
Abstract
According to one of the currently developed mechanisms of self-healing of epoxy protective coatings, an important role in the process is played by the diffusion migration stage of the composition components to the crack network defects, on the one hand, by swelling of the pore walls, on the other hand, and by their adhesive interaction, on the third. PART 3.1; 3.2, and 3.3 of this chapter summarize the results of our investigations performed at the Laboratory of Structural and Morphological Research of the Institute of Physical Chemistry of the Russian Academy of Sciences concerning the translational mobility of epoxy oligomers, epoxy-amine adducts as model systems for investigation of curing processes of epoxy oligomers and phase equilibrium and structure formation during curing of epoxy compositions. It seems that the presented experimental and methodological material will make it possible to significantly advance our understanding of the details of the self-curing mechanism.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Epoxy oligomer
- Adduct
- Epoxy curing
- Epoxy matrix modification
- Phase equilibrium
- Interdiffusion
- Self-diffusion
- Interdiffusion
- Epoxy oligomers
- Epoxy polymers
- Translational mobility of the macromolecules
- Activation energy
- Pulsed magnetic field gradient
- Interferometry
1 Translational Mobility of Epoxy Oligomers. Influence of Molecular Weight and Thermal Prehistory
The results of the study of self-diffusion and interdiffusion in melts of individual epoxy oligomers and their mixtures of different molecular weights are summarized. The results on the concentration dependences of interdiffusion coefficients and partial coefficients of self-diffusion and diffusion activation energies are presented for the first time. The effect of end groups on the parameters of molecular weight dependence of transfer coefficients of oligomers diffusion is described. In the investigated molecular weight range of Eos, there is a linear dependence of \(D - M^{b}\) with \(b\), depending on the temperature: for T < 433 K − \(b\) = 2.4 and for T > 433 K \(b\) = 2.0. This dependence differs from the previously obtained data for several polymer melts at M < Mcr, for which in the oligomeric region \(b\) < 2.
1.1 Introduction
It is known [1, 2] that the problem related to the influence of the molecular weight (MW) and molecular weight distribution (MWR) of oligomers and polymers on the translational mobility of their macromolecules has several solutions. The simplest solution to this problem can be analyzed in the framework of the Einstein-Stokes model. According to this model, the translational diffusion coefficient of any molecule and macromolecule is unambiguously related to their geometric dimensions R and friction coefficient by the following relation:
Usually, for spherical non-solvated molecules and ellipsoidal or wand-like particles, it is written as
respectively [3]. Here, \(k\) is the Boltzmann constant, \(\nu\) is the molar volume of the link, \(N_{{\text{A}}}\) is the Avogadro number; \(\eta_{{\text{O}}}\) is the effective viscosity or microviscosity of the matrix related to the local friction coefficient experienced by the moving molecule in this or that diffusion medium; \(M\) is the molecular weight of diffusing molecules or macromolecules; \(\varphi\) is the form factor, and \(\chi \left( p \right)\) is the function characterizing the degree of particle asymmetry.
These relations are widely enough used in the practice of physicochemical research. For example, in [4], monomer and solvent diffusion coefficients are used to calculate the monomer friction coefficients of polymers and oligomers. In [2], the MW of spherical, ellipsoidal, and bubble-shaped non-solvated particles is determined by Eq. (2) and the diffusion coefficient of macromolecules in a viscous solvent. In [5], using the viscosity coefficients of oligomer melts, the values of their partial diffusion coefficients are estimated using Eq. (1).
Therefore, it follows that if the diffusion process in a binary system in which the molecules of component “1” are used as a diffusant, the effect of the molecular weight of the oligomers that form the diffusion medium (let us denote it by index “2”) will manifest itself in the value of the diffusion coefficient through its monomer friction coefficient, which, in turn, is related to the local mobility of the oligomer chain fragments.
If the interdiffusion process is realized in another region of solutions, when oligomers of different MW are the diffusant and another oligomer or polymer is the diffusion medium, i.e., \(\zeta_{1} = {\text{const}}\), ,, the influence of the MW of the oligomers on (\(D_{21}\) (2 → 1)) is associated with a change in the contour length of the molecules, on the one hand, and a change in their conformational state, on the other hand.
From this point of view, polymer self-diffusion ((\(D_{22}^{*}\))) takes a special place in the study of translational mobility of macromolecules since two parameters simultaneously change when comparing diffusion coefficients of oligomers and polymers of different MW: both the monomer friction coefficient and the size of the diffusing molecule.
Thus, to characterize the influence of MW oligomers on their translational mobility to the full extent, it is necessary to obtain detailed information on the diffusion properties of oligomer solutions and melts: to describe the concentration and temperature dependences of interdiffusion and self-diffusion coefficients and to determine activation energies characterizing elementary acts of motion of macromolecule fragments and chains as a whole.
It should be noted that, at present, epoxide oligomer (EO) melts are most often considered associative liquids, which naturally impose specific difficulties on the interpretation of the experimental data. However, the nature of associates, the morphology, the thermal stability, and the kinetic stability are still open. The data obtained by methods of turbidity spectrum, NMR, light scattering, infrared spectroscopy, and viscometers are ambiguous and contradictory. First of all, it concerns the results of rheological investigations, which are frequently used as basic information for the formation of representations about the structural organization of EO melts [5,6,7,8]. According to Aleman’s data [9, 10], viscous flow activation energy (Eη) of epoxy oligomers measured in the temperature interval from 400 to 450 K is constant, E = 130 kJ/mol, and slightly depends on the molecular mass of oligomers. In [11, 12], the Eη was measured in a slightly larger temperature range from 350 to 450 K, decreases with increasing temperature from 90 to 45 kJ/mol, and changes continuously with changes in the molecular weight of EO. In [13, 14], the extreme dependence of Eη of the viscous flow with temperature was described. The ambiguity of these results encourages researchers to continuously search for new information on the properties of epoxy oligomers melts. In this connection in the present work, we studied the self and mutual diffusion of epoxy oligomers in a wide range of temperatures and molecular weights.
1.2 Experimental Section
The article summarizes the results of research on epoxy oligomers of different brands: domestic—ED-20, ED-40, ED-44, ED-49, and ED-5 and resins of foreign firms—Epikote 1009—Shell (USA), Araldite GT 6610, Ciba (Switzerland), YD-128—Toto Kasei Co. (Japan), and DER-664 by DAU Chemical. Along with individual EOs, their mixtures were also studied. The molecular weights of oligomers were determined by gel permeation chromatography (Table 1).
The self-diffusion coefficients of EO molecules were measured in the temperature range from 353 to 483 K by NMR with a pulsed magnetic field gradient (IGMP NMR), with a proton resonance frequency of 60 MHz and the amplitude of the maximum magnetic field gradient of 56 Tl/m. A two-pulse stimulated echo sequence technique was used in the range of diffusion times from 3 ms to 1 s [17, 18]. The accuracy of the \(D^{*}\) measurement was 10%.
Interdiffusion was studied by the optical wedge method on the ODA-2 interferometer (IPC RAS Moscow, Russia). The measurement technique did not differ from the traditional one [19, 20]. A higher molecular weight EO was placed in a wedge-shaped capillary of a diffusion cell and brought into optical contact with the surface of metalized quartz plates at T ≥ Tg. The assembled cell was placed in a thermostatted cuvette preheated to the preset temperature. Then, the cell was filled with liquid oligomers. The moment of contact of oligomers was observed visually on the monitor screen and considered the beginning of the interdiffusion process. The resulting interference pattern characterizing the concentration distribution profile in the interdiffusion zone was used to plot the composition distribution curves and calculate the interdiffusion coefficients by the Matano-Boltzmann method. Microphotographs illustrating the interdiffusion process are presented in Sects. 1.2 and 1.3.
1.3 About the Course of Mass Transfer Processes
A study of the self-diffusion process in melts of epoxy oligomers and their mixtures has shown that two dependences of diffusion damping \(A - {\text{g}}^{2}\) are observed for these compounds: exponential and no exponential (Fig. 1). We observed the second, non-exponential one, as a rule, for EO mixtures with bi- or polymodal MWR and, as a consequence, a set of exponential modes with self-diffusion coefficients \(D_{i}^{*}\):

Magnetization amplitude dependence on the magnetization field gradient for EO-2 (1) and EO-2/Epikote 1009 mixture (2) at 413 K. The diffusion time is 150 ms
where \(P_{i}\) is the population of components with self-diffusion coefficients \(D_{i}^{*}\).
The no exponential course of diffusion damping was also described earlier in [21,22,23]. The authors of these studies made various assumptions about the possible reasons for this fact. For example, in [21], the no exponential form of diffusion damping was attributed to the presence of macromolecule associations in EO melts, which, as the authors believed, arise due to entanglements between macromolecules and have finite lifetimes. Other papers [22, 23] associated c with molecular-mass heterogeneity of industrial EO grades.
The analysis of experimental data on diffusion in binary epoxy systems, the components of which differ from each other only by molecular masses, showed that all the studied systems in the temperature range from Tg to Tg + 180 K, where the Tll-transition temperature is also situated [13], belong to the systems with unlimited mutual solubility of components. The spontaneous mixing of the epoxy oligomers brought in contact follows the purely diffusive regularities; that is, each isoconcentration plane situated inside the diffusion zone with respect to any reference system (Hittorff, laboratory, mass-fixed, or volume-fixed) moves with a constant rate in the \(X - t^{1/2}\) coordinate system, where \(X\) is the coordinate of the plane and \(t\) is the observation time. It is of fundamental importance that the kinetic constants of the mixing process are well reproduced when the figurative point of the system moves both up and down the temperature scale. This means, first, that no changes in the molecular weight characteristics of the components in the studied temperature range occur in the EO melts during the process, in contrast to the effects in EO-elastomer systems described in [24, 25]. Second, the associative nature of EO melts does not appear in traditional diffusion experiments. This is possible when the diffusion relaxation time of the local concentration gradient is longer or comparable to the lifetime of associates of EO macromolecules.
1.4 Concentration Dependence of Diffusion Coefficients
Typical concentration dependences of interdiffusion coefficients \(D_{{\text{V}}} - \varphi_{1}\) for some binary EO systems are shown in Figs. 2, 3 and 4. It can be seen that \(D_{{\text{V}}} - \varphi_{1}\) dependences are uniform. The value of \(D_{{\text{V}}}\) changes smoothly and monotonically at the transition from one component to another, i.e., from one limiting value of the coefficient of interdiffusion \(D_{12} \equiv D_{{\text{V}}}\) at \(\varphi_{1} \to 0\), to another \(D_{21} \equiv D_{{\text{V}}}\) at \(\varphi_{1} \to 1\). Here, index “2” denotes a high-molecular-weight oligomer and index “1” denotes a low-molecular-weight oligomer.

Concentration dependence of interdiffusion coefficients in the system EO-2/EO-4 (a) EO-2/EO-9 (b) at temperatures 353 (1), 373 (2), 393 (3); 413 (4), 423 (5), 433 (6), 443 (7), 463 (8), 483 K (9), and 495 K (10)

Dependences of the partial self-diffusion coefficients of EO-8 (1) and EO-1 (2) and mutual diffusion coefficient (3) on the EO-1/EO-8 system composition at 433 K

Dependences of interdiffusion coefficients on the composition of C-systems EO-9/EO-1 (1), EO-9/EO-2 (2); EO-9/EO-4 (3); and EO-9/EO-5 (4) at 493 K
In this case, the difference in the values of the limiting diffusion coefficients \(\Delta D = {\text{lg}}\left[ {D_{12} /D_{21} } \right]\), characterizing the intensity of the change in the translational mobility of molecules with composition, decreases from 2.0 at 353 K to 0.3 at 473 K. In some systems with close MW values of the components, the concentration dependence of \(D_{{\text{V}}}\) degenerates.
Analysis of the effect of temperature on the character of the concentration dependences of the interdiffusion coefficient in EO mixtures allowed us to distinguish two temperature regions:
– (region I) T < 443 K, which is characterized by a marked increase in \(D_{{\text{V}}}\) with a change in system composition,
– (region II) 443 < T < 503 K, where \(D_{{\text{V}}}\) weakly changes with a change in concentration.
Figure 4 shows the data for the case when the diffusion medium is the same (Epikote 1009) and diffusants are different: DGEBA, ED-20, E40, and E44. As the MW of the diffusant M1 increases or the M2/M1 ratio decreases (M2 corresponds to the molecular weight of EPIKOTE 1009), the concentration dependence of \(D_{V}\) (1) degenerates against the background of a general decrease in the absolute values of diffusion coefficient.
1.5 Temperature Dependence of Diffusion Coefficients
Notably, in the analysis of the experimental data on the self-diffusion coefficients of melts, the translational mobility of molecules in ED melts, and mutual diffusion has been studied in the temperature range, which covers the areas from the glass transition temperature to the temperature above the ll-transition. Note that earlier measurements were performed only for some low-molecular-weight oligomers—ED-20, ED-16, and DGEBA [26].
Figure 5 shows the temperature dependences of self-diffusion coefficients in the coordinates of the Arrhenius equation:

Temperature dependences of self-diffusion coefficients of EO: 1 and 1′—DGEBA, 2—ED-20 and YD-128, 3—E40, 4—E44, 5—DER, 6—Araldite 6610, 7—Epikote 1009
where \(E^{*}\) is the apparent activation energy of self-diffusion.
One can see that the coefficient values for EO-2 and YD-128 coincide since their molecular masses have close values, while for DGEBA, they lie below the \(D^{*}\) values of the oligomers whose mass is greater than that of DGEBA. For comparison, the dotted line shows the \(D^{*}\) values of DGEBA extracted from [19]. It can be seen that these values are close to those obtained by us.
The self-diffusion coefficients of EO decrease with increasing molecular weight over the entire temperature range. The intensity of the decrease is different for temperatures near Tg and Tll. For all epoxy oligomers, the Arrhenius dependences can be approximated by two linear (first approximation) sections characterized by different slope angles, \(E^{*}\) and \(E^{**}\). The first section corresponds to the temperature range from 353 to 423–433 K (\(E^{*}\)) and the second from 433 to 493 K (\(E^{**}\)). It should be noted that the breakpoint temperature (\(T^{*}\) 433 K) is close to the ll-transition temperature in epoxy oligomers identified by infrared spectroscopy and relaxation spectrometry [28].
This character of the dependence is reproduced by repeated cycles of “rise and fall” temperature. Special experiments have shown that the appearance of a kink in the \(lgD^{*} - 1/T\) dependence is associated neither with chemical reactions (self-crosslinking), which could take place at high temperature nor with polymer degradation. On the one hand, this was confirmed by refractometric studies [29] and, on the other hand, by gel chromatography data. The chromatograms of epoxy compounds preheated at 473 K coincide with those obtained for EO not subjected to heat treatment. In addition, according to DTA data, thermal decomposition of EO is observed at temperatures above 573 K.
Table 2 and Fig. 6 present the values of activation energy calculated from the dependence \(lgD^{*} - 1/T\) for each of the indicated temperature intervals. The values of \(D^{*} \left( {T_{{\text{g}}} } \right)\) obtained by extrapolation of the first part of this dependence to \(T_{{\text{g}}}\) are also given there. It can be seen that \(D^{*} \left( {T_{{\text{g}}} } \right)\) are quite close for all EO regardless of their molecular weight and, on average, reach ~12 × 10–11 cm2/s.

Dependence of the activation energy of EO self-diffusion on the molecular weight for T < 433 K (1) and T > 433 K (2)
For most of the systems studied, with the exception of those cases where the diffusion media were DGEBA and ED-20, a single trend of changes in the DE diffusion coefficients with temperature is observed (Fig. 7).

Temperature dependences of the limiting coefficient of interdiffusion of EO-2 a in matrices EO-5 (1), EO-6 (2), EO-8 (3), and EO-9 (4); and b marginal coefficient of interdiffusion of oligomers of EO-5 (1), EO-6 (2), EO-8 (3), and EO-9 (4) in the matrix EO-2
1.6 Interdiffusion of Epoxy Oligomers
For most investigated systems, except for the cases when the diffusion media were DGEBA and ED-20, the same tendency of changes of DE diffusion coefficients with temperature was observed. It can be seen that the temperature dependence of the limiting interdiffusion coefficient \(D_{12}\) can also be approximated by two linear sections characterized by different slope angles, namely EI (correlation coefficient 0.98) and EII (correlation coefficient 0.96). As in the case of self-diffusion, the first section corresponds to the temperature region 353–433 K and the second to the interval 455–493 K. It should be noted that the position of the kink in the temperature dependences on the coefficient of interdiffusion of EO (455 K) is also close to the temperature of Tll-transition in EO (433 K), that is, this temperature does not depend on the way of measuring the translational mobility of oligomers but is a characteristic value of the matrix in which mass transfer occurs.
Numerical values of apparent activation energies EI and EII are given in Table 3. Their concentration dependence is shown in Fig. 8. For all systems, regardless of the EO being a diffusant, EI > EII. In this case, if the EI value depends on concentration and occupies an intermediate position between the activation energies of self-diffusion of components EII ≤ EI ≤ E*22, then EII is practically independent of the co-stock and satisfies the condition EII ≅ E*11 ≅ E*22.

Concentration dependences of apparent activation energies of interdiffusion in the EO-2/EO-9 system (1.3) and EO-2/EO-5 system (2.4) at temperatures T < Tll (1, 2) and T > Tll (3, 4)
Figure 8 shows that EI, as E*, is a function of the molecular weight of the diffusion medium, increasing slightly in the transition from ED-20 to EPIKOTE 1009. For EII, this dependence is weakly expressed.
A comparison of the activation energies of EII translational mobility with the activation energies characterizing the segmental and group mobility of oligomer macromolecules showed that it is close to the activation energy and α-transitions ~71.06 and 50.16 kJ/mol, respectively, and agrees with the activation energy—transitions ~16.72 kJ/mol [30]. It means that in the temperature range T < 438 K, large-scale motions of macromolecule fragments forming the diffusion medium are responsible for the translational mobility of epoxy oligomers molecules. At T > 433 K—small-scale movements of macromolecule fragments and monomer units (CH3−, OH-groups).
It appears that such a temperature dependence of diffusion coefficients in ED melts is related to the formation of “hole vacancies” of extended sizes in diffusion media at high temperatures (Tg + 150) in the so-called free volume flicker clusters whose lifetime exceeds the time of the diffusion jump of the oligomer molecule (displacement of its center of gravity). Obviously, under these conditions, the contribution to the activation energy of the work of micro-cavity formation becomes minimal, and the value of the activation energy is determined only by the energy costs associated with overcoming the intermolecular interaction and the friction forces in the movement of the molecule along the surface of the free volume cluster. At low temperatures (T < Tll), the main contribution to the mechanism of translational motion of EO molecules in melts is related to the formation of micro-cavity of necessary size near them.
1.7 The Influence of the Molecular Weight of Oligomers
According to the existing theories of translational mobility of macromolecules [18, 19], two groups of systems are distinguished when considering the dependence of self-diffusion coefficients on molecular mass. The first refers to monodisperse or close to monodisperse melts, while the second group refers to systems with a broad molecular-mass distribution. For the first group of systems for which the theoretical analysis is described in [24, 25], the relations \(D^{*} \cong M^{ - 1}\) at M < Mcr and \(D^{*} \cong M^{ - 2}\) at M > Mcr. Mcr is the critical molecular mass at which a net of physical links between macromolecules develops.
However, the results of experimental studies are different. For example, \(D^{*} \cong M^{ - 1.7}\) was obtained for PEG and PDMS in [3], and both \(D^{*} \cong M^{ - 1}\) and \(D^{*} \cong M^{ - 2.7}\) were found for polystyrene melts [3]. A detailed analysis of the molecular weight dependence of the self-diffusion coefficient of PEG melts is given in a monograph [32]. According to these authors, the dependence of \(D^{*}\) on Mn has two characteristic regions and is described by the relation \(D^{*} \cong M_{n}^{ - b}\), where b ~ 1 for Mn ≤ 103 and b ~ 2 for Mn > 3 × 103.
The systematic experimental investigations have not been practically carried out for the second group of systems, though these systems are of the most significant interest. There are only separate attempts to analyze the translational mobility of these systems within the concept of the self-diffusion and cooperative diffusion coefficient spectra [32, 33].
Before proceeding to the analysis of the diffusion characteristics of EO with different molecular weight distributions, we note that we will refer to the data on the diffusion of individual EO to their weight-average molecular weight Mw, and we will choose the equation as the basis for their relationship with the translational mass transfer coefficients:
where \(D_{0}\) is the diffusion coefficient corresponding to the diffusion coefficient of the monomer. In works [1,2,3], \(D_{0}\) is identified with diffusion coefficients of “monomer,” “segment in diffusion medium,” “monomer friction coefficient,” and friction coefficient of blob with the environment.
The complex nature of the temperature dependence of the self-diffusion coefficients of epoxy oligomers required the analysis of the dependence of \(D^{*} - M_{{\text{w}}}\) for different temperatures. The following intervals were chosen: T < 433 K (393 K, 413 K) and T > 433 K (453, 473 K).
It has been established (Fig. 9) that in the investigated range of molecular masses, linear dependence of \({\text{lg}}D - {\text{lg}}M_{{\text{w}}}\) (the correlation coefficient is 0.97) with a temperature-dependent slope: for T < 433 K b = 2.4, and for T > 433 K b = 2.0 really takes place. This clearly differs from the data obtained earlier for a number of polymer melts at M < Mcr, for which, in the oligomeric region, b < 2.

Dependence of the limiting coefficients of interdiffusion (1–3) and self-diffusion (4) on the molecular weight of EO. T = 393 K (a) and 493 K (b). The arrows indicate the self-diffusion coefficients and the MW of the oligomers forming the diffusion media
It should be noted that, for epoxide oligomers, in this temperature interval, one can observe anomalies in the dependence of Newton viscosity on the molecular weight: b varies from 2.7 (403 K) to 3.5 (343 K).
Figure 9 shows some dependences of \(D_{{{\text{ij}}}}\) on the molecular mass Mi of EO diffusing into the medium of EO with a constant molecular mass Mj. Three cases are given: when the MW of the diffusing molecules is less than the MW of the diffusing medium (Mi < Mj); when Mi > Mj, and when the Mj of the diffusing medium is within the MW interval corresponding to the homologous series of diffusing EO molecules. It is seen that for all cases at different temperatures, the experimental points in the coordinates lie on straight lines (the correlation coefficient is 0.98), crossing the dependences at the points corresponding to the MW and self-diffusion coefficient of EO molecules that compose the diffusion medium (arrows mark these points in Fig. 9).
The exponent of Eq. (6), the value of which is commonly used to determine the diffusion mechanism [31, 34, 35], varies in the rather narrow range of 0.90 ± 0.06. Importantly, it does not depend on temperature, in contrast to the factor b calculated from self-diffusion data. This means that the \(M_{{\text{w}}}^{ - b}\) multiplier in Eq. (6) is really a structural characteristic of diffusing unchained molecules, whose mono-dimensional friction coefficient is determined by \(D_{0}\).
We experimentally determined the multiplier \(D_{0}\) as the limiting diffusion coefficient of ED-20. Dependences of \(D_{0}\) on MW of EO molecules forming the diffusion medium Mcr are shown in Fig. 10. As Mcr increases, the monomeric diffusion coefficient decreases, asymptotically approaching some constant value of \(D_{0\infty }\) ≅ \(D_{0}\) at Mcr → ∞. This type of \(D_{0} - M_{{{\text{cr}}}}\) dependence is observed at all temperatures and for any other diffusant. The specificity of each diffusant is evident in the values of \(D_{0}\) and Mcr. Thus, for ED-20, it is 1 × 104, for dibutyl phthalate Mcr = 7 × 103, for cyclohexanone—6 × 103.

The dependence of the diffusion coefficients of EO-2 (1–4) in epoxy oligomers on the MW of the oligomer medium. T = 393 K (1), 433 K (2), 443 K (3), and 494 K (4)
Experimental dependences \(D_{0} - M_{{{\text{cr}}}}\) at degrees of polymerization of epoxy oligomer N > 5 are satisfactorily described by the empirical equation
where \(k\) is a constant (Fig. 11). We used these dependences in determining \(D_{0\infty }\).

Dependence of diffusion coefficients of EO-2 in epoxy oligomers on their degree of polymerization in the coordinates of Eq. (7). T = 393 K (1), 433 K (2), and 494 K (3)
In the whole range of polymerization degrees (N), the dependences \(D_{0} - M_{{{\text{cr}}}}\) are described by the equation:
\(f_{\infty }\) is the free volume fraction of EO at \(M \to \infty\), B is a constant close to unity; a is a constant related to the local free volume of links and end groups of macromolecules). Note that the coefficient of thermal expansion of the free volume of polyhydroxy ethers calculated (under assumption B ≅ 1) along the segments \(f_{\infty } /B\), segmented by straight lines on the ordinate axis, is (5 ± 0.1) × 10–4 and is close to the standard value of 4.8 × 10–4 [2, 4].
Thus, the presented experimental material clearly shows that the monomeric diffusion coefficient (friction) \(D_{0}\) depends on the MW of the diffusion medium. It is obvious that this effect should be taken into account when interpreting the dependence of the self-diffusion coefficient on MW. Therefore, it can be stated that Eq. (8) is valid for the analysis of translational mobility of unchained macromolecules only when the MW of diffusing molecules changes and the MW of macromolecules forming the diffusion medium remains unchanged. Probably, in this case, it can be assumed that for unchained molecules b ≅ 1.
When describing self-diffusion in oligomer melts, relation (7) should be written in the form:
which takes into account explicitly the contribution to the self-diffusion coefficient made by end groups, whose concentration continuously changes during the transition from one polymer homologue to another.
1.8 Conclusion
Coefficients of mutual and self-diffusion of epoxy oligomers of different molecular weights were determined by NMR spectroscopy (spin-echo) and optical interferometry. The dependences of diffusion coefficients on temperature and concentration of binary systems are presented. It was found for the first time that for all EO, the Arrhenius temperature dependences of the coefficients of the self and interdiffusion can be approximated by two linear sections characterized by different activation energies—EI and EII. The first section corresponds to the temperature range from 353 to 423–433 K, and the second from 433 to 493 K. The kink temperature (T* ≅ 433 K) of the Arrhenius temperature dependences is close to the temperature ll-transition of EO identified by infrared spectroscopy and relaxation spectrometry.
It has been established that in the investigated range of molecular masses there is a linear dependence \(D \cong M^{b}\) with a temperature-dependent slope angle: for T < 433 K – b ≅ 2.4, and for T > 433 K – b ≅ 2.0. This clearly differs from the previously obtained data for a number of polymer melts at M < Mcr, for which in the oligomeric region b < 2.
2 Epoxy-Amine Adducts as Model Systems for Investigation of Curing Processes of Epoxy Oligomers
A method for studying the process of obtaining polymer composite materials through the use of special objects—adducts, partially cured epoxy resins—is proposed. The paper describes the method of obtaining adducts on the basis of industrial epoxy resin ED-20 and curing agent—diaminodiphenylsulfone. Preliminary studies have established the mutual solubility of these components, the rate of their mutual mixing, and the temperature range of the chemical reaction. The dependence of the molecular weight characteristics of the obtained adducts on the degree of conversion as well as their glass transition temperatures was traced. The obtained adducts were combined with thermoplastics—polyetherimide and polymethylmethacrylate—and the mixing processes were observed by optical interferometry. The evolution of phase state diagrams of thermoplastic-reactoplastic mixtures and changes in interdiffusion coefficients in these systems during the chemical curing reaction were traced in this work.
2.1 Introduction
At the present time, many methods of obtaining materials based on epoxy resins with self-healing properties have been described in the literature. At the same time, the recovery process of the epoxy matrix after a crack has sprouted in it can occur by different mechanisms, for example, both thorough wetting of the fracture surface by the adhesive released from the previously introduced microcapsules and through the realization of complex chemical reactions or interdiffusion processes between the composite material components [36,37,38,39,40,41,42,43,44]. In the latter case, a number of difficulties arise for the study since the mass transfer processes take place practically in the matrix, which is in the glassy state. In our opinion, this complexity can be circumvented by investigating the behavior of the system during the chemical curing reaction at different stages.
One of the effective experimental approaches allows to study of the change of translational mobility of components during curing (formation of the spatial mesh of reactoplastic), tracing the evolution of boundary curves of the phase diagram, and evaluating the kinetics and mechanism of mutual diffusion, and solubility at different stages of structure formation is related to the study of mass transfer in the epoxy oligomer adducts (aER)—thermoplastics systems. It is assumed that a set of adducts with different conversion rates (α) allows adequate modeling of the process of formation of spatially cross-linked structures of composite binders at different stages of the reactoplastic curing process.
In the present work, we summarize the results of our studies of the kinetics of hardener dissolution in a dian epoxy oligomer; the partial translational diffusion coefficients are determined and their changes with changes in the oligomer curing degree are traced; the apparent activation energies of interdiffusion are calculated. Note that in our experiments, thermoplastics act as macromolecular probes whose partial mass transfer coefficients will make it possible to estimate the influence of the parameters of the spatially cross-linked structures on their translational migration in oligomer solutions at different curing stages.
2.2 Objects and Methods of Research
The following objects were used as objects of research: ED-20 dian epoxy resin (Mn = 380 g/mol), curing agent—diaminodiphenylsulfone (DADPS), thermoplastic polymers: polyetherimide (PEI, Ultem 1000, Mw = 62,000 g/mol) and polymethylmethacrylate (PMMA, Aldrich, Mw = 996,000 g/mol); epoxy oligomer adducts (aER) of various degrees of conversion (α).
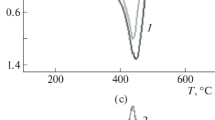
When synthesizing adducts with different degrees of conversion of epoxy groups, the ratio of the component oligomers: hardener was taken as a multiple of the stoichiometric ratio 10:3 by mass [16]. So, for an adduct corresponding to the conversion degree α = 0.1, for 100 mass parts of the oligomer, there were 3 mass parts of the hardener, for α = 0.2 − 6 mass parts, etc. After the components were combined and thoroughly mixed, the mixtures were heated and thermostatted for 6 h at 453 K. The complete conversion of reactive groups during the curing reaction was confirmed by infrared spectroscopy. Preliminary measurements by differential scanning calorimetry (Netzsch Phoenix DCS 204F1) determined the optimum temperature interval for curing EO (433–453 K), selected high-temperature curing agent DADPS (Fig. 12).

Curing thermogram of DADPS epoxy oligomer at stoichiometric ratio
It was assumed that each of the epoxy groups of the oligomer would react with the terminal-NH2 DADPS (Fig. 13), the amount of which is sufficient to form, eventually, a continuous network of chemical bonds in the mixture.

The reaction of the interacting functional groups of the epoxy oligomer with the amine hardener (a) and macromolecule growth (b)
The content of epoxy and amine groups during the curing of each adduct was determined by infrared spectroscopy in the frequency range of 400–4000 cm–1 using an IFS 66v Fourier infrared spectrophotometer with a high-temperature cell. After careful mixing of the components, the compositions were applied to a KBr tablet, which was covered with the same tablet on top, thus forming an adduct layer with a thickness of ~20 μm. The tablets were placed in the spectrometer cell holder, and IR spectra were recorded at curing temperature every 15 min. The spectra were processed using a standard OPUS program. The main absorption bands, the intensity of which changes during curing, are the broad band of valence vibrations of hydroxyl groups in the region of 3200–3700 cm–1, and bands of strain vibrations of the epoxy cycle and primary amino groups in the regions of 880–950 cm–1 and 1629 cm–1, respectively. Absorption bands [45] at 1510 cm–1 and 1374 cm–1, caused by valent vibrations of the >C=C< bond in the aromatic ring and strain vibrations of methyl groups at the carbon atom, respectively, were used as “internal standards.” Based on the values of the optical density of the epoxide group absorption bands and the “internal standards” in accordance with the Lambert–Beer law, the concentrations of epoxide groups in the curing amino-epoxide compositions were calculated.
Molecular weight characteristics of aER were determined by gel permeation chromatography. The samples of epoxy oligomers and oligomer adducts were analyzed on a Waters (USA) high-pressure gel chromatograph with a 264 nm UV detector and Styragel HR 4E columns 300 mm long and 7.8 mm in diameter. Tetrahydrofuran was used as the solvent. The elution rate was 1 ml/min, polymer concentration in the solvent was 1 mg/ml, and the sample volume was 10 μl. Column and injector thermostat temperature was set to 298 K. Calibration of the chromatographic system was carried out with reference polystyrene samples from “Waters” company, which have monodisperse molecular weight distribution (Mw/Mn = 1.03). Samples of reference polystyrene and the analyzed polymer were dissolved in THF at room temperature and then transferred to the injector of the chromatograph.
Figure 14 shows a typical chromatogram of the initial epoxy oligomer ED-20. It can be seen that it is formally polymodal. On the chromatogram, at least three molecular fractions A1, A2, and A3 can be distinguished. However, the proportion of fractions A2 and A3 is small, so processing the chromatogram by standard methods gives the value of the molecular weight distribution equal to 1.09, which allows us to refer to the oligomer chosen for the study as monodisperse.

The molecular weight distribution of ED-20 epoxy oligomer
The chromatograms of the synthesized adducts are shown in Fig. 15. We can see that the number and proportion of peaks falling on higher molecular weight fractions increase with growth α, i.e., the polydispersity of the adducts increases. At the same time, peaks corresponding to initial oligomer fractions decrease both in intensity and area, indicating a decrease in the number of initial epoxy units. It was found that the growing peaks correspond to MW multiples of the initial oligomer MW.

Molecular weight characteristics of aER. Conversion degrees α: a 0, b 0.05, c 0.10, d 0.20, e 0.30
At low conversion rates (α ≤ 0.30), an increase in the MW of the initial oligomer occurred due to the reaction of attachment of the hardener molecules and lengthening of the MW of the initial oligomer (Fig. 13). At higher (α ≥ 0.35), the formation of the so-called microgel structures occurred, and the appearance of which was accompanied by an increase in pressure of the elution solution when feeding it into the chromatographic column, which did not allow us to obtain correct information on the MW aER in this way (Fig. 16).

Dependence of the average molecular weight of aER on the degree of conversion. Region I corresponds to the formation of long-branched macromolecules of aER, and region II corresponds to the formation of microgel particles
Additional information on the evolution of the molecular weight characteristics of the adducts was obtained by chromatogram decomposition in the ORIDGEN 7 program. It was found that the content of the monomeric fraction of EE in the adduct decreased from 76 to 25 wt%, the dimer from 14 to 6 wt%. Whereas the tetramer content in aER with a conversion rate of 0.1 reaches 24 wt%. It is interesting to note that the tetramer content in samples with conversion rates of 0.1–0.3 changes little, whereas, in the adducts, the share of molecular fractions with M = 3150 appears and grows. The greatest changes in molecular weight characteristics occur at α ≥ 0.3. It is under these conditions that microgels and mesh fragment particles appear. In Fig. 16, this state of the adducts, which can be called transitional, is indicated by the dotted line. A further increase in the content of the hardener in the mixtures and, consequently, the degree of conversion led to the formation of a high-molecular-weight “tail” in the chromatograms as a result of the reaction of copolymerization of di- and trimers.
Additional information about the MW of the adducts of various degrees of hardening, including α ≥ 0.35, was obtained using their glass transition temperatures, which were determined by DSC. Figure 17 shows typical thermograms of the temperature dependences of the heat capacity of aERs in their glass transition region.

DSC thermograms of the adducts. Conversion degrees α: ED-20—0% (1), 0.2 (2), 0.6 (3), and stoichiometric component ratio (4)
It can be seen that the Cp–T dependences ∆ are S-shaped, and the aER glass transition temperature increases and reaches its maximum value at a sharp α → 1. Increase in the glass transition temperature (Fig. 18) is associated with the formation of a spatial bonding network. For this area of adducts, the MW aER value calculated from the Nielsen equation [46] characterizes the value of the molecular mass of the reactant between the nodes of the mesh (see Table 4).

Dependence of the glass transition temperature of aER-DADPS on the degree of conversion
Subsequently, these values of the molecular weight characteristics of the adducts were used to analyze the behavior of polymer-oligomer mixtures at various stages of the chemical curing reaction.
To study solubility, interdiffusion, and construction of phase diagrams in binary systems thermoplastic-ER and thermoplastic-aER, we used the optical wedge interferometer ODA-2 method (IPC RAS). The methodology of the experiments did not differ from the traditional one [19]. A pre-pressed thermoplastic film was placed inside the interference cell, heated in a thermal furnace to a temperature higher than the thermoplastic glass transition temperature, and brought into optical contact with the surface of glass plates of the wedge capillary. After the assembled cell was thermostatted at the experimental temperature, a diffusant (epoxy oligomer or adduct) was introduced into the wedge gap between the plates. The moment of contact between the components was observed visually on the monitor screen and was taken as the beginning of the interdiffusion process. Interferograms of interdiffusion zones were recorded periodically at intervals of ~110 min depending on the diffusion rate and resolution of interference fringes. Studies of interdiffusion were carried out in isothermal mode. For plotting phase state diagrams, a step mode of temperature rise and fall with step 20/30 K in the temperature range 293–553 K was used. At each stage, the system was thermostatted from 20 to 60 min, depending on its lability. The concentration distribution profiles obtained by the Matano-Boltzmann method were used to calculate the interdiffusion coefficients; the positions of points on the binodal curves of the phase state diagrams were determined by concentration jumps at the interphase boundaries.
Phase and relaxation transition temperatures in homopolymers and decomposition temperatures were obtained by differential scanning calorimetry (Netzsch Phoenix DCS 204F1) at scanning speeds of 10 K/min.
2.3 Phase Equilibrium and Interdiffusion in the Epoxy Oligomer–Curing Agent System
In the production technology of composite materials based on thermosetting binders, the correct choice of temperature-time homogenization mode of the epoxy-hardener mixture and curing parameters of the composition is of great importance [26]. Naturally, it is of interest to determine under what conditions the dissolution process of hardener particles in the oligomer melt occurs and what kinetic constants describe this process.
The figure shows typical interferograms obtained by combining ER with DADPS both in the isothermal holding mode, used to obtain data on mass transfer processes (Fig. 19). The step heating/cooling mode is used to build a phase state diagram (PSD) of the system. It was found that the phase equilibrium of the ER/DADPS system is characterized by a phase state diagram with crystalline equilibrium (Fig. 20). In accordance with the obtained data at low temperature, there is a partial dissolution of DADPS. In this case, near the crystal surface, the concentration of the saturated solution is established, which changes with temperature. At T ≥ Tm (Fig. 20b), the system is homogenized. The interferograms of the interdiffusion zone are characterized by a continuous change in the refractive index in the transition from one component to another.

Phase state diagram of the ED-20−DADPS system. Areas I, II, and III are the true solution-melt, crystalline state of DADPS, and chemical reaction, respectively. The diagram was constructed according to the compositions of coexisting phases established near the phase boundary (Fig. 20), determined by the interferometric method

Interferograms of interdiffusion zones of the DADPS−ED-20 system at temperatures: a 363 K (T ≤ Tm), b and c 453 K (explanations in the text)
Along with the component’s dissolution in the diffusion zone at elevated temperatures, the curing reaction of ED-20 proceeds, manifested in the interferograms by thickening the interference bands in the mixing region and an increase in the total number of bands in the system (Fig. 20c). We were able to identify two states of the system, which are characterized by different kinetic parameters of the diffusion of the ingredients (Fig. 21). At the initial part of the mixing process, a linear dependence of the position of the isoconcentration planes coordinate X on time t1/2 is observed, indicating a diffusion mechanism of mixing. In the course of further mass-exchange processes, transfer rates slow down due to copolymerization of the components, growth of the molecular weight of oligomers, and then practically stop due to the formation of a network of spatial bonds, which is reflected in the change in the slope angles of the dependencies X − t1/2.

Kinetic dependences of the movement of isoconcentration planes in the system DADPS−ED-20 at temperatures of 363 K (1, 1’), 373 K (2, 2’), 413 K (3, 3’), and 453 K (4, 4’). I and II are the diffusion regions of DADPC in ER and ER in DADPS, respectively
The calculated values of diffusion coefficients for the extreme concentrations of the systems (dissolution of DADPS in ER and ER in DADPS) are shown in Fig. 22. The apparent activation energies calculated from the values presented are 24.1 and 92.8 kJ/mol for the diffusion of DADPS into ER and ER into DADPS, respectively.

Temperature dependences of coefficients of mutual diffusion of DADPS in ER (1) and ER in DADPS (2)
Thus, it can be argued that the dissolution kinetics of DADPS in ER obeys diffusion patterns up to the temperature of the beginning of the chemical reaction of the epoxy oligomer curing.
2.4 Phase Equilibria and Interdiffusion in Adducts of Epoxy Oligomers
Figure 23 shows typical interferograms illustrating the spontaneous formation of interdiffusion zones during conjugation of thermoplastics with aERs of different compositions. It was found that at low degrees of conversion, α ≤ 0.15, which corresponds to the initial stages of curing of epoxy oligomers, in the whole temperature-concentration range, the blends are homogeneous at any ratio of components despite the gradient nature of compositions distribution.

Interferograms of interdiffusion zones at 453 K of the PMMA—aER system α = 0.1 (a) and α = 0.2 (b), where I is the interdiffusion zone, P is the phase boundary
As the degree of conversion increases, which is identical to the increase in the molecular weight of oligomers and the formation of branched molecular structures, the same structural and morphological changes are observed in all systems, regardless of the nature of the homopolymer monomer links. Starting from a certain degree of transformation of epoxy groups in the diffusion zone, enriched with aER, a phase boundary appears, separating the areas of dissolution of the adduct in the thermoplastic and thermoplastic in the adduct.
Increasing the degree of curing leads, on the one hand, to the formation of a phase boundary in the region of more concentrated solutions (Fig. 24b) for both PEI and aER, and, on the other hand, to a shift in the homogenization temperature (binodal dome) to a higher temperature region. Note that for such systems, two optical boundaries are identified on the concentration distribution profile.

Interferograms of interdiffusion zones at 293 K (a, c) and 493 K (b, d) of the PEI–aER system: α = 0.1 (a, b), (0.2 c, d), where I is the interdiffusion zone, P is the phase boundary, O is the optical boundary
The moment of phase boundary formation was most clearly recorded in the PEI–aER system. It was found that regardless of how the interdiffusion process was organized—under isothermal conditions, stepwise increase and/or decrease in temperature, as the temperature approached 373 K in the mixture of PEI with aER in the diffusion zone enriched with adduct (α = 0.1), a phase boundary appeared (Fig. 24), separating the regions of aER solutions in PEI from aER solutions of PEI. As the temperature increases, the phase boundary degenerates, and the diffusion gradient zone is again in the single-phase region. This means that the epoxy oligomer does not form a continuous network of spatial bonds at this degree of curing (Fig. 25).

Interferograms of interdiffusion zones at 373 K of the PEI–aER system α = 0.1 (a) and at 408 K α = 0.2 (b), where O is the optical boundary, I is the interdiffusion zone, P is the phase boundary
It should be noted that in the PEI–aER system up to a conversion degree of 0.3, the formation and dissolution of heterogeneous structures occur as a result of cyclic cooling and heating, while starting from α > 0.35, a network of chemical bonds is formed in the aER, which prevents dissolution of thermoplastic in aER and promotes swelling of the adduct. Fundamental changes in the PEI–aER system occur for adducts with α > 0.3. In this case, a phase boundary is formed, and the solubilities of PEI in aER and aER in PEI decrease sharply. Nevertheless, the composition distribution profiles in the interdiffusion zone are reproduced in the heating–cooling cycles. At α > 0.35, a network of chemical bonds is formed in the ER adduct, which prevents the dissolution of thermoplastic macromolecules in the epoxy polymer. The sol fraction of the adduct continues to dissolve in PEI.
Note that the processes of interdiffusion in partially cured epoxy oligomers have a common character both with thermoplastics and linear oligomers, solvents, and plasticizers. Specificity is observed only in quantitative values of diffusion constants and compositions of coexisting phases.
Thus, in all of the systems studied, partial compatibility of the components occurs mainly due to the dissolution of the adduct fraction in the thermoplastic, while the solubility of the thermoplastic in aER is negligible. Note that this information is of fundamental importance when modifying epoxy oligomers with thermoplastics and thermoplastics with epoxy resins. An increase in the degree of conversion leads to an expansion of the heterogeneous region, mainly due to a decrease in the solubility of the adduct in the thermoplastic and a decrease in the sol fraction in the epoxy polymer composition.
We should add to the above that in the mixtures of epoxy oligomer adducts with thermoplastics, the movement of isoconcentration planes occurred by the diffusion mechanism in strict accordance with the law X − t1/2 (Fig. 26).

Kinetics of motion of the PEI front in aER (I) and aER in PEI (II) α: −0.2 (1, 2), 0.1 (3, 4) in diffusion coordinates. The temperature of the experiments was 453 K
Binodal curves of phase state diagrams of systems for adducts with different degrees of conversion of epoxy groups were plotted according to the compositions of coexisting phases (Figs. 27 and 28). It can be seen that all partially cured mixtures are characterized by amorphous stratification diagrams. The increase in solubility with increasing temperature allows them to be attributed to the class of systems with UCST. It was experimentally possible to fix UCST directly only in the PEI-aER system at conversion degrees 0.1 and 0.2.

Phase state diagrams of the PEI-aER system during curing of ED-20. (1, 2)—binodal curves at different stages of the curing reaction. Conversion degree: 1—0.1, 2—0.2, the dashed line shows glass transition temperature of PEI–aER mixtures. Areas in the diagram: I—homogeneous, II—heterogeneous, III—vitrified

Phase state diagrams of PMMA-aER system during curing of ED-20. (1, 2)—binodal curves at different stages of the curing reaction. Conversion degree: 1—0.2, 2—0.3, the temperature of glass transition of PMMA–aER mixtures is shown with a dashed line. Areas in the diagram: I—homogeneous, II—heterogeneous, III—vitrified, IV—zone of thermodegradation of components
As was shown earlier, as the degree of conversion; hence the molecular weight of the adduct increases, the mutual solubility of polymers deteriorates mainly due to a decrease in the solubility of aER in the thermoplastic. This fact is clearly illustrated by isothermal cross-sections of phase state diagrams. In this case, the two-phase state regions of polymer mixtures are within the zone bounded by solubility isotherms. Extrapolation of these dependences to α → 1 shows that the system disintegrates into coexisting phases with compositions close to the pure components when fully solidified (Fig. 29). The dome of solubility isotherms, to which the critical parameters correspond (critical concentration and degree of conversion and, consequently, molecular weight at a particular temperature) is constructed taking into account that the initial systems are either completely compatible, as in the case of PMMA mixtures, or partially compatible as in the mixture of PEI with initial ER, where already at 293 K phase boundary formation was observed.

Isothermal cross-sections of DPS of the system aER-PEI (dependence of the solubility of components in the system aER–PEI on the molecular weight of aER) at temperatures: 1—293, 2—313, and 3—353 K. Regions: I—homogeneous, II—heterogeneous. Gray rhombuses indicate the calculated critical points
Figures 30, 31 and 32 show the concentration dependences of the mutual diffusion coefficients for mixtures of PEI and PMMA with aER of various degrees of conversion. For comparison, the concentration dependences of diffusion coefficients corresponding to the systems with initial linear oligomers are plotted.

Concentration dependences of interdiffusion coefficients (1–3) of the PEI–aER system at 453 K, where 1 corresponds to the initial ER, and 2 and 3 to ER adducts with α = 0.1 and 0.2

Concentration dependences of interdiffusion coefficients (1–4) of the PMMA–aER system at 453 K, where 1 corresponds to the initial aER, and 2, 3, and 4 to the ER adducts with α = 0.1, 0.2, and 0.3, respectively

Molecular weight dependences of reduced diffusion coefficients characterizing the rate of penetration of macromolecules of oligomers (1) and adducts (2) into the volume of thermoplastics, determined at 453 K. (Schematic representation of macromolecules is taken from [8], some additional points are taken from [15]). Structures: a flexible ball, b statistical long branches, c sparsely cross-linked, d densely cross-linked
One can see that the general tendency of change of interdiffusion coefficients with composition in spatially cross-linked systems is similar to linear solutions of oligomers and thermoplastics. All concentration curves Dv − φ are convex, and the values of interdiffusion coefficients change smoothly with a change in the composition of solutions. As the degree of conversion increases, the absolute values of interdiffusion coefficients in the region of true solutions of the diagram decrease. It is interesting to note that the transition from linear oligomers to spatially cross-linked adducts at α > 0.35 is accompanied by a decrease in their translational diffusion coefficients by 4 decimal orders of magnitude, from 10–7 to 10–11 cm/s2. As the temperature increases, the diffusion coefficients increase, and the range of solution compositions within which they can be determined expands.
A specific feature of this class of systems is the behavior of translational diffusion coefficients in the region of dilute thermoplastic solutions in adducts of epoxy oligomers. This peculiarity is related to the fact that in this temperature-concentration region, there are boundary curves, zones of two-phase state and labile solutions, and domes of binodales. The appearance of heterogeneous temperature-concentration zones at certain degrees of conversion leads, for quite understandable reasons [35], to a break in the concentration dependences of interdiffusion coefficients and a decrease in their values in the region of compositions close to those of coexisting phases, in the region of the so-called Frenkel heterophase states.
Note that all of the diffusion coefficient dependences shown in Figs. 30 and 31 and concentration dependences contain very limited information about the limiting partial diffusion coefficients of thermoplastics in epoxy oligomers adducts (the right branch of the binodal curve is adjacent to the adduct axis).
At the end of this section, we tried to perform a comparative analysis of the translational diffusion coefficients of linear epoxy oligomers and their adducts. For this purpose, we used the dependences of the translational diffusion coefficients on the values of the molecular masses of the macromolecules of linear oligomers and their adducts (Fig. 32).
One can see that in the entire MW range, the diffusion coefficients of linear oligomers are higher than the translational diffusion coefficients of adducts. This effect is related, as it seems to us, to the branching of the adduct macromolecules. The nonlinearity of the dependences excludes any traditional analysis [2] in terms of conformational behavior of the pre-converted epoxy oligomer macromolecules during their diffusion in the volume of thermoplastics.
2.5 Conclusion
Thus, we can expect that in the thermoplastic solutions we studied, three stages of the process will be realized in the formation of the reactoplastic phase structure: first, the stage of evolution or “migration of the binodal curve,” or rather the dome of the binodal curve, into the temperature-concentration region, where the figurative point of a given system is located; responsible for this stage is the kinetics of the increase in the molecular weight of the initial oligomer; secondly, the stage of solution macro-dissolution, formation, and growth of disperse phase particles of thermoplastic saturated with epoxy oligomer adducts; and finally, the stage of secondary phase decomposition occurs in the area of high degrees of transformation of the epoxy oligomer. At this stage, we should expect the formation of nanosized particles of dispersed phase distributed in the volume of thermoplastic macrophase and at their periphery in the matrix of the cured thermoplastic.
3 Phase Equilibrium and Structure Formation During Curing of Epoxy Compositions
Consideration of self-healing of epoxy curing materials should include the peculiarities of creating modern structural materials associated with the widespread use of a modification of thermoset resins with thermoplastic polymers. The phase structure created as a result of the formation of a spatial network of chemical bonds in the modified systems can not only determine the final performance characteristics but also influence the principles and processes of self-healing.
3.1 Introduction
A modern and, apparently, long-term trend in the creation of new structural polymeric materials is the use of mixed compositions based on thermoset resins modified with thermoplastics: polysulfone, polyethersulfone, polyetherimides, etc. [47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65]. The creation of new engineering plastics based on such polymer blends is of fundamental importance and determines the ways of development of structural polymeric materials with high mechanical and adhesive properties, thermal and electrical properties, chemical resistance, and good technological characteristics.
Now, it is impossible to consider blends only as a direction in polymer modification to expand the range of existing material grades and applications. In fact, polymer–polymer compositions form their own class of materials with diverse, sometimes specific properties and specific structural-morphological and phase organization, providing their wide application.
In order to obtain composite materials possessing a complex of required physical and mechanical characteristics, it is necessary to form a given phase structure. For the controlled course of the processes of “self-assembly of macromolecules” into micro- and macro-dimensional phase formations initiated by curing reactions of thermoset resins, information on phase equilibria, diffusion transfer coefficients, the evolution of boundary curves of phase diagrams at different stages of mesh structure formation becomes in demand [62,63,64,65,66,67].
The analysis of the technology of engineering structural materials in general and on the basis of thermo- and thermosetting plastics mixtures, in particular, showed that the synthesis of structural materials is multistage, and the stages are separated in space and time. Traditionally, there is a stage of binder preparation, i.e., mixing the components; a stage of prepreg formation, i.e., impregnation of the fiber filler with the binder; and, finally, a stage of binder curing in a given product. It is at this stage that the main chemical and phase transformations take place, and the required structural and morphological organization of the material as a whole is formed.
At first sight, it seems that each stage of synthesis technology is connected with different physical and chemical processes; so, mixing—with the kinetics of dissolution of polymer particles in the oligomer, impregnation—with the viscous flow of solution in the porous structure of fiber filler and its adsorption interaction with filler surface, curing—with the kinetics of the reaction of spatial grid formation in a binder and so on. However, from the fundamental point of view, the most important thing is the information about phase diagrams, translational diffusion coefficients, thermodynamic parameters of interaction of components at different stages of preparation, and chemical transformation of the reactive component. It is this information, as will be shown below, that allows one to quantitatively describe and predict all the structural-morphological transformations occurring with mixtures, solutions, and dispersions during the entire technological cycle of the preparation of structural materials based on thermo- and thermosetting plastics in a general form.
3.2 Phase Equilibria and Interdiffusion in the Epoxy Oligomers—Thermoplastics Systems
3.2.1 Dissolution Kinetics
Among the methods for studying the kinetics of polymer dissolution, the simplest informative method is the optical wedge or multi-beam microinterferometry [19, 65, 68]. This method is used to record the kinetics of changes in the distribution profile of the refractive index (concentration) spontaneously occurring in the interface zone of the oligomer and thermoplastic phases. Figure 33 shows typical interferograms of interdiffusion zones of homogeneous (Figs. 33a and 34a), two-phase amorphously stratified (Fig. 33b), and two-phase crystallizing mixtures (Fig. 34b). It can be seen that at temperatures above the critical and melting temperatures, the concentration distribution profiles are determined in the entire range of compositions from one component to the other. At temperatures below the melting point of the thermoplastic (in this case, polyethylene glycol (PEG)), only a portion of the concentration profile corresponding to the dissolution of crystallites in the oligomer is observed (Fig. 34b). At temperatures below the upper critical solution temperature (UCST) in the diffusion zone, there is a phase interface separating oligomer solutions in thermoplastic from thermoplastic solutions in the oligomer. Concentrations corresponding to compositions of coexisting phases are established near the interface for systems with amorphous and crystalline equilibrium. Special thermokinetic studies of the concentration distribution in the diffusion zones have shown that the compositions of coexisting phases do not depend on the observation time and are quantitatively reproduced in the heating and cooling modes. This indicates the equilibrium and reversibility of the boundary concentrations measured in this way.

Interferograms of interdiffusion zones of ED-20–PSU (a) and E44–PSU (b) systems at 220 °C. V—diffusion zone, P—phase boundary, I—diffusion zone of EO in PSU, II—diffusion zone of PSU in EO. Diffusion time 64 min. E44–EO with Mn = 1.9 kDa

Interferograms of interdiffusion zones of the E44–PEG systems at 120 (a) and 40 °C (b). Mn PEG = 10 kDa. The notations correspond to Fig. 33
The character of the concentration profile shows that the movement of isoconcentration planes within the interdiffusion zones obeys the law X(φ) − √t, which indicates a diffusion mechanism of mixing epoxy oligomers (EO) with thermoplastics at all temperatures and regardless of the phase and physical state of polymers.
Phase diagrams
Binodal curves and liquidus lines of phase diagrams for mixtures of thermoplastics with EO of different molecular weights were constructed using the temperature dependences of the compositions of coexisting phases. Using the methods for analyzing phase equilibria described in [69], generalized diagrams of the phase and physical states of the systems were constructed using fragments of the binodal curves (Fig. 35).

Generalized phase diagrams of EO—thermoplastic systems: E44–PSU (a), ED-20–PS MnPS = 35 kDa (b), E49–PVA (c), E44–PEG (d); 1—binodal curve, 2—spinodal curve, 3—change of glass transition temperature by Fox equation, 4—phase glass transition temperature, 5—liquidus. Areas: I—true solutions (solutions-melts), II—metastable states, III—labile solutions zone, IV—glassy state, V—thermal destruction, VI—crystalline state
The influence of the molecular weight (MW) of epoxy oligomers on their compatibility with thermoplastics is most clearly demonstrated by isothermal cross-sections of phase diagrams (Fig. 36). In the coordinates φi − 1/Mn (φi is the composition of the ith coexisting phase), the experimental points form two lines framing the two-phase state region of the systems and intersecting at the critical point. It is shown that this construction of the phase diagrams makes it possible to determine the critical composition of the mixture and the molecular weight of the oligomer and polymer.

Dependence of component solubility in the systems: E44–PSU on the molecular weight of EO at 1–100, 2–250 °C (a) and ED-20–PS on the molecular weight of PS at 1–80, 2–120 °C. The arrows indicate the molecular weights of loss of compatibility at these temperatures
As the MW of the components increases for all systems, the heterogeneous region expands. It is important that complete loss of solubility of thermoplastic in epoxy oligomer occurs earlier than oligomer in thermoplastic.
The position of the critical point of the phase diagrams with amorphous stratification we estimated using Alexeev’s diameter. It was found that the critical concentration (φcr), as the molecular weight of EO or thermoplastic increases, shifts to the region of compositions enriched with a lower molecular component in full accordance with the equation
following from the classical Flory–Huggins-Scott theory of polymer solutions [47]. Here, ri is the degree of polymerization of the ith component. It is shown that using experimentally found value φcr and having information about the molecular mass of one of the components, it is possible to solve the inverse problem—to estimate the value of the degree of polymerization and, consequently, the molecular weight of the second unknown component.
Based on the compositions of the coexisting phases, we calculated the paired parameters of the interaction of the components, the temperature dependences of which we used to calculate the critical temperatures (the calculation scheme is shown in Fig. 37). The critical value of χ was calculated using the equation

Temperature dependences of the paired interaction parameter for the EO–PSU systems. EO: 1—1.9, 2—2.9, 3—5.1 kDa. The arrows show the UCST
We also used the values of χ extrapolated to high temperatures to construct a dome of phase diagrams in the region of upper critical solution temperatures (dotted curves in Fig. 35a).
It was found that the dependence of the critical temperature and the Flory–Huggins parameter on the molecular weights of the components for all the systems studied are linear.
3.2.2 Interdiffusion
The concentration dependences of interdiffusion coefficients (Dv) (Fig. 38) are determined by two parameters: the distance of figurative points of the systems from the critical temperature and the ratio of the molecular weights of the components. Figurative point is a point on the phase diagram corresponding to the investigated system of a certain composition at a certain temperature. Thus, at a temperature above UCST and the molecular weight of the oligomer is much less than the molecular weight of the thermoplastic, the change in the diffusion coefficient with a change in the composition occurs according to the curves with a maximum (Fig. 38a). Near the critical point, a minimum appears on the concentration dependences, the position of which coincides with the position of UCST (Fig. 38b). A further decrease in temperature (below the UCST) leads to the appearance of a break in the concentration dependence of interdiffusion coefficients. At these temperatures, a decrease in the coefficient is observed as the solution composition approaches the binodal curve (Fig. 38c). The reasons for such behavior of the diffusion coefficients are associated with a change in the chemical potential of the solutions as the figurative point of the system approaches the spinodal concentration.

Concentration dependences of interdiffusion coefficients for the systems: E40–PSU (a), ED-20–PS MnPS = 35 kDa (b), E40–PS (c), E44–PEG MnPEG = 10 kDa (d). Diffusion temperatures are indicated next to the corresponding curves. Dotted lines are binodal curves of phase diagrams. ED-20 (MW = 0.36 kDa), E40 (MW = 0.48 kDa)
An increase in MW of the components generally leads to a decrease in Dv, except for the EO–PS system, wherein the area of concentrated and moderately concentrated solutions of polystyrene in the epoxy oligomer, the diffusion coefficients do not depend on MW of the high-molecular-weight component.
In the region of dilute and semi-dilute solutions, the translational mobility of dissolved polystyrene macromolecules in epoxy oligomer is described by the equation Dv = D0M−b with exponent b = 0.5, which indicates a small deviation of the system from Θ-conditions.
The increase in the molecular weight of polyglycols also leads to a decrease in the interdiffusion coefficient. In this case, on the dependence lgDv − lgMn, there is a kink, and the index of degree changes from 0.5 to 1, which indicates the formation of a mesh of meshes in the solution. For PEG, the molecular weight between the meshing nodes is ≈5000, which agrees with the literature data.
The temperature dependences of the coefficients of interdiffusion in the coordinates of the Arrhenius equation are linear throughout the temperature range studied. The exception is the system E44–PEG (φEO → 1), for which a kink is observed at the temperature of the Tll-transition. The concentration dependences of the apparent activation energies of interdiffusion (Fig. 39) are located between the activation energies of self-diffusion of the diffusion system components.

Concentration dependences of apparent activation energies (E) of interdiffusion of the systems: ED-20–PSU, ED-20–PS, E44–PEG. Dotted line marks E of self-diffusion of individual components
Thus, it has been established that all initial systems are characterized either by amorphous stratification diagrams with the UCST or by crystalline equilibrium. Generalized phase and physical diagrams have been constructed, which allow predicting a priori the structural-morphological and relaxation state of mixtures of any composition in a wide range of temperatures and molecular weights of the components. It is shown that the mechanism of mixing epoxy oligomers with thermoplastics is diffusive. Diffusion coefficients in the region of temperatures where operations of mixing of components and prepreg production are traditionally performed vary in the range of values from 10–7 to 10–8 cm2 s−1. Using these values of diffusion coefficients, it is also possible to estimate a priori the time of diffusion relaxation—the dissolution of the thermoplastic particle in the epoxy oligomer matrix.
Note that in [63, 65], the authors discuss the problem of the influence of mutual solubility of oligomers and polymers on the structure and phase state of the cured composition in sufficient detail. In the framework of the information described above, this problem is reduced to the determination of the position of the figurative point on the temperature-concentration field of the phase diagram.
3.3 Phase Equilibria and Interdiffusion in the Systems Thermoplastics—Epoxy Oligomers Adducts
This section summarizes the results of modeling studies in which an attempt was made to obtain information about the evolution of phase equilibria and translational mobility of components during the formation of spatially cross-linked structures in mixtures of thermoplastics with thermoset resins. A set of epoxy oligomer adducts (ad.EO, aEO) of different degrees of conversion (α) obtained in ED-20 curing reactions with diamine curing agent was used as one of the mixture components whose content in the reaction system was less than stoichiometric. In each case, the completeness of the amino-group conversion was identified by infrared spectroscopy. At present, this method of modeling curing reactions is generally accepted in the practice of polymer materials science.
3.3.1 Mixing Kinetics
During the transition from linear EO to their adducts in the interdiffusion zones, which spontaneously arise at the conjugation of thermoplastic and adduct phases and characterize the process of their dissolution (Fig. 40), one observes the same type of concentration-morphological changes. First, starting from a certain degree of precursor conversion, there appears a clearly identifiable phase boundary separating the areas of diffusion dissolution of the adduct in thermoplastic (I) and thermoplastic in partially cured EO (II). Second, it was found that before the formation of spatially cross-linked structures in adducts (α < αgel, here αgel is the degree of precursor transformation corresponding to gel formation), the phase interface is observed only in a limited temperature range. As a rule, for this adduct state, it degenerates when the critical solution temperature of the components is reached in the process of increasing temperature. After the formation of gel structures (gel) in EO adducts, however, the phase interface remains unchanged at all temperature and time conditions of the interdiffusion process up to the temperatures of epoxy polymer decomposition. Thus, by scanning the interdiffusion zone by temperature, the points of true gel formation in the matrices of EO adducts were determined. It was found that, within the error range, the gel varies in the range from 0.35 to 0.45 and depends little on the nature of the thermoplastic, i.e., it is a characteristic of the cured oligomer.

Interferogram of the interdiffusion zones of the system aEO–PEG. Adduct conversion rate α = 0.65, process temperature T = 100 °C, observation time 64 min. P—phase boundary, I—diffusion zone of aEO into thermoplastic, II—diffusion zone of thermoplastic into aEO. The curing agent is DETA
Thirdly, an increase in the degree of conversion of the adduct α > αgel leads to a decrease in solubility of both thermoplastic macromolecules in the cross-linked precursor phase and the sol fraction of ad.EO in the polymer matrix. At high conversion rates α > 0.7, zone I degenerates. For such adducts in the interdiffusion zone, only the phase interface and the area of dissolution of thermoplastic macromolecules in the spatially cross-linked phase of the epoxy polymer are observed. Obviously, in this case, we can speak only about “one-side” diffusion or about the swelling of cross-linked EO in the thermoplastic melt.
It has been noted that the described processes of interdiffusion in EO adducts are of a general character both for their mixtures with thermoplastics and solvents, plasticizers, and oligomers. The specificity of the systems is manifested only in quantitative parameters of the process: the size of interdiffusion zones, the rate of movement of isoconcentration planes, numerical values of compositions of coexisting phases, and the initial phase state of the mixture.
Of principal importance for all these systems is the fact that the compositions of coexisting phases, which are established near the interphase boundaries, and the isothermal kinetics of the movement of isoconcentration planes in the adduct and thermoplastic phases are quantitatively reproduced in the stepwise temperature increase and decrease modes. This allows us to speak, first, about the reversibility of the information obtained and, second, to use it to construct generalized phase equilibrium diagrams of the adduct-thermoplastic systems.
It should also be noted that in EO adducts, the sizes of interdiffusion zones change with time in strict accordance with the X − √t law, i.e., the diffusion mechanism of mixing the polymers brought into contact is also realized in these systems at different degrees of curing.
3.3.2 Phase Diagrams
Binodal curves of amorphous separation diagrams for mixtures of ad.EO with PSU, PS, PVA, and amorphous-crystalline equilibrium diagrams for ad.EO–PEG mixtures, modeling the phase separation at different stages of the precursor crosslinking (Fig. 41a–d), were plotted using the compositions of coexisting phases. It can be seen that the solubility of ad.EO in thermoplastics, as well as in the initial linear systems, increases with increasing temperature, i.e., these systems also belong to the kind of systems with UCST. However, it is possible to directly register the UCST position only for adducts with a low degree of conversion, up to 0.25. At α > 0.3, the UCST of the systems appears in the region of thermodegradation of the epoxy component. It is interesting to note that the nature of the curing agent has little effect on the position of the binodal curves of the adherent-epoxy-thermoplastic systems.

Phase diagrams of aEO–PSF (a), aEO–PEG (b), aEO–PS (c), and aEO–PVA (d) systems. 1—liquidus lines, 2—binodal curves. The curing agent is DETA. Dotted line marks the calculated phase diagrams. Numbers in the curves correspond to the degree of conversion of the adduct. I and II—regions of true solutions and heterogeneous state, respectively. The arrows indicate the direction of the evolution of the phase diagram with increasing degree of conversion
As the degree of transformation of EO increases, the size of the heterogeneous state region in the amorphous stratification diagrams increases, and the mutual solubility of the components of the mixtures decreases. This is manifested to a greater extent at the position of the left branch of the binodal curve, corresponding to the solubility of ad.EO in thermoplastics. The solubility of homopolymers in ad.EO (the right branch of the binodal curve) changes to a much lesser extent.
Obviously, this fact should be taken into account when choosing the conditions of structure formation in these temperature-concentration regions of the phase diagrams.
The change in the mutual solubility of adducts in thermoplastics as a function of the conversion degree is most pronounced in the isothermal sections of the phase diagrams (Fig. 42). The dome of solubility isotherms (dotted lines), to which the critical system parameters (αcr, φcr, UCST) correspond, is constructed taking into account the fact that the initial systems are either completely or partially compatible. It can be seen that the critical value of the degree of adduct transformation corresponding to the beginning of the phase separation of the solutions is significantly lower than the gelation degree. This means that the phase separation of the precursor and thermoplastic adduct solutions may begin before a continuous network of cross-links is formed in the precursor and curing agent phase. In fact, this is the specifics of the intermolecular interaction between epoxy oligomers and polymers.

Isothermal cross-sections of phase diagrams for systems of aEO with PSU (a) and PEG (b)
Extrapolation of these dependencies to α → 1 shows that at the content of the curing agent in the compositions close to stoichiometric (at 100% degree of conversion of epoxy and amine groups), one should expect complete stratification of the system into coexisting phases whose composition is close to the cured epoxy polymer and thermoplastic.
The above data on the solubility diagrams of precursor adducts and linear thermoplastics can be used to estimate their average molecular weight. Using the method of critical compositions [see Eq. (10)] and numerical values of the parameters of the critical points of specific systems, the molecular weight of the adducts of epoxy oligomers was calculated (Fig. 43). One can see that as the degree of conversion increases, the effective molecular weight of ad.EO increases quite rapidly, reaching a value of ≈40 kDa during gelation. It is interesting to note that the numerical values of the effective molecular weight of ad.EO do not depend on the nature and molecular weight of the reference polymer, i.e., thermoplastic. The proposed technique makes it possible to obtain quantitative information on the molecular weight of adducts, which is so necessary for theoretical analysis of the evolution of phase equilibria in curing reactions.

Dependence of the effective molecular weight of the adducts on the degree of conversion. Systems: 1—aEO–PVA, 2—aEO–PS
The obtained fragments of phase diagrams of the systems ad.EO—thermoplastics can be used for predicting the UCST of cured epoxy polymers in the region of high temperatures, where the operation of engineering structural plastics is possible. For this purpose, it is possible to take advantage of the construction of temperature dependences of the dimensions of the sections connecting the intersection points of the isotherms with the binodal curves ∆φ(T) (corresponding to the distance between the compositions of coexisting phases) and their extrapolation to the zero value of ∆φ (Fig. 44). Figure 45 shows that the critical temperatures thus obtained change with changes in the degree of precursor transformation according to linear dependences, which can be extrapolated in the region of low and high degrees of curing, and low and high temperatures.

Temperature dependence for aEO–PS systems: 1—0.07, 2—0.22 и 3—0.29

Dependence of critical temperatures on the degree of conversion for mixtures of aEO with PEG (1), PSU (2), and PS (3)
3.3.3 Interdiffusion
Since the area of compositions with high precursor content is of the greatest interest when modifying thermoset resins, particularly epoxy polymers, we focused our attention on determining the so-called limiting diffusion coefficients characterizing the translational mobility of dissolved thermoplastic macromolecules in a partially cured oligomer matrix (Fig. 46). One can see that as spatially cross-linked structures form in the epoxy oligomer matrix, homopolymer diffusion coefficients decrease. The most intense decrease of thermoplastics diffusion coefficients occurs in the region of gel formation in the epoxy oligomer matrix. For PS, PSU, and PVA macromolecules, diffusion coefficients reach values 10–11–10–12 cm2 s−1, typical for translational mobility of macromolecules near the glass transition temperature [47].

Dependence of the relative thermoplastic diffusion coefficient into the EO adduct on the degree of conversion for aEO with PS (1), PVA (2), PSU (3), and PEG (4) systems
Some difference in the dependences of thermoplastic diffusion coefficients on the degree of curing of the oligomer is related to the position of the figurative point of the system relative to the “expanding” binodal curve. According to the phase diagrams (Fig. 41), during the curing of the epoxy oligomer, the binodal curve gradually shifts toward the infinitely diluted thermoplastic solutions in the epoxy oligomer adduct, approaching the figurative point of the system located on the isoconcentration plane, by the movement of which we determined the diffusion coefficient value. This leads to an increase in the contribution of thermodynamic non-ideality of the solution and, as a consequence, to a drop in the diffusion coefficient.
Thus, it can be argued that the phase disintegration of EO solutions, curing agent, and linear polymers, initiated by the reactions of formation of the spatial bonding network in the epoxy oligomer matrix, in the growth and formation of thermoplastic disperse phase particles, will occur in conditions with continuously decreasing translation diffusion coefficient of macromolecules and continuously increasing oversaturation both in the particle volume and in the dispersion environment surrounding it.
3.4 Structure Formation During Curing of Mixtures of Epoxy Oligomers with Thermoplastics
In contrast to the previous chapters, which analyzed the phase and diffusion characteristics of blends of thermoplastics with epoxy oligomers and epoxy polymers whose chemical nature remained unchanged during the measurement process, this section presents the results of structural-morphological, optical, rheological, thermochemical, and phase studies of the same systems, but recorded during the formation of cross-linked structures in the epoxy oligomer matrix. In this section, we tried to answer the question of how widely the results of “static” measurements can be used in the transition to the kinetics of the structure formation process initiated by chemical reactions of the formation of spatial reticulated structures.
Despite the obvious importance of quantitative information on the phase equilibria of the initial systems and systems at different stages of their transformations, only fragments of phase diagrams (mainly in the region of the binodal curve dome) are found in the literature, and the proposed models of boundary curve evolution are based not on specific experimental data but are created by analogy with epoxy-rubber systems. Such models have been proposed mainly for systems with amorphous stratification, characterized either by UCST (Fig. 47a) or low critical solution temperature (LCST) (Fig. 47b) [65]. Obviously, in the form presented in Fig. 47, they allow us to speak only about the most general trends in changes in the phase state of the reaction mixture. It is impossible to obtain comprehensive information on the changes in the compositions and volumes of the coexisting phases, dispersity and nature of the inclusion particles and matrix phase, state of the interphase transition zones, and presence of macro- and microcomposite inhomogeneities in the system from the materials of such studies.

Evolution models of the boundary curves of phase diagrams with UCST and LCST during curing of thermoset: 1 and dashed lines—model binodal curves, 2 and dotted lines—concentration dependences of glass transition temperature for mixtures of PSU and PES with adducts. The arrows indicate the directions of the shift of the binodal curves with an increasing degree of conversion [65]
Below, by the example of two systems characterized by amorphous PSU–EO stratification and complex amorphous-crystalline PEG–EO equilibrium, the current state of this area of phase transformations of polymer systems is considered.
3.4.1 General Remarks
The starting point for building a model of the structure formation process in our case is the information of the phase diagrams of the EO–PSU, ad.EO–PSU, PEG–EO, and PEG–ad.EO systems and the experimental fact of constancy of the selected mixture compositions and reaction temperature during the entire curing process.
As an example, Fig. 48 shows in the temperature-concentration field of the diagram the position of some figurative points of mixtures of EO with PSU, which further investigated the kinetics of structure formation during curing of ED-20. It can be seen that at the beginning of the process, the initial mixtures of all selected compositions (5, 10, and 15 wt.% PSU) are in the area of true solutions of the phase diagram. Estimates from the dependences of the critical temperatures on the degree of conversion (Fig. 45) showed that their distance from the UCST reaches ~200 °C.

Phase diagrams of ad.ED-20–PSU systems. α: 1—0.29; 2—0.33; 3—0.5; 4—0.17; 5—0.63. The curing agent is PEPA. Dotted line indicates the calculated phase diagrams.
 —figurative points of the systems under study. I and II are the regions of true solutions and heterogeneous state, respectively. The arrows indicate the directions of phase diagram evolution with increasing α
—figurative points of the systems under study. I and II are the regions of true solutions and heterogeneous state, respectively. The arrows indicate the directions of phase diagram evolution with increasing α
When spatially cross-linked structures are formed in such solutions as a result of the interaction of functional groups of the oligomer and curing agent molecules, the thermodynamic compatibility of the components deteriorates [63], which manifests itself in the approach of the binodal curves to the figurative points of the mixtures (shown by arrows in Fig. 48). At the degree of transformation ~0.2, the dome of the binodal curve reaches and then crosses the process isotherm (160 °C). Thus, figurative points of given compositions appear under binodal curves in areas of labile and metastable states, which, naturally, leads PSF solutions in EO to stratification. The specific mechanism of delamination—spinodal or nucleation—depends on the ratio of the critical point composition to the given mixture composition.
Further growth of the mesh density in the epoxy oligomer matrix will be accompanied by a shift of the left branch of the binodal curve to the region of dilute solutions of EO reaction products in PSU and the right branch to the ordinate of the epoxy three-dimensional polymer. Obviously, the degree of supersaturation of such solutions with an increasing degree of oligomer conversion should increase, thus creating the necessary conditions for further (secondary) phase separation of mixtures. It is essential that these processes occur in the heterogeneous region of the phase diagram.
It can be assumed with a high degree of probability that after the system reaches the glass transition of the epoxy matrix, the chemical processes of formation of spatially cross-linked structures will slow down and, as a consequence, the phase separation rate will decrease.
The phase diagram (Fig. 48) also follows the trend of changes in the compositions of the coexisting phases of the matrix and dispersed inclusions, the proportion of the dispersed phase, which can be estimated by the lever rule. It is interesting to note that, since the right branch of the binodal curve shifts insignificantly with increasing degree of conversion, the main changes in the phase structure should be expected in changes in the composition of dispersed phases.
In contrast to the system considered above, mixtures of EO and PEG in the initial state are characterized only by crystalline equilibrium; more precisely, one can assume that the UCST for the solutions of these polymers is below the liquidus line. The situation is different for the adducts of epoxy oligomers. The formation of a network of chemical bonds leads to the fact that UCST is higher than Tm, and the phase diagram of such systems is characterized by a complex amorphous-crystalline equilibrium (Fig. 49). Obviously, this imposes a certain imprint on the patterns of phase transformations, which can proceed differently in different zones of the phase diagram. Thus, Fig. 49 shows that mixtures containing 15 wt.% PEG at all temperatures (20, 60, 80, and 120 °C) at the beginning of the process are in the region of true polymer melt solutions. After the beginning of the curing reaction, the binodal curve crosses the liquidus line, the region of the two-phase state of the mixtures increases in size, and the right branch of the binodal curve gradually approaches the figurative point of the given mixture. Finally, it crosses it, which creates conditions for amorphous stratification of the system.

Phase diagrams of the ad.ED-20–PEG systems. 1, 2’, 3’, 4'—liquidus lines. 2, 3, 4—binodal curves. The curing agent is DETA. α: 1—0; (2, 2’)—0.43; (3, 3’)—0.58; (4, 4’)—0.65.
 –figurative points of the systems; I, II, III—regions of true solutions, amorphous stratification, and crystalline state, respectively
–figurative points of the systems; I, II, III—regions of true solutions, amorphous stratification, and crystalline state, respectively
It is interesting to note that in the region of low temperatures (below the PEG melting point), one should expect a phase disintegration associated with the intersection of figurative points with the liquidus line, i.e., with the release of the PEG crystal phase also initiated by the chemical crosslinking reaction of linear oligomer molecules.
3.4.2 Kinetics of Structure Formation
The methods of photocolorimetry, viscosity, and DSC revealed that in all systems at different curing temperatures, the optical density of the mixtures changes according to S-curves (Figs. 50 and 51). Regardless of the phase nature of the disperse phase particles (particles of amorphous PSU or crystallites of PEG), the following is clearly identified: the induction period τind, the stage of a rapid increase in turbidity (the phase separation of the solution itself), and the stage of the steady state of the system when the optical density of mixtures does not change.

Kinetic dependences of relative optical density of curing system EO-20–PSU with PSU = 10 wt.% and Tcur: 1—80, 2—100, 3—120, and 4—160 °C (a) and Tcur = 80 °C and PSU: 1—5, 2—10, and 3—15 wt.% (b)

Kinetic dependences of the relative optical density of the curing system ED-20–PEG with PEG = 15 wt.% and Tcur: 1—20, 2—40, 3—60, 4—80, and 5—120 °C. The curing agent is MFDA
The parameters of the kinetic curves are significantly influenced by the temperature and the thermoplastic content of the composition. As a rule, with increasing temperature and concentration of PSU and PEG, the duration of the induction period decreases.
The duration of the induction period can be considered as a characteristic kinetic parameter of the system since, during this time, such molecular-chemical changes occur in the solutions, which lead to a shift of the dome of the binodal curve to the area of the figurative points of the mixtures. Indeed, the temperature dependence of τind, like any kinetic parameter, is described by the exponential dependence τind = Bexp(E/RT), where B is a constant and E is the effective activation energy, reflecting the set of different phenomena associated with the beginning of the phase separation of the mixture. The experimental points for all the systems studied in ln τind − 1/T form a single straight line (Fig. 52), and the effective activation energy of the process calculated from these data of 48 kJ mol−1 (PSU), 42 kJ mol−1 (PEG), and 45 kJ mol−1 (PVA) is close to the activation energy of curing of MFDA epoxy oligomers, which according to various authors varies in the range from 45 to 62 kJ mol−1 [3]. These facts clearly indicate a single mechanism of structure formation in mixtures of epoxy polymer—thermoplastic, the limiting stage of which is the kinetics of change in the molecular weight characteristics of the epoxy oligomer during its curing.

Temperature dependence of the induction period time of phase separation of the EO-PSU system in the coordinates of the Arrhenius equation. PSU concentration: 1—5, 2—10, and 3—15 wt.%
Figures 53 and 54 compare kinetic curves of changes in the optical density of solutions, the relative viscosity of mixtures, and the degree of conversion calculated from DSC data. It can be seen that the course of the dependence η/η0(t), presented in the coordinates of the equation η/η0 = Rtn, is similar to that described for many other curing oligomeric compositions in the monograph [3]. The viscosity growth curve can be approximated by three linear sections with different values of n. Following the concept outlined in [3], it can be assumed that in the initial rather short section (n ≈ 0.5), linear growth of the molecular weight of macromolecules prevails. At the second stage (n ≈ 3), the process of formation of fragments of three-dimensional structures begins to play a major role, as a result of which the system rather quickly reaches the point of gel formation (t*) (at t > t* n ≈ 10).

Dependence of relative optical density (1), relative viscosity (2), and relative degree of conversion (3) on curing time for ED-20–PSU system with a concentration of PSU = 15 wt.% and Tcur = 120 °C. See details in the text

Kinetic curves of changes in relative optical density (1–3) and relative degree of conversion (1’–3’) for ED-20–PEG system. Concentration of PEG = 15 wt.%, Tcur: 1 and 1’—20, 2 and 2’—40, 3 and 3’—60 °C
Comparison of kinetic curves of changes in optical density, viscosity, and degree of conversion clearly shows that the phase disintegration of the EO–PSU system occurs already at the first stage of the process at αcr < 0.15. Gel formation occurs at αgel ≈ 0.4, when the system is in the two-phase state. A change in the composition of the mixtures affects the length of the induction period. Thus, at the PSU content of more than 10% (corresponds to the critical composition of the system), the phase disintegration occurs already at the first stage of the process, whereas in mixtures with a small PSU content of ~5% (φ < φcr)—near the gelation point.
Thus, the onset time of phase separation, and hence the molecular-kinetic state of the system, is determined by the radius of curvature of the binodal curve dome, the critical composition, and the difference between the curing temperature and the critical temperature of the initial system. The difference in these temperatures determines the degree of conversion of the onset of phase separation. The higher this difference, the higher the influence of the chemical bonding network on the formation and growth of dispersed phase particles.
It is known that under isothermal conditions, the kinetics of mesh formation in EO can be satisfactorily described in the coordinates of the Avrami equation [70]
where X is the fraction of the substance transferred to the new phase during time t and k is the parameter that takes into account all temperature-dependent terms. It was shown that the Avrami degree index (n) in the EO—curing agent systems varies in the range from 0.95 to 2.0, depending on the nature of the reagents and reaction conditions [26]. The authors of these works attributed the obtained result to the nucleation of microgel structures in the oligomer during its curing, which formed a continuous network of transverse bonds in the final product.
An attempt was made to analyze the kinetics of curing epoxy oligomer in a mixture with thermoplastics according to DSC data and the kinetics of phase separation of the same systems according to changes in optical density (Figs. 55 and 56) in the framework of Avrami approach. It can be seen from the figures that the experimental points are satisfactorily described by Eq. (12).

Kinetics of change in the degree of conversion in the coordinates of the Avrami equation of the system ED-20–PEG. Concentration of PEG = 15 wt.%, Tcur: 60 (1), 40 (2), 20 °C (3)

Kinetics of changes in optical density in the coordinates of the Avrami equation of the system ED-20–PEG. Concentration of PEG = 15 wt.%, Tcur = 60 °C
It is interesting to note that the kinetic curves of changes in the degree of conversion are characterized by the Avrami degree indices close to n = 1 and are not sensitive to changes in temperature, mixture composition, or the type of phase separation of the system. This fact complements the results obtained above on the effective activation energies of the curing process calculated from the extent of the induction period of the optical density change curves. Now, it can be stated that the mechanism of the chemical reaction of epoxy oligomer curing with amine curing agent does not change when transitioning to mixtures of EO with thermoplastics.
The Avrami degree indices for the phase separation curves obtained from optical density measurements vary from 1.7 to 5.8. The correlation coefficient reaches values of 0.96.
The kinetic curves of changes in optical density reflect the total process associated with both the formation of the chemical bond network and the formation of the dispersed structure of samples. Nevertheless, it can be argued that the kinetics of the structure formation process in mixtures of epoxy oligomers with thermoplastics can be quantitatively described within a unified approach accepted in the chemistry and physics of mesh polymers [26].
3.4.3 Structure of Cured Systems
Typical SEM images illustrating the structural and morphological organization of the cured epoxy polymer-thermoplastic systems are shown in Figs. 57 and 58.

Morphology of ED-20–PSU system. Chipping of the sample. Concentration of PSU: 5 (a), 10 (b), and 15 wt.% (c)

Morphology of ED-20–PSU blend. Chipping of the sample. Concentration of PSU 10 wt.%. The insets show fragments of the spectra of the characteristic X-ray emission of the microphases
It can be seen that all of them are characterized by a “matrix-inclusion” type structure. The specifics of each system are related to the size of the particles, their size distribution, the volume fraction of the dispersed phase, and the mutual arrangement of the particles of the dispersion medium.
At low thermoplastic content (less than 5 wt.%), practically for all compositions, uniform distribution of spherical polymer particles over the volume of cured epoxy polymer is observed (Fig. 57a). Particle distribution curves are unimodal; the average particle size varies in the range of 0.5–2 μm. According to X-ray microanalysis data for EO–PSU mixtures, the matrix consists mainly of epoxy polymer and inclusion—polysulfone.
A fundamentally different structure is formed in compositions with a high content of thermoplastics. For example, in mixtures with PSF content of more than 10 wt.% (Fig. 57b) at low magnifications, it is clearly seen that the system is macro heterogeneous and, despite the relatively small thermoplastic content, is in the region of phase inversion. Elemental analysis of the cross-sectional chip surface of each macrophase showed that the “dark” phase (by secondary electron contrast) is depleted in PSU (6%), while the “light” phase is enriched in it (20%).
Under higher magnification, we can see that the “dark” macrophase (Fig. 58a) has a structure similar to that of the mixture shown in Fig. 57a. The “light” macrophase (Fig. 58b) contains the dispersed phase in the form of spherical particles. Local microanalysis showed that the spherical inclusions contained mostly epoxy polymer, while the dispersed medium was polysulfone (≈94%).
Such a complex multilevel structure of the mixture makes it possible to reconstruct the evolution of phase separation during the curing of the epoxy oligomer. For this purpose, information on the compositions of the coexisting phases for the macro- and microlevels was plotted on the phase diagram (Fig. 59). It can be seen that the phase containing 5 wt.% PSU is near the right branch of the binodal curve, where the continuous phase is the epoxy polymer, and the PSU-enriched phase is the inclusions. In contrast, the mixture containing more than 10 wt.% PSU is in the middle region of compositions, which coincides with the region of phase inversion. The presence of extended macro phases for this system indicates that their formation took place in conditions of low viscosity and high translational mobility of components (Dv 3 × 10–7 cm2 s−1), which determined a high rate of phase growth (estimated time of formation of particles with a radius of 50 μm in these conditions ~200 s), that is, this structure was formed at the early stages of phase separation. Indeed, the ratio of macrophase volumes calculated from microphotographs and determined from compositions of coexisting phases and figurative point of the system coincide (Figs. 60 and 61).

Schematic of the phase separation of the EO–PSU system during curing.
 –figurative points of the initial systems (5, 10, and 15 wt.% PSU),
–figurative points of the initial systems (5, 10, and 15 wt.% PSU),
 —concentrations of coexisting phases in the initial stages of curing (at loss of mobility),
—concentrations of coexisting phases in the initial stages of curing (at loss of mobility),
 —concentrations of coexisting phases at the end of curing. 1—Suggested binodal curve of loss of mobility, 2—Binodal curve of epoxy polymer-PSU. The points were obtained by processing of morphological patterns, according to local X-ray microanalysis, and by shifting the glass transition temperature of phases enriched with PSU and epoxy polymer
—concentrations of coexisting phases at the end of curing. 1—Suggested binodal curve of loss of mobility, 2—Binodal curve of epoxy polymer-PSU. The points were obtained by processing of morphological patterns, according to local X-ray microanalysis, and by shifting the glass transition temperature of phases enriched with PSU and epoxy polymer

Morphology of ED-20—PVA blend. Chipping of the sample. Concentration of PVA: 15 (a) and 5 wt.% (b), Tcur = 30 °C

Morphology of ED-20–PVA mixture. Chipping of the sample. Concentration of PVA: 15 wt.%, Tcur = 60 °C. a—in the volume, b—in the near-surface layers
The phase diagrams (Figs. 48 and 59) show that the compositions of the coexisting phases on the isotherm change differently during curing. The right branch of the binodal curve (solubility of PSU in the epoxy adduct matrix) changes little, while the left branch (solubility of EO adducts in the PSU matrix) changes significantly. This leads to the fact that in the macrophase formed in the initial stages of phase separation and enriched with PSU, supersaturation rapidly grows, which causes secondary phase separation.

Particle size distribution in the ED-20–PVA mixture cured at 30 °C. Concentration of PVA: 5 (a), 15 wt.% (b)
This phase separation occurs at a relatively low translational mobility of macromolecules in the epoxy polymer network (Dv 10–9 cm2 s−1), which suppresses particle growth (the estimated time of particle formation with a radius of 5 µm under these conditions is ~1500 s) and leads to the formation of dispersed spherical microphase of small size (up to 1 µm), enriched in epoxy polymer with component concentrations close to the right branch of the binodal curve and dispersion medium enriched in PSU (94%).
Similar structures were obtained in the compositions with PVA and PEG. It was found that in mixtures with a small PVA content (less than 15%), structures characterized by a bimodal distribution of particle sizes were observed on the surface (Figs. 60 and 61). At that, small particle fractions (~0.2–2 μm), as a rule, are concentrated at the periphery of large ones (~30–40 μm). Specific features of this system should include the fact of non-uniform distribution of the dispersed phase across the sample. As a rule, the surface layer is enriched with particles of the dispersed phase in comparison with the volume. As we approach the sample surface, structures with bimodal particle size distribution are more often found (Fig. 61b). All this allows us to assume that the formation of gradient structures (from the phase and concentration points of view) is associated with the processes of macro-dissociation of mixtures caused, probably, by “displacement” of thermoplastic by the network of chemical bonds forming in the matrix (Fig. 62).
The secondary phase separation is most clearly manifested in the EO–PEG system (Fig. 63). It can be seen that there are large phase formations of rounded shape in the matrix. Around these particles, there are concentrated microphase separations whose size is two decimal orders of magnitude smaller than the size of macroparticles. This bimodal particle size distribution (Fig. 64) is most often observed at low curing temperatures (below the PEG melting point).

Morphology of ED-20–PEG mixture. Surface of the sample. Concentration of PEG = 15 wt.%. Tcur = 20 °C

Particle size distribution in the ED-20–PEG mixture cured at 20 °C. Concentration of PEG = 15 wt.%
In Fig. 63b, there is an interesting fragment of the secondary phase separation localized near the interfacial boundary of a large particle. The microparticles are located exactly along the perimeter of the macroparticle at some small distance from its surface. Along with that, there are observed particles of intermediate size, which are distant from the interphase boundary of the large particle. It can be concluded that the phase separation proceeds in several stages. Initially, large particles are formed, then at a greater distance from the smaller particles, and, finally, at large degrees of conversion, the smallest ones are formed.
Summarizing the experimental material on the phase behavior of mixtures of epoxy oligomers and polymers with thermoplastics obtained by us and described in the literature, we can unequivocally state that the mechanism of formation of heterogeneous structures is determined by the mutual positioning of critical points of solutions-melts of linear oligomers and their adducts with thermoplastics and figurative points of specific mixtures on the phase diagram field, on the one hand, and the value of translation diffusion coefficients of macromolecules of thermoplastics dissolved initially in epoxy.
It follows from the generalized phase diagrams that during the curing process, the UCST and binodal curves change their position so that the heterogeneous state region increases in size and the figurative points of specific mixtures fall into the two-phase state zone. At this stage of the structure formation process, phase separation and particle growth occur under conditions of high partial translational mobility of thermoplastic molecules. According to the formal theory of phase transformations, the growth rate of particles and the distance between them are determined by the diffusion coefficient and supersaturation in the middle of the distance coordinate (Fig. 65). The specificity of systems during the curing process is that both thermodynamic (compositions of coexisting phases) and kinetic (diffusion coefficients) parameters are continuously changing as a result of the formation of the chemical bonding network.

Schematic of the secondary phase separation in the epoxy-thermoplastic mixture matrix and the concentration distribution profiles occurring at different curing stages. Double arrows indicate zones of diffusion outflow. I and II—dispersion particles formed during primary and secondary phase decomposition, respectively
This leads to a gradual imbalance between the growth of concentration supersaturation at the periphery of the particles and the flow of thermoplastic macromolecules to the growing particle. In other words, the growth rate of supersaturation exceeds the diffusion rate of concentration flux to the particles of the dispersed phase.
At the same time, the balance between the growth of supersaturation with respect to the dissolved epoxy oligomer in the thermoplastic and the rate of its flow to the inner surface of the particle is also disturbed inside the particle. Thus, both in the dispersed phase and in the dispersion medium, conditions for secondary phase separation are created.
At this stage of phase separation, the formation of dispersed phase particles whose composition corresponds to the composition of coexisting phases on the phase diagrams is localized in one case in the matrix, but near the surface of the macroparticle, in the second case inside the macroparticle. Note that the secondary phase separation inside the particles, in contrast to the matrix, also depends on their sizes since the diffusion relaxation time is related to this parameter. It can be expected that under these conditions (inside the particles of dispersed phases), it will be realized at large degrees of conversion when the diffusion coefficients reach extremely low values.
Thus, the proposed mechanism of phase decomposition of mixtures of reactive oligomers with thermoplastics qualitatively explains the whole set of experimental data obtained. Naturally, the specific parameters of the phase structure of the mixtures are determined by the specific numerical values of the diffusion coefficients, the UCST position, the activation energy, and the reaction constants of the formation of the mesh structures.
References
Pascault, J.P., Williams, R.J.J.: Epoxy Polymers: New Materials and Innovations, p. 387, 1st edn. Wiley-VCH (2010). ISBN-13: 978-3527324804
Tsvetkov, V.N., Eskin, V.E., Frenkel, S.Ya.: Structure of Macromolecules in Solutions, p. 720. Nauka, Moscow (1964) (In Russian)
Malkin, A.Ya., Isayev, A.I.: Rheology. Concepts, methods and applications. Appl. Rheol. 16(5), 240–241 (2006). https://doi.org/10.1515/arh-2006-0039
Ferry, J.D.: Viscoelastic Properties of Polymers, p. 499. John Wiley and Sons, Inc., New York (1961)
Nechitailo, L.G., Gerasimov, I.G., Batog, A.E., Zaitsev, Y.S.: Dependence epoxydiane oligomer viscosity on temperature and molecular mass. Ukr. Khim. Zh. 53(9), 997–1000 (1988)
Gerasimov, I.G., Nechitailo, L.G., Batog, A.E., Zaitsev, Y.S.: Isothermal relaxation of viscosity of epoxydian oligomers. Ukr. Khim. Zh. 55(10), 1093–1095 (1989)
Zhavoronok, E.S.: Reactive Rubber-Epoxy Compositions, p. 163. Ph.D. Thesis, Frumkin Institute of Physical Chemistry and Electrochemistry Russian Academy of Sciences (IPCE RAS), Moscow, Russia (2002)
Irzhak, V.I.: Architecture of Polymers, p. 368. Nauka, Moscow (2012). ISBN 978-5-02-038459-0 (In Russian)
Aleman, J.V.: Flow properties of epoxide prepolymers. J. Polym. Sci. Part A Polym. Chem. 18(8), 2567–2575 (1980). https://doi.org/10.1002/pol.1980.170180815
Aleman, J.V.: Flow instabilities of epoxide prepolymers. Polym. Eng. Sci. 23(4), 177–182 (1983). https://doi.org/10.1002/pen.760230402
Markevich, M.A., Rytov, B.L., Vladimirov, L.B., Shashkin, D.P., Shiryaev, P.A., Solovev, A.G.: Structural organization in epoxydiane oligomers and polymers. Vysokomol. Soedin. Ser. A 28(8), 1595–1602 (1986)
Markevich, M.A.: Viscosity of epoxydiane oligomer solutions. Vysokomolekulyarnye Soedineniya Seriya B. 27(11), 838–842 (1985)
Ghijsels, A., Groesbeek, N., Raadsen, J.: Temperature-dependence of the zero-shear melt viscosity of oligomeric epoxy-resins. Polymer 25(4), 463–466 (1984). https://doi.org/10.1016/0032-3861(84)90203-9
Venediktov, E.A., Rozhkova, E.P.: Kinetic relationships of metal filling of epoxy resin by in situ reduction of silver nitrate. Russ. J. Appl. Chem. 86(6), 928–932 (2013). https://doi.org/10.1134/S1070427213060244
Budylin, N.Yu.: Phase Equilibria and Interdiffusion in Reactoplastics—Thermoplastics Systems, p. 180. Ph.D. Thesis, Frumkin Institute of Physical Chemistry and Electrochemistry Russian Academy of Sciences (IPCE RAS), Moscow, Russia (2014)
Kochnova, Z.A., Zhavoronok, E.S., Chalykh, A.E.: Epoxy Resins and Hardeners: Industrial Products, p. 200. OOO “Paint-Media”, Moscow (2006). ISBN 5-902904-03-X (In Russian)
Lantsov, V.M., Pakter, M.K., Irzhak, V.I., Abdrakhmanova, L.A., Abramova, Y.I., Zakirov, I.N., Vasilev, G.I., Paramonov, Y.N., Zaitsev, Y.S.: Kinetic Heterogeneity of epoxide oligomers. Vysokomol. Soedin. Ser. A 29(11), 2297–2301 (1987)
Maklakov, A.I., Skirda, V.D., Fatkulin, N.F.: Self-diffusion in solutions and melts of polymers, p. 222. Publishing House of Kazan State University, Kazan (1987) (In Russian)
Chalykh, A.E., Zagaitov, A.L., Korotchenko, D.P.: Optical Diffusiometer, p. 34. IFKh RAN: Moscow, Russia (1996) (In Russian)
Malkin, A., Ascadsky, A., Kovriga, V., Chalykh, A.E.: Experimental Methods of Polymer Physics, vol. 520. MIR: Moscow, Russia (1983)
Zakirov, I.N., Irzhak, V.I., Lantsov, V.M., Rozenberg, B.A.: On the nature of large-scale mobility in dense network epoxide polymers. Vysokomol. Soedin. Ser. A 30(5), 915–921 (1988)
Koike, T.: Melt viscosity at glass-transition temperature for some epoxide oligomers. J. Appl. Polym. Sci. 56(9), 1183–1186 (1995). https://doi.org/10.1002/app.1995.070560919
Lantsov, V.M., Pakter, M.K., Abdrakhmanova, L.A., Zakirov, I.N., Paramonov, Y.M., Zaitsev, Y.S.: Study of molecular mobility of epoxy-amine polymers by the impulse NMR method. Vysokomol. Soedin., Ser. A 28(5), 1047–1051 (1986)
Irzhak, V.I., Mezhikovskii, S.M.: Structural aspects of polymer network formation upon curing of oligomer systems. Russ. Chem. Rev. 78(2), 165–194 (2009). https://doi.org/10.1070/RC2009v078n02ABEH003896
Lu, M., Kim, S.: Kinetics and thermal characterization of epoxy-amine systems. J. Appl. Polym. Sci. 71(14), 2401–2408 (1999). https://doi.org/10.1002/(SICI)1097-4628(19990404)71:14%3c2401::AID-APP12%3e3.0.CO;2-C
Khozin, V.G.: Reinforcing Epoxy Resins, p. 446. House of Press, Kazan (2004). ISBN 5-94259-143-1 (In Russian)
Askadskii, A.A., Bartenev, G.M., Belyakov, A.P., Pastukhov, A.V.: Various forms of molecular mobility and relaxation transitions in epoxydian polymer. Vysokomolekulyarnye Soedineniya Seriya B. 30(11), 868–873 (1998). (In Russian)
Chalykh, A.E., Zhavoronok, E.S., Kolesnikova, E.F., Kostina, Y.V., Bondarenko, G.N.: Kinetics of curing of diane and aliphatic epoxy oligomer blends: IR-spectroscopy study. Polym. Sci. Ser. B 53(7–8), 466–475 (2011). https://doi.org/10.1134/S156009041108001X
Shmaliy, O.N., Diffusion in Epoxy Oligomers, p. 144. Ph.D. Thesis, Institute of Physical Chemistry Russian Academy of Sciences (IPC RAS), Moscow, Russia (1995)
Bartenev, G.M., Shut, N.I., Danilenko, G.D., Askadskii, A.A., Pastukhov, V.V.: Relaxation properties of a linear epoxy polymer. Vysokomolekulyarnye Soedineniya Seriya A Seriya B. 35(9), A1498–A1506 (1993)
Skirda, V.D., Sevriugin, V.A., Maklakov, A.I.: Peculiarities of translational mobility of macromolecules in melts and solutions of polymers. Dokl. Akad. Nauk SSSR 269(3), 638–640 (1983)
Tanner, J.E., Liu, K.J., Anderson, J.E.: Proton magnetic resonance self-diffusion studies of poly(ethylene oxide) and polydimethylsiloxane solution. Macromolecules 4(5), 586–588 (1971). https://doi.org/10.1021/ma60023a014
Kumagai, J., Watanabe, H., Miyasaka, K., Hata, T.: Diffusion measurement of tritium labeled polystyrene in polymer bulk—effect of molecular-weight on diffusion-coefficient. J. Chem. Eng. Jpn. 12(1), 1–4 (1979). https://doi.org/10.1252/jcej.12.1
de Gennes, P.-G.: Scaling Concepts in Polymer Physics, p. 324. Cornell University Press, London (1979)
Chalykh, A.E.: Diffusion in Polymer Systems, p. 312. Chemistry Publishing House, Moscow, Russia (1987) (In Russian)
Paolillo, S., Bose, R.K., Santana, M.H., Grande, A.M.: Intrinsic self-healing epoxies in polymer matrix composites (PMCs) for aerospace applications. Polymers 13, 201 (2021). https://doi.org/10.3390/polym13020201
Peterson, A.M., Jensen, R.E., Palmese, G.R.: Room-temperature healing of a thermosetting polymer using the Diels-Alder reaction. Appl. Mater. Interf. 2(4), 1141–1149 (2010). https://doi.org/10.1021/am9009378
Bai, N., Saito, K., Simon, G.P.: Synthesis of a diamine cross-linker containing Diels-Alder adducts to produce self-healing thermosetting epoxy polymer from a Widely used epoxy monomer. Polym. Chem. 4, 724–730 (2013). https://doi.org/10.1039/c2py20611k
Billiet, S., Camp, V.W., Hillewaere, X., Rahier, H., Du Prez, F.E.: Development of optimized autonomous self-healing systems for epoxy materials based on maleimide chemistry. Polymer 53(12), 2320–2326 (2012). https://doi.org/10.1016/j.polymer.2012.03.061
Wang, S., Urban, M.W.: Self-healing polymers. Nature Rev. Mater. 5, 562–583 (2020). https://doi.org/10.1038/s41578-020-0202-4
Toohey, K.S., Hansen, C.J., Lewis, J.A., White, S.R., Sottos, N.R.: Delivery of two-part self-healing chemistry via microvascular networks. Adv. Func. Mat. 19, 1399–1405 (2009). https://doi.org/10.1002/adfm.200801824
McIlroy, D.A., Blaiszik, B.J., Caruso, M.M., White, S.R., Moore, J.S., Sottos, N.R.: Microencapsulation of a reactive liquid-phase amine for self-healing epoxy composites. Macromolecules 43, 1855–1859 (2010). https://doi.org/10.1021/ma902251n
Reddy, K.R., El-Zein, A., Airey, D.W., Alonso-Marroquin, F., Schubel, P., Manalo, A.: Self-healing polymers: synthesis methods and applications. Nano-Struct. Nano-Obj. 23 (2020). https://doi.org/10.1016/j.nanoso.2020.100500
Zhao, Y.H., Vuluga, D., Lecamp, L., Burel, F.: Photoinitiated thiol–epoxy addition for the preparation of photoinduced self-healing fatty coatings. RSC Adv. 6, 32098–32105 (2016). https://doi.org/10.1039/c6ra03693g
Bellamy, L.: The Infra-red Spectra of Complex Molecules, p. 433. Springer, Dordrecht (1975). ISBN 978-0-412-13850-8
Nielsen, L.E.: Mechanical Properties of Polymers and Composites, vol. 2, p. 556. Marcel Dekker (1974). ISBN 978-0-8247-6208-7
D.R. Paul, C.B. Bucknall, Polymer Blends. John Wiley & Sons, Inc.: Hoboken, NJ, USA (2000)
Farooq, U., Teuwen, J., Dransfeld, C.: Toughening of epoxy systems with interpenetrating polymer network (IPN): a review. Polymers 12 (2020). https://doi.org/10.3390/polym12091908
Korokhin, R.A., Solodilov, V.I., Zvereva, U.G., Solomatin, D.V., Gorbatkina, Y.A., Shapagin, A.V., Lebedeva, O.V., Bamborin, M.Y.: Epoxy polymers modified with polyetherimide. Part II: Physicomechanical properties of modified epoxy oligomers and carbon fiber reinforced plastics based on them. Polym. Bull. 77, 2039–2057 (2020). https://doi.org/10.1007/s00289-019-02841-9
Kishi, H., Tanaka, S., Nakashima, Y., Saruwatari, T.: Self-assembled three-dimensional structure of epoxy/polyethersulphone/silver adhesives with electrical conductivity. Polymer 82, 93–99 (2016). https://doi.org/10.1016/j.polymer.2015.11.043
Chen, Z.G., Luo, J., Huang, Z., Cai, C.Q., Tusiime, R., Li, Z.Y., Wang, H.X., Cheng, C., Liu, Y., Sun Z.Y., et al.: Synergistic toughen epoxy resin by incorporation of polyetherimide and amino groups grafted MWCNTs. Compos. Commun. 21 (2020). https://doi.org/10.1016/j.coco.2020.100377
Tretyakov, I.V., Vyatkina, M.A., Cherevinskiy, A.P., Solodilov, V.I., Shapagin, A.V., Korokhin, R.A., Budylin, N.Y., Kireinov, A.V., Gorbatkina, Y.A.: Effect of polyethersulfone on the properties of epoxy resin and wound unidirectional glass fiber reinforced plastics based on it. Bull. Russ. Acad. Sci. Phys. 85, 876–880 (2021). https://doi.org/10.3103/s1062873821080293
Sun, Z.Y., Xu, L., Chen, Z.G., Wang, Y.H., Tusiime, R., Cheng, C., Zhou, S., Liu, Y., Yu, M.H., Zhang, H.: Enhancing the mechanical and thermal properties of epoxy Resin via blending with thermoplastic polysulfone. Polymers 11 (2019). https://doi.org/10.3390/polym11030461
Korokhin, R.A., Solodilov, V.I., Gorbatkina, Y.A., Shapagin, A.V.: Rheological and physicomechenical properties of epoxy-polyetherimide compositions. Mech. Compos. Mater. 51, 313–320 (2015). https://doi.org/10.1007/s11029-015-9502-y
Jiang, M.Q., Liu, Y., Cheng, C., Zhou, J.L., Liu, B.H., Yu, M.H., Zhang, H.: Enhanced mechanical and thermal properties of monocomponent high performance epoxy resin by blending with hydroxyl terminated polyethersulfone. Polym. Test. 69, 302–309 (2018). https://doi.org/10.1016/j.polymertesting.2018.05.039
Solodilov, V.I., Korokhin, R.A., Gorbatkina, Y.A., Kuperman, A.M.: Comparison of fracture energies of epoxypolysulfone matrices and unidirectional composites based on them. Mech. Compos. Mater. 51, 177–190 (2015). https://doi.org/10.1007/s11029-015-9488-5
Mimura, K., Ito, H., Fujioka, H.: Improvement of thermal and mechanical properties by control of morphologies in PES-modified epoxy resins. Polymer 41, 4451–4459 (2000). https://doi.org/10.1016/s0032-3861(99)00700-4
Shapagin, A.V., Budylin, N.Y., Chalykh, A.E.: Regulation of a phase structure at the interface in epoxy-polysulfone systems. Russ. Chem. Bull. 67, 2172–2177 (2018). https://doi.org/10.1007/s11172-018-2350-x
Won, J.S., Lee, J.E., Park, J.K., Lee, M.Y., Kang, S.H., Lee, S.G.: Cure behavior and toughness properties of polyethersulfone/multifunctional epoxy Resin blends. Polym. Korea 43, 60–68 (2019). https://doi.org/10.7317/pk.2019.43.1.60
Brantseva, T.V., Solodilov, V.I., Antonov, S.V., Gorbunova, I.Y., Korokhin, R.A., Shapagin, A.V., Smirnova, N.M.: Epoxy modification with poly(vinyl acetate) and poly(vinyl butyral). I. Structure, thermal, and mechanical characteristics. J. Appl. Polym. Sci. 133 (2016). https://doi.org/10.1002/app.44081
Chistyakov, E.M., Terekhov, I.V., Shapagin, A.V., Filatov, S.N., Chuev, V.P.: Curing of epoxy Resin DER-331 by Hexakis(4-acetamidophenoxy)cyclotriphosphazene and properties of the prepared composition. Polymers 11, 12 (2019). https://doi.org/10.3390/polym11071191
Rosetti, Y., Alcouffe, P., Pascault, J.P., Gerard, J.F., Lortie, F.: Polyether sulfone-based epoxy toughening: from micro- to nano-phase separation via PES end-chain modification and process engineering. Materials 11 (2018). https://doi.org/10.3390/ma11101960
Chalykh, A.E., Gerasimov, V.K., Bukhteev, A.E., Shapagin, A.V., Kudryakova, G.K., Brantseva, T.V., Gorbatkina, Y.A., Kerber, M.L.: Compatibility and phase structure evolution in polysulfone-curable epoxy oligomer blends. Polym. Sci. Ser. A 45, 676–685 (2003)
Korokhin, R.A., Shapagin, A.V., Solodilov, V.I., Zvereva, U.G., Solomatin, D.V., Gorbatkina, Y.A.: Epoxy polymers modified with polyetherimide. Part I: rheological and thermomechanical characteristics. Polym. Bull. 78, 1573–1584 (2021). https://doi.org/10.1007/s00289-020-03174-8
Shapagin, A.V., Budylin, N.Y., Chalykh, A.E., Solodilov, V.I., Korokhin, R.A., Poteryaev, A.A.: Phase equilibrium, morphology, and physico-mechanics in epoxy–thermoplastic mixtures with upper and lower critical solution temperatures. Polymers 13, 35 (2020). https://doi.org/10.3390/polym13010035
Surendran, A., Joy, J., Parameswaranpillai, J., Anas, S., Thomas, S.: An overview of viscoelastic phase separation in epoxy based blends. Soft. Matter. 16, 3363–3377 (2020). https://doi.org/10.1039/c9sm02361e
Zhang, Y.W., Shen, Y.C., Shi, K.X., Wang, T.W., Harkin-Jones, E.: Constructing a filler network for thermal conductivity enhancement in epoxy composites via reaction-induced phase separation. Compos. Part A Appl. Sci. Manuf. 110, 62–69 (2018). https://doi.org/10.1016/j.compositesa.2018.04.009
Nikulova, U.V., Chalykh, A.E.: Phase Equilibrium and Interdiffusion in Poly(Vinyl Methyl Ether)-Water System. Polymers 12 (2020). https://doi.org/10.3390/polym12112445
Chalykh, A.E., Gerasimov, V.K., Mikhailov, Y.M.: Phase State Diagrams of Polymer Systems. Yanus-K: Moscow, Russia (1998) (In Russian)
Mandelkern, L.: Crystallization of Polimers. McGraw-Hill book company, New York-San-Franciscko-Toronto-London (1964)
Acknowledgements
Part of this work was supported by the Ministry of Science and Higher Education of the Russian Federation (project 122011300052-1), and another one was supported by Russian Foundation for Basic Research (project No. 20-03-00722).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Chalykh, A.E., Budylin, N.Y., Shapagin, A.V. (2023). Diffusion in Epoxy Oligomers and Polymers. In: Hameed, N., Capricho, J.C., Salim, N., Thomas, S. (eds) Multifunctional Epoxy Resins. Engineering Materials. Springer, Singapore. https://doi.org/10.1007/978-981-19-6038-3_3
Download citation
DOI: https://doi.org/10.1007/978-981-19-6038-3_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-6037-6
Online ISBN: 978-981-19-6038-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)