
Abstract
Metabolic network has been intensely studied and considered as fundamental biological phenomena that participate in numerous healthy and pathological processes, especially in tumorigenesis and cancer development. One hallmark of cancer cells is adaptive metabolic reprogramming which is highlighted by reduced oxidative phosphorylation and enhanced aerobic glycolysis. The metabolic reprogramming is accompanied by accumulated oxidative stress which is another important hallmark of cancer. Simultaneously, redox signaling is emerging as a critical aspect that maintains proper cellular functions, and redox imbalance is thought to be a hallmark of various diseases. Growing body of evidence has suggested an intrinsic link between metabolic network and redox biology. Redox status can change the shift of metabolic flux through regulating the expression, function, or subcellular distribution of metabolic enzymes. The alteration of metabolic flux can in turn modulate the redox status, which controls a wide range of cellular processes. Therefore, uncovering the intricate mechanisms underlying redox regulation of metabolic enzymes will lead to a deep understanding of tumorigenesis and tumor progression and facilitate the translational medicine.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Metabolic abnormalities are widely identified in cancer cells and reported to be closely linked with the tumorigenesis and tumor development [1]. Cancer cells are prone to utilize glycolysis to fulfil the substance requirement for rapid proliferation even in the presence of oxygen [2]. This phenomenon is known as aerobic glycolysis or Warburg effect [3]. These metabolic alterations are associated with oncogenic mutations and activation of pro-proliferation signaling pathway, leading to malignant transformation of cancer [4]. Therefore, targeting metabolic abnormalities holds great potential for cancer therapy [5]. However, since metabolic enzymes are also utilized by normal cells, targeting these metabolic enzymes may lead to side effect and thus impedes its application. On account of this, new insight is urgently needed to improve our understanding in the regulatory mechanism of metabolic reprogramming. Emerging evidence suggests that oxidative stress induced by oncogenic mutation or metabolic reprogramming is involved in the regulation of metabolic reprogramming through modulating the expression, function, or subcellular localization of metabolic enzymes [6]. Intriguingly, some metabolic enzymes are redox sensors which could be oxidized under oxidative stress [7]. These oxidative modifications are occurred on the free cysteine residues of metabolic enzymes, leading to alteration of the function and subcellular localization of metabolic enzymes, thus influencing cell fate [8,9,10]. Although some questions have not been fully understood, this field holds the potential to be a research hot spot that leads toward translational medicine. In this review, we will discuss the bidirectional regulation of redox homeostasis and metabolic abnormalities, as well as the potential therapeutic strategies derived.
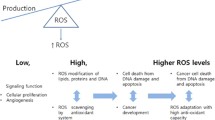
Oxidative stress is exerted by accumulated reactive oxygen species (ROS) which include non-radical molecules such as hydrogen peroxide (H2O2) and free radicals such as superoxide (O2•−) and (HO•) [11]. These oxygen-containing chemical species are principally produced in the mitochondria, the endoplasmic reticulum (ER), and the peroxisomes [12, 13]. Mitochondrion is the major organelle that produces superoxide through the electron leakage from the electron transport chain (ETC) [14]. The ER is another ROS-producing compartment in which electrons transfer from the FADH2 to oxygen and generate ROS [15]. The peroxisome is considered to play dual roles in regulating ROS production: one is to scavenge ROS by catalase (CAT) which decomposes H2O2 to H2O and the other is to produce ROS through β-oxidation of fatty acids [16,17,18] (Fig. 11.1). Moreover, some mitochondrial metabolic enzymes including glycerol-3-phosphate dehydrogenase (GPDH), flavoprotein-ubiquinone oxidoreductase (FQR), 2-ketoglutarate dehydrogenase (KGDH), and pyruvate dehydrogenase (PDH) can directly promote the conversion of O2 to superoxide [6, 19, 20]. In addition, other metabolic-associated enzymes such as NADPH oxidases (NOXs) and xanthine oxidases (XOs) also contribute to the generation of ROS [21]. Furthermore, glycolytic enzymes such as phosphoglycerate mutase family member 5 (PGAM5) are reported to influence the production of ROS [22] (Fig. 11.1). Taken together, these evidences suggest that these metabolic enzymes might be another leading cause of ROS generation.

The major sources of ROS production. Mitochondria, peroxisomes, and endoplasmic reticulum (ER) are major sources for ROS generation. In mitochondria, O2 can be converted to superoxide, while in peroxisome, ROS can be produced through β-oxidation of fatty acids. ER favors the formation of protein disulfide bond and increases ROS levels. Besides, metabolic enzymes such as PGAM5 and GPDH can also increase ROS level
To maintain cellular redox homeostasis, cells are developed with nimble antioxidant system. The transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) is the master regulator of the transcription of numerous ROS-detoxifying enzymes such as glutathione peroxidase (GPX), thioredoxin (TXN), thioredoxin reductase 1 (TXNRD1), and peroxiredoxin 1 (PRDX1) [23]. Moreover, the metabolic byproducts including glutathione (GSH) and NADPH are important antioxidant metabolites and essential for redox homeostasis maintenance [24]. GSH is the most abundant non-enzymatic antioxidant molecule within the cell. GSH can be oxidized to GSSG, and the ratio of GSH/GSSG indicates the cellular reductive potential. GSH is regenerated from GSSG by glutathione reductase (GSR) with the consumption of NADPH [25, 26].
Oxidative stress has long been considered as a key pathological factor, especially in cancers [27]. Excessive ROS may oxidize and damage the intracellular targets such as DNA, protein, and lipid, which is conventionally regarded as the pivotal reasons for the occurrence of many diseases such as cancer and aging [28]. However, recent studies have revealed that moderate levels of ROS can serve as a second messenger, which is indispensable for physiological processes [29]. Appropriate level of ROS, together with antioxidant systems, coordinates reduction-oxidation (redox) signaling to maintain intracellular redox homeostasis and proper functions of organisms [30, 31]. Indeed, redox imbalance has been found to be involved in a variety of diseases, indicating a possible therapeutic strategy by redox resetting [32,33,34].
Redox regulation of protein function is under extensive study, although it’s still far from resolved. Reduced form of thiol groups (-SH) in cysteine can undergo oxidative modification, thereby generating S-hydroxylated derivatives (-SOH), which may then produce disulfides (-s-s-). It allows the conformation changes and the protein-protein interaction, leading to function alterations. On the contrary, the oxidized form of protein can also be reduced by cellular antioxidant systems such as Trx reductase and Grx reductase [35, 36]. Therefore, the redox regulation can serve as molecular switches that control the function of some proteins. Importantly, a number of metabolism-related proteins were found to undergo redox modifications, suggesting that the redox regulation of some metabolic enzymes is critical for the metabolic alteration [6].
11.1 Redox Regulation of Metabolic Enzymes in Cancer Cells
11.1.1 AMPK
5’-AMP-activated protein kinase (AMPK) is a well-known energy sensor which is activated to regulate cellular metabolism under energy stress [37]. Our previous work has demonstrated that PRKAA, a catalytic subunit of AMPK, was activated by hepatitis B virus (HBV) infection-induced oxidative stress, indicating that AMPK is a potential target for redox modification [38]. Furthermore, emerging evidence has proven that AMPK is sensitive to ROS-mediated oxidative posttranslational modifications (oxPTMs). Zmijewski et al. reported that the kinase activity of AMPK can be stimulated under H2O2 exposure through S-glutathionylation at Cys299 and Cys304. In this circumstance, AMPK activation may result from S-glutathionylation-induced conformational change [39] (Fig. 11.2). Intriguingly, oxidation at Cys130 and Cys174 of AMPKα can disrupt the interaction between AMPK and AMPK kinases (AMPKK), thus attenuating the activity of AMPK. In contrast, thioredoxin-1 (Trx1) can cleave the disulfide bond between Cys130 and Cys174 to protect AMPK from oxidation-induced inactivation [40]. These studies imply that redox regulation of AMPK is especially sophisticated, which may be essential for the establishment of the circuit between cellular redox states and metabolism. Whether the subtle redox regulation of AMPK activity is correlated with cancer initiation and progression remains to be further explored.

The overview of redox modification in several metabolic enzymes. Under oxidative stress, several metabolic enzymes can undergo oxidative modification on their reduced thiol group in cysteine residues (GAPDH, PKM2, LDHA, IDH, AMPK, etc.). The oxidative modification of these metabolic enzymes may alter their metabolic activities and thus affect cell fate
11.1.2 GAPDH
It has been reported for a long time that GAPDH is one of the major targets in response to oxidative stress [41]. In 1994, Ravichandran and colleagues found that GAPDH can be S-thiolated with a decrease in enzymatic activity during the respiratory burst in blood monocytes [42]. Consistently, in yeast cells, S-thiolation of GAPDH may serve as an adaptive response during oxidative stress to protect cells from irreversible oxidation [43]. More in-depth studies revealed that Cys152 is an active site of GAPDH and can be oxidatively modified (Fig. 11.2). Oxidized GAPDH interacts with different proteins to exert moonlighting functions beyond metabolism [44]. Interestingly, Nakajima et al. found that the amyloid-like aggregation of GAPDH triggered by oxidative stress can lead to cell death, in which Cys152 plays a critical role. This study indicates the importance of GAPDH redox regulation in brain damage and some types of neuroblastoma [45]. Furthermore, in KRAS and BRAF mutant colorectal cancer cells, the S-glutathionylation level of GAPDH was demonstrated to be significantly higher during oxidative stress which resulted from high-dose vitamin C treatment. S-glutathionylation inhibits the activity of GAPDH and subsequent glycolysis, contributing to ATP depletion and cell death [46]. This study demonstrates that redox regulation of GAPDH plays an essential role in the homeostasis of cancer cell metabolism, thus determining cell fate. Taken together, GAPDH has been recognized as the critical mediator of oxidative stress responses. Redox regulation of GAPDH in cancer is now under wide investigation.
11.1.3 PKM2
The glycolytic enzyme PKM2 catalyzes the dephosphorylation reaction of phosphoenolpyruvate to pyruvate, which is accompanied by net ATP production [47]. PKM2 is upregulated in several types of cancers, and its oxidation form plays an important part in tumor progression. In human lung cancer cells, the enzymatic activity of PKM2 is inhibited through Cys358 oxidation, which rewires glycolysis flux to the oxidative pentose phosphate pathway (PPP) (Fig. 11.2). This diversion is especially critical for ROS detoxification through generating reducing potential, leading to lung cancer formation and progression [48]. This example illustrates that the redox regulation of PKM2 is necessary for cancer cell to withstand oxidative stress. In addition, oxidative stress can induce mitochondrial translocation of PKM2, where it phosphorylates Bcl2 to prevent the ubiquitin-mediated degradation of Bcl2. These events protect glioma cells from oxidative stress-induced apoptosis, which further highlights the importance of PKM2 in cancer cell adaption to high levels of ROS [49]. A recent study has reported that tetrameric PKM2 oxidized at its Cys423 can bind with and suppress the transcriptional activity of p53, thus inhibiting apoptosis. The high oxidation state of PKM2 is critical for its regulatory role in p53 activity [50] (Fig. 11.2). Thus, PKM2 is one of the typical enzymes sensitive to redox regulation, which is a key regulator in metabolic-redox circuits.
11.1.4 LDHA
LDHA catalyzes the conversion of pyruvate to L-lactate with the production of NADH in the last step of glycolysis. According to our recent work, LDHA translocates to the nucleus in response to human papilloma virus (HPV)-mediated ROS accumulation, which is accompanied by ROS-induced tetramer-to-dimer transition of LDHA (Fig. 11.2). Nuclear LDHA acquires a non-canonical enzymatic activity to produce α-hydroxybutyrate, which promotes the interaction between LDHA and DOT1L, and triggers histone H3K79 hypermethylation. This epigenetic alteration finally activates antioxidant responses and maintains cellular redox homeostasis. The non-canonical function of LDHA triggered by oxidative stress is crucial to HPV-induced cervical cancer progression [51]. This finding indicates that the redox status of LDHA is a potential biomarker for cervical cancer and that LDHA may have the potential to become a therapeutic target.
11.1.5 IDH
IDH catalyzes the conversion of isocitrate to α-ketoglutarate (α-KG) and carbon dioxide, which includes oxidation of isocitrate and the following decarboxylation. The point mutations of IDH have been widely documented in cancer, especially in gliomas and glioblastomas [52, 53]. Mutated IDH catalyzes the conversion of α-KG to 2-hydroxyglutarate (2-HG), which competitively suppresses α-KG-dependent enzymes, thus resulting in the global alterations of methylation levels [54]. It has been reported that IDH is a potential target of oxPTMs. Mitochondrial NADP(+)-dependent isocitrate dehydrogenase (IDPm) can be glutathionylated at Cys269 during oxidative stress, which leads to a decrease in IDPm activity (Fig. 11.2). The inactivated IDPm can also be reactivated through deglutathionylation, indicating that IDPm plays a crucial role in maintaining mitochondrial redox balance [55]. Moreover, a recent study on intracranial patient-derived xenografts of gliomas found that IDH mutations trigger metabolic landscape alteration, which affects cellular oxidative stress pathways [56]. This finding further highlights the regulatory role of IDH in redox hemostasis, in which oxPTMs of IDH may be involved. More interestingly, a competitive inhibitor of IDH, oxalomalate, has been proven to inhibit metastatic melanoma through ROS-dependent signaling pathways [57]. This finding suggests that IDH can act as a promising therapeutic target in clinical cancer management.
11.2 Targeting the Metabolic Abnormalities in Cancer Cell
As metabolism-derived cofactors are critical in regulating epigenetics, which is involved in tumorigenesis and cancer progression, it is feasible to target these metabolic cofactors or enzymes to interfere with chromatin modifications to benefit clinical outcomes. The most straightforward way to affect the chromatin modifications on the basis of metabolites adjustment is to deplete or replenish cellular metabolic cofactors such as SAM, α-KG, FAD, acetyl-CoA, NAD+, and lactate. Multiple agents designed to inhibit the production or recycling of these metabolites are now under intensive investigations for cancer treatment. For example, 3-deazaneplanocin A (DZNep), which could inhibit the activity of SAH hydrolase and disrupt the recycling of SAM thus influencing the methylation of DNA and histones, has been found to induce apoptosis and reduce proliferation in AML cells and breast cancer cells, respectively [58, 59]. Moreover, quinoxaline compound 1-(2,3-di(thiophen-2-yl) quinoxalin-6-yl)-3-(2-methoxyethyl) urea could inhibit the activity of acyl-CoA synthetase short-chain family member 2 (ACSS2), leading to decreased acetate incorporation into both lipid and histones [60]. In addition, modifying the ratio between epigenetics-associated metabolites and their competitive metabolites (α-KG/2-HG, SAM/SAH, acetyl-CoA/CoA, etc.) could also influence the chromatin modifications. Moreover, biosynthesis of epigenetics-associated metabolites at different subcellular compartments could lead to specific alteration of chromatin modifications by changing the concentrations of local cofactors [61].
Glucose and glutamine are essential biomolecules which provide cancer cells with energy and intermediates to fulfil the requirement of rapid proliferation. Several compounds are developed for targeting the glucose or glutamine addiction in cancer cells. For example, 2-deoxy-D-glucose (2-DG), known as a glucose analog, could inhibit glycolysis and induce proliferation arrest in several cancer types [62]. Moreover, inhibition of LDHA could prevent the regeneration of NAD+ which is essential for glycolysis and thus suppress tumor growth. Gossypol is a LDHA inhibitor which shows antitumor effect in several solid cancer types and is now under clinical trials [63, 64]. Metformin, known as a reference drug for type II diabetes treatment, could reduce cancer risk through the inhibition of mitochondrial complex I [65]. In addition, pyruvate dehydrogenase kinase (PDK) is overexpressed in several cancer types and promotes glycolytic phenotype [66]. PDK inhibitor dichloroacetate (DCA) can inhibit aerobic glycolysis and restrain tumor growth, indicating that targeting highly glycolytic tumors holds great potential for cancer therapy. Glutamine can be converted to α-KG to fuel TCA, during which glutaminase (GLS) and glutamate dehydrogenase (GDH) are required. Therefore, targeting GLS or GDH may benefit cancer therapy through inhibiting glutamine addiction in cancer cells. BPTES and CB-839 are two established GLS inhibitors for restricting the supply of α-KG to the TCA, and CB-839 is now under clinical studies in several cancers. Furthermore, aminooxyacetate (AOA) is an important aminotransferase inhibitor which exhibits potent antitumor effect in several cancer types including lung adenocarcinoma through targeting glutamine metabolism [67].
11.3 Modulating ROS for Cancer Therapy
The fact that some metabolic enzymes are regulated by cellular redox state highlights the promise of modulating the redox status for cancer therapy. For instance, as mentioned above, HPV16 E7 induces intracellular ROS accumulation, resulting in the translocation of LDHA in the nucleus where it gets a non-canonical function to generate α-HB and epigenetically regulate antioxidant responses [51]. Hence, ROS modulators that have been specifically developed to disable nuclear translocation of LDHA may be a promising choice to prevent and treat cervical cancer.
GSH, as discussed above, is an important free radical scavenger and detoxifying molecule in living cells [68]. GSH can maintain a reduced environment of subcellular compartments such as mitochondria, nucleus, and cytosol. Ten percent of synthesized GSH is found in mitochondria, where GSH can eliminate excessive ROS and protect cells from apoptosis. The high ratio of GSH/GSSG in the nucleus ensures the synthesis of deoxyribonucleotides and preserves the sulfhydryl groups of proteins for the biosynthesis of proper nucleic acids and DNA repair. In contrast, a low ratio of GSH/GSSG leads to an oxidized environment and enhanced addition reaction of disulfide bonds to the nascent proteins [69]. Moreover, reduced GSH can conjugate with many chemotherapeutic drugs such as cisplatin and doxorubicin, via a glutathione S-transferases (GST)-catalyzed reaction, resulting in increased clearance of these drugs [70, 71]. The efflux of chemotherapeutic drugs is often mediated by multidrug resistance protein 1 (MRP1), leading to cellular detoxification and apoptosis resistance [72]. Cancer cells are equipped with higher levels of GSH, which can be utilized for selectively targeting cancer cells. It has been reported that a doxorubicin-loaded glutathione-responsive cyclodextrin nanosponges (GSH-NS) could release doxorubicin in cancer cells which possess high GSH levels, resulting in higher antitumor efficacy and less cytotoxicity in normal cells [73]. Moreover, buthionine sulfoximine (BSO) is a potent inhibitor of GSH synthesis via inhibiting the activity of glutamate-cysteine ligase (GCL). BSO is now under intensive studies and has been used in clinic practice.
SAH, the byproduct of methyl transfer reaction, can be hydrolyzed to homocysteine and adenosine by SAH hydrolase (SAHH) [74]. Homocysteine is involved in the transsulfuration pathway to produce cysteine, a critical precursor and rate-limiting factor of GSH synthesis [75]. As GSH is the major antioxidant molecule to buffer ROS, the diversion of homocysteine from methionine regeneration into the cysteine production is important to maintain cellular redox homeostasis [76]. Conceivably, the direction (methionine/SAM regeneration or GSH biosynthesis) that homocysteine follows decides the fate of a cell by influencing the cellular redox states and epigenetic events [77]. Since several types of cancer cells often exhibit high ROS levels and DNA hypo-methylation, cancer cells are prone to utilize homocysteine to regenerate methionine during tumorigenesis. Therefore, targeting the recycle of homocysteine may be promising for tumor suppression.
Targeting the synthesis of GSH and NADPH or using pharmacological ROS inducers including As2O3, β-phenylethyl isothiocyanate (PEITC), and doxorubicin (Dox) constitutes the strategy of increasing ROS for anticancer therapy [78]. Importantly, scavenging ROS also exhibits favorable anticancer effect. For example, vitamin E is an important dietary antioxidant which reduces ROS levels through reacting with free radicals. Intriguingly, it has been reported that intake of vitamin E can reduce the risk of liver cancer, indicating that clearance of ROS by dietary supplementation of antioxidant holds great potential for preventing tumorigenesis [79]. However, this hypothesis is facing challenges since supplementation of β-carotene, or vitamin E, cannot reduce the risk of lung cancer [80]. The comprehensive understanding of involved regulatory mechanisms is urgently needed for facilitating the early intervention of tumorigenesis.
11.4 Conclusion
Metabolic network and redox homeostasis are closely intertwined with each other and constitute an orchestrated regulatory loop to participate in tumorigenesis and tumor development. Thus, targeting the vulnerabilities of metabolism and redox imbalance in cancer cells holds great potential for selectively killing malignant cells. However, our understanding regarding the complicated metabolic-redox loop still lacks, and more efforts should be paid to achieve effective approaches. More importantly, since the roles of ROS are multifaceted, it should be discreet to consider the application of ROS modulators (scavengers or inducers) in different cancer types. Furthermore, compounds targeting global metabolism, or redox homeostasis may elicit unexpected side effects on normal tissues, and more specific delivery system may be required for favorable therapeutic outcomes.
References
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21:297–308.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47.
Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–8.
Tarrado-Castellarnau M, de Atauri P, Cascante M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget. 2016;7:62726–53.
Vernieri C, Casola S, Foiani M, Pietrantonio F, de Braud F, Longo V. Targeting cancer metabolism: dietary and pharmacologic interventions. Cancer Discov. 2016;6:1315–33.
Wang K, Jiang J, Lei Y, Zhou S, Wei Y, Huang C. Targeting metabolic-redox circuits for cancer therapy. Trends Biochem Sci. 2019;44:401–14.
Bigelow DJ, Squier TC. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim Biophys Acta. 1703;2005:121–34.
Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–77.
Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev. 2013;113:4633–79.
Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014;2:123–39.
Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;
Parvez S, Long MJC, Poganik JR, Aye Y. Redox signaling by reactive electrophiles and oxidants. Chem Rev. 2018;118:8798–888.
Lau AT, Wang Y, Chiu JF. Reactive oxygen species: current knowledge and applications in cancer research and therapeutic. J Cell Biochem. 2008;104:657–67.
O’Malley J, Kumar R, Inigo J, Yadava N, Chandra D. Mitochondrial stress response and cancer. Trends Cancer. 2020;
Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249.
Sandalio LM, Rodriguez-Serrano M, Romero-Puertas MC, del Rio LA. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell Biochem. 2013;69:231–55.
Bonekamp NA, Volkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: struggling for balance. Biofactors. 2009;35:346–55.
Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 1763;2006:1755–66.
Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–21.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13.
Magnani F, Mattevi A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr Opin Struct Biol. 2019;59:91–7.
Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene. 2015;34:5796–806.
Lei XG, Zhu JH, Cheng WH, Bao Y, Ho YS, Reddi AR, et al. Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol Rev. 2016;96:307–64.
He L, He T, Farrar S, Ji L, Liu T, Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. 2017;44:532–53.
Mironczuk-Chodakowska I, Witkowska AM, Zujko ME. Endogenous non-enzymatic antioxidants in the human body. Adv Med Sci. 2018;63:68–78.
Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Asp Med. 2009;30:1–12.
Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–47.
Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol Sci. 2017;38:592–607.
Wang J, Lu Q, Cai J, Wang Y, Lai X, Qiu Y, et al. Nestin regulates cellular redox homeostasis in lung cancer through the Keap1-Nrf2 feedback loop. Nat Commun. 2019;10:5043.
Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–62.
Dawane JS, Pandit VA. Understanding redox homeostasis and its role in cancer. J Clin Diagn Res. 2012;6:1796–802.
Liu H, Liu X, Zhang C, Zhu H, Xu Q, Bu Y, et al. Redox imbalance in the development of colorectal cancer. J Cancer. 2017;8:1586–97.
Pastore S, Korkina L. Redox imbalance in T cell-mediated skin diseases. Mediat Inflamm. 2010;2010:861949.
Banhegyi G, Mandl J, Csala M. Redox-based endoplasmic reticulum dysfunction in neurological diseases. J Neurochem. 2008;107:20–34.
Meyer Y, Buchanan BB, Vignols F, Reichheld JP. Thioredoxins and glutaredoxins: unifying elements in redox biology. Annu Rev Genet. 2009;43:335–67.
Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–36.
Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–908.
Xie N, Yuan K, Zhou L, Wang K, Chen HN, Lei Y, et al. PRKAA/AMPK restricts HBV replication through promotion of autophagic degradation. Autophagy. 2016;12:1507–20.
Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem. 2010;285:33154–64.
Shao D, Oka S, Liu T, Zhai P, Ago T, Sciarretta S, et al. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014;19:232–45.
Nicholls C, Li H, Liu JP. GAPDH: a common enzyme with uncommon functions. Clin Exp Pharmacol Physiol. 2012;39:674–9.
Ravichandran V, Seres T, Moriguchi T, Thomas JA, Johnston RB Jr. S-thiolation of glyceraldehyde-3-phosphate dehydrogenase induced by the phagocytosis-associated respiratory burst in blood monocytes. J Biol Chem. 1994;269:25010–5.
Shenton D, Grant CM. Protein S-thiolation targets glycolysis and protein synthesis in response to oxidative stress in the yeast Saccharomyces cerevisiae. Biochem J. 2003;374:513–9.
Hwang NR, Yim SH, Kim YM, Jeong J, Song EJ, Lee Y, et al. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem J. 2009;423:253–64.
Nakajima H, Amano W, Kubo T, Fukuhara A, Ihara H, Azuma YT, et al. Glyceraldehyde-3-phosphate dehydrogenase aggregate formation participates in oxidative stress-induced cell death. J Biol Chem. 2009;284:34331–41.
Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–6.
Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–80.
Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–83.
Liang J, Cao R, Wang X, Zhang Y, Wang P, Gao H, et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017;27:329–51.
Saleme B, Gurtu V, Zhang Y, Kinnaird A, Boukouris AE, Gopal K, et al. Tissue-specific regulation of p53 by PKM2 is redox dependent and provides a therapeutic target for anthracycline-induced cardiotoxicity. Sci Transl Med. 2019;11
Liu Y, Guo JZ, Liu Y, Wang K, Ding W, Wang H, et al. Nuclear lactate dehydrogenase A senses ROS to produce alpha-hydroxybutyrate for HPV-induced cervical tumor growth. Nat Commun. 2018;9:4429.
Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–98.
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 2009;30:7–11.
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30.
Kil IS, Park JW. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem. 2005;280:10846–54.
Fack F, Tardito S, Hochart G, Oudin A, Zheng L, Fritah S, et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol Med. 2017;9:1681–95.
Kim SH, Kim H, Lee JH, Park JW. Oxalomalate suppresses metastatic melanoma through IDH-targeted stress response to ROS. Free Radic Res. 2019;53:418–29.
Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–63.
Conte M, Altucci L. Molecular pathways: the complexity of the epigenome in cancer and recent clinical advances. Clin Cancer Res. 2012;18:5526–34.
Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell. 2014;159:1591–602.
Meier JL. Metabolic mechanisms of epigenetic regulation. ACS Chem Biol. 2013;8:2607–21.
Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Trans Med. 2012;4:124ra27.
Van Poznak C, Seidman AD, Reidenberg MM, Moasser MM, Sklarin N, Van Zee K, et al. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: a phase I/II clinical trial. Breast Cancer Res Treat. 2001;66:239–48.
Xie H, Yin J, Shah MH, Menefee ME, Bible KC, Reidy-Lagunes D, et al. A phase II study of the orally administered negative enantiomer of gossypol (AT-101), a BH3 mimetic, in patients with advanced adrenal cortical carcinoma. Investig New Drugs. 2019;37:755–62.
Kamarudin MNA, Sarker MMR, Zhou JR, Parhar I. Metformin in colorectal cancer: molecular mechanism, preclinical and clinical aspects. J Exp Clin Cancer Res. 2019;38:491.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85.
Wang X, Min S, Liu H, Wu N, Liu X, Wang T, et al. Nf1 loss promotes Kras-driven lung adenocarcinoma and results in Psat1-mediated glutamate dependence. EMBO Mol Med. 2019;11
Benhar M, Shytaj IL, Stamler JS, Savarino A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J Clin Invest. 2016;126:1630–9.
Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217:2291–8.
Cole SP. Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol. 2014;54:95–117.
Cole SP, Deeley RG. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol Sci. 2006;27:438–46.
Wiese M, Stefan SM. The A-B-C of small-molecule ABC transport protein modulators: from inhibition to activation-a case study of multidrug resistance-associated protein 1 (ABCC1). Med Res Rev. 2019;39:2031–81.
Daga M, Ullio C, Argenziano M, Dianzani C, Cavalli R, Trotta F, et al. GSH-targeted nanosponges increase doxorubicin-induced toxicity “in vitro” and “in vivo” in cancer cells with high antioxidant defenses. Free Radic Biol Med. 2016;97:24–37.
Xiao Y, Xia J, Cheng J, Huang H, Zhou Y, Yang X, et al. Inhibition of S-Adenosylhomocysteine hydrolase induces endothelial dysfunction via epigenetic regulation of p66shc-mediated oxidative stress pathway. Circulation. 2019;139:2260–77.
Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–83.
Aldini G, Altomare A, Baron G, Vistoli G, Carini M, Borsani L, et al. N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. Free Radic Res. 2018;52:751–62.
Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 2015;22:861–73.
Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50–64.
Zhang W, Shu XO, Li H, Yang G, Cai H, Ji BT, et al. Vitamin intake and liver cancer risk: a report from two cohort studies in China. J Natl Cancer Inst. 2012;104:1173–81.
Alpha-Tocopherol BCCPSG. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029–35.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81821002, 81790251, and 82003098), National Key Research and Development Project (2020YFA0509400, 2020YFC2002705), and Guangdong Basic and Applied Basic Research Foundation (2019B030302012).
Disclosure Statement
No competing financial interests exist.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Jiang, J., Huang, C. (2021). Redox Regulation of Metabolic Enzymes in Cancer. In: Huang, C., Zhang, Y. (eds) Oxidative Stress. Springer, Singapore. https://doi.org/10.1007/978-981-16-0522-2_11
Download citation
DOI: https://doi.org/10.1007/978-981-16-0522-2_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0521-5
Online ISBN: 978-981-16-0522-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)