 4 patients in the first remission who received mAb 3F8 + subcutaneous GM-CSF + cis-retinoic acid (Cheung et al. 2012a; Cheung et al. 2014). In these studies, patients with the FCGR2A (R/R) genotype had a better outcome (Cheung et al. 2006). 131I-labeled mAb 3F8 showed anticancer efficacy in human neuroblastoma xenografts in mice (Cheung et al. 1986). A phase I trial of 131I-labeled mAb 3F8 given by intrathecal injection was conducted in 3 patients with recurrent metastatic central nervous system neuroblastoma. Extended survival (43, 41, and 33 months) of these patients was observed (Kramer et al. 2010). However, 131I-labeled mAb 3F8 at dose levels of 4 mCi/kg/day × 5 days resulted in primary hypothyroidism (Bhandari et al. 2010). Thus, to avoid potential adverse effects, protection of the thyroid gland will be needed for therapy of human neuroblastoma with 131I-labeled mAb 3F8.
2.2 Chimeric Anti-GD2 Monoclonal Antibody
To reduce the induction of human antimouse antibodies, a chimeric mAb ch14.18 (Fig. 1a) was generated by fusing the variable regions of mAb 14G2a with constant regions of human IgG1-k (Gillies et al. 1989). The mAb ch14.18 triggered CDC and ADCC reactions to kill neuroblastoma cells in vitro and suppressed experimental liver metastasis in an NXS2 neuroblastoma animal model in vivo (Barker et al. 1991; Zeng et al. 2005). Moreover, the mAb ch14.18 displays 50–100-fold greater ADCC activity than mAb 14G2a (Mueller et al. 1990). In phase I studies, when compared to mAb 14G2a, mAb ch14.18 has a longer half-life (66.6 ± 27.4 h vs. 18.3 ± 11.8 h) (Handgretinger et al. 1995; Uttenreuther-Fischer et al. 1995a, b) but similar toxicity profiles (Handgretinger et al. 1995; Yu et al. 1998). The German Collaborative Neuroblastoma Study NB97 showed mydriasis and accommodation deficiency in 10/85 patients with high-risk neuroblastoma who received mAb ch14.18 treatment, but these ocular symptoms were reversible with time (Kremens et al. 2002). Notably, some patients with refractory/relapsed neuroblastoma in the phase I studies of mAb ch14.18 showed complete response (CR) and partial response (PR) (Handgretinger et al. 1995; Yu et al. 1998). To further enhance the anticancer efficacy, mAb ch14.18 was combined with granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-2, both of which enhance ADCC (Hank et al. 1990; Tarr 1996). Of 17 refractory/recurrent neuroblastoma, 5 CRs and 3 stable diseases (SDs) were observed in a pilot study of mAb ch14.18 + GM-CSF (Yu et al. 1995). Subsequently, a phase II study of mAb ch14.18 + GM-CSF showed 2 CRs, 2 PRs, and 1 mixed response (Yu et al. 1997). Subsequently, the MTD of mAb ch14.18 in combination with GM-CSF and IL-2 was 25 mg/m2/d for 4 days when administered following consolidation therapy and stem cell transplant (Gilman et al. 2009). The toxicity of the regimen of mAb ch14.18 + GM-CSF + IL-2 was manageable and reversible (Hank et al. 1990). Consequently, a randomized phase III trial (COG study ANBL0032) of mAb ch14.18 with GM-CSF, interleukin-2, and isotretinoin was conducted in children with high-risk neuroblastoma after completing stem-cell transplantation and radiotherapy. Two-year event-free survival and overall survival rates were superior in patients treated with immunotherapy, that is, ch14.18 (dinutuximab) + GM-CSF + IL-2 + isotretinoin, than those treated with isotretinoin only (66 ± 5% vs. 46 ± 5%, P = 0.01 and 86 ± 4% vs. 75 ± 5%, P = 0.02, respectively) (Yu et al. 2010). Based on this data, US FDA and European medicines agency approved dinutuximab for the treatment of high-risk neuroblastoma in 2015. Since then, treatment with dinutuximab has become a standard of care for high-risk neuroblastoma. This is the first successful immunotherapy targeting a tumor-associated glycolipid antigen. Recently, a long-term follow-up (median 9.97 years) of the COG ANBL0032 study showed that mAb ch14.18 + GM-CSF + IL-2 + isotretinoin significantly improved the 5-year EFS (57% vs. 46%) and OS (73% vs. 57%) of patients with high-risk neuroblastoma (Yu et al. 2021, 2022), although the benefits have decreased due to late relapses. Notably, immune correlative analyses revealed that patients with high affinity FCGR3A genotype, and KIR2DL2/C1 or KIR3DL1/HLA-B-Bw4-T80 genotypes had better outcome with anti-GD2 immunotherapy (Erbe et al. 2017, 2018; Desai et al. 2022).
To improve the efficacy of dinutuximab by combining with chemotherapy, COG conducted a randomized phase II selection design trial in patients with relapsed/refractory neuroblastoma. Eligible patients were randomly assigned to irinotecan and temozolomide plus either temsirolimus or dinutuximab + GM-CSF. Irinotecan–temozolomide–dinutuximab-GMCSF (I/T/DIN/GM-CSF) showed superior antitumor activity with 9/17 (53%) objective responses, versus 1/18 (6%) patients treated with I/T/temsirolimus (Mody et al. 2017). The trial was expanded to recruit additional 36 patients nonrandomly assigned to I/T/DIN/GM-CSF. In total, 22/53 patients showed objective responses (41·5%) (Mody et al. 2020). These findings provided the rationales for the ongoing development of a COG phase III study of combining dinutuximab with induction chemotherapy in high-risk neuroblastoma.
In view of the limited access of mAb ch14.18 produced by murine myeloma SP2/0 cells in Europe, the International Society of Paediatric Oncology European Neuroblastoma Group (SIOPEN) uses mAb ch14.18 generated by Chinese hamster ovary cells, designated as mAb ch14.18/CHO (dinutuximab beta) for clinical investigations. As compared to mAb ch14.18/SP2/0, similar toxicology and comparable pharmacokinetics were observed in phase I bridging study of mAb ch14.18/CHO in patients with neuroblastoma (Ladenstein et al. 2013). Subsequently, a randomized SIOPEN study of mAb ch14.18/CHO with or without IL2 showed similar efficacy for high-risk neuroblastoma (Ladenstein et al. 2018), but the combination of IL-2 with mAb ch14.18/CHO resulted in more toxicities than mAb ch14.18/CHO alone. In 2017, mAb ch14.18/CHO received regulatory approval by the European Medicines Agency, and designated as Dinutuximab-beta.
2.3 Humanized Anti-GD2 Antibody
Humanization of murine antibodies can reduce immunogenicity, enhance effector function, and increase half-life in the serum. A humanized mAb 14.18 (hu14.18) antibody was generated by CDR grafting of mAb ch14.18 V regions onto variable regions of human IgG1 heavy and kappa light chains (Metelitsa et al. 2002). In addition, a point mutation K322A at the Fc region was introduced to reduce complement activation, which causes neuropathic pain induced by fixation of C5a complement on anti-GD2-binding neuron (Sorkin et al. 2010). Furthermore, YB2/0 rat myeloma cells, which have low fucosylation activity leading to increased ADCC (Niwa et al. 2004), were used to produce the mAb hu14.18K322A (Fig. 1a). Indeed, the mAb hu14.18K322A displayed ADCC activity but induced negligible CDC and less allodynia than mAb ch14.18 in a rat model (Sorkin et al. 2010). A phase I clinical trial of mAb hu14.18K322A showed that the maximum tolerated dose was 60 mg/m2/d for 4 days (Navid et al. 2014). Reversible ocular toxicities were seen in patients treated with mAb hu14.18K322A (Tse et al. 2015). A phase II study of mAb hu14.18K322A in combination with 6 courses of chemotherapy was conducted in patients with newly diagnosed high-risk neuroblastoma. After the first two chemo-immunotherapy cycles, an impressive 67% PR or better responses were noted in 42/63 patients (Furman et al. 2019). At the end-of-induction 60 of 62 patients (97%) had a partial response or better. Updated results of the induction chemotherapy with hu14.18K322A have demonstrated that 3-year EFS and OS were 73.7% and 86%, respectively (Furman et al. 2022). In addition, treatment of children with recurrent/refractory neuroblastoma with hu14.18K322A in combination with chemotherapeutic agents and haploidentical NK cell therapy resulted in 3 CRs, 1 very good partial response, 4 PRs, and 5 stable diseases (Federico et al. 2017).
In 2012, a humanized mAb 3F8 (hu3F8) was generated, and its binding activity to GD2 was similar to murine mAb 3F8. When compared to murine mAb 3F8, mAb hu3F8 had more potent ADCC but lower CDC activity (Cheung et al. 2012a, b). Based on in silico assisted design, a D32H (located at the L-CDR1) mutation was introduced in mAb hu3F8 to enhance the binding affinity to GD2. Although the binding affinity and ADCC of mAb hu3F8D32H were increased in vitro, its antitumor efficacy in IMR-32-bearing mice was not further improved (Zhao et al. 2015). A phase I trial of mAb hu3F8 in combination with GM-CSF for patients with resistant neuroblastoma demonstrated reversible neuropathic pain, low immunogenicity, and prolonged progression-free survival (Cheung et al. 2017; Kushner et al. 2018). Two single-arm, open-label trials (NCT03363373 and NCT01757626) were conducted to evaluate the antitumor efficacy of mAb hu3F8 in patients with relapsed/refractory neuroblastoma in the bone or bone marrow. The overall response rate for NCT03363373 and NCT01757626 was 45% and 34%, respectively (FDA 2020). These findings led to the FDA approval for mAb hu3F8 (naxitamab) for the treatment of relapsed/refractory high-risk neuroblastoma in 2020.
2.4 Cytokine-Conjugated GD2-Specific Antibodies
Another strategy to enhance anticancer efficacy is to link an antibody with cytokine to generate immunocytokine fusion protein, thereby inducing high concentration of cytokine in the tumor microenvironment to stimulate cellular immune responses against cancer cells. A fusion protein of hu14.18 and IL-2, hu14.18-IL2 (Fig. 1a), prolonged survival of NXS2-bearing mice with the involvement of NK cells and T cells (Neal et al. 2004a, b). Furthermore, upon binding to tumor cells, hu14.18-IL2 remained on the surface of tumor cells for a prolonged time, leading to the recruitment of NK cells to kill tumor cells (Buhtoiarov et al. 2011). In a phase I trial of patients with recurrent/refractory neuroblastoma, MTD of hu14.18-IL2 (EMD 273063) was 12 mg/m2/day with expected toxicities similar to the combination of anti-GD2 and IL-2, and antitumor activity was observed in three patients (Osenga et al. 2006). In a phase II study, hu14.18-IL2 resulted in 5 CR (5/23) for patients with disease evaluable by 123I-metaiodobenzylguanidine (MIBG) imaging and/or bone marrow histology, while patients with measurable disease had no responses (Shusterman et al. 2010). Moreover, the mismatch between KIR and KIR-ligand was associated with better clinical response to hu14.18-IL2 therapy (Delgado et al. 2010).
2.5 Bispecific Antibody
It is an attractive therapeutic approach to combine fragments of two monoclonal antibodies to make a bispecific antibody that binds to two different antigens. Many bispecific antibody formats against GD2 have been generated and evaluated in vitro and in vivo. Generated by electrofusion of hybridoma cells, a trifunctional bispecific antibody TRBs07 was shown to redirect T cells to kill GD2-expressing melanoma cell lines (Ruf et al. 2004). Surek (anti-GD2 17A2 x anti-mouse CD3 Me361 with an intact Fc) (Fig. 1a), a trifunctional bispecific antibody generated by the TRION antibody platform technology, could not only prolong the survival of mice but also induce a long-term memory response to tumor cells (Eissler et al. 2012; Ruf et al. 2012). Interestingly, subcutaneous and intravenous injection of the Surek showed similar antitumor efficacy in mice; however, only subcutaneously injected Surek reduced CTLA-4 expression on CD4+ and CD8+ T cells (Deppisch et al. 2015). Subsequently, the anti-mouse CD3 in Surek was replaced with an antihuman CD3 to generate an Ektomun (anti-GD2 x anti-human CD3). Upon coculture of human neuroblastoma cells with PBMCs, Ektomun could activate effector cells to release proinflammatory cytokines and exert cytotoxic effects (Zirngibl et al. 2021). Another bispecific antibody anti-CD3 x anti-GD2 (3F8BiAb) generated by chemical heteroconjugation could redirect T cells to target neuroblastoma cells expressing GD2 (Yankelevich et al. 2012). A humanized hu3F8-BsAb showed GD2-specific cytotoxicity to neuroblastoma cells in vitro and anticancer efficacy in vivo (Xu et al. 2015). An MDX-260 BsAb prepared by crosslinking F(ab’) fragments of mAb 7A4 (anti-GD2) and mAb 22 (anti-FcγRI) using bis-maleimide could trigger neutrophils to kill tumor in vitro (Michon et al. 1995).
A 5F11-BsAb (Fig. 1a) has been generated by genetic engineering with a gene construct comprised of VH-VL of anti-GD2 mAb 5F11, VHS44C and VLA100C mutations to stabilize disulfide bond, (GGGGS)3 linker to link anti-huCD3 scFv, and an affinity maturation mutation VHP104Y. In IMR-32-bearing BALB/cA-Rag2−/− Il2rg−/− mice where human PBMCs were used as effector cells, the 5F11-BsAb effectively suppressed tumor growth (Cheng et al. 2015). However, renal clearance of 5F11-BsAb was rapid due to its small size. Thus, to prevent the rapid renal clearance and enhance the potency of 5F11-BsAb, a dimerization domain (residues 1-32) of human hepatocyte nuclear factor 1α (HNF1α) was added to the C-terminus of the 5F11-BsAb to generate 5F11-HDD-BsAb (Fig. 1a). The HNF1α dimerization domain could increase molecular size and the avidity of 5F11-BsAb, without introducing immunogenicity. Surface plasmon resonance assay showed that 5F11-HDD-BsAb had a better affinity to GD2 than 5F11-BsAb, while both had a similar binding affinity to CD3. Moreover, the 5F11-HDD-BsAb significantly reduced tumor growth and prolonged survival in the IMR-32-bearing animal model. Notably, serum pharmacokinetic analysis revealed that 5F11-HDD-BsAb displayed a significantly longer half-life than 5F11-BsAb (Ahmed et al. 2015).
3 Adoptive Cellular Therapies
Adoptive cellular therapy (ACT) is a rapidly emerging immunotherapy approach, fueled by the FDA approval of two CAR T-cell therapies in 2017, one for the treatment of children with acute lymphoblastic leukemia (ALL) and the other for adults with advanced lymphomas (Mullard 2021). CAR is a synthetic receptor that combines a tumor-antigen-specific single-chain variable fragment with a transmembrane domain and an intracellular signaling domain (Rafiq et al. 2020). Upon binding to cancer cells by single-chain variable fragment, the intracellular signaling domain of CAR can transduce a signal to activate CAR T cells. Thus, CAR T cells can eliminate cancer cells in a major histocompatibility complex class I–independent fashion. Several GD2-directed CAR-T therapeutics are under evaluation in clinical trials. Epstein-Barr virus-specific T lymphocytes expressing a CAR specific for the GD2 (Fig. 1b, GD2 CAR T) have been generated and used for the treatment of patients with high-risk neuroblastoma. Tumor necrosis or complete remission was observed in 2 and 1 patients, respectively, at 6 weeks after treatment (Pule et al. 2008). Moreover, a long-term follow-up showed that the patients with persistence of GD2-CAR-T cells (>6 weeks) had a prolonged time to progression (Louis et al. 2011). Furthermore, tumor cells infected with oncolytic viruses to express RANTES and IL-15 ectopically could enhance the antitumor function of the GD2-CAR-T cells (Nishio and Dotti 2015). Another anti-GD2-BB-ζ CAR T cells (Fig. 1b), which comprised of scFv of mAb 126 (anti-GD2 IgM), the transmembrane domain of human CD8a hinge, human 4-1BB, and signaling domain of human CD3-ζ, killed neuroblastoma cells in vitro and suppressed tumor growth SH-SY5Y-bearing NOD.scid mice (Prapa et al. 2015). In a phase I clinical trial, six patients with refractory/recurrent neuroblastoma showed stable disease 6 months after treatment with 4SCAR-GD2 T cells (Fig. 1b), which contained intracellular signaling domain of CD28/4-1BB/CD3ζ-iCasp9, with an inducible caspase 9 gene in the anti-GD2-CAR construct (Yu et al. 2021, 2022). Adverse events associated with the 4SCARGD2 T-cell therapy, which included cytokine release syndrome and neuropathic pain, are manageable (Yu et al. 2021, 2022). In addition, the frequency of MDSC in human peripheral blood is a negative predictor of response to GD2-CAR-T (14G2a scFv-CD28-4-1BB-CD3ζ) cell therapy (Tumino et al. 2021).
In addition to CAR-T cells, different forms of ACT are being actively pursued. Natural killer T (NKT) cells, which are a sublineage of T cells with Vα24 invariant T cell receptors, play a role in neuroblastoma (Metelitsa et al. 2004; Song et al. 2007; Liao et al. 2021). A G28BBz-CAR construct, which consisted of mAb 14G2a scFv, CD28 transmembrane domain, CD28 endodomain, 4-1BB endodomain, and ζ chain endodomain (Fig. 1b), was used to transfect primary human NKT cells to generate CAR.GD2 NKT cells. The CAR.GD2 NKT cells could infiltrate into the tumor site to exhibit potent cytotoxic activity (Heczey et al. 2014). Another CAR.GD2 NKT is generated using CAR construct (GD2.28z.15), which consisted of mAb 14G2a scFv, CD8 hinge and transmembrane regions, CD28 endodomain, CD3ζ signaling domain, 2A sequence of the equine rhinitis A virus, and IL-15 (Fig. 1b). It showed superior antitumor activity in CHLA-255 neuroblastoma-bearing mice (Xu et al. 2019). Thus, a first-in-human phase I trial of GD2.28z.15 CAR NKTs for children with relapsed/refractory neuroblastoma was conducted. An interim analysis of the first 3 patients showed that the GD2.28z.15 CAR NKTs could expand in vivo and migrate to tumor sites, and one patient had a partial response (Heczey et al. 2020). Thus, GD2-CAR T/NKT cell therapy will likely bring hope for the treatment of neuroblastoma in the near future. Nevertheless, it should be cautioned that it is still early days for CAR T cells and other forms of ACT. Whether they will ever be as effective against solid tumors as hematological malignancies remains to be demonstrated.
4 Cancer Vaccine
4.1 Racotumomab Vaccine
The majority (85%) of neuroblastoma expressed N-glycolyl GM3 (NeuGcGM3), regardless of NMYC amplification (Scursoni et al. 2011). NeuGcGM3 was undetectable in healthy human tissues (Irie et al. 1998), making it a potential target for neuroblastoma therapy. An anti-idiotype murine mAb racotumomab (also known as 1E10) (Vazquez et al. 1998) against anti-NeuGcGM3 mAb P3 (Vazquez et al. 1995) has been generated. In B16 melanoma- and F3II mammary tumor-bearing animal models, vaccination with racotumomab decreased tumor growth (Vazquez et al. 2000). Treatment of neuroblastoma patients with racotumomab showed grade 1–2 toxicity at the injection site and induction of IgG and IgM against NeuGcGM3 in the phase I trial (Cacciavillano et al. 2015). A phase II of racotumomab for children with high-risk neuroblastoma is ongoing (NCT02998983). Importantly, progression-free and overall survival were significantly prolonged in patients with non-small cell lung cancer who were vaccinated with racotumomab in a randomized phase II/III trial (Alfonso et al. 2014). Racotumomab was approved for the treatment of patients with recurrent/advanced non-small cell lung cancer in Cuba and Argentina in 2013.
4.2 GD2 Vaccines
In general, carbohydrate or glycolipid molecular including GD2 are poor antigens for inducing immune responses. Several approaches have been applied to enhance the immunogenicity of GD2. For example, conjugation of GD2 to keyhole limpet hemocyanin (KLH) protein derived from the Megathura crenulato, which is a T-cell-dependent antigen, can stimulate the immune system to produce antibodies (Swaminathan et al. 2014). The use of a potent immunostimulatory adjuvant, such as QS21 isolated from the Quillaja saponaria, can activate dendritic cells and CD169+ macrophages to elicit strong immune responses. Another adjuvant, β-glycans, which are yeast cell wall carbohydrates, can activate immune cells and enhance ADCC (Cheung and Modak 2002; Dambuza and Brown 2015). GD2-KLH and GD3-KLH vaccines were produced by chemically crosslinking KLH to either GD2 or GD3. In a phase I trial, children with high-risk neuroblastoma in second or later remission were treated with GD2-KLH, GD3-KLH, a QS-21 equivalent OPT-821, and b-glucans. This regimen did not cause major toxicity and induced antibody responses against GD2/GD3 in 12 of 15 patients (Kushner et al. 2014). Subsequently, a phase II of the GD2-KLH/GD3-KLH/OPT-821/b-glucan in patients with high-risk neuroblastoma in remission was conducted. Multivariable analyses showed anti-GD2 IgG1 titer > = 150 ng/mL by week 8 could contribute to favorable PFS and OS (Cheung et al. 2021).
4.3 GD2 Mimotope
GD2 mimotopes are carbohydrate-mimetic peptides that stimulate immune response to induce GD2-specific T cells and antibodies. Using a decamer phage display library, Forster-Waldl et al. obtained GD2 peptide mimotopes that could compete for binding to mAb ch14.18 (Forster-Waldl et al. 2005). To enhance immunogenicity, the GD2 peptide mimotopes were linked to KLH. The GD2 mimotope-KLH vaccine could elicit IgG antibodies to GD2 (Riemer et al. 2006). Another GD2 mimotope 47-LDA obtained from X(15) phage display library had increased mimicry to GD2 by molecular modeling. The 47-LDA-encoded plasmid DNA vaccine could induce GD2 cross-reactive IgG antibody response in vivo (Bolesta et al. 2005). Moreover, the 47-LDA DNA vaccine could induce cytotoxic T lymphocyte response to prolong survival of syngeneic neuroblastoma NXS2-bearing mice (Wierzbicki et al. 2008). In addition, computer modeling of mAb ch14.18 was used to design a GD2 mimicry MD (C-DGGWLSKGSW-C). A pSA-MA vector encoding for T-cell helper epitope and GD2-mimicking MD peptide was generated and transfected into S. typhimurium SL7207. Induction of tumor protective immunity was observed in NXS2-bearing mice, which were immunized by oral gavage of SL7207 carrying the pSA-MA vectors. Notably, the vaccine could increase NK cell activity, IFN-γ secretion, CD4+ T cells, and CD8+ T cells (Fest et al. 2006). In summary, GD2 mimotopes can induce not only humoral response but also cellular response, providing strong evidence for further development.
4.4 Anti-GD2 Idiotype Monoclonal Antibody
Danish immunologist Niels Kaj Jerne conceived the idiotypic network theory that antibodies (termed “Ab2” antibody) react to antigen-specific antibodies (termed “Ab1” antibody) (Jerne 1974). Anti-idiotypic (anti-Id) antibodies, which belong to Ab2, can bind to the idiotope of another antibody. An idiotope is a unique region within the Fv region of an antibody. Thus, the antigen-binding domain of anti-Id antibodies can structurally resemble the antigen epitope. The mAb 14G2a was used to generate an anti-idiotype antibody mAb 1A7 (also known as TriGem), which mimics the GD2 antigen (Saleh et al. 1993). Besides, DNA vaccines encoding scFv of mAb 1A7 could induce immune sera against GD2 in vivo (Zeytin et al. 2000).
A phase I clinical trial required 47 patients with advanced melanoma to study the toxicity and immune response of TriGem. Minimal toxicity was observed, including mild fever, chills, and local reaction at the injection site. Induction of anti-GD2 IgG antibody (Ab3) was found in 40 patients (85.1%, 40/47). Notably, one complete response and 12 stable diseases occurred in 47 patients with TriGem therapy (Foon et al. 1998, 2000).
In addition, 31 patients with high-risk neuroblastoma in first or subsequent complete remission or very good partial remission after frontline therapy were enrolled in a clinical trial of mAb 1A7 vaccine mixed with QS21 (Yu et al. 2001). Adverse reactions were transient fever, chills, serum sickness, and local reactions. Anti-mAb 1A7 was detected in all patients. Immune sera from some patients lysed human neuroblastoma cells in the ADCC and CDC reactions. At a median follow-up of 6.8 years, 16/21 patients (76.1%) remained in first remission with no evidence of disease (Yu et al. 2001).
Other idiotypic antibodies mimicking GD2 were generated. The mAb 4B5 was generated by fusing murine myeloma cell line Ag8 with peripheral blood lymphocytes of a patient who received mAb 14G2a treatment (Saleh et al. 1993). A clinical trial of human mAb 4B5 + GM-CSF was conducted in patients with stage III/IV melanoma (NCT00004184), but no results have been published. In addition, the mAb 3F8 was used to generate rat anti-3F8-idiotypic antibodies C2D8, Idio-2, AIG4, C2H7, C4E4, and A2A6. These anti-Id antibodies could inhibit the binding of mAb 3F8 to GD2 and induce GD2-specific antibodies in mice (Cheung et al. 1993). So far, these anti-Id antibodies have not been tested in a clinical trial.
In summary, immunotherapy with anti-Id GD2 vaccines can induce humoral responses to GD2-expressing tumor cells. Its therapeutic efficacy awaits verification in randomized clinical trials.
5 O-Acetyl GD2-Specific Antibody
A 9(7)-O-acetyl transferase can add an O-acetyl ester to the external sialic acid of GD2 to form an O-acetyl GD2 (Sjoberg and Varki 1993). An O-acetyl GD2-specific mAb 8B6 was generated by screening antibodies that bind to alkali-labile ganglioside O-acetyl GD2 after immunizing A/J mice with LAN-1 cells (Cerato et al. 1997). The mAb 8B6 could bind to many human tumor tissues, including neuroblastoma, melanoma, small cell line cancer, and renal tumors, but not pain nerve fibers (Alvarez-Rueda et al. 2011). Moreover, a human-mouse chimeric mAb c.8B6 not only exhibited ADCC and CDC activity in vitro but also reduced neuroblastoma liver metastasis in vivo. Importantly, the mAb ch14.18-inducing allodynia in a rat model was not observed in animals treated with the mAb c.8B6 (Terme et al. 2014). In addition, the mAb 8B6 could induce apoptosis and cell cycle arrest to inhibit the growth of GD2-expressing cancer cells (Cochonneau et al. 2013), as well as targeting O-acetyl GD2+ breast cancer stem cells to suppress tumor growth in vivo (Cheng et al. 2021). These promising preclinical findings provide a strong rationale for developing the O-acetyl GD2-targeting immunotherapeutics.
6 Immune Checkpoint Inhibitors
Blockade of immune checkpoint molecules cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1) has shown remarkable therapeutic efficacy in some cancer patients. A few clinical trials of immune checkpoint inhibitors in neuroblastoma are now underway. Expression of programed death-ligand 1 (PD-L1) in clinical specimens of neuroblastoma has been reported to range from 18.9% to 35% in several studies (Zuo et al. 2020) (Saletta et al. 2017). Another study showed 25% (5/20) and 65% (13/20) of neuroblastoma at diagnosis and after 5-cycles of chemotherapy, respectively, expressed PD-L1 (Shirinbak et al. 2021). The clinical relevance of PD-L1 expression to the efficacy of anti-PD-1/PD-L1 in neuroblastoma remains to be determined. In preclinical studies, mAb PD-1 alone and mAb PD-L1 alone had no effect on tumor progression in syngeneic mice bearing with PD-L1-expressing Neuro2a and NXS2 neuroblastoma cells (Rigo et al. 2017). In a phase 1-2 trial of nivolumab (3 mg/kg) in children with measurable neuroblastoma, five of ten patients had stable disease (Davis et al. 2020). In another phase I clinical trial of ipilimumab, one neuroblastoma patient had stable disease for 2 months after single dose of ipilimumab at 10 mg/kg (Merchant et al. 2016). To improve the efficacy of ICI inhibitor in neuroblastoma, anti-GD2 in combination with PD-1 blockade was shown to significantly reduce tumor volume in A/J mice bearing neuroblastoma NXS2-HGW cells (Siebert et al. 2017). Based on this preclinical finding, two patients with refractory neuroblastoma were treated with the combination of Nivolumab and dinutuximab beta. One patient attained complete remission for 6 months, and the other showed complete disappearance of soft tissue lesions and regression of skeletal lesions (Ehlert et al. 2020). Another checkpoint molecule CD47, which is a potent “don’t eat me” signal to prevent phagocytosis by macrophages, is a potential therapeutic target for cancer immunotherapy. Combination of CD47 blockade and anti-GD2 showed significant antitumor efficacy in TH-MYCN syngeneic neuroblastoma model (Theruvath et al. 2022). In another study, inhibition of CSF-1R+ myeloid cells with a tyrosine kinase inhibitor BLZ945 was shown to improve the anticancer efficacy of blocking antibodies against both PD-1 and PD-L1 in spontaneous neuroblastoma TH-MYCN mice (Mao et al. 2016). These findings suggest that neuroblastoma patients may benefit from ICI in combination with immunotherapy or other immune-modulating agents.
7 Conclusions
Improved outcomes in high-risk neuroblastoma patient after postconsolidation immunotherapy with dinutuximab have fueled further interest in refining immunotherapy against neuroblastoma. Strategies that aim for ameliorating the toxicities of the dinutuximab, improving efficacy by combination with chemotherapy or ICI, or developing new immunotherapeutics targeting GD2, O-acetyl GD2, GD3, NeuGcGM3 are flourishing. These endeavors will likely further improve the outcome of neuroblastoma.
References
Ahmed M, Cheng M, Cheung IY, Cheung NK (2015) Human derived dimerization tag enhances tumor killing potency of a T-cell engaging bispecific antibody. Onco Targets Ther 4:e989776. https://doi.org/10.4161/2162402X.2014.989776
Alfonso S, Valdes-Zayas A, Santiesteban ER, Flores YI, Areces F, Hernandez M, Viada CE, Mendoza IC, Guerra PP, Garcia E, Ortiz RA, de la Torre AV, Cepeda M, Perez K, Chong E, Hernandez AM, Toledo D, Gonzalez Z, Mazorra Z, Crombet T, Perez R, Vazquez AM, Macias AE (2014) A randomized, multicenter, placebo-controlled clinical trial of racotumomab-alum vaccine as switch maintenance therapy in advanced non-small cell lung cancer patients. Clin Cancer Res 20:3660–3671. https://doi.org/10.1158/1078-0432.CCR-13-1674
Alvarez-Rueda N, Desselle A, Cochonneau D, Chaumette T, Clemenceau B, Leprieur S, Bougras G, Supiot S, Mussini JM, Barbet J, Saba J, Paris F, Aubry J, Birkle S (2011) A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PLoS One 6:e25220. https://doi.org/10.1371/journal.pone.0025220
Amler LC, Schwab M (1989) Amplified N-myc in human neuroblastoma cells is often arranged as clustered tandem repeats of differently recombined DNA. Mol Cell Biol 9:4903–4913. https://doi.org/10.1128/mcb.9.11.4903-4913.1989
Barker E, Mueller BM, Handgretinger R, Herter M, Yu AL, Reisfeld RA (1991) Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res 51:144–149
Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, Guerra R, Sahin AA, Marini FC, Hortobagyi G, Mani SA, Andreeff M (2012) Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest 122:2066–2078. https://doi.org/10.1172/JCI59735
Bhandari S, Cheung NK, Kushner BH, Kramer K, Modak S, Larson SM, Yeh S, Heller G, Sklar CA (2010) Hypothyroidism after 131I-monoclonal antibody treatment of neuroblastoma. Pediatr Blood Cancer 55:76–80. https://doi.org/10.1002/pbc.22452
Biswas S, Biswas K, Richmond A, Ko J, Ghosh S, Simmons M, Rayman P, Rini B, Gill I, Tannenbaum CS, Finke JH (2009) Elevated levels of select gangliosides in T cells from renal cell carcinoma patients is associated with T cell dysfunction. J Immunol 183:5050–5058. https://doi.org/10.4049/jimmunol.0900259
Bolesta E, Kowalczyk A, Wierzbicki A, Rotkiewicz P, Bambach B, Tsao CY, Horwacik I, Kolinski A, Rokita H, Brecher M, Wang X, Ferrone S, Kozbor D (2005) DNA vaccine expressing the mimotope of GD2 ganglioside induces protective GD2 cross-reactive antibody responses. Cancer Res 65:3410–3418. https://doi.org/10.1158/0008-5472.CAN-04-2164
Buhtoiarov IN, Neal ZC, Gan J, Buhtoiarova TN, Patankar MS, Gubbels JA, Hank JA, Yamane B, Rakhmilevich AL, Reisfeld RA, Gillies SD, Sondel PM (2011) Differential internalization of hu14.18-IL2 immunocytokine by NK and tumor cell: impact on conjugation, cytotoxicity, and targeting. J Leukoc Biol 89:625–638. https://doi.org/10.1189/jlb.0710422
Cacciavillano W, Sampor C, Venier C, Gabri MR, de Davila MT, Galluzzo ML, Guthmann MD, Fainboim L, Alonso DF, Chantada GL (2015) A phase I study of the anti-idiotype vaccine racotumomab in neuroblastoma and other pediatric refractory malignancies. Pediatr Blood Cancer 62:2120–2124. https://doi.org/10.1002/pbc.25631
Cerato E, Birkle S, Portoukalian J, Mezazigh A, Chatal JF, Aubry J (1997) Variable region gene segments of nine monoclonal antibodies specific to disialogangliosides (GD2, GD3) and their O-acetylated derivatives. Hybridoma 16:307–316
Cheng M, Ahmed M, Xu H, Cheung NK (2015) Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int J Cancer 136:476–486. https://doi.org/10.1002/ijc.29007
Cheng JY, Hung JT, Lin J, Lo FY, Huang JR, Chiou SP, Wang YH, Lin RJ, Wu JC, Yu J, Yu AL (2021) O-acetyl-GD2 as a therapeutic target for breast cancer stem cells. Front Immunol 12:791551. https://doi.org/10.3389/fimmu.2021.791551
Cheung NK, Modak S (2002) Oral (1-->3),(1-->4)-beta-D-glucan synergizes with antiganglioside GD2 monoclonal antibody 3F8 in the therapy of neuroblastoma. Clin Cancer Res 8:1217–1223
Cheung NK, Landmeier B, Neely J, Nelson AD, Abramowsky C, Ellery S, Adams RB, Miraldi F (1986) Complete tumor ablation with iodine 131-radiolabeled disialoganglioside GD2-specific monoclonal antibody against human neuroblastoma xenografted in nude mice. J Natl Cancer Inst 77:739–745
Cheung NK, Lazarus H, Miraldi FD, Abramowsky CR, Kallick S, Saarinen UM, Spitzer T, Strandjord SE, Coccia PF, Berger NA (1987) Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J Clin Oncol 5:1430–1440
Cheung NK, Canete A, Cheung IY, Ye JN, Liu C (1993) Disialoganglioside GD2 anti-idiotypic monoclonal antibodies. Int J Cancer 54:499–505
Cheung NK, Kushner BH, Yeh SD, Larson SM (1998) 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: a phase II study. Int J Oncol 12:1299–1306. https://doi.org/10.3892/ijo.12.6.1299
Cheung NK, Sowers R, Vickers AJ, Cheung IY, Kushner BH, Gorlick R (2006) FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J Clin Oncol 24:2885–2890. https://doi.org/10.1200/JCO.2005.04.6011
Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, Modak S (2012a) Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol 30:3264–3270. https://doi.org/10.1200/JCO.2011.41.3807
Cheung NK, Guo H, Hu J, Tassev DV, Cheung IY (2012b) Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. Onco Targets Ther 1:477–486
Cheung NK, Cheung IY, Kramer K, Modak S, Kuk D, Pandit-Taskar N, Chamberlain E, Ostrovnaya I, Kushner BH (2014) Key role for myeloid cells: phase II results of anti-G(D2) antibody 3F8 plus granulocyte-macrophage colony-stimulating factor for chemoresistant osteomedullary neuroblastoma. Int J Cancer 135:2199–2205. https://doi.org/10.1002/ijc.28851
Cheung IY, Kushner BH, Modak S, Basu EM, Roberts SS, Cheung NV (2017) Phase I trial of anti-GD2 monoclonal antibody hu3F8 plus GM-CSF: impact of body weight, immunogenicity and anti-GD2 response on pharmacokinetics and survival. Onco Targets Ther 6:e1358331. https://doi.org/10.1080/2162402X.2017.1358331
Cheung IY, Cheung NV, Modak S, Mauguen A, Feng Y, Basu E, Roberts SS, Ragupathi G, Kushner BH (2021) Survival impact of anti-GD2 antibody response in a phase II ganglioside vaccine trial among patients with high-risk neuroblastoma with prior disease progression. J Clin Oncol 39:215–226. https://doi.org/10.1200/JCO.20.01892
Cochonneau D, Terme M, Michaud A, Dorvillius M, Gautier N, Frikeche J, Alvarez-Rueda N, Bougras G, Aubry J, Paris F, Birkle S (2013) Cell cycle arrest and apoptosis induced by O-acetyl-GD2-specific monoclonal antibody 8B6 inhibits tumor growth in vitro and in vivo. Cancer Lett 333:194–204. https://doi.org/10.1016/j.canlet.2013.01.032
Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, Garaventa A, Castel V, Matthay KK, Force IT (2009) The international neuroblastoma risk group (INRG) classification system: an INRG task force report. J Clin Oncol 27:289–297. https://doi.org/10.1200/JCO.2008.16.6785
Dambuza IM, Brown GD (2015) C-type lectins in immunity: recent developments. Curr Opin Immunol 32:21–27. https://doi.org/10.1016/j.coi.2014.12.002
Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, Minard CG, Voss S, Berg SL, Weigel BJ, Mackall CL (2020) Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol 21:541–550. https://doi.org/10.1016/S1470-2045(20)30023-1
Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, Kim K, Shusterman S, Gillies SD, Reisfeld RA, Yang R, Gadbaw B, DeSantes KB, London WB, Seeger RC, Maris JM, Sondel PM (2010) Genotypes of NK cell KIR receptors, their ligands, and Fc-gamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res 70:9554–9561. https://doi.org/10.1158/0008-5472.CAN-10-2211
Deppisch N, Ruf P, Eissler N, Neff F, Buhmann R, Lindhofer H, Mocikat R (2015) Efficacy and tolerability of a GD2-directed trifunctional bispecific antibody in a preclinical model: subcutaneous administration is superior to intravenous delivery. Mol Cancer Ther 14:1877–1883. https://doi.org/10.1158/1535-7163.MCT-15-0156
Desai AV, Gilman AL, Ozkaynak MF, Naranjo A, London WB, Tenney SC, Diccianni M, Hank JA, Parisi MT, Shulkin BL, Smith M, Moscow JA, Shimada H, Matthay KK, Cohn SL, Maris JM, Bagatell R, Sondel PM, Park JR, Yu AL (2022) Outcomes following anti-GD2 antibody-based post-consolidation therapy after cessation of randomization on ANBL0032: a report from the children’s oncology group. J Clin Oncol
Doronin II, Vishnyakova PA, Kholodenko IV, Ponomarev ED, Ryazantsev DY, Molotkovskaya IM, Kholodenko RV (2014) Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer 14:295. https://doi.org/10.1186/1471-2407-14-295
Durbas M, Horwacik I, Boratyn E, Kamycka E, Rokita H (2015) GD2 ganglioside specific antibody treatment downregulates PI3K/Akt/mTOR signaling network in human neuroblastoma cell lines. Int J Oncol 47:1143–1159. https://doi.org/10.3892/ijo.2015.3070
Ehlert K, Hansjuergens I, Zinke A, Otto S, Siebert N, Henze G, Lode H (2020) Nivolumab and dinutuximab beta in two patients with refractory neuroblastoma. J Immunother Cancer 8(1):e000540. https://doi.org/10.1136/jitc-2020-000540
Eissler N, Ruf P, Mysliwietz J, Lindhofer H, Mocikat R (2012) Trifunctional bispecific antibodies induce tumor-specific T cells and elicit a vaccination effect. Cancer Res 72:3958–3966. https://doi.org/10.1158/0008-5472.CAN-12-0146
Erbe AK, Wang W, Reville PK, Carmichael L, Kim K, Mendonca EA, Song Y, Hank JA, London WB, Naranjo A, Hong F, Hogarty MD, Maris JM, Park JR, Ozkaynak MF, Miller JS, Gilman AL, Kahl B, Yu AL, Sondel PM (2017) HLA-Bw4-I-80 isoform differentially influences clinical outcome as compared to HLA-Bw4-T-80 and HLA-A-Bw4 isoforms in rituximab or dinutuximab-based cancer immunotherapy. Front Immunol 8:675. https://doi.org/10.3389/fimmu.2017.00675
Erbe AK, Wang W, Carmichael L, Kim K, Mendonca EA, Song Y, Hess D, Reville PK, London WB, Naranjo A, Hank JA, Diccianni MB, Reisfeld RA, Gillies SD, Matthay KK, Cohn SL, Hogarty MD, Maris JM, Park JR, Ozkaynak MF, Gilman AL, Yu AL, Sondel PM (2018) Neuroblastoma patients’ KIR and KIR-ligand genotypes influence clinical outcome for dinutuximab-based immunotherapy: a report from the children’s oncology group. Clin Cancer Res 24:189–196. https://doi.org/10.1158/1078-0432.CCR-17-1767
FDA (2020) FDA grants accelerated approval to naxitamab for high-risk neuroblastoma in bone or bone marrow Journal 2022:U.S. Food & Drug
Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, Meagher M, Shafer A, Ng CY, Leung W, Janssen WE, Wu J, Mao S, Brennan RC, Santana VM, Pappo AS, Furman WL (2017) A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin Cancer Res 23:6441–6449. https://doi.org/10.1158/1078-0432.CCR-17-0379
Fest S, Huebener N, Weixler S, Bleeke M, Zeng Y, Strandsby A, Volkmer-Engert R, Landgraf C, Gaedicke G, Riemer AB, Michalsky E, Jaeger IS, Preissner R, Forster-Wald E, Jensen-Jarolim E, Lode HN (2006) Characterization of GD2 peptide mimotope DNA vaccines effective against spontaneous neuroblastoma metastases. Cancer Res 66:10567–10575. https://doi.org/10.1158/0008-5472.CAN-06-1158
Foon KA, Sen G, Hutchins L, Kashala OL, Baral R, Banerjee M, Chakraborty M, Garrison J, Reisfeld RA, Bhattacharya-Chatterjee M (1998) Antibody responses in melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. Clin Cancer Res 4:1117–1124
Foon KA, Lutzky J, Baral RN, Yannelli JR, Hutchins L, Teitelbaum A, Kashala OL, Das R, Garrison J, Reisfeld RA, Bhattacharya-Chatterjee M (2000) Clinical and immune responses in advanced melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. J Clin Oncol 18:376–384
Forster-Waldl E, Riemer AB, Dehof AK, Neumann D, Bramswig K, Boltz-Nitulescu G, Pehamberger H, Zielinski CC, Scheiner O, Pollak A, Lode H, Jensen-Jarolim E (2005) Isolation and structural analysis of peptide mimotopes for the disialoganglioside GD2, a neuroblastoma tumor antigen. Mol Immunol 42:319–325. https://doi.org/10.1016/j.molimm.2004.07.011
Furman WL, Federico SM, McCarville MB, Shulkin BL, Davidoff AM, Krasin MJ, Sahr N, Sykes A, Wu J, Brennan RC, Bishop MW, Helmig S, Stewart E, Navid F, Triplett B, Santana VM, Bahrami A, Anthony G, Yu AL, Hank J, Gillies SD, Sondel PM, Leung WH, Pappo AS (2019) A phase II trial of Hu14.18K322A in combination with induction chemotherapy in children with newly diagnosed high-risk neuroblastoma. Clin Cancer Res 25:6320–6328. https://doi.org/10.1158/1078-0432.CCR-19-1452
Furman WL, McCarville B, Shulkin BL, Davidoff A, Krasin M, Hsu CW, Pan H, Wu J, Brennan R, Bishop MW, Helmig S, Stewart E, Navid F, Triplett B, Santana V, Santiago T, Hank JA, Gillies SD, Yu A, Sondel PM, Leung WH, Pappo A, Federico SM (2022) Improved outcome in children with newly diagnosed high-risk neuroblastoma treated with chemoimmunotherapy: updated results of a phase II study using hu14.18K322A. J Clin Oncol 40:335–344. https://doi.org/10.1200/JCO.21.01375
Furukawa K, Takamiya K, Furukawa K (2002) Beta1,4-N-acetylgalactosaminyltransferase--GM2/GD2 synthase: a key enzyme to control the synthesis of brain-enriched complex gangliosides. Biochim Biophys Acta 1573:356–362
Gillies SD, Lo KM, Wesolowski J (1989) High-level expression of chimeric antibodies using adapted cDNA variable region cassettes. J Immunol Methods 125:191–202
Gilman AL, Ozkaynak MF, Matthay KK, Krailo M, Yu AL, Gan J, Sternberg A, Hank JA, Seeger R, Reaman GH, Sondel PM (2009) Phase I study of ch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: a report from the children’s oncology group. J Clin Oncol 27:85–91. https://doi.org/10.1200/JCO.2006.10.3564
Handgretinger R, Anderson K, Lang P, Dopfer R, Klingebiel T, Schrappe M, Reuland P, Gillies SD, Reisfeld RA, Neithammer D (1995) A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur J Cancer 31A:261–267
Hank JA, Robinson RR, Surfus J, Mueller BM, Reisfeld RA, Cheung NK, Sondel PM (1990) Augmentation of antibody dependent cell mediated cytotoxicity following in vivo therapy with recombinant interleukin 2. Cancer Res 50:5234–5239
Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, Gao X, Guo L, Yvon E, Hicks J, Liu H, Dotti G, Metelitsa LS (2014) Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 124:2824–2833. https://doi.org/10.1182/blood-2013-11-541235
Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, Ghatwai N, Dakhova O, Liu B, Raveh-Sadka T, Chauvin-Fleurence CN, Xu X, Ngai H, Di Pierro EJ, Savoldo B, Dotti G, Metelitsa LS (2020) Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med 26:1686–1690. https://doi.org/10.1038/s41591-020-1074-2
Heiner JP, Miraldi F, Kallick S, Makley J, Neely J, Smith-Mensah WH, Cheung NK (1987) Localization of GD2-specific monoclonal antibody 3F8 in human osteosarcoma. Cancer Res 47:5377–5381
Horwacik I, Durbas M, Boratyn E, Wegrzyn P, Rokita H (2013) Targeting GD2 ganglioside and aurora A kinase as a dual strategy leading to cell death in cultures of human neuroblastoma cells. Cancer Lett 341:248–264. https://doi.org/10.1016/j.canlet.2013.08.018
Horwacik I, Durbas M, Boratyn E, Sawicka A, Wegrzyn P, Krzanik S, Gorka A, Drozniak J, Augustyniak E, Kowalczyk A, Rokita H (2015) Analysis of genes involved in response to doxorubicin and a GD2 ganglioside-specific 14G2a monoclonal antibody in IMR-32 human neuroblastoma cells. Acta Biochim Pol 62:423–433. https://doi.org/10.18388/abp.2015_1035
Irie A, Koyama S, Kozutsumi Y, Kawasaki T, Suzuki A (1998) The molecular basis for the absence of N-glycolylneuraminic acid in humans. J Biol Chem 273:15866–15871. https://doi.org/10.1074/jbc.273.25.15866
Jerne NK (1974) Towards a network theory of the immune system. Ann Immunol (Paris) 125C:373–389
Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, Titze U, Leuchte K, Landmeier S, Hotfilder M, Dirksen U, Hardes J, Gosheger G, Juergens H, Rossig C (2012) The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer 106:1123–1133. https://doi.org/10.1038/bjc.2012.57
Kolter T, Proia RL, Sandhoff K (2002) Combinatorial ganglioside biosynthesis. J Biol Chem 277:25859–25862. https://doi.org/10.1074/jbc.R200001200
Kowalczyk A, Gil M, Horwacik I, Odrowaz Z, Kozbor D, Rokita H (2009) The GD2-specific 14G2a monoclonal antibody induces apoptosis and enhances cytotoxicity of chemotherapeutic drugs in IMR-32 human neuroblastoma cells. Cancer Lett 281:171–182. https://doi.org/10.1016/j.canlet.2009.02.040
Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Smith-Jones P, Zanzonico P, Humm JL, Xu H, Wolden SL, Souweidane MM, Larson SM, Cheung NK (2010) Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neuro-Oncol 97:409–418. https://doi.org/10.1007/s11060-009-0038-7
Kremens B, Hero B, Esser J, Weinel P, Filger-Brillinger J, Fleischhack G, Graf N, Gruttner HP, Niemeyer C, Schulz A, Wickmann L, Berthold F (2002) Ocular symptoms in children treated with human-mouse chimeric anti-GD2 mAb ch14.18 for neuroblastoma. Cancer Immunol Immunother 51:107–110. https://doi.org/10.1007/s00262-001-0259-x
Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK (2014) Phase I trial of a bivalent gangliosides vaccine in combination with beta-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res 20:1375–1382. https://doi.org/10.1158/1078-0432.CCR-13-1012
Kushner BH, Cheung IY, Modak S, Basu EM, Roberts SS, Cheung NK (2018) Humanized 3F8 anti-GD2 monoclonal antibody dosing with granulocyte-macrophage Colony-stimulating factor in patients with resistant neuroblastoma: A phase 1 clinical trial. JAMA Oncol 4:1729–1735. https://doi.org/10.1001/jamaoncol.2018.4005
Ladenstein R, Weixler S, Baykan B, Bleeke M, Kunert R, Katinger D, Pribill I, Glander P, Bauer S, Pistoia V, Michon J, Garaventa A, Lode HN (2013) Ch14.18 antibody produced in CHO cells in relapsed or refractory stage 4 neuroblastoma patients: a SIOPEN phase 1 study. MAbs 5:801–809. https://doi.org/10.4161/mabs.25215
Ladenstein R, Potschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, Laureys G, Brock P, Michon JM, Owens C, Trahair T, Chan GCF, Ruud E, Schroeder H, Beck Popovic M, Schreier G, Loibner H, Ambros P, Holmes K, Castellani MR, Gaze MN, Garaventa A, Pearson ADJ, Lode HN (2018) Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol 19:1617–1629. https://doi.org/10.1016/S1470-2045(18)30578-3
Ladisch S, Becker H, Ulsh L (1992) Immunosuppression by human gangliosides: I. Relationship of carbohydrate structure to the inhibition of T cell responses. Biochim Biophys Acta 1125:180–188
Liang YJ, Ding Y, Levery SB, Lobaton M, Handa K, Hakomori SI (2013) Differential expression profiles of glycosphingolipids in human breast cancer stem cells vs. cancer non-stem cells. Proc Natl Acad Sci U S A 110:4968–4973. https://doi.org/10.1073/pnas.1302825110
Liao YM, Hung TH, Tung JK, Yu J, Hsu YL, Hung JT, Yu AL (2021) Low expression of IL-15 and NKT in tumor microenvironment predicts poor outcome of MYCN-non-amplified neuroblastoma. J Pers Med 11(2):122. https://doi.org/10.3390/jpm11020122
Lin JJ, Huang CS, Yu J, Liao GS, Lien HC, Hung JT, Lin RJ, Chou FP, Yeh KT, Yu AL (2014) Malignant phyllodes tumors display mesenchymal stem cell features and aldehyde dehydrogenase/disialoganglioside identify their tumor stem cells. Breast Cancer Res 16:R29. https://doi.org/10.1186/bcr3631
Liu B, Wu Y, Zhou Y, Peng D (2014) Endothelin A receptor antagonism enhances inhibitory effects of anti-ganglioside GD2 monoclonal antibody on invasiveness and viability of human osteosarcoma cells. PLoS One 9:e93576. https://doi.org/10.1371/journal.pone.0093576
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK (2011) Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118:6050–6056. https://doi.org/10.1182/blood-2011-05-354449
Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R (2016) Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin Cancer Res 22:3849–3859. https://doi.org/10.1158/1078-0432.CCR-15-1912
Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, Delbrook C, Lodish M, Bishop R, Wolchok JD, Streicher H, Mackall CL (2016) Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin Cancer Res 22:1364–1370. https://doi.org/10.1158/1078-0432.CCR-15-0491
Metelitsa LS, Gillies SD, Super M, Shimada H, Reynolds CP, Seeger RC (2002) Antidisialoganglioside/granulocyte macrophage-colony-stimulating factor fusion protein facilitates neutrophil antibody-dependent cellular cytotoxicity and depends on FcgammaRII (CD32) and Mac-1 (CD11b/CD18) for enhanced effector cell adhesion and azurophil granule exocytosis. Blood 99:4166–4173
Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, Groshen S, Wilson SB, Seeger RC (2004) Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med 199:1213–1221. https://doi.org/10.1084/jem.20031462
Michon J, Moutel S, Barbet J, Romet-Lemonne JL, Deo YM, Fridman WH, Teillaud JL (1995) In vitro killing of neuroblastoma cells by neutrophils derived from granulocyte colony-stimulating factor-treated cancer patients using an anti-disialoganglioside/anti-Fc gamma RI bispecific antibody. Blood 86:1124–1130
Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, Parisi MT, Servaes SE, Diccianni MB, Sondel PM, Bender JG, Maris JM, Park JR, Bagatell R (2017) Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol 18:946–957. https://doi.org/10.1016/S1470-2045(17)30355-8
Mody R, Yu AL, Naranjo A, Zhang FF, London WB, Shulkin BL, Parisi MT, Servaes SE, Diccianni MB, Hank JA, Felder M, Birstler J, Sondel PM, Asgharzadeh S, Glade-Bender J, Katzenstein H, Maris JM, Park JR, Bagatell R (2020) Irinotecan, temozolomide, and dinutuximab with GM-CSF in children with refractory or relapsed neuroblastoma: A report from the children’s oncology group. J Clin Oncol 38:2160–2169. https://doi.org/10.1200/JCO.20.00203
Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, Mestdagh P, van Sluis P, Valentijn LJ, van Nes J, Broekmans M, Haneveld F, Volckmann R, Bray I, Heukamp L, Sprussel A, Thor T, Kieckbusch K, Klein-Hitpass L, Fischer M, Vandesompele J, Schramm A, van Noesel MM, Varesio L, Speleman F, Eggert A, Stallings RL, Caron HN, Versteeg R, Schulte JH (2012) LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet 44:1199–1206. https://doi.org/10.1038/ng.2436
Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, Devoto M, Maris JM (2008) Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455:930–935. https://doi.org/10.1038/nature07261
Mueller BM, Romerdahl CA, Gillies SD, Reisfeld RA (1990) Enhancement of antibody-dependent cytotoxicity with a chimeric anti-GD2 antibody. J Immunol 144:1382–1386
Mujoo K, Kipps TJ, Yang HM, Cheresh DA, Wargalla U, Sander DJ, Reisfeld RA (1989) Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res 49:2857–2861
Mullard A (2021) FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discov 20:166. https://doi.org/10.1038/d41573-021-00031-9
Murray JL, Cunningham JE, Brewer H, Mujoo K, Zukiwski AA, Podoloff DA, Kasi LP, Bhadkamkar V, Fritsche HA, Benjamin RS et al (1994) Phase I trial of murine monoclonal antibody 14G2a administered by prolonged intravenous infusion in patients with neuroectodermal tumors. J Clin Oncol 12:184–193
Navid F, Sondel PM, Barfield R, Shulkin BL, Kaufman RA, Allay JA, Gan J, Hutson P, Seo S, Kim K, Goldberg J, Hank JA, Billups CA, Wu J, Furman WL, McGregor LM, Otto M, Gillies SD, Handgretinger R, Santana VM (2014) Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J Clin Oncol 32:1445–1452. https://doi.org/10.1200/JCO.2013.50.4423
Neal ZC, Imboden M, Rakhmilevich AL, Kim KM, Hank JA, Surfus J, Dixon JR, Lode HN, Reisfeld RA, Gillies SD, Sondel PM (2004a) NXS2 murine neuroblastomas express increased levels of MHC class I antigens upon recurrence following NK-dependent immunotherapy. Cancer Immunol Immunother 53:41–52. https://doi.org/10.1007/s00262-003-0435-2
Neal ZC, Yang JC, Rakhmilevich AL, Buhtoiarov IN, Lum HE, Imboden M, Hank JA, Lode HN, Reisfeld RA, Gillies SD, Sondel PM (2004b) Enhanced activity of hu14.18-IL2 immunocytokine against murine NXS2 neuroblastoma when combined with interleukin 2 therapy. Clin Cancer Res 10:4839–4847. https://doi.org/10.1158/1078-0432.CCR-03-0799
Nishio N, Dotti G (2015) Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Onco Targets Ther 4:e988098. https://doi.org/10.4161/21505594.2014.988098
Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, Matsushima K, Ueda R, Hanai N, Shitara K (2004) Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res 64:2127–2133. https://doi.org/10.1158/0008-5472.can-03-2068
Osenga KL, Hank JA, Albertini MR, Gan J, Sternberg AG, Eickhoff J, Seeger RC, Matthay KK, Reynolds CP, Twist C, Krailo M, Adamson PC, Reisfeld RA, Gillies SD, Sondel PM, Children’s Oncology Group (2006) A phase I clinical trial of the hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: a study of the children’s oncology group. Clin Cancer Res 12:1750–1759. https://doi.org/10.1158/1078-0432.CCR-05-2000
Prapa M, Caldrer S, Spano C, Bestagno M, Golinelli G, Grisendi G, Petrachi T, Conte P, Horwitz EM, Campana D, Paolucci P, Dominici M (2015) A novel anti-GD2/4-1BB chimeric antigen receptor triggers neuroblastoma cell killing. Oncotarget 6:24884–24894. https://doi.org/10.18632/oncotarget.4670
Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, Yvon E, Weiss HL, Liu H, Rooney CM, Heslop HE, Brenner MK (2008) Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14:1264–1270. https://doi.org/10.1038/nm.1882
Rafiq S, Hackett CS, Brentjens RJ (2020) Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol 17:147–167. https://doi.org/10.1038/s41571-019-0297-y
Reis CA, Osorio H, Silva L, Gomes C, David L (2010) Alterations in glycosylation as biomarkers for cancer detection. J Clin Pathol 63:322–329. https://doi.org/10.1136/jcp.2009.071035
Riemer AB, Forster-Waldl E, Bramswig KH, Pollak A, Zielinski CC, Pehamberger H, Lode HN, Scheiner O, Jensen-Jarolim E (2006) Induction of IgG antibodies against the GD2 carbohydrate tumor antigen by vaccination with peptide mimotopes. Eur J Immunol 36:1267–1274. https://doi.org/10.1002/eji.200535279
Rigo V, Emionite L, Daga A, Astigiano S, Corrias MV, Quintarelli C, Locatelli F, Ferrini S, Croce M (2017) Combined immunotherapy with anti-PDL-1/PD-1 and anti-CD4 antibodies cures syngeneic disseminated neuroblastoma. Sci Rep 7:14049. https://doi.org/10.1038/s41598-017-14417-6
Ruf P, Jager M, Ellwart J, Wosch S, Kusterer E, Lindhofer H (2004) Two new trifunctional antibodies for the therapy of human malignant melanoma. Int J Cancer 108:725–732. https://doi.org/10.1002/ijc.11630
Ruf P, Schafer B, Eissler N, Mocikat R, Hess J, Ploscher M, Wosch S, Suckstorff I, Zehetmeier C, Lindhofer H (2012) Ganglioside GD2-specific trifunctional surrogate antibody Surek demonstrates therapeutic activity in a mouse melanoma model. J Transl Med 10:219. https://doi.org/10.1186/1479-5876-10-219
Saleh MN, Khazaeli MB, Wheeler RH, Dropcho E, Liu T, Urist M, Miller DM, Lawson S, Dixon P, Russell CH et al (1992) Phase I trial of the murine monoclonal anti-GD2 antibody 14G2a in metastatic melanoma. Cancer Res 52:4342–4347
Saleh MN, Stapleton JD, Khazaeli MB, LoBuglio AF (1993) Generation of a human anti-idiotypic antibody that mimics the GD2 antigen. J Immunol 151:3390–3398
Saletta F, Vilain RE, Gupta AK, Nagabushan S, Yuksel A, Catchpoole D, Scolyer RA, Byrne JA, McCowage G (2017) Programmed death-ligand 1 expression in a large cohort of pediatric patients with solid tumor and association with clinicopathologic features in neuroblastoma. JCO Precis Oncol 1:1–12. https://doi.org/10.1200/PO.16.00049
Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA (1984) Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res 44:5914–5920
Scursoni AM, Galluzzo L, Camarero S, Lopez J, Lubieniecki F, Sampor C, Segatori VI, Gabri MR, Alonso DF, Chantada G, de Davila MT (2011) Detection of N-glycolyl GM3 ganglioside in neuroectodermal tumors by immunohistochemistry: an attractive vaccine target for aggressive pediatric cancer. Clin Dev Immunol 2011:245181. https://doi.org/10.1155/2011/245181
Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D (1985) Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 313:1111–1116. https://doi.org/10.1056/NEJM198510313131802
Shirinbak S, Chan RY, Shahani S, Muthugounder S, Kennedy R, Hung LT, Fernandez GE, Hadjidaniel MD, Moghimi B, Sheard MA, Epstein AL, Fabbri M, Shimada H, Asgharzadeh S (2021) Combined immune checkpoint blockade increases CD8+CD28+PD-1+ effector T cells and provides a therapeutic strategy for patients with neuroblastoma. Onco Targets Ther 10:1838140. https://doi.org/10.1080/2162402X.2020.1838140
Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, Reynolds CP, Kimball J, Albertini MR, Wagner B, Gan J, Eickhoff J, DeSantes KB, Cohn SL, Hecht T, Gadbaw B, Reisfeld RA, Maris JM, Sondel PM (2010) Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a children’s oncology group (COG) phase II study. J Clin Oncol 28:4969–4975. https://doi.org/10.1200/JCO.2009.27.8861
Siebert N, Zumpe M, Juttner M, Troschke-Meurer S, Lode HN (2017) PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD2 antibody ch14.18/CHO. Onco Targets Ther 6:e1343775. https://doi.org/10.1080/2162402X.2017.1343775
Sjoberg ER, Varki A (1993) Kinetic and spatial interrelationships between ganglioside glycosyltransferases and O-acetyltransferase(s) in human melanoma cells. J Biol Chem 268:10185–10196
Song L, Ara T, Wu HW, Woo CW, Reynolds CP, Seeger RC, DeClerck YA, Thiele CJ, Sposto R, Metelitsa LS (2007) Oncogene MYCN regulates localization of NKT cells to the site of disease in neuroblastoma. J Clin Invest 117:2702–2712. https://doi.org/10.1172/JCI30751
Sorkin LS, Otto M, Baldwin WM 3rd, Vail E, Gillies SD, Handgretinger R, Barfield RC, Ming Yu H, Yu AL (2010) Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain 149:135–142. https://doi.org/10.1016/j.pain.2010.01.024
Svennerholm L, Bostrom K, Fredman P, Jungbjer B, Lekman A, Mansson JE, Rynmark BM (1994) Gangliosides and allied glycosphingolipids in human peripheral nerve and spinal cord. Biochim Biophys Acta 1214:115–123
Swaminathan A, Lucas RM, Dear K, McMichael AJ (2014) Keyhole limpet haemocyanin - a model antigen for human immunotoxicological studies. Br J Clin Pharmacol 78:1135–1142. https://doi.org/10.1111/bcp.12422
Tarr PE (1996) Granulocyte-macrophage colony-stimulating factor and the immune system. Med Oncol 13:133–140. https://doi.org/10.1007/BF02990841
Terme M, Dorvillius M, Cochonneau D, Chaumette T, Xiao W, Diccianni MB, Barbet J, Yu AL, Paris F, Sorkin LS, Birkle S (2014) Chimeric antibody c.8B6 to O-acetyl-GD2 mediates the same efficient anti-neuroblastoma effects as therapeutic ch14.18 antibody to GD2 without antibody induced allodynia. PLoS One 9:e87210. https://doi.org/10.1371/journal.pone.0087210
Theruvath J, Menard M, Smith BAH, Linde MH, Coles GL, Dalton GN, Wu W, Kiru L, Delaidelli A, Sotillo E, Silberstein JL, Geraghty AC, Banuelos A, Radosevich MT, Dhingra S, Heitzeneder S, Tousley A, Lattin J, Xu P, Huang J, Nasholm N, He A, Kuo TC, Sangalang ERB, Pons J, Barkal A, Brewer RE, Marjon KD, Vilches-Moure JG, Marshall PL, Fernandes R, Monje M, Cochran JR, Sorensen PH, Daldrup-Link HE, Weissman IL, Sage J, Majeti R, Bertozzi CR, Weiss WA, Mackall CL, Majzner RG (2022) Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat Med 28:333–344. https://doi.org/10.1038/s41591-021-01625-x
Tong W, Maira M, Gagnon M, Saragovi HU (2015) Ligands binding to cell surface ganglioside GD2 cause Src-dependent activation of N-methyl-D-aspartate receptor signaling and changes in cellular morphology. PLoS One 10:e0134255. https://doi.org/10.1371/journal.pone.0134255
Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, Coze C, Philip N, Frebourg T, Munnich A, Lyonnet S, Delattre O, Amiel J (2004) Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet 74:761–764. https://doi.org/10.1086/383253
Tsao CY, Sabbatino F, Cheung NV, Hsu JC, Villani V, Wang X, Ferrone S (2015) Anti-proliferative and pro-apoptotic activity of GD2 ganglioside-specific monoclonal antibody 3F8 in human melanoma cells. Onco Targets Ther 4:e1023975. https://doi.org/10.1080/2162402X.2015.1023975
Tse BC, Navid F, Billups CA, O’Donnell T, Hoehn ME (2015) Ocular abnormalities in patients treated with a novel anti-GD2 monoclonal antibody, hu14.18K322A. J AAPOS 19:112–115. https://doi.org/10.1016/j.jaapos.2014.11.005
Tsuchida T, Saxton RE, Morton DL, Irie RF (1987) Gangliosides of human melanoma. J Natl Cancer Inst 78:45–54
Tumino N, Weber G, Besi F, Del Bufalo F, Bertaina V, Paci P, Quatrini L, Antonucci L, Sinibaldi M, Quintarelli C, Maggi E, De Angelis B, Locatelli F, Moretta L, Vacca P, Caruana I (2021) Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J Hematol Oncol 14:191. https://doi.org/10.1186/s13045-021-01193-0
Uttenreuther-Fischer MM, Huang CS, Reisfeld RA, Yu AL (1995a) Pharmacokinetics of anti-ganglioside GD2 mAb 14G2a in a phase I trial in pediatric cancer patients. Cancer Immunol Immunother 41:29–36
Uttenreuther-Fischer MM, Huang CS, Yu AL (1995b) Pharmacokinetics of human-mouse chimeric anti-GD2 mAb ch14.18 in a phase I trial in neuroblastoma patients. Cancer Immunol Immunother 41:331–338
Vazquez AM, Alfonso M, Lanne B, Karlsson KA, Carr A, Barroso O, Fernandez LE, Rengifo E, Lanio ME, Alvarez C et al (1995) Generation of a murine monoclonal antibody specific for N-glycolylneuraminic acid-containing gangliosides that also recognizes sulfated glycolipids. Hybridoma 14:551–556. https://doi.org/10.1089/hyb.1995.14.551
Vazquez AM, Perez A, Hernandez AM, Macias A, Alfonso M, Bombino G, Perez R (1998) Syngeneic anti-idiotypic monoclonal antibodies to an anti-NeuGc-containing ganglioside monoclonal antibody. Hybridoma 17:527–534. https://doi.org/10.1089/hyb.1998.17.527
Vazquez AM, Gabri MR, Hernandez AM, Alonso DF, Beausoleil I, Gomez DE, Perez R (2000) Antitumor properties of an anti-idiotypic monoclonal antibody in relation to N-glycolyl-containing gangliosides. Oncol Rep 7:751–756. https://doi.org/10.3892/or.7.4.751
Wierzbicki A, Gil M, Ciesielski M, Fenstermaker RA, Kaneko Y, Rokita H, Lau JT, Kozbor D (2008) Immunization with a mimotope of GD2 ganglioside induces CD8+ T cells that recognize cell adhesion molecules on tumor cells. J Immunol 181:6644–6653
Wu ZL, Schwartz E, Seeger R, Ladisch S (1986) Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res 46:440–443
Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK (2015) Retargeting T cells to GD2 pentasaccharide on human tumors using bispecific humanized antibody. Cancer Immunol Res 3:266–277. https://doi.org/10.1158/2326-6066.CIR-14-0230-T
Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, Jin J, Courtney AN, Liu B, Di Pierro EJ, Hicks J, Barragan GA, Ngai H, Chen Y, Savoldo B, Dotti G, Metelitsa LS (2019) NKT cells co-expressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin Cancer Res 25:7126–7138. https://doi.org/10.1158/1078-0432.CCR-19-0421
Yankelevich M, Kondadasula SV, Thakur A, Buck S, Cheung NK, Lum LG (2012) Anti-CD3 x anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr Blood Cancer 59:1198–1205. https://doi.org/10.1002/pbc.24237
Yu AL, Uttenreuther-Fischer MM, Kamps A (1995) Combined use of a human mouse chimeric anti-GD2 (ch14.18) and GM-CSF in the treatment of refractory neuroblastoma. Antibody Immunocon Radiopharm 8:12
Yu AL, Batova A, Alvarado C, Rao VJ, Castleberry RP (1997) Usefulness of a chimeric anti-GD2 (ch14.18) and GM-CSF for refractory neuroblastoma: a POG phase II study. PROC ASCO 16:1846
Yu AL, Uttenreuther-Fischer MM, Huang CS, Tsui CC, Gillies SD, Reisfeld RA, Kung FH (1998) Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol 16:2169–2180
Yu AL, Eskenazi A, Strother D (2001) A piot study of anti-idiotype monoclonal antibody as tumor vaccine in patients with high risk neuroblastoma. Proc Am Soc Clin Oncol 20(abstr 1470):18s
Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, Shimada H, Grupp SA, Seeger R, Reynolds CP, Buxton A, Reisfeld RA, Gillies SD, Cohn SL, Maris JM, Sondel PM (2010) Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 363:1324–1334. https://doi.org/10.1056/NEJMoa0911123
Yu AL, Gilman AL, Ozkaynak MF, Naranjo A, Diccianni MB, Gan J, Hank JA, Batova A, London WB, Tenney SC, Smith M, Shulkin BL, Parisi M, Matthay KK, Cohn SL, Maris JM, Bagatell R, Park JR, Sondel PM (2021) Long-term follow-up of a phase III study of ch14.18 (dinutuximab) + cytokine immunotherapy in children with high-risk neuroblastoma: COG study ANBL0032. Clin Cancer Res 27:2179–2189. https://doi.org/10.1158/1078-0432.CCR-20-3909
Yu L, Huang L, Lin D, Lai X, Wu L, Liao X, Liu J, Zeng Y, Liang L, Zhang G, Wang B, Wu Z, Tao S, Liu Y, Jiao C, Chang LJ, Yang L (2022) GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J Cancer Res Clin Oncol 148(10):2643–2652. https://doi.org/10.1007/s00432-021-03839-5
Yuki N, Yamada M, Tagawa Y, Takahashi H, Handa S (1997) Pathogenesis of the neurotoxicity caused by anti-GD2 antibody therapy. J Neurol Sci 149:127–130
Zeng Y, Fest S, Kunert R, Katinger H, Pistoia V, Michon J, Lewis G, Ladenstein R, Lode HN (2005) Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol Immunol 42:1311–1319. https://doi.org/10.1016/j.molimm.2004.12.018
Zeytin HE, Tripathi PK, Bhattacharya-Chatterjee M, Foon KA, Chatterjee SK (2000) Construction and characterization of DNA vaccines encoding the single-chain variable fragment of the anti-idiotype antibody 1A7 mimicking the tumor-associated antigen disialoganglioside GD2. Cancer Gene Ther 7:1426–1436. https://doi.org/10.1038/sj.cgt.7700240
Zhao Q, Ahmed M, Guo HF, Cheung IY, Cheung NK (2015) Alteration of electrostatic surface potential enhances affinity and tumor killing properties of anti-ganglioside GD2 monoclonal antibody hu3F8. J Biol Chem 290:13017–13027. https://doi.org/10.1074/jbc.M115.650903
Zirngibl F, Ivasko SM, Grunewald L, Klaus A, Schwiebert S, Ruf P, Lindhofer H, Astrahantseff K, Andersch L, Schulte JH, Lode HN, Eggert A, Anders K, Hundsdoerfer P, Kunkele A (2021) GD2-directed bispecific trifunctional antibody outperforms dinutuximab beta in a murine model for aggressive metastasized neuroblastoma. J Immunother Cancer 9(7):e002923. https://doi.org/10.1136/jitc-2021-002923
Zuo S, Sho M, Sawai T, Kanehiro H, Maeda K, Yoshida M, Tsukada R, Nomura M, Okuyama H (2020) Potential role of the PD-L1 expression and tumor-infiltrating lymphocytes on neuroblastoma. Pediatr Surg Int 36:137–143. https://doi.org/10.1007/s00383-019-04616-9
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Hung, JT., Yu, A.L. (2023). Immunotherapy of Neuroblastoma Targeting GD2 and Beyond. In: Furukawa, K., Fukuda, M. (eds) Glycosignals in Cancer. Springer, Singapore. https://doi.org/10.1007/978-981-19-7732-9_10
Download citation
DOI: https://doi.org/10.1007/978-981-19-7732-9_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-7731-2
Online ISBN: 978-981-19-7732-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)