
Abstract
Spinal muscular atrophy (SMA) is the most common genetic cause of infant mortality. Homozygous deletion of Survival Motor Neuron (SMN) 1 causes the disease in 95% of cases, but severity varies with the copy number of SMN2. The nearly identical sequences of the SMN genes make differentiation between them difficult, and current methodologies such as multiplex ligation-dependent probe amplification (MLPA) and real-time PCR (RT-PCR) present limitations for diagnosis. In this report, we analyse SMA samples of known copy number to compare new emerging technologies (Linked-Reads, Cytoscan array and next generation sequencing) against MLPA. MLPA analysis was the only method able to report the known copy numbers of the samples, making it still the most reliable means of SMA carrier screening and diagnosis. However, the three new methodologies are still emerging technologies for routine diagnostics compared to MLPA, and with further optimisation, they could greatly improve SMA carrier screening and diagnosis.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
- Spinal muscular atrophy (SMA)
- SMN1
- SMN2
- Linked-Reads
- Cytoscan (CYT) array
- Whole genome sequencing (WGS)
- Multiplex ligation-dependent probe amplification (MLPA)
1.1 Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive motor neuron disease with an annual incidence of about 1 in 6000 to 1 in 10,000 live births, and the carrier frequency is as high as 1 in 40 [1]. 95% of SMA patients are homozygous for SMN1 deletion. SMN2 partially compensates for SMN1 loss, lowering the severity of SMA. However, when two SMN1 genes are carried in cis, this results in a silent carrier (i.e. a carrier that does not express the SƒMA phenotype) [2, 3]. Further complicating diagnosis, SMN1 and SMN2 are nearly identical inversions [4], differing by only five base pairs: c.835−45G>A, c.840C>T, c.*3+100A>G, c.*3+214A>G and c.*248A>G [5]. Therefore, there is a need to (1) differentiate between SMN1 and SMN2, (2) call copy number (CN) of SMN1 and SMN2 and (3) determine the phase of SMN1 and SMN2 for SMA clinical classification, prognosis, carrier identification and diagnosis [6]. Current methodologies such as multiplex ligation-dependent probe amplification (MLPA) and real-time PCR (RT-PCR) present some limitations. MLPA is unable to determine the phase of SMN1 and SMN2, resulting in the inability to identify silent carriers with 2 SMN1 on one chromosome but none on the other, and hence, false negative results in silent carriers. SMN genes are unevenly amplified in RT-PCR, which leads to inaccurate results. Therefore, in order to overcome these limitations, we evaluated new emerging methods—Linked-Reads, Cytoscan (CYT) array and whole genome sequencing (WGS)—against MLPA, the most commonly used method for carrier screening and diagnosis for SMA, by their ability to: 1. differentiate between SMN1 and SMN2, 2. determine the CN of SMN1 and SMN2, 3. locate structural variants in SMN1 and SMN2 and 4. phase alleles.
1.1.1 Hypothesis
We hypothesise that Linked-Reads, CYT array and WGS can overcome the limitations of current methods in determining patient or carrier status by differentiating between the two almost identical SMN genes, as well as calling CN in trans.
1.2 Methods
1.2.1 Sample Information
A total of six anonymised data sets from SMA patients with known SMN1 and SMN2 CN were provided by the National University of Singapore, Department of Paediatrics.
1.2.2 Technologies
-
1.
Linked-Reads
DNA was sheared and put through size selection. The ChromiumTM system was then used for automated barcoded library construction. The barcoded libraries were then sequenced using Illumina Whole Exome Sequencing (WES). The data obtained was visualised on Loupe, a genome browser by 10× Chromium Platform designed for visualisation of Linked-Reads data [7]. The BAM file obtained was also visualised on Integrative Genome Browser (IGV).
-
2.
CYT Array
Gene probes were deposited on a chip. cDNA, labelled with either green or red fluorescence, was generated from mRNA extracted. cDNA complementary base pairs with probes on the chip were analysed by fluorescence emission. The data obtained was visualised on Chromosome Analysis Suite (ChAS). Manual guides provided by Thermo Fisher Scientific Inc. were used in aiding the usage of software [8].
-
3.
WGS
Patient DNA was sequenced through whole genome sequencing (WGS) at 40× read depth. The BAM data obtained was visualised on IGV. User guides provided by the Broad Institute (2018) were used in aiding the usage of software. Genome Reference Consortium Human Build 37 (hg19) was used as reference for WGS samples.
-
4.
MLPA
DNA strands were denatured to separate the strands and hybridised with probes. The right probe oligo contains a stuffer sequence which is used to identify DNA pieces. The DNA was then amplified through PCR. MLPA amplicons are separated by length using capillary electrophoresis [9]. The measured fluorescence was visualised as a peak pattern and used to quantify each probe. CN was determined using probe ratio.
-
5.
CN calling using SMN/mean read depth ratio
The c.840C>T site on exon 7 is the critical difference between SMN1 and SMN2. Additionally, we were provided with the read depths of SMN1 and SMN2 exons 7 and 8 for each of our samples. Thus, using protocols modified from [10, 11], we determined the CN for our Linked-Reads and WGS samples by comparing the read depth of SMN1 and SMN2 exons 7 and 8 against each sample’s overall mean read depth using the following formula.
1.3 Results
1.3.1 Linked-Reads
Linked-Reads sequencing generates reads with an integrated barcode which traces the reads back to the original DNA molecule [12]. This allowed the reads to be mapped to the SMN1 and SMN2 genes, as shown from the read coverage in Fig. 1.1. The coverage of the c.840C site on SMN1 and c.840T site on SMN2 was also verified on IGV, confirming that Linked-Reads was able to differentiate between the SMN genes.

a SMN1 and SMN2 on sample 300,097. b, c SMN2 (left), SMN1 (right). Green bar in the coverage track indicates read depth for the region. Genes and their exons are identified and labelled in the genes track
Structural variants (SVs) can be detected through calls and candidates recognised by Linked-Reads, where calls meet the higher-quality call threshold than candidates and occur in unambiguous regions of the reference genome. However, no SVs were called by Linked-Reads in our samples. A deletion of exon 7 in SMN1 was observed in Sample 300,099 when viewing the reads in IGV, corresponding to the known SMN1 CN of 0.
CN was also calculated by comparing the read depths of exons 7 and 8 in SMN1 and SMN2 against each sample’s mean read depth. As seen in Table 1.1, there are discrepancies between the calculated and known CN, indicating that observed read depths underestimate the actual CN. This discrepancy could be due to difficulties in sequencing for the following reasons: 1. SMN1 and SMN2 genes are part of a 500 kb highly repetitive inverted duplication on chromosome 5, making it difficult to determine the organisation of this genomic region [13]; 2. high GC level of 54% in SMN1 and SMN2 [14], leading to a poor coverage of reads and less complete assembly; 3. low input DNA mass of 0.4–0.5 ng, which was below the recommended range of 1–3 ng, affecting the performance of sequencing.
Linked-Reads is able to phase alleles by assembling long reads from short reads, creating a phase block by utilising continuous reliable heterozygous variants (phasing quality > 23) to connect the reads [15]. However, in this analysis, reads in SMN1, SMN2 and their flanking regions in samples 300,097–30,099 were not assigned to either haplotype as there were insufficient single-nucleotide variants (SNVs) [15] present in our samples that were informative for Long Ranger to determine phase blocks.
1.3.2 CYT Array
Both SMN genes are labelled as SMN1 and SMN2 simultaneously by ChAS (Fig. 1.2), showing that ChAS is unable to differentiate between SMN1 and SMN2.

SMN1 and SMN2 annotation in ChAS
Mean weighted Log2 ratio and smooth signal values were calculated by ChAS and used to determine the CN. Log2 ratio indicates gain or loss in genetic material, with a ratio of 0 indicating a CN of 2. The Log2 ratios of SMN1 and SMN2 in CYT34 and CYT221 are close to 0, indicating that CN for both genes in both samples is 2. Smooth signal is a smoothed calibrated estimate which can represent non-integer CN. It uses the Gaussian function to reduce noise within the array, thus allowing for a more accurate CN to be determined. The smooth signal values of SMN1 and SMN2 in CYT34 and CYT221 are also close to 2, corroborating the Log2 ratio calculated CN of both samples. However, there is a discrepancy between the calculated and the known CN (Table 1.2), as Cytoscan is unable to differentiate between highly homologous regions such as SMN1 and SMN2 [15].
1.3.3 WGS
WGS was able to identify the SMN1 and SMN2 genes, as indicated by the presence of reads in these regions. Reads that align with the reference sequence are displayed in grey. However, WGS was not able to differentiate between the two genes well. Out of the five base pair differences in the SMN2 genes, a misalignment of SMN1 reads to SMN2 was observed in four of these regions in sample NGS-1108 (Fig. 1.3). Critically, at the crucial c.840 site, no reads were observed in SMN1, but reads were mapped to C (24 reads) and T (17 reads), respectively, in SMN2 (Fig. 1.3). This misalignment reflects WGS’s inability to distinguish between homologous regions such as the SMN genes [16] (Table 1.3).

a c.840C on SMN1 of sample NGS-1108. No reads were observed. b c.840T on SMN2 of sample NGS-1108. Bar is coloured in proportion to the read count of each base. Cytosine is in blue, and thymine is in red
1.3.4 MLPA
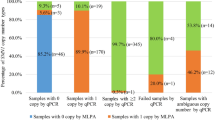
SMN1 and SMN2 genes were identified and differentiated by MLPA (Table 1.4) using probes specific to SMN1 and SMN2 exons 7 and 8. CNs were deduced from probe ratios provided by the manufacturer [9]. Sample O221 had a single copy of SMN1 exon 7 and exon 8 and is, therefore, a carrier of SMA. Sample O34 was detected to have two copies of SMN1, which indicates that the patient is unaffected. However, this is only true when the SMN1 genes occur in trans. As MLPA does not phase alleles, it cannot confirm that O34 is not a silent carrier. Sample O34 has two copies of SMN2 exon 7, but only one copy of SMN2 exon 8. This indicates a deletion of SMN2 exon 8 in sample O34. Hence, the second copy of SMN2 is not a full functional gene. MLPA can be considered a reliable tool for determining CN, as the obtained CN was consistent with the known CN (Table 1.4).
1.3.5 Comparison of Linked-Reads, CYT Array and WGS with MLPA
These newly emerging methods—Linked-Reads, CYT array and WGS—were compared against MLPA. Linked-Reads is capable of differentiating between the highly homologous SMN1 and SMN2 genes, a critical factor which measures up to MLPA and sets it apart from CYT array and WGS. Linked-Reads identification of molecules is more reliable than CYT array as it uses different “identification codes” for each molecule [12], whereas CYT array uses microarray analysis, which utilises probes [17] that are similar for the highly homologous SMN1 and SMN2 genes. This is also a drawback of WGS as reads from SMN1 may misalign to a highly homologous SMN2 gene during sequence assembly due to short read length.
In this analysis, we used a simple method of estimating CN using Linked-Reads and WGS data by comparing the read depths of SMN exons 7 and 8 to each sample’s mean read depth, due to the lack of access to sophisticated computational pipelines or software (such as those described by [18]) for determining CN. Our method gives an approximate estimation which would have to be confirmed either computationally or through wet experiments. Nevertheless, as we expect CN to be either in terms of deletion or between two to four, the exact value of the increased read depth can be rounded off. While Linked-Reads was able to detect a deletion of exon 7 in SMN1 corresponding to sample 300,099’s SMN1 CN of 0, the CNs of SMN1 and SMN2 could not be accurately determined using Linked-Reads and WGS data, unlike in MLPA.
Due to the lack of heterozygous SNVs in close proximity within SMN regions, Linked-Reads was not able to determine the haplotype of SMN1 and SMN2 in our samples, which is important for identifying silent carriers of SMA. However, if there were adequate heterozygous SNVs in SMN regions, Linked-Reads would be able to determine phase blocks and resolve haplotypes [19], giving it a huge advantage over the other three methods.
1.4 Conclusion
Considering Linked-Reads’ ability to differentiate between SMN1 and SMN2 genes, identify SNPs, and its potential ability to identify SVs and phase alleles to determine haplotypes, Linked-Reads can be viewed as a possible tool for carrier screening and diagnosis as it presents the ability to overcome limitations of MLPA and RT-PCR, which is the inability to phase SMN1 and SMN2 and uneven amplification of genes, respectively. Although Linked-Reads and WGS were not able to call SMN gene CNs accurately in this analysis, further work can be done to optimise the technology to be up to par with MLPA’s ability to call CN. To overcome the limitation of low read depth, normalisation of read counts can be done to account for GC bias [19], and DNA input mass can be increased to 1 ng per library [20].
References
MedlinePlus (2020). SMN1 gene—survival of motor neuron 1, telomeric. U.S. National Library of Medicine. https://medlineplus.gov/genetics/gene/smn1/
Prior, T. W., Nagan, N., Sugarman, E. A., Batish, S. D., & Braastad, C. (2011). Technical standards and guidelines for spinal muscular atrophy testing. Genetics in Medicine, 13(7), 686–694. https://doi.org/10.1097/GIM.0b013e318220d523
Jones, K., & Livingston, A. (2019). Spinal Muscular Atrophy and the Difficult SMN1 Gene. https://www.thermofisher.com/blog/behindthebench/spinal-muscular-atrophy-and-the-difficult-smn1-gene/
National Center for Biotechnology Information (NCBI) (2020, December 6). SMN1 survival of motor neuron 1, telomeric [Homo sapiens (human)]. U.S. National Library of Medicine. https://www.ncbi.nlm.nih.gov/gene/6606
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H. M., & McPherson, J. D. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Human Molecular Genetics, 8(7), 1177–1183. https://doi.org/10.1093/hmg/8.7.1177
Chen, X., Sanchis-Juan, A., French, C. E., Connell, A. J., Delon, I., Kingsbury, Z., Chawla, A., Halpern, A. L., Taft, R. J., Bentley, D. R., Butchbach, M. E. R., Raymond, F. L., Eberle, M. A., & BioResource, N. I. H. R. (2020). Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genetics in Medicine, 22(5), 945–953. https://doi.org/10.1038/s41436-020-0754-0
Zhang, L., Zhou, X., Weng, Z., & Sidow, A. (2019). Assessment of human diploid genome assembly with 10× linked-reads data. GigaScience, 8(giz141). https://doi.org/10.1093/gigascience/giz141
Thermo Fisher Scientific Inc. (2020). Chromosome analysis suite (ChAS) v4.2. https://assets.thermofisher.com/TFS-Assets/GSD/Handbooks/ChAS_4.2_Manual.pdf
MRC-Holland. (n.d.). Principle of MLPA. https://www.mrcholland.com/technology/mlpa/technique
Ogino, S., Gao, S., Leonard, D. G. B., Paessler, M., & Wilson, R. B. (2003). Inverse correlation between SMN1 and SMN2 copy numbers: Evidence for gene conversion from SMN2 to SMN1. European Journal of Human Genetics, 11(3), 275–277. https://doi.org/10.1038/sj.ejhg.5200957
Tattini, L., D’Aurizio, R., & Magi, A. (2015). Detection of genomic structural variants from next-generation sequencing data. Frontiers in Bioengineering and Biotechnology, 3, 92. https://doi.org/10.3389/fbioe.2015.00092
Chen, Z., Pham, L., Wu, T.-C., Mo, G., Xia, Y., Chang, P., Porter, D., Phan, T., Che, H., Tran, H., Bansal, V., Shaffer, J., Belda-Ferre, P., Humphrey, G., Knight, R., Pevzner, P., Pham, S., Wang, Y., & Lei, M. (2019). Ultra-low input single tube linked-read library method enables short-read NGS systems to generate highly accurate and economical long-range sequencing information for de novo genome assembly and haplotype phasing. BioRxiv, 852947.https://doi.org/10.1101/852947
Ott, A., Schnable, J. C., Yeh, C.-T., Wu, L., Liu, C., Hu, H.-C., Dalgard, C. L., Sarkar, S.
Berkner, S., & Lipps, G. (2007). An active nonautonomous mobile element in Sulfolobus islandicus REN1H1. Journal of Bacteriology, 189(5), 2145–2149. https://doi.org/10.1128/JB.01567-06
10x Genomics. (n.d.). What is the difference between a phase set in the long ranger VCF and a phase block in the loupe browser? https://kb.10xgenomics.com/hc/en-us/articles/360001336291-What-is-the-difference-between-a-phase-set-in-the-Long-Ranger-VCF-and-a-phase-block-in-the-Loupe-Browser
Mandelker, D., Schmidt, R. J., Ankala, A., McDonald Gibson, K., Bowser, M., Sharma, H., Duffy, E., Hegde, M., Santani, A., Lebo, M., & Funke, B. (2016). Navigating highly homologous genes in a molecular diagnostic setting: A resource for clinical next-generation sequencing. Genetics in Medicine, 18(12), 1282–1289. https://doi.org/10.1038/gim.2016.58
Nature Education (2014). Microarray. https://www.nature.com/scitable/definition/microarray-202/
Lutgen, D., Ritter, R., Olsen, R.-A., Schielzeth, H., Gruselius, J., Ewels, P., García, J.T., Shirihai, H., Schweizer, M., Suh, A., Burri, R. (2020). Linked‐read sequencing enables haplotype‐resolved resequencing at population scale. https://doi.org/10.1111/1755-0998.13192
Chouvarine, P., Wiehlmann, L., Losada, P. M., DeLuca, D. S., & Tümmler, B. (2016). Filtration and normalization of sequencing read data in whole-metagenome shotgun samples. PLoS ONE, 11(10), e0165015. https://doi.org/10.1371/journal.pone.0165015
Ruhno, C., McGovern, V. L., Avenarius, M. R., Snyder, P. J., Prior, T. W., Nery, F. C., Muhtaseb, A., Roggenbuck, J. S., Kissel, J. T., Sansone, V. A., Siranosian, J. J., Johnstone, A. J., Nwe, P. H., Zhang, R. Z., Swoboda, K. J., & Burghes, A. (2019). Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Human Genetics, 138(3), 241–256. https://doi.org/10.1007/s00439-019-01983-0
Acknowledgements
We would like to thank Professor Lai Poh San, Dr Liu Chun Ping and Ms Grace Tan Li Xuan from the Department of Paediatrics for their guidance and National University Singapore (NUS) for the valuable learning opportunity.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Sim, J., Xie, J., Tan, G.L.X., Liu, C.P., Lai, P.S. (2022). Spinal Muscular Atrophy: Evaluation of New Emerging Methods for Carrier Screening and Diagnosis. In: Guo, H., Ren, H., Wang, V., Chekole, E.G., Lakshmanan, U. (eds) IRC-SET 2021. Springer, Singapore. https://doi.org/10.1007/978-981-16-9869-9_1
Download citation
DOI: https://doi.org/10.1007/978-981-16-9869-9_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-9868-2
Online ISBN: 978-981-16-9869-9
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)