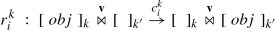This chapter presents the Infobiotics Workbench (IBW), an integrated software suite developed for computational systems biology. The tool is built upon stochastic P systems, a probabilistic extension of P systems, as modelling framework. The platform utilizes computer-aided modelling and analysis of biological systems through simulation, verification, and optimization. IBW allows modelling and analyzing not only cell-level behavior but also multicompartmental population dynamics. This enables comparing between macroscopic and mesoscopic interpretations of molecular interaction networks and investigating temporospatial phenomena in multicellular systems. These capabilities make IBW a useful, coherent, and comprehensive in silico tool for systems biology research.
The modelling and analysis of biological systems using computational approaches alternative to mathematical methods have been the focus of many recent studies since these approaches can reveal more information about system behavior. Various computational formalisms have been introduced and studied in this context, including state transition systems [32], rule-based systems [33], Petri nets [68], and process algebra [59].
Membrane computing is a popular subfield of rule-based systems. Due to its affinity with the functioning and structure of living cells, it has been utilized in modelling and analysis of a number of biological systems [12, 44, 49, 50, 65].
In membrane computing, where models are called P systems
, computations represent biological processes that take place within compartments of a living cell. Membrane structures mimic cell structures of living organisms, where compartments contain multisets of objects that evolve by the execution of a set of rules.
Stochastic P systems [64] are a probabilistic variant of P systems, where reaction rates are obtained from elementary rate constants according to the law of mass action kinetics. Stochastic P systems offer a suitable, intuitive, and amenable modeling framework for biological and chemical systems, where the inherent noise that exists in stochastic dynamics of small copy number of systems cannot be properly captured by more traditional mathematical methods. The reaction rules with associated rate constants translate directly and without additional input into probabilistic transitions of the continuous time Markov process that defines the stochastic model.
The Infobiotics Workbench (IBW) is an integrated software suite built upon stochastic P systems models. The platform utilizes computer-aided modelling and analysis of biological systems through a number of important features:
Modelling Language
IBW features a domain-specific language, where individual cells are represented by stochastic P systems. The language also allows specifications of multicellular populations distributed over various geometric surfaces, such as lattices.
Simulation
IBW implements a native stochastic simulator that enables molecular populations to be visualized over cellular populations in space and time. The results can be viewed in different formats, including time series, histograms, and 3D surface plots.
Verification
IBW has a verification component used for validating biological properties. Using powerful probabilistic model checking tools, the platform enables inferring novel system information through formal probabilistic queries and exhaustive analysis of all possible system behaviors.
Optimization
The optimization engine permits optimization parameters by estimating the rate constants in order to converge model dynamics toward laboratory observations. It also optimizes model structures by changing the composition of rule sets managing potential state transitions in compartments to generate alternative reaction networks recreating target dynamics more accurately.
IBW allows modelling and analysis of not only cell-level behavior but also multicompartmental population dynamics. This enables comparing between macroscopic and mesoscopic interpretations of molecular interaction networks and investigating temporospatial phenomena in multicellular systems.
This chapter is divided into the following sections: a presentation of the stochastic P systems, a description of IBW’s key features, two case studies where we illustrate using the IBW features, a short description of a related tool used for qualitative analysis, and finally, a presentation of the next-generation infobiotics for synthetic biology.
4.2 Stochastic P Systems
In IBW, each cell is represented by a stochastic P system (Definition 4.1). The definitions given in this section are borrowed from [12].
Definition 4.1
A stochastic P system (SP system) is a probabilistic variant of P systems, whose semantics is given by a tuple:
O is a finite set of objects that specify the entities that are part of the system (such as genes, RNAs, proteins, etc.);
L = {l1, …, ln} is a finite set of labels that name compartments (such as cells, nucleus, cytoplasm, etc.);
μ is a membrane structure containing n ≥ 1 membranes that define the regions or compartments;
Mi = (li, wi, si), for each 1 ≤ i ≤ n, is the initial configuration of the membrane i (defining a compartment or a region), where li ∈ L is the membrane label, wi ∈ O∗ is a finite multiset of objects, and si is a finite set of strings over O;
\(R_{l_{k}}=\{r_{1}^{l_{k}},\dots ,r_{m_{l_{k}}}^{l_{k}}\}\), for each 1 ≤ k ≤ n, are a set of multiset rewriting rules
that describe molecular interactions, for example, complex formation and gene regulation. Here, each set of rewriting rules \(R_{l_{k}}\) are linked to the corresponding compartment identified by the label lk. The multiset rewriting rules are defined as:
(4.2)
where o1, o2 and \(o_{1}^{\prime },o_{2}^{\prime }\) are multisets of objects (that might be empty), over O, representing molecular species that are consumed/produced in corresponding molecular reactions. The label l (linked to the square brackets) specifies the compartment where the interaction takes place. When such a rule is applied, the contents of the membrane with label l change by replacing the objects o2 with \(o_{2}^{\prime }\). The contents of the outside membrane also change by replacing the objects o1 with \(o_{1}^{\prime }\). The stochastic constant \(c_{i}^{l_{k}}\) is used to compute the rule propensity (i.e., probability and time required to apply the rule [23]).
Definition 4.1 provides the formal specification for an individual cell. Many biological systems are multicompartmental in nature, that is, they have spatial characteristics in that molecule exchanges between adjacent cells determine overall phenotypes. However, this type of structures cannot be defined by stochastic P systems as these systems have only hierarchical (nested) membrane structures that do not capture multicompartments. Therefore, stochastic P systems should be complemented with a spatial framework. Here, we define such a framework as a two-dimensional geometric lattice, which consists of a population of cells represented by SP systems. Rules moving objects from one cell to another on the lattice are associated with a vector describing where to place these molecules. This geometric extension of stochastic P systems is called lattice population P systems (LPP systems for short)
[64].
To capture the spatial distribution of cells forming colonies and tissues, we define a finite point lattice or grid with regularly distributed points [56] that can describe possible spatial geometries in Fig. 4.1. The spatial distribution of cells is defined by a finite point lattice, Definition 4.2.
Given B = {v1, …, vn} a list of linearly independent basis vectors, \(o\in \mathbb {R}^{n}\) a point referred to as origin, and a list of integer bounds \((\alpha _{1}^{min},\alpha _{1}^{max},\)\(\dots ,\alpha _{n}^{min},\alpha _{n}^{max})\), a finite point lattice generated by:
Given a finite point lattice, generated by Lat, where the coefficients {αi : i = 1, …, n} uniquely identify each point \(x=o+\sum _{i=1}^{n}\alpha _{i}v_{i}\in P(Lat)\), hence denoted as x = (α1, …, αn).
LPP systems allow the distribution of instances of stochastic P systems representing cells on a lattice according to Definition 4.3.
Definition 4.3
A lattice population P (LPP) system is a formal specification of a set of geometrically organized cells, denoted by the following tuple:
Lat defines a finite point lattice in \(\mathbb {R}^{n}\) (typically n = 2) as in Definition 4.2 describing the geometry of cellular population.
SP1, …, SPp are SP systems as in Definition 4.1 representing different cell types in the population.
Pos : P(Lat) →{SP1, …, SPp} is a function that distributes different instances of SP systems SP1, …, SPp over the lattice points.
\(T_{k}=\{r_{1}^{k},\dots ,r_{n_{k}}^{k}\}\) for each 1 ≤ k ≤ p is a finite set of translocation rules included in the skin membrane of the corresponding SP system
SPk, allowing the interchange of objects between different SP systems located in different geometrical locations. The translocation rules are specified as follows:
(4.6)
where obj is a multiset of objects, v is a vector in \(\mathbb {R}^{n}\), and \(c_{i}^{k}\) is the stochastic constant. When a translocation rule is applied in the skin membrane of an SP system SPk located at the point p (Pos(p) = SPk), the objects obj are removed from this membrane and placed in the skin membrane of \(SP_{k'}\) located at the point p + v, \(Pos(\mathbf {p}+\mathbf {v})=SP_{k'}\).
In system biology, there are cases where molecular reaction networks can be divided into modules, each of which performs a specific task [27]. It has been shown some modules, called motifs, appear recurrently in transcriptional networks. Motifs carry out particular functions like response acceleration and noise filtering [2].
In order to capture the modularity in LPP systems, hence to be able to model motifs, we have introduced P system modules [12], defined as follows:
Definition 4.4
A P system module, Mod, is defined using three finite ordered sets of variables O = {O1, …, Ox}, C = {C1, …, Cy}, and Lab = {L1, …, Lz} (where O, C and Lab represent objects, stochastic kinetic constants, and compartment labels, respectively). Modules contain a finite set of rewriting rules that have the same form in Eq. (4.2):
In order to specify and manipulate LPP system models, we have introduced LPP XML [12], a set of machine-readable data formats closely mirroring our formal definitions. LPP XML allows us to define LPP system models which consist of stochastic P system modules with initial multisets and instantiations of rules and a geometric lattice and distribution of stochastic P systems over the lattice.
The LPP XML formats are very convenient for software implementation, but writing, reading, and manipulating models in XML by hand is a very cumbersome task with syntax obscuring information. Hence, to utilize this process, we have defined a user-friendly DSL (domain-specific language), called LPP DSL. IBW implements a parser that directly reads LPP DSL files and automatically converts them into XML.
The LPP formalism permits the reuse of some components:
Inter-model reuse: Modules (in libraries), stochastic P systems, and lattices are put into different files that can be used and referred from multiple LPP system models.
Intra-model reuse: Multiple SP systems can reside within each LPP system, utilizing the model construction of homogeneous or heterogeneous bacterial colonies or tissues.
Intra-submodel reuse: Modules of rules can be parameterized and instantiated multiple times within an SP system using different instantiations.
P systems modules can be made more or less abstract by parameterizing different elements, such as species and stochastic rate constants. Motifs, corresponding to the topology of the underlying biological network, can be specified by modules that are made fully abstract by representing all components as parameters. In this scenario, parameter names should point out what role their values will play in the module.
4.3 Software Description
The Infobiotics Workbench (IBW) [30] is an integrated in silico platform built upon lattice population P (LPP) systems models [11, 12]. IBW has several functionalities. It allows simulating LPP models using a custom-built stochastic simulator, mcss, and provides a user-friendly dashboard to visualize the simulation experiments in various formats. The dashboard uses adjustable editor views, allowing to edit and run model files easily.
The platform features a model checking component, pmodelchecker, that permits users verify temporospatial dynamic system properties using probabilistic or statistical model checking. IBW also offers parameter and model structure optimization using evolutionary algorithms via POptimizer.
The users can perform experiments using the integrated dashboard or individual components separately outside the workbench. IBW makes the flow of information between different components seamless and easy by passing parameter files and model files through different components (see Fig. 4.2 [12]).
Fig. 4.2
Summary of the data flow between different components of IBW. Information is passed as files: parameters (.params) and models (.sbml, .lpp or .xml). Various intermediary files are generated: simulation data (.h5) and verification data (.psm). The results can be exported in various formants: tabulated data (.csv), image (.jpg,.png,.eps), and videos (.avi, .mpg)
The Infobiotics Workbench features a custom-built simulation platform, mcss (multicompartmental stochastic simulation)
, comprising two types of quantitative simulations: deterministic numerical approximation with standard solvers and stochastic simulation using Gillespie algorithms [23]
. mcss extends the baseline Gillespie method with multicompartmental stochastic algorithms [63] that relies on compartmentalized nature of lattice population P systems models. The algorithm uses queues that store the next rule to execute in each compartment in the heap and only recalculates the reaction propensities in a compartment where a rule is fired. This approach significantly improves performance by reducing the simulation time for models that consist of a large number of compartments.
IBW features a very user-friendly simulation dashboard (see Fig. 4.3) [12]. The simulation environment allows tweaking various simulation parameters, for example, number of runs, time points, and intervals, concentration units, and species to be displayed. The results can be displayed as time series and histograms. System population dynamics can also be observed as surface plotting functions in 3D by selecting a subset of compartments. The results can be exported in common data formats (e.g., csv) for manipulating by third-party software.
The simulation dashboard has a number of features to make the simulation experiments simple, customizable, and reproducible. Users can: (i) select a subset of (or all) entries, multiple, species, and compartments; (ii) filter species or sort them in alphabetical order; (iii) filter compartments or sort them by their geometric positions on the lattice; (iv) adjust simulation time points and intervals; (v) set data and display units (species concentrations as molecules, moles, or concentrations; compartment volumes as liters, milliliter, microliters, and nanoliters; and time points as seconds, minutes, or hours); (vi) select whether species’ amounts in each compartment over the selected runs should be averaged for obtaining approximate results; (vii) get an estimated memory requirement for each simulation experiment to predict how fast the experiment can be carried out; (viii) export the selected and rescaled datapoints in various data formats (.csv, .xls, .npz); and (ix) plot results for selected runs and compartments as time series or histograms, which allows making exact (combined) or relative (stacked/tiled) comparisons of the temporal behavior of different molecular species of same/different compartments based on specific, several, or averaged over many simulation runs. (x) export plots as images for further comparison with experimental observations (see Fig. 4.4) [12]. The figure toolbar enables zooming, panning, and subplot configuration and (xi) visualize the system dynamics at real-time in 2D space using 3D heat-mapped meshes or surface plots to capture the dynamic distribution of selected species over time (see Fig. 4.5) [12]. Surfaces plots provide an intuitive means of qualitative evaluation of population level dynamics that may (cautiously) be compared to laboratory observations.
Surface plots illustrating dynamic expression patterns for two proteins. Users can progress time either by moving the time point index slider forward or backward or by pressing the Play/Pause button
Formal methods have been used in systems biology in order to better understand system behavior. As a complementary approach to simulation, formal verification is a method which exhaustively analyzes all possible system behaviors, which cannot be done via simulation, to evaluate the correctness of systems. It allows inferring “more novel information about system properties” [44].
Model checking [14]
, an algorithmic verification approach, is used to verify whether a model with a finite structure satisfies certain system properties. Model checking requires a formal system model and a formal specification, expressed in a logical notation [34,35,36,37,38,39]. It then evaluates the formal specification against all possible behaviors of the system model, which are computed by enumerating all possible sequence of traces.
Model checking has been widely utilized in computing and engineering applications for the last two decades in verifying various systems, for example, safety-critical systems [40], concurrent systems [3], distributed systems [69], network protocols [42], stochastic systems [41], multi-agent systems [1, 47], pervasive systems [4, 43, 48], and swarm robotics [45, 46] as well as some engineering applications [57, 58]. Due to its novel features to infer information about system behavior, there is a growing interest to apply this technique in systems biology [8, 9]. Recently, it has been applied to analysis of various biological systems [21, 49, 49, 50, 52, 65].
Probabilistic model checking
is a stochastic extension of classical model checking complemented with quantitative techniques to verify properties about the likelihood of the observation of certain behavior. However, they require a probabilistic state machine (such as Discrete-Time Markov Chains (DTMCs), Continuous-Time Markov Chains (CTMCs)) or Markov Decision Processes (MDPs) in a dedicated syntax. System properties are written as probabilistic logical statements, often probabilistic logics: CSL(Continuous Stochastic Logic) [5] for CTMCs and PCTL(Probabilistic Computation Tree Logic) [26] for DTMCs and MDPs. A probabilistic model checker then automatically verifies if the system model satisfies the property using some analytical methods.
The Infobiotics Workbench features a verification module, called pmodelchecker
, which integrates two third-party probabilistic models checkers Prism [28]
and MC2 [15]
. Properties of stochastic P system models are written as probabilistic logic formulas and automatically verified using either Prism or MC2. pmodelchecker extends the verification capability to multicompartments so as to verify LPP system models.
pmodelchecker supports both exact (i.e., numerical) and approximate (i.e., statistical) model checking methods
. To perform exact probabilistic model checking, LPP systems are automatically converted into the reactive modules specification, from which Prism is executed. In this approach, the full state space is generated and each property is verified against all states of the model, which is usually computationally very demanding. The approximate probabilistic model checking does not require generating all system states. Instead, simulations are run up to a specified maximum number of runs or a confidence threshold (defined by users), and properties are verified against the simulation traces instead of the system model. To perform approximate probabilistic model checking, users can either (i) call Prism’s discrete event simulator or (ii) run MC2 using previous simulation results or running new simulations.
The pmodelchecker dashboard provides an interface for both Prism and MC2 (see Fig. 4.6) [12]. Users can adjust verification parameters for each model checker, accordingly. The dashboard allows loading multiple formulas from a file and selecting a specific formula that can be edited or removed. Users can also add a new formula using the respective buttons.
The pmodelchecker dashboard features a result view which displays the outcome of a model checking experiment (see Fig. 4.7) [12]. The results can be displayed in 2D if the probability of a property in question is compared against one selected variable, or the results can be displayed in 3D if the probability is checked against two variables. The dashboard allows performing queries that depend on several variables by enabling the choice of variables so that the results of n-dimensional queries to be viewed in a consistent manner.
The correct reproduction of cellular behavior depends on the accuracy of kinetic rate constants used in both deterministic and stochastic models. Unfortunately, well-characterized rate constants are not often available in many systems, and those that are known for some models use artificial values that are obtained from similar systems. One possible solution to this problem is using parameter optimization to estimate the rate constants in order to fit model dynamics to laboratory observations.
For this purpose, IBW features the POptimizer component, which optimizes models in two ways:
1.
Numerical model parameters: The stochastic kinetic constants
linked to each rule can be tweaked to fit the given target.
2.
Model structure: The composition and structure of the rule sets managing possible state transitions occurring in compartments can be changed to generate alternative reaction networks recreating the target dynamics more accurately.
Both of these optimization steps aim to minimize the distance between the stochastically simulated and user-provided quantities of molecular species at every target time point, quantitatively evaluating the fitness of candidate models and automatically discriminating between them.
POptimizer
searches both parameter and structure spaces using well-known population-based optimization algorithms: Covariance Matrix Adaptation Evolution Strategies (CMA-ES) [25], Estimation of Distribution Algorithms (EDA), Differential Evolution (DE) [67], and genetic algorithms (GA) [24]. The current version of the optimization process is limited to single compartment models because multicompartmental structures significantly increase the algorithmic complexity. This is mainly due to the fact that simulating many copies of the cells at those compartments would increase the computational cost and makes it difficult to provide accurate target data. Hence, model optimization is generally feasible for smaller models, which can then be reintegrated, provided they can be decoupled.
POptimizer implements a genetic algorithm [13, 62] to produce candidate models. This is initially done by random choice and then by mutating the fittest models of the previous round, performing several runs of parameter optimization steps on each model to ensure that the candidate models have fair chance of fitting the target behavior before using the final fitness function to choose the next generation.
The result of an optimization process is the fittest model generated, and the outcome is displayed at the dashboard. POptimizer also allows a visual comparison of the quantities of each species for target and the optimized models (see Fig. 4.8) [12].
In this section, we will illustrate using the IBW features in two case studies. In the first case study, we will use the pulse generator system [10], consisting of a bacterial colony that displays a propagation behavior of a wave of gene expression. The second case study is a genetic circuit, repressilator.
4.4.1 Pulse generator
The pulse generator system [10] synthesizes a signalling molecule AHL, triggering the production of the green fluorescent protein (GFP). The system exhibits a propagation behavior, that is, the propagation of the GFP expression along the bacterial colony (see Fig. 4.11 and 4.12) [12]. The system consists of two different bacterial strains, sender cells and pulsing cells (see Fig. 4.9) [50], which work as follows:
“Sender cells contain the gene luxI from Vibrio fischeri. This gene codifies the enzyme LuxI responsible for the synthesis of the molecular signal 3OC12HSL (AHL). The luxI gene is expressed constitutively under the regulation of the promoter PLtetO1 from the tetracycline resistance transposon.”
Fig. 4.9
The sender and pulsing cells of the pulse generator.
“Pulsing cells contain the luxR gene from Vibrio fischeri that codifies the 3OC12HSL receptor protein LuxR. This gene is under the constitutive expression of the promoter PluxL. It also contains the gene cI from lambda phage codifying the repressor CI under the regulation of the promoter PluxR that is activated upon binding of the transcription factor LuxR_3OC12. Finally, this bacterial strain carries the gene gfp that codifies the green fluorescent protein under the regulation of the synthetic promoter PluxPR combining the Plux promoter (activated by the transcription factor LuxR_3OC12) and the PR promoter from lambda phage (repressed by the transcription factor CI).”
The sender and pulsing bacterial strains are distributed along a lattice, where the sender cells are located at one end of the lattice, and the pulsing cells are placed at the rest of the lattice (see Fig. 4.10).
As discussed in Sect. 4.2, IBW accepts lattice population systems as input. The pulse generator system is captured by an LPP model, representing a bacterial colony over a rectangular lattice, which distributes the sender cells at one end of the lattice and the pulsing cells over the rest of the lattice. The LPP model contains two stochastic P systems models, one for each different cell type. The first SP model represents the stochastic behavior of the sender cell, capturing the production of the signal 3OC6-HSL (AHL). The second model represents the pulsing cell, capturing the production of GFP protein as a response to the signal 3OC6-HSL (AHL). In both SP models, the reaction rules govern the regulation of the corresponding promoters used in the sender and pulsing cells. The complete stochastic model of the pulse generator example (written in LPP) is available in the IBW website [60].
Simulation
The IBW simulation dashboard visualizes the system behavior via time series, histogram, or surface plotting functions. Users are able to choose species they want to simulate over a subset of datapoints. Below, we present a set of simulation experiments [12, 44, 50].
Figure 4.11 shows the propagation of a pulse of GFP over a single pulsing cell using time series. Figure 4.12 illustrates the spatial propagation over a bacterial colony using 3D animation. The propagation of the GFP protein continues through pulsing cells until the concentration level drops to 0.
Figure 4.13 shows the signalling molecule signal3OC6 amount, the number of molecules, over time, suggesting that the pulsing cells located further away from the sender cells produce lower concentrations of GFP.
These experiments suggest IBW’s stochastic simulation algorithms allow users to generate realistic trajectories of molecular dynamics that can be compared to laboratory data.
Verification
IBW’s pmodelchecker component allows users to perform verification using two third-party probabilistic model checkers Prism and MC2 to infer more information about system behavior.
Below, we present a set of verification experiments [50] based on probabilistic model checking. Here, we consider a lattice of size 2 × 6. The sender cells are positioned to the initial 2 × 2 segment of the lattice, followed by the pulsing cells that are distributed to the rest (2 × 4 ) of the lattice (see Fig. 4.10).
In the following, we show the informal representation of queries (i.e., system requirements to be verified) and their corresponding translations to the language that pmodelchecker accepts as input.
Query 1
“What is the probability thatGFPconcentration at row n ∈{3, 4, 5, 6} exceeds 100 at the time instant T?”
The verification results are illustrated in Fig. 4.14a.
Fig. 4.14
Quantitative analysis using probabilistic model checking. Row n denotes the nth row of the pulsing cells in the lattice and T denotes time. (a) Prob. of GFP exceeds threshold (Prop. 1). (b) Prob. of relative GFP (Prop. 2). (c) Expected GFP protein (Prop. 3). (d) Expected signal3OC6 (Prop. 4)
“What is the probability thatGFPconcentration at row n ∈{3, 4, 5} stays greater thanGFPconcentration at row 6 until the time instant T whereGFPconcentration at row 6 exceedsGFPconcentration at row n?”
The corresponding verification results are shown in Fig. 4.14d.
Figure 4.14a,c confirm the propagation of a pulse of GFP, whose concentration first increases in the rows near to the sender cells and then gradually drops to zero. The rows distant from the sender cells exhibit a similar behavior with some delay, which is proportional to the distance between the row and the sender cells. Figure 4.14d shows that pulsing cells located further away from the sender cells produce lower concentrations of GFP.
These results show verification, by means of formal queries, can provide more novel information about the system behavior and dynamics, complementary to simulation.
4.4.2 Repressilator
The repressilator is a genetic circuit [17] used as a canonical example in some P system models [19].
The system contains three genes codifying the corresponding repressors: the operon lactose repressor, lacI; the repressor from the tetracycline transposon, tetR; and a repressor from the λ phage virus, cI. These three genes are linked in a gene regulatory network in such a way that lacI represses the expression of tetR; the tetR gene then represses cI. Finally, cI represses the expression of lacI to close the cycle.
Modelling
The repressilator system is captured as a stochastic P system. The molecular interactions within the stochastic P system are defined in a modular manner. The bacterial colony is modelled by a lattice population system over a rectangular lattice. This is done by distributing the copies of this cell type over the points of a rectangular lattice. The complete stochastic model of the repressilator system is available in the IBW website [61].
Simulation
Figure 4.15 shows the system evolution over time for the LacI, CI, and TetR proteins, confirming that the circuit generates oscillations of these repressor molecules based on the order they are connected within the regulatory network.
The oscillations significantly differ in amplitude and frequency due to stochastic effects. Therefore, different cells in the lattice might exhibit different oscillatory behavior, not necessarily synchronous (as illustrated in Fig. 4.15).
The asynchronous oscillatory behavior in different cells can be better observed using the population dynamics. Figure 4.16 shows the spatiotemporal evolution of LacI, CI, and TetR in the entire colony carrying the repressilator.
Fig. 4.16
Spatiotemporal evolution of the CI and TetR proteins in the colony
Below, we show two queries used to calculate the probability of having more or fewer than 300 proteins of LacI, CI, and TetR simultaneously over different time points of the evolution.
Query 1
“What is the probability thatLacI, CI, andTetRcan simultaneously be below 300 molecules?”
Similarly, the verification results for t = 20, 000…40, 000 (with increments of 5000) are zero.
The results obtained in both scenarios suggest that these three proteins cannot be above or below 300 molecules simultaneously, confirming oscillating behavior.
4.5 KPWorkbench: A Qualitative Analysis Tool
We have illustrated how IBW facilitates the quantitative analysis of biological systems using stochastic P systems. However, in some cases, quantitative analysis might not be needed if, for example, we only want to observe the detection of molecular species rather than measuring their concentration. In such cases, we can only rely on qualitative analysis where we can apply some abstraction methods to reduce the model complexity. One typical abstraction method is removing kinetic constants from a stochastic model. In this way, we can obtain much simpler nondeterministic models that can be used for detecting the existence of molecular species.
A nondeterministic model captures all interactions included in its stochastic counterpart but in a rather symbolic and qualitative way in that it removes more precise quantitative aspects of the system. All possible system pathways are still contained in the nondeterministic model but as exact molecular concentrations are not necessary for these models. In certain circumstances, the multisets are bounded, even restricted to one or two elements, describing their presence rather than their molecular concentrations.
In order to facilitate the qualitative modelling, we have introduced kernel P systems [22], a non-probabilistic variant of stochastic P systems, which mimic biological membranes without any quantitative information. Kernel P systems allow building nondeterministic models, which are used for qualitative analyses where molecular concentrations are not necessary or a chain of reactions already analyzed can be replaced by some abstractions mimicking their behavior through simpler rewriting mechanisms.
The expressive power and efficiency of kP systems have been illustrated by a number of representative case studies [49, 50, 58]. In this respect, we have also introduced a modelling language, called kP–Lingua, allowing one to write kP system models. The theoretical aspects of the methods and techniques developed for kP systems have been discussed in [6, 7, 16, 20].
We have also developed the kPWorkbench platform [53] (available and downloadable from its website [54]), which allows modelling and analysis of membrane systems through various computational approaches, including modelling, simulation, agent-based high-performance simulation [51], and verification. To simplify verification queries, we have introduced a user-friendly property language based on natural language statements. These unique features allow kPWorkbench to support the non-probabilistic modelling and analysis of membrane systems using various computational approaches. The usability and novelty of our approach have been illustrated by some case studies from systems and synthetic biology [49, 50] to some engineering problems [57, 58].
4.6 Next-Generation Infobiotics for Synthetic Biology
Systems biology mainly focuses on studying existing organisms. In computational biology, there is a growing trend to study biological phenomena that do not exist in nature. To this end, synthetic biology, aiming to design new biological entities, is emerging rapidly. As DNA sequencing and synthesis technology get cheaper and become easy to reach [55], the scale and complexity of engineered biology systems will grow. Moreover, rapidly emerging biotechnology is accelerating the adoption of synthetic biology across various disciplines including computing science as well as industrial applications.
In line with these advances, synthetic biology introduces new challenges difficult for existing tools and approaches to address. It is well known that most of synthetic biology models are complex, with a rich combinatorics of biochemical interactions and certain motifs occurring.
Although IBW provides a good tool support for systems biology, and it can be utilized for some small-scale synthetic biology systems, it cannot address the challenges imposed by synthetic biology. The IBW language allows modelling systems at a relatively high abstraction level but does not provide any support for further refinements at the DNA level, which is a requirement of synthetic biology, where different operations at that level have to be specified. Also, the simulation and verification processes that are normally efficient for systems biology can be very cumbersome depending on the complexity of synthetic systems.
In an attempt to provide a robust tool support for synthetic biology, we have developed a new version of Infobiotics Workbench [31] that can assist synthetic biologists in an informed, iterative workflow of system specification, verification, simulation, and biocompilation. This new version of IBW features a unique domain-specific language, called IBL (Infobiotics Language), offering a combined grammar for modelling, verification, and biocompilation statements rather than relying upon individual complex formalisms for each computational aspect. This novel approach offers seamless interoperability across different tools as well as compatibility with common data exchange formats, for example, SBOL (Synthetic Biology Open Language) [18]
and SBML (Systems Biology Markup Language) [29]
, and eliminates the need of manual translations for stand-alone applications.
The new IBW also significantly improves the efficiency of computational processes so as to cope with scaling-up demand of synthetic biology. The platform implements a new simulation module, incorporating all the variants of Gillespie’s stochastic simulation algorithms (SSAs) complemented with prediction tool that selects the best performing SSA using machine learning algorithms. The simulation algorithms are also speeded up via parallel implementation and executed on cloud-based GPU clusters.
The verification queries use natural language statements, which are embedded within the IBL language. This makes IBL easy to use and intuitive for nonexperts. The verification process relies on statistical model checking approach [66], which significantly improves model checking times. This allows verifying queries for large systems in seconds rather than hours.
IBW also features a biocompilation module that allows automated compilation of a specified synthetic circuit into eventual genetic sequence information and import from/export to standard data exchange formats.
These unique features make IBW a very useful in silico tool for synthetic biology.
4.7 Conclusion
In this chapter, we have presented the Infobiotics Workbench, a computer-aided in silico design suite for systems biology. We have provided an overview of the platform’s important features: (a) a domain-specific language, where individual cells are represented by stochastic P systems and multicellular populations are represented by lattice population P systems; (b) a multicellular stochastic simulator that enables molecular populations to be visualized over cellular populations in space and time using a variety of visualization formats; (c) a verification component that validates biological properties using probabilistic model checking; and (d) an optimization engine that optimizes model parameters and model structures.
We have shown the usability and applicability of the platform with two case studies: pulse generator and repressilator. For each case study, we have discussed the respective modelling, along with its simulation and verification results.
We have also provided a brief overview of the new version of Infobiotics Workbench [31] developed to address the challenges and requirements of synthetic biology by providing an informed, iterative workflow of system specification, verification, simulation, and biocompilation.
References
H. Abbink, R. van Dijk, T. Dobos, M. Hoogendoorn, C. Jonker, S. Konur, P.P. van Maanen, V. Popova, A. Sharpanskykh, P. van Tooren, J. Treur, J. Valk, L. Xu, P. Yolum, Automated support for adaptive incident management, in Proceedings of the 1st International Workshop on Information Systems for Crisis Response and Management (ISCRAM’04) (Brussels, Belgium, 2004), pp. 153–170
R. Alur, K. McMillan, D. Peled, Model-checking of correctness conditions for concurrent objects. Inf. Comput. 160(1–2), 167–188 (2000). https://doi.org/10.1006/inco.1999.2847
M. Arapinis, M. Calder, L. Denis, M. Fisher, P. Gray, S. Konur, A. Miller, E. Ritter, M. Ryan, S. Schewe, C. Unsworth, R. Yasmin, Towards the verification of pervasive systems. Electron. Commun. EASST 22, 1–15 (2009). https://doi.org/10.14279/tuj.eceasst.22.315
C. Baier, B Haverkort, H. Hermanns, J.P. Katoen, Model-checking algorithms for continuous-time markov chains. IEEE Trans. Software Eng. 29, 524–541 (2003). https://doi.org/10.1109/TSE.2003.1205180
M.E. Bakir, F. Ipate, S. Konur, L. Mierlă, I. Niculescu, Extended simulation and verification platform for kernel P systems, in Membrane Computing (CMC 2014), ed. by M. Gheorghe, G. Rozenberg, A. Salomaa, P. Sosík, C. Zandron. Lecture Notes in Computer Science, vol. 8961 (2014), pp. 158–178. https://doi.org/10.1007/978-3-319-14370-5_10
M.E. Bakir, S. Konur, M. Gheorghe, I. Niculescu, F. Ipate, High performance simulations of kernel P systems, in Proceedings of the 2014 IEEE International Conference on High Performance Computing and Communications, 2014 IEEE 6th International Symposium on Cyberspace Safety and Security, 2014 IEEE 11th International Conference on Embedded Software and System (HPCC,CSS,ICESS) (2014), pp. 409–412. https://doi.org/10.1109/HPCC.2014.69
M.E. Bakir, M. Gheorghe, S. Konur, M. Stannett, Comparative analysis of statistical model checking tools, in Membrane Computing (CMC 2016), ed. by A. Leporati, G. Rozenberg, A. Salomaa, C. Zandron. Lecture Notes in Computer Science, vol. 10105 (2017), pp. 119–135. https://doi.org/10.1007/978-3-319-54072-6_8
M.E. Bakir, S. Konur, M. Gheorghe, N. Krasnogor, M. Stannett, Automatic selection of verification tools for efficient analysis of biochemical models. Bioinformatics 34(18), 3187–3195 (2018). https://doi.org/10.1093/bioinformatics/bty282
S. Basu, Y. Gerchman, C.H. Collins, F.H. Arnold, R. Weiss, A synthetic multicellular system for programmed pattern formation. Nature 434 (2005), 1130–1134. https://doi.org/10.1038/nature03461
J. Blakes, J. Twycross, F.J. Romero-Campero, N. Krasnogor, The Infobiotics Workbench: an integrated in silico modelling platform for systems and synthetic biology. Bioinformatics 27(23), 3323–3324 (2011). https://doi.org/10.1093/bioinformatics/btr571
J. Blakes, J. Twycross, S. Konur, F.J. Romero-Campero, N. Krasnogor, M. Gheorghe, Infobiotics workbench: a P systems based tool for systems and synthetic biology, in Applications of Membrane Computing in Systems and Synthetic Biology. Series Emergence, Complexity and Computation, Chapter 7 (2014), pp. 1–41. https://doi.org/10.1007/978-3-319-03191-0_1
H. Cao, F.J. Romero-Campero, S. Heeb, M. Cámara, N. Krasnogor, Evolving cell models for systems and synthetic biology. Syst. Synth. Biol. 4(1), 55–84 (2010). https://doi.org/10.1007/s11693-009-9050-7
R. Donaldson, D. Gilbert, A Monte Carlo model checker for probabilistic LTL with numerical constraints. Res. Rep. (2008), TR-2008-282. Department of Computing Science, University of Glasgow
C. Dragomir, F. Ipate, S. Konur, R. Lefticaru, L. Mierlă, Model checking kernel P systems, in Membrane Computing (CMC 2013), ed. by A. Alhazov, S. Cojocaru, M. Gheorghe, Y. Rogozhin, G. Rozenberg, A. Salomaa. Lecture Notes in Computer Science, vol. 8340 (2013), pp. 151–172. https://doi.org/10.1007/978-3-642-54239-8_12
M.B. Elowitz, S. Leibler, A synthetic oscillatory network of transcriptional regulators. Nature 403(6767), 335–338 (2000). https://doi.org/10.1038/35002125
M. Galdzicki, K.P. Clancy, E. Oberortner, M. Pocock, J.Y. Quinn, C.A. Rodriguez, R. Nicholas, M.L. Wilson, L. Adam, J.C. Anderson, The synthetic biology open language (SBOL) provides a community standard for communicating designs in synthetic biology. Nat. Biotechnol. 32(6), 545–550 (2014). https://doi.org/10.1038/nbt.2891
M. Gheorghe, V. Manca, F.J. Romero-Campero, Deterministic and stochastic P systems for modelling cellular processes. Nat. Comput. 9(2), 457–473 (2009). https://doi.org/10.1007/s11047-009-9158-4
M. Gheorghe, S. Konur, F. Ipate, L. Mierlă, M.E. Bakir, M. Stannett, An integrated model checking toolset for kernel P systems, in Membrane Computing (CMC 2015), ed. by G. Rozenberg, A. Salomaa, J.M. Sempere, C. Zandron. Lecture Notes in Computer Science, vol. 9504 (2015), pp. 153–170. https://doi.org/10.1007/978-3-319-28475-0_11
M. Gheorghe, S. Konur, F. Ipate, Kernel P systems and stochastic P Systems for modelling and formal verification of genetic logic gates, in Advances in Unconventional Computing, ed. by A. Adamatzky. Series Emergence, Complexity and Computation, vol. 22 (2017), pp. 661–675. https://doi.org/10.1007/978-3-319-33924-5_25
M. Gheorghe, R. Ceterchi, F. Ipate, S. Konur, R. Lefticaru, Kernel P systems: from modelling to verification and testing. Theor. Comput. Sci. 724, 45–60 (2018). https://doi.org/10.1016/j.tcs.2017.12.010
D. Gillespie, A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 22(4), 403–434 (1976). https://doi.org/10.1016/0021-9991(76)90041-3
A. Hinton, M. Kwiatkowska, G. Norman, D. Parker, Prism: a tool for automatic verification of probabilistic systems, in Tools and Algorithms for the Construction and Analysis of Systems, 12th International Conference, TACAS 2006 Held as Part of the Joint European Conferences on Theory and Practice of Software, ETAPS 2006, Vienna, Austria, March 25—April 2, 2006. Lecture Notes in Computer Science, vol. 3920 (2006), pp. 441–444. https://doi.org/10.1007/11691372_29
M. Hucka, A. Finney, H.M. Sauro, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19(4), 524–531 (2002). https://doi.org/10.1093/bioinformatics/btg015
S. Konur, A decidable temporal logic for events and states, in Proceedings of the Thirteenth International Symposium on Temporal Representation and Reasoning (TIME’06), Budapest, 2006 (2006), pp. 36–41. https://doi.org/10.1109/TIME.2006.1
S. Konur, An interval logic for natural language semantics, in Proceedings of the Seventh Conference on Advances in Modal Logic, Nancy, France, 9–12 September 2008, ed. by C. Areces, R. Goldblatt (2008), pp. 177–191
S. Konur, A survey on temporal logics for specifying and verifying real-time systems. Front. Comput. Sci. 7(3), 370–403 (2013). https://doi.org/10.1007/s11704-013-2195-2
S. Konur, Towards light-weight probabilistic model checking. J. Appl. Math. 2014, Article ID 814159, 1–15 (2014). https://doi.org/10.1155/2014/814159
S. Konur, M. Fisher, Formal analysis of a VANET congestion control protocol through probabilistic verification, in Proceedings of the 2011 IEEE 73rd Vehicular Technology Conference (VTC Spring), Yokohama, 2011 (2011), pp. 1–5. https://doi.org/10.1109/VETECS.2011.5956327
S. Konur, M. Gheorghe, A property-driven methodology for formal analysis of synthetic biology systems, in IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 12(2), 360–371 (2015). https://doi.org/10.1109/TCBB.2014.2362531
S. Konur, C. Dixon, M, Fisher, Formal verification of probabilistic swarm behaviours, in Swarm Intelligence (ANTS 2010), ed. by M. Dorigo et al. Lecture Notes in Computer Science, vol. 6234 (2010), pp. 440–447. https://doi.org/10.1007/978-3-642-15461-4_42
S. Konur, C. Dixon, M. Fisher, Analysing robot swarm behaviour via probabilistic model checking. Rob. Auton. Syst. 60(2), 199–213 (2012). https://doi.org/10.1016/j.robot.2011.10.005
S. Konur, M. Fisher, S. Schewe, Combined model checking for temporal, probabilistic, and real-time logics. Theor. Comput. Sci. 503, 61–88 (2013). https://doi.org/10.1016/j.tcs.2013.07.012
S. Konur, M. Fisher, S. Dobson, S. Knox, Formal verification of a pervasive messaging system. Formal Aspects Comput. 26(4), 677–694 (2014). https://doi.org/10.1007/s00165-013-0277-4
S. Konur, M. Gheorghe, C. Dragomir, F. Ipate, N. Krasnogor, Conventional verification for unconventional computing: a genetic XOR gate example. Fundam. Inform. 134, 97–110 (2014). https://doi.org/10.3233/FI-2014-1093
S. Konur, M. Gheorghe, C. Dragomir, L. Mierlă, F. Ipate, N. Krasnogor, Qualitative and quantitative analysis of systems and synthetic biology constructs using P systems. ACS Synth. Biol. 4(1), 83–92 (2015). https://doi.org/10.1021/sb500134w
S. Konur, M. Kiran, M. Gheorghe, M. Burkitt, F. Ipate, Agent-based high-performance simulation of biological systems on the GPU, in Proceedings of the 2015 IEEE 17th International Conference on High Performance Computing and Communications, 2015 IEEE 7th International Symposium on Cyberspace Safety and Security, and 2015 IEEE 12th International Conference on Embedded Software and Systems, New York, NY (2015), pp. 84–89. https://doi.org/10.1109/HPCC-CSS-ICESS.2015.253
S. Konur, H. Fellermann, L.M. Mierlă, D. Sanassy, C. Ladroue, S. Kalvala, M. Gheorghe, N. Krasnogor, An integrated in silico simulation and biomatter compilation approach to cellular computation, in Advances in Unconventional Computing, ed. by A. Adamatzky. Series Emergence, Complexity and Computation, vol. 23 (2017), pp. 655–676. https://doi.org/10.1007/978-3-319-33921-4_25
P. Kuhn, K. Wagner, K. Heil, M. Liss, N. Netuschil, Next generation gene synthesis: from microarrays to genomes. Eng. Life Sci. 17(1), 6–13 (2017). https://doi.org/10.1002/elsc.201600121
R. Lefticaru, S. Konur, Ü. Yildirim, A. Uddin, F. Campean, M. Gheorghe, Towards an integrated approach to verification and model-based testing in system engineering, in Proceedings of the 2017 IEEE International Conference on Internet of Things (iThings) and IEEE Green Computing and Communications (GreenCom) and IEEE Cyber, Physical and Social Computing (CPSCom) and IEEE Smart Data (SmartData), Exeter, 2017 (2017), pp. 131–138. https://doi.org/10.1109/iThings-GreenCom-CPSCom-SmartData.2017.25
R. Lefticaru, M.E. Bakir, S. Konur, M. Stannett, F. Ipate, Modelling and validating an engineering application in kernel P systems, in Membrane Computing (CMC 2017), ed. by M. Gheorghe, G. Rozenberg, A. Salomaa, C. Zandron. Lecture Notes in Computer Science, vol. 10725 (2018), pp. 183–195. https://doi.org/10.1007/978-3-319-73359-3_12
F.J. Romero-Campero, H. Cao, M. Camara, N. Krasnogor, Structure and parameter estimation for cell systems biology models, in Proceedings of the 10th Annual Conference on Genetic and Evolutionary Computation (GECCO ’08), Atlanta, GA, USA, July 12–16, 2008 (2008), pp. 331–339. https://doi.org/10.1145/1389095.1389153
F.J. Romero-Campero, J. Twycross, M. Cámara, M. Bennett, M. Gheorghe, N. Krasnogor, Modular assembly of cell systems biology models using P systems. Int. J. Found. Comput. Sci. 20(3), 427–442 (2009). https://doi.org/10.1142/S0129054109006668
F.J. Romero-Campero, J. Twycross, H. Cao, J. Blakes, N. Krasnogor, A multiscale modeling framework based on P systems, in Membrane Computing (WMC 2008), ed. by D.W. Corne, P. Frisco, Gh. Păun, G. Rozenberg, A. Salomaa. Lecture Notes in Computer Science, vol. 5391 (2009), pp. 63–77. https://doi.org/10.1007/978-3-540-95885-7_5
D. Sanassy, H. Fellermann, N. Krasnogor, S. Konur, L. Mierlă, M. Gheorghe, C. Ladroue, S. Kalvala, Modelling and stochastic simulation of synthetic biological Boolean gates, in Modelling and Stochastic Simulation of Synthetic Biological Boolean Gates, 2014 IEEE International Conference on High Performance Computing and Communications, 2014 IEEE 6th International Symposium on Cyberspace Safety and Security, 2014 IEEE 11th International Conference on Embedded Software and Syst (HPCC,CSS,ICESS), Paris, 2014 (2014), pp. 404–408. https://doi.org/10.1109/HPCC.2014.68
K. Sen, M. Viswanathan, G. Agha, Statistical model checking of black-box probabilistic systems, in Computer Aided Verification (CAV 2004), ed. by R. Alur, D.A. Peled. Lecture Notes in Computer Science, vol. 3114 (2004), pp. 202–215. https://doi.org/10.1007/978-3-540-27813-9_16
R. Storn, K. Price, Differential evolution: a simple and efficient heuristic for global optimization over continuous spaces. J. Global Optim. 11, 341–359 (1997). https://doi.org/10.1023/A:1008202821328
F.J.W. Symons, Introduction to numerical Petri nets, a general graphical model of concurrent processing systems. Aust. Telecommun. Res. 14(1), 28–32 (1980)
M. Yabandeh, Model checking of distributed algorithm implementations, Ph.D. thesis, IC (2011). École Polytechnique Fédérale de Lausanne. https://doi.org/10.5075/epfl-thesis-4858
Zhang, G. et al. (2021). Infobiotics Workbench: An In Silico Software Suite for Computational Systems Biology.
In: Membrane Computing Models: Implementations. Springer, Singapore. https://doi.org/10.1007/978-981-16-1566-5_4

 (4.2)
(4.2)

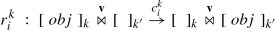 (4.6)
(4.6)