
Abstract
Sphingolipids (SL) are a class of chemically diverse lipids that have important structural and physiological functions in eukaryotic cells. SL entail a long chain base (LCB) as the common structural element, which is typically formed by the condensation of l-serine and long chain acyl-CoA. This condensation is the first and the rate-limiting step in the de novo SL synthesis and catalyzed by the enzyme serine palmitoyltransferase (SPT). Although palmitoyl-CoA is the preferred substrate, SPT can also metabolize other acyl-CoAs, thereby forming a variety of LCBs, which differ in structures and functions. The mammalian SPT enzyme is composed of three core subunits: SPTLC1, SPTLC2, and SPTLC3. Whereas SPTLC1 and SPTLC2 are ubiquitously expressed, SPTLC3 expression is restricted to a few specific tissues. The SPTLC1 subunit is essential and can associate with either SPTLC2 or SPTLC3 to form an active enzyme. Depending on the stoichiometry of the SPTLC2 and SPTLC3 subunits, the spectrum of SPT products varies. While SPTLC1 and SPTLC2 primarily form C18 and C20 LCBs, the combination of SPTLC1 and SPTLC3 produces a broader spectrum of LCBs. Genetic and population based studies have shown that SPTLC3 expression and function are associated with an altered plasma SL profile and an increased risk for cardio-metabolic diseases. Animal and in vitro studies showed that SPTLC3 might be involved in hepatic and cardiac pathology and could be a therapeutic target for these conditions.
Here we present an overview of the current data on the role of SPTLC3 in normal and pathological conditions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

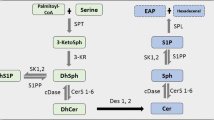

Keywords
- Serine palmitoyltransferase
- Serine palmitoyltransferase subunit 3 (Sptlc3)
- Sphingolipid biosynthesis
- Sphingolipids
- Metabolic diseases
4.1 The Serine Palmitoyltransferase and Long Chain Base Synthesis
Sphingolipids (SL) are an essential class of lipids and found in some prokaryotes (Bacteroides, Sphingomonads) and all eukaryotes [1]. Structurally, SL are defined by the presence of long chain amino-hydroxy alkanes also called long chain bases (LCB). The LCB formation is the first and rate-limiting step in the de novo SL synthesis and catalyzed by the serine palmitoyltransferase (SPT). Typically, SPT condenses an activated fatty acid with l-serine in a PLP dependent reaction. LCBs can vary in the number of carbons, hydroxylation as well as in their saturation state [2]. Some LCB structures are specific to certain species [3]. Most variations in the LCB spectrum arise due to the broad and variable substrate specificity of the SPT enzyme although some LCB modifications are also introduced downstream of SPT. SPT metabolizes acyl-CoA’s of variable structure and length, and the formation of odd and even numbered LCBs as well as methyl-branched structures was reported across species [3]. The 18-carbon dihydroxy amino alkene, sphingosine (SO, d18:1) is the most abundant LCB in mammals including humans. However, plants and yeast generally contain the saturated trihydroxy derivate, phytosphingosine (PhytoSO, t18:0), whereas shorter LCBs (C14 and C16) are commonly found in insects (Drosophila) while C. elegans forms iso-branched C17 LCBs [4, 5]. Under conditions of serine deficiency, SPT can also metabolize L-alanine and glycine as alternative substrates, which results in the formation of a non-canonical and neurotoxic class of 1-deoxysphingolipids [6].
SPT belongs to the family of pyridoxal 5-phosphate (PLP)-dependent a-oxoamine synthases (POAS) similar to the 5-amino levulinic acid synthase that is involved in heme metabolism. In contrast to other members of this family, which are mostly soluble cytosolic proteins, SPT is an integral membrane protein and located at the outer membrane of the endoplasmic reticulum (ER) [7, 8]. In mammals, the SPT core subunits are encoded by the three genes SPTLC1, SPTLC2, and SPTLC3. A minimally functional SPT enzyme consists of the SPTLC1 subunit in conjunction with either SPTLC2 or SPTLC3 [9]. Accessory subunits (ORMDL3, ssSPTa, ssSPTb) that modulate activity and substrate specificity have been identified in yeasts and eukaryotes (Fig. 4.1). In response to cellular SL levels, a conserved family of integral ER membrane proteins (ORMDL1-3) controls SPT activity and SL de novo synthesis [10]. Budding yeast encodes for two isoforms (Orm1 and Orm2), while mammals express three ORM orthologues (ORMDL1-3). The two accessory small proteins, ssSPTa and ssSPTb are specific to mammals and reported to regulate the synthesis of C18 and C20 LCBs [9, 11]. Here, ssSTPa promotes the use of palmitoyl-CoA forming C18 LCBs, while the presence of ssSPTb enhances the activity with stearoyl-CoA forming a C20 LCB. The expression of ssSPTb appears to be specific to neuronal tissue and a pathologically increased synthesis of C20 LCBs due to a gain-of-function mutation in the ssSPTb gene causes neurodegeneration and macular defects in mouse models [12].

SPTLC1-SPTLC2 and SPTLC1-SPTLC3 protein structure: Overlay of SPTLC1-SPTLC2 and an SPTLC1-SPTLC3 based dimer. The SPTLC1-SPTLC3 structure was modeled in silico using the recently published cryo EM structure of the SPT complex [7, 8]. Closely located to the residues Met-28 and Tyr-27 is a helical domain. The charge distribution of this helix differs considerably between SPTLC2 and SPTLC3. Residues Tyr-27 and Glu-485 in SPTLC2 and His-477 in SPTLC3 (upper right) can be assumed to form a gate to the long chain base binding site. At physiological pH, Glu-48 in SPTLC2 is negatively charged and the gate is expected to be open due to a repulsion between the hydrophobic Tyr-27 and Glu-485. In contrast, Tyr-27 and the neutral His-477 of SPTLC3 build hydrogen bonds and form a closer gate. At acidic conditions, the repulsion of the then positively charged His-477 and Tyr-27 is expected to bring SPTLC3 into a more open conformation compared to physiological pH. Further differences are to be found in the charge distribution over the entire helix (upper right). The residue Lys-488 of SPTLC2 is replaced by Glu-480 in SPTLC3, reversing the charge in this area. The residue Asn-490 of SPTLC2 is replaced by Lys-482 in SPTLC3, introducing a positive charge at the very beginning of the helix. Models were generated by the SWISS-MODEL server using the automated mode and selecting the model with 100% coverage and the best global model quality estimate (GMQE) and qualitative model energy analysis (QMEAN) [48,49,50,51,52]
In contrast, SPTLC3 appears to form a rather large spectrum of non-canonical LCBs in mammals especially in humans. SPTLC3 was cloned more than a decade ago [13] but despite a significant association with different metabolic traits in several genome-wide studies, its physiological function remains largely unknown. However, recent reports shed new light on its function and relevance.
4.2 Evolution of SPT and Structure
As an essential metabolic enzyme, SPT has evolved from simpler to more complex forms of life. This relates to gene copy numbers, structural heterogeneity of the enzyme as well as function. The bacterial SPT from Sphingomonas paucimobilis is a soluble homodimer and transcribed from a single gene [14], whereas in yeast (S. cerevisiae), SPT is a heteromeric and membrane bound enzyme that is encoded by the two genes LCB1 and LCB2 [1]. Arabidopsis encodes for a homologue of SPTLC1 and for two homologues of SPTLC2 (AtLCB2a and AtLCB2b) [15]. AtLCB2a/b are 405 amino acid long proteins that share 95% identity and appear to be ubiquitously expressed and functionally redundant in Arabidopsis [16]. A third, SPTLC2 related but truncated coding sequence (At3g48790) is present in tandem with AtLCB2a. This truncated form is likely the result of a gene duplication event and lacks 140 residues of the N-terminus. However, the gene product was not yet detected at protein level.
Human SPTLC3 is also slightly shorter in length than SPTLC2 (552 vs. 562 amino acids, respectively) and shares about 68% overall identity with SPTLC2. The two subunits are rather conserved in their central regions and than at their N- and C-terminal ends [13].
Both SPTLC2 and SPTLC3 bear a conserved PLP binding motif with a central Lysine residue that is absent in SPTLC1. This lysine is essential for the transferase reaction, as PLP forms a Schiff’s base with the amino acid substrate, creating an external aldimine (Fig. 4.1). Therefore, SPTLC2 and 3 are considered to be the catalytically active subunits, whereas SPTLC1 appears to be important for protein stability, membrane insertion, and serves as a focal point to control enzyme activity.
The cryo EM structure of the human SPT complex (SPTLC1, SPTLC2, ORMDL3, and ssSPTa) was published recently in two independent studies [7, 8]. The structures provide a good insight into the mechanisms of substrate recognition, catalytic activity, and enzyme regulation. They show that SPTLC1 and 2 are both involved in the recognition of the acyl-CoA head group, whereas the acyl-tail is mostly stabilized by residues from SPTLC2. The amino acid environment around the acyl-CoA binding pocket is important for activity, as mutations of these SPTLC2 residues either decrease or completely block enzyme activity [8]. In addition, a single amino acid residue from ssSPTa (Met28) appears to participate in the acyl chain coordination [8]. The large hydrophobic methionine protrudes into the acyl binding pocket of SPTLC2, acting as a plug that defines the possible length of the acyl chain that can be accommodated within the pocket [8]. The corresponding amino acid in the human subunit ssSPTb is Val25 which leaves more room in the acyl binding pocket. Consequently, it was demonstrated that the exchange of Met28 to valine increases the affinity of SPT enzyme towards the bigger stearoyl-CoA [8, 17, 18].
Based on the published structure data we generated an in silico model of an SPTLC1-SPTLC3 dimer (Fig. 4.1). In comparison, the SPTLC1-SPTLC3 dimer showed a different configuration of the substrate binding pocket that likely allows more variability in the biding of different acyl-CoA structures. The biggest differences was seen at the interface to ssSPTa, in close vicinity to the residues ssSPTa Met28 and Tyr27 (Fig. 4.1 lower left). The structural changes around Met28 can be expected to influence the substrate binding properties of the enzyme. A not yet solved aspect is the role of the flexible protein termini that could not be resolved in the recently published structures [7, 8]. They may interact with lipids or other proteins influencing substrate binding and product spectrum.
4.3 Role of SPTLC3 in Sphingolipid Metabolism
The two subunits SPTLC1 and SPTLC2 are expressed ubiquitously and the relative mRNA levels vary little between tissues. In contrast, SPTLC3 is specifically expressed in certain tissues and mRNA levels between these tissues vary significantly. SPTLC3 shows moderate expression in liver, significant expression in skin, and very high expression levels in placenta and trophoblasts [13]. The cellular and physiological reasons for this tissue specificity are not yet understood, but it is reasonable that SPTLC3 expression is highest in those tissues that benefit most from a broad LCB spectrum. This might explain the significantly high expression in skin, which has a highly complex arrangement of SL and derivatives that contribute to the hydrophobic barrier that prevents the body from transdermal water loss. However, SPTLC3 expression in human placenta and trophoblast cells are many-fold higher than in skin [13] but it is currently not clear whether such a broad spectrum of SLs is of funtional importance for the placenta. Unfortunately, SPTLC3 knockout (KO) models that could address these questions are not yet available.
In Arabidopsis, the deletion of either AtLCB2a or AtLCB2b is well tolerated with no observable consequences on plant growth [16]. Both forms appear to generate similar LCB profiles and can complement LCB auxotrophy in SPT deficient yeast cells [16]. However, the mammalian SPTLC2 and SPTLC3 subunits appear to diverge functionally. The earliest indications to functional divergence between the two enzymes came from the fact that in vitro activity of SPTLC3 but not of SPTLC2 is inhibited in the presence of Triton-X-100 (0.2%) [12]. This detergent sensitivity is unlikely a result of the disruption of SPTLC1-SPTLC3 interaction, which is efficiently maintained even at higher detergent concentrations (0.5%) [9]. The effect is likely related to non-covalent structural modification of the enzyme as the activity of the soluble SPT from S. paucimobilis is also inhibited by the detergent [13], perhaps through interference with the acyl-CoA binding site.
Unlike human SPTLC2 and Arabidopsis AtLCB2a/b, SPTLC3 only partially rescues LCB auxotrophy in LCB1/LCB2 deficient yeast cells [11]. In animals, systemic SPTLC2 deficiency is lethal, whereas SPTLC3 deficient models have not yet been reported. A conditional KO of SPTLC2 in cardiomyocytes causes cardiac dysfunction, myopathy, and fibrosis in mice [19]. In response, SPTLC3 expression is induced in KO cells but this does not compensate for the SPTLC2 deficiency [19]. These differences between the two subunits could be attributed to different activities. Whereas the combination of SPTLC1/SPTLC2 and ssSPTa preferentially metabolizes palmitoyl-CoA (Fig. 4.2a, c), the combinations of SPTLC1/SPTLC3 showed a broader affinity for fatty acids of variable length (Fig. 4.2b, c). The length of the metabolized acyl-CoA’s ranges from myristic (C14) to stearic acid (C18), including acyl-CoA’s with even and odd carbon chains. The human SPTLC3 is effectively metabolizing penta (C15)- and heptadecanoic (C17) acid to generate odd chain 17- and 19-carbon LCBs but has poor activity with nonadecanoic (C19) acid. The presence of the ssSPTb subunit shifts this pattern and increases activity of both SPTLC2 and SPTLC3 towards stearoyl-CoA forming C20 LCBs although the capacity to form these LCBs is still higher for SPTLC3 [9]. In addition, human SPTLC3 also forms an anteiso branched omega-3-methyl LCB (meC18SO) which uses anteiso-methyl-palmitate as a substrate. Branched chain fatty acids (BCFAs) are formed from branched chain amino acids (BCAA) such as Ile, Leu, and Val. In fact, stable isotope tracing showed that primarily Ile contributes to the formation of both, odd chain C17SO and branched meC18SO. Anteiso-methyl-palmitate is generated from the intermediate precursor molecule, 2-methyl-butyryl-CoA, whereas pentadecanoic (C15) acid is formed by the elongation of propionyl-CoA, an end-product of the BCAA catabolic pathway. Interestingly, BCAA were shown to be significantly elevated in metabolic diseases such as T2DM [20] suggesting that also C17SO and meC18SO levels might be increased in these conditions.

SPT subunit composition and the resulting LCB product spectrum: (a) SPTLC1 + 2 and (b) SPTLC1 + 3 based SPT enzyme. The specificity of each subunit combination is shown through the inset-overlap (c). Colors in the insert correspond to the protein subunit in the structures above. The SPTLC1 + SPTLC2 based enzyme generates primarily C18 (d18:0) and to a lesser extend C20 (d20:0) based LCBs. In contrast, the SPTLC3 subunit (top right and large-brown inset below) forms a broader range of LCBs in the range of C16–C20. The synthesis of C16, C17, and the omega-3-methyl sphingosine is exclusive to the SPTLC3 subunit, whereas the activity of the SPTLC2 and 3 overlaps concerning the formation of C18 and C20 LCBs. In combination with SPTLC2, the small subunit ssSPTa (blue protein top left/blue square inset) increases C18 LCB synthesis, whereas ssSPTb increases the formation of C20 LCBs (purple top right for both subunits). ORMDL3 interacts with SPTLC1 and thereby controls enzyme activity
Interestingly, the formation of meC18SO seems to be specific to humans as meC18SO is present in human plasma and in purified human lipoproteins but was absent in the plasma of mice [9]. However, it is not clear whether this is because of intrinsic differences in the activity of human and mouse SPTLC3 or differences in the substrate availability [21]. The availability of relevant fatty acid substrates such as pentadecanoic acid and anteiso-methyl-palmitate is a significant factor determining the overall LCB spectrum in SPTLC3 expressing cells.
The physiological function of SPTLC3 derived SLs is currently not known. SLs with different LCB and N-acyl profiles might affect the biophysical properties of membranes. It has been shown that the length of the LCB in ceramides is inversely associated with the amount needed to induce lateral segregation in vitro [22]. In these experiments, the LCB chain length had a stronger effect on segregation than the N-linked acyl chain. In addition, phosphorylated atypical LCBs might alter sphingosine-1-phoshphate (S1P) and S1P-receptor (S1PR) mediated signaling pathways. For example, it was reported that S1PR activation is S1P-alkyl chain length dependent [23]. Additionally, C20S1P was recently shown to diminish signaling from SP1R2 [24].
4.4 SPTLC3 in Metabolic Diseases
SNPs in SPTLC3 have been significantly associated with several lipid traits in genome-wide association studies (GWAS) and repeatedly reported as significant influencers of circulating SL levels [25,26,27,28,29,30,31]. Sphingolipids, ceramides, and sphingomyelins are independent markers for cardiovascular disease [32] and plasma ceramides were shown to be relevant biomarkers to assess the severity of cardiovascular dysfunction [33, 34]. Tabassum et al. showed that SNP rs364585 in the SPTLC3 locus was associated with reduced plasma ceramides and a decreased risk for intracerebral hemorrhage [29]. Animal studies have shown that the SPTLC3 dependent ceramides formation is increased in the heart of mice on high fat diet (HFD). The addition of C16SA (d16:0) but not of C18SA (d18:0) was toxic to cultured cardiomyocytes in vitro and linked to a non-apoptotic pathway [35].
SNPs in the SPTLC3 locus also appear to be associated with changes in other lipids classes in liver and plasma. Mirkow et al. showed that the SNP rs168622 was associated with an elevated SPTLC3 expression and increased hepatic lipid content [36], whereas other studies linked SNPs in SPTLC3 with altered plasma LDL-C levels [25, 28, 31]. This association with plasma cholesterol could emanate from an intimate intertwining of the SL and cholesterol metabolism, co-transport in plasma, and their alliance in establishing functional eukaryotic membrane microdomains. Sphingomyelin and ceramide levels regulate cholesterol synthesis by modulating SREBP activation and HMG-CoA reductase activity [37]. Similarly, sphingomyelin sequestration of cholesterol within cellular membranes could set off futile cycles of synthesis and unloading.
However, given the fact that SPTLC3 generated SL account for a relatively small fraction of total SL in human plasma, these strong associations are rather surprising.
Metabolically, it was shown that SPTLC3 expression is influenced by a variety of factors. Shah et al. [38] reported a significant increase in SPTLC3 expression in epididymal adipose tissues of mice on HFD with a modest decrease in SPTLC2 expression. Others reported induction of hepatic SPTLC3 expression in mice on HFD [39,40,41]. SPTLC3 expression was also associated with insulin resistance in adipose [38] hepatic tissues [39, 41] and upregulated in steatotic livers of mice supplemented with homocysteine [42]. This points to the fact that steatosis, irrespective of its nature, induces the expression of SPTLC3. In addition, SPTLC3 expression appears to be related to plasminogen activator inhibitor 1 (PAI1) as PAI1 deficiency decreased SPTLC3 expression in mice on HFD [38]. Similarly, cannabinoid receptor 1 (CB1R) antagonists were reported to decrease hepatic SPTLC3 expression and improve insulin sensitivity in mice on HFD [39]. Polyunsaturated fatty acids and sulforaphane also reduced SPTLC3 expression, prevented from steatosis, and improved insulin sensitivity [41, 42]. Inhibition of SL synthesis by myriocin or siRNA mediated downregulation of SPTLC3 levels appears to induce similar response in vitro indicating that SPTLC3 could be a therapeutic target in metabolic conditions.
The association of SPTLC3 with cardio-metabolic conditions in animal models is further supported by a recent clinical study including 2′302 ethnically Chinese Singaporean participants and a long-term follow-up of 11 years. Lipidomics analysis showed a significant correlation of C16SO based sphingolipids with obesity and diabetes [43]. Unfortunately, the authors did not include C17SO and meC18SO based SL in their analysis but speculate on altered SPTLC3 function in response to metabolic conditions. It will be important to see whether obesity and insulin resistance correlate with increased hepatic SPTLC3 expression in humans as it was shown for mice.
The evidence that SPTLC3 related SNPs are determining tissue or plasma SL levels and also involvement in cardio-metabolic conditions is strong, but information on how these associations translate to enzymatic function and pathology is missing.
4.5 SPTLC3 in Other Diseases
Several mutations in SPTLC1 and SPTLC2 cause the rare peripheral neuropathy HSAN1 [6]. The HSAN1 mutations shift the substrate preference of SPT towards L-alanine and glycine, leading to the formation of an atypical class of neurotoxic 1-deoxySL [6]. Plasma 1-deoxySLs are also increased under conditions of serine deficiency and in this context involved in the course of the rare retinopathy (macular telangiectasia type 2) and cancer [44, 45]. However, this alternative activity with L-alanine and glycine seems to be specific for SPTLC2 and was not yet reported for SPTLC3.
Recently, another group of mutations in SPTLC1 was associated with early onset of amyotrophic lateral sclerosis (ALS) [46]. In contrast to the SPT-HSAN1 mutations, the SPT-ALS mutations cluster in exon 2 of SPTLC1 and result in a pathologically increased SL formation.
So far, no mutations in SPTLC3 have been associated with either HSAN1 or ALS. However, Gonzaga-Jauregui et al. reported a male patient (30 years) with a SPTLC3p.W150R variant, which was predicted to be loss of function mutation. This SPTLC3 variant was associated with a sensory neuropathy, deformation, and atrophy of the extremities and some bulbar involvement [47]. The authors showed that suppressing SPTLC3 expression in zebrafish causes defects in axons of motor neurons. Expression of human wild type SPTLC3 but not of the SPTLC3 p.W150R variant suppressed these defects [47]. Structurally, the mutation is replacing a hydrophobic residue (W150) with a positively charged arginine that might impair the binding of the hydrophobic substrate (Fig. 4.1, lower right). This indicates that SPTLC3 activity is also relevant to neuronal developmental and might contribute to C20 LCB formation in neuronal tissue, in particular as C20 LCB synthesis by SPTLC3 is greatly enhanced in presence of ssSPTb [9].
Taken together, it appears that SPTLC3 expression is involved in the generation of cell/tissue specific SL profiles. SPTLC3 activity might be modulated genetically but also by substrate availability and the presence of other components of the SPT complex. However, the mechanistic details of how SPTLC3 generated SLs affect metabolism, membrane integrity, and cell signaling need to be addressed in future work.
References
Harrison, P. J., Dunn, T. M., & Campopiano, D. J. (2018). Sphingolipid biosynthesis in man and microbes. Natural Product Reports, 35(9), 921–954.
Carreira, A. C., et al. (2019). Mammalian sphingoid bases: Biophysical, physiological and pathological properties. Progress in Lipid Research, 75, 100988.
Pruett, S. T., et al. (2008). Biodiversity of sphingoid bases (“sphingosines”) and related amino alcohols. Journal of Lipid Research, 49(8), 1621–1639.
Fyrst, H., Herr, D. R., Harris, G. L., & Saba, J. D. (2004). Characterization of free endogenous C14 and C16 sphingoid bases from Drosophila melanogaster. Journal of Lipid Research, 45(1), 54–62.
Hannich, J. T., Mellal, D., Feng, S., Zumbuehl, A., & Riezman, H. (2017). Structure and conserved function of iso-branched sphingoid bases from the nematode Caenorhabditis elegans. Chemical Science, 8(5), 3676–3686.
Lone, M. A., Santos, T., Alecu, I., Silva, L. C., & Hornemann, T. (2019). 1-Deoxysphingolipids. Biochimica et Biophysica Acta—Molecular and Cell Biology of Lipids, 1864(4), 512–521.
Wang, Y., et al. (2021). Structural insights into the regulation of human serine palmitoyltransferase complexes. Nature Structural & Molecular Biology, 28(3), 240–248.
Li, S., Xie, T., Liu, P., Wang, L., & Gong, X. (2021). Structural insights into the assembly and substrate selectivity of human SPT–ORMDL3 complex. Nature Structural & Molecular Biology, 28(3), 249–257.
Lone, M. A., et al. (2020). Subunit composition of the mammalian serine-palmitoyltransferase defines the spectrum of straight and methyl-branched long-chain bases. Proceedings of the National Academy of Sciences of the United States of America, 117(27), 15591–15598.
Davis, D., Kannan, M., & Wattenberg, B. (2018). Orm/ORMDL proteins: Gate guardians and master regulators. Advances in Biological Regulation, 70, 3–18.
Han, G., et al. (2009). Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proceedings of the National Academy of Sciences of the United States of America, 106(20), 8186–8191.
Zhao, L., et al. (2015). Elevation of 20-carbon long chain bases due to a mutation in serine palmitoyltransferase small subunit b results in neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America, 112(42), 12962–12967.
Hornemann, T., Richard, S., Rutti, M. F., Wei, Y., & von Eckardstein, A. (2006). Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. The Journal of Biological Chemistry, 281(49), 37275–37281.
Ikushiro, H., Hayashi, H., & Kagamiyama, H. (2001). A water-soluble homodimeric serine palmitoyltransferase from Sphingomonas paucimobilis EY2395T strain. Purification, characterization, cloning, and overproduction. The Journal of Biological Chemistry, 276(21), 18249–18256.
Teng, C., et al. (2008). Serine palmitoyltransferase, a key enzyme for de novo synthesis of sphingolipids, is essential for male gametophyte development in Arabidopsis. Plant Physiology, 146(3), 1322–1332.
Dietrich, C. R., et al. (2008). Loss-of-function mutations and inducible RNAi suppression of Arabidopsis LCB2 genes reveal the critical role of sphingolipids in gametophytic and sporophytic cell viability. The Plant Journal, 54(2), 284–298.
Harmon, J. M., et al. (2013). Topological and functional characterization of the ssSPTs, small activating subunits of serine palmitoyltransferase. The Journal of Biological Chemistry, 288(14), 10144–10153.
Kimberlin, A. N., et al. (2013). Arabidopsis 56-amino acid serine palmitoyltransferase-interacting proteins stimulate sphingolipid synthesis, are essential, and affect mycotoxin sensitivity. Plant Cell, 25(11), 4627–4639.
Lee, S. Y., et al. (2012). Cardiomyocyte specific deficiency of serine palmitoyltransferase subunit 2 reduces ceramide but leads to cardiac dysfunction. The Journal of Biological Chemistry, 287(22), 18429–18439.
Lynch, C. J., & Adams, S. H. (2014). Branched-chain amino acids in metabolic signalling and insulin resistance. Nature Reviews. Endocrinology, 10(12), 723–736.
Wallace, M., et al. (2018). Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nature Chemical Biology, 14(11), 1021–1031.
Al Sazzad, M. A., Yasuda, T., Murata, M., & Slotte, J. P. (2017). The long-chain sphingoid base of ceramides determines their propensity for lateral segregation. Biophysical Journal, 112(5), 976–983.
Troupiotis-Tsailaki, A., et al. (2017). Ligand chain length drives activation of lipid G protein-coupled receptors. Scientific Reports, 7(1), 2020.
Vutukuri, R., et al. (2020). S1P d20:1, an endogenous modulator of S1P d18:1/S1P2-dependent signaling. The FASEB Journal, 34(3), 3932–3942.
Hicks, A. A., et al. (2009). Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genetics, 5(10), e1000672.
Illig, T., et al. (2010). A genome-wide perspective of genetic variation in human metabolism. Nature Genetics, 42(2), 137–141.
Demirkan, A., et al. (2012). Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. PLoS Genetics, 8(2), e1002490.
Willer, C. J., et al. (2013). Discovery and refinement of loci associated with lipid levels. Nature Genetics, 45(11), 1274–1283.
Tabassum, R., et al. (2019). Genetic architecture of human plasma lipidome and its link to cardiovascular disease. Nature Communications, 10(1), 4329.
Cresci, S., et al. (2020). Genetic architecture of circulating very-long-chain (C24:0 and C22:0) ceramide concentrations. Journal of Lipid and Atherosclerosis, 9(1), 172–183.
McGurk, K. A., et al. (2021). Heritability and family-based GWAS analyses of the N-acyl ethanolamine and ceramide plasma lipidome. Human Molecular Genetics, 30(6), 500–513.
Choi, R. H., Tatum, S. M., Symons, J. D., Summers, S. A., & Holland, W. L. (2021). Ceramides and other sphingolipids as drivers of cardiovascular disease. Nature Reviews. Cardiology, 18(10), 701–711.
Poss, A. M., et al. (2020). Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. The Journal of Clinical Investigation, 130(3), 1363–1376.
Hilvo, M., et al. (2020). Development and validation of a ceramide- and phospholipid-based cardiovascular risk estimation score for coronary artery disease patients. European Heart Journal, 41(3), 371–380.
Russo, S. B., Tidhar, R., Futerman, A. H., & Cowart, L. A. (2013). Myristate-derived d16:0 sphingolipids constitute a cardiac sphingolipid pool with distinct synthetic routes and functional properties. The Journal of Biological Chemistry, 288(19), 13397–13409.
Mirkov, S., Myers, J. L., Ramirez, J., & Liu, W. (2012). SNPs affecting serum metabolomic traits may regulate gene transcription and lipid accumulation in the liver. Metabolism, 61(11), 1523–1527.
Gulati, S., Liu, Y., Munkacsi, A. B., Wilcox, L., & Sturley, S. L. (2010). Sterols and sphingolipids: Dynamic duo or partners in crime? Progress in Lipid Research, 49(4), 353–365.
Shah, C., et al. (2008). Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. The Journal of Biological Chemistry, 283(20), 13538–13548.
Cinar, R., et al. (2014). Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology, 59(1), 143–153.
Yoshimine, Y., et al. (2015). Hepatic expression of the Sptlc3 subunit of serine palmitoyltransferase is associated with the development of hepatocellular carcinoma in a mouse model of nonalcoholic steatohepatitis. Oncology Reports, 33(4), 1657–1666.
Teng, W., et al. (2019). Sulforaphane prevents hepatic insulin resistance by blocking serine palmitoyltransferase 3-mediated ceramide biosynthesis. Nutrients, 11(5), 1185.
Dong, Y. Q., et al. (2017). Omega-3 PUFA ameliorates hyperhomocysteinemia-induced hepatic steatosis in mice by inhibiting hepatic ceramide synthesis. Acta Pharmacologica Sinica, 38(12), 1601–1610.
Chew, W. S., et al. (2019). Large-scale lipidomics identifies associations between plasma sphingolipids and T2DM incidence. JCI Insight, 5(13), e126925.
Gantner, M. L., et al. (2019). Serine and lipid metabolism in macular disease and peripheral neuropathy. The New England Journal of Medicine, 381(15), 1422–1433.
Muthusamy, T., et al. (2020). Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature, 586(7831), 790–795.
Mohassel, P., et al. (2021). Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nature Medicine, 27(7), 1197–1204.
Gonzaga-Jauregui, C., et al. (2015). Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Reports, 12(7), 1169–1183.
Waterhouse, A., et al. (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303.
Bienert, S., et al. (2017). The SWISS-MODEL repository-new features and functionality. Nucleic Acids Research, 45(D1), D313–D319.
Guex, N., Peitsch, M. C., & Schwede, T. (2009). Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis, 30(Suppl 1), S162–S173.
Studer, G., et al. (2020). QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics, 36(6), 1765–1771.
Bertoni, M., Kiefer, F., Biasini, M., Bordoli, L., & Schwede, T. (2017). Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Scientific Reports, 7(1), 10480.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive licence to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Lone, M.A., Bourquin, F., Hornemann, T. (2022). Serine Palmitoyltransferase Subunit 3 and Metabolic Diseases. In: Jiang, XC. (eds) Sphingolipid Metabolism and Metabolic Disease. Advances in Experimental Medicine and Biology, vol 1372. Springer, Singapore. https://doi.org/10.1007/978-981-19-0394-6_4
Download citation
DOI: https://doi.org/10.1007/978-981-19-0394-6_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-0393-9
Online ISBN: 978-981-19-0394-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)