
Abstract
Autism spectrum disorder (ASD) development is a highly multifaceted process as evidenced by the complexity of the factors involved in the etiology of ASD, including genetic and nongenetic factors. Several forms of ASD result from genetic alterations in genes that regulate the process of protein synthesis. A growing body of evidence suggests that abnormal synaptic protein synthesis might contribute to ASD and ASD-like clinical features. Several reports of different mutated genes responsible for ASD cases and genetic models have emerged, revealing dysregulation of many crucial signaling pathways. In this chapter, the authors summarize the various factors described to contribute to ASD, both genetic and nongenetic, and their association with WNT, SHH, RA, FGF, and BMP/TGF-β signaling pathways. In addition, the authors discuss the scope for additional research for a better understanding of the pathophysiology of ASD in the context of disrupted signaling pathways, which could help open the doors to identify possible gene targets and novel therapeutic strategies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1.1 Introduction
Autism spectrum disorder (ASD) encompasses a heterogenous set of multifactorial challenges in neurodevelopment, classified according to three fundamental features; compromised social correspondence skills, delayed dialect development, and raised stereotyped alternately tedium practices (Mohn et al. 2014; Golden et al. 2017; Abrahams and Geschwind 2008; Geschwind 2008; Sudhof 2008; Zoghbi 2003). ASD is believed to occur due to the complicated processes that involve numerous gene-environment interactions, as evidenced by the association of multiple elements (genetic and nongenetic). People with autism are recognized to have several traits, such as hampered social interactions, communication impairment, and increased tedium behaviors, implying that aberrant signaling pathways during brain development resulted in the disturbance of specific neural circuits in ASD (Styles et al. 2020; Al-Dewik et al. 2020). The disruption of multiple critical signaling pathways, including WNT (Bae and Hong 2018; Kalkman 2012; Mulligan and Cheyette 2017), BMP (Zhang et al. 2017; Kashima et al. 2016; Li et al. 2016; Sajan et al. 2011), SHH (Halepoto et al. 2015; Patel et al. 2017), and retinoic acid (RA) (Niculae and Pavăl 2016), has been discovered in research using genetic models in ASD individuals. While direct evidence has not been found, indirect evidence of abnormal FGF or TGF-β signaling in ASD exists (Ansari et al. 2017; Chen et al. 2017; Iwata and Hevner 2009). The success of therapy treatments is severely constrained due to a lack of data on the etiology of ASD and the mechanisms involved. In this chapter the authors aim to summarize current knowledge on the various ASD-associated factors (genetic and nongenetic), and their interactions with signaling pathways that are frequently altered in ASD. This chapter also discusses what additional research could be conducted to gain better insights into altered pathways in ASD.
1.2 Principal Signaling Pathways
1.2.1 Altered WNT Signaling in ASD
WNT signaling involves the secretion of cysteine-rich glycolipoproteins that are WNT receptors themselves. The WNT signaling pathway controls important developmental and regulatory processes, including embryonic development and tissue homeostasis, through regulating receptors, such as frizzled (FZD), when the receptors interact with the WNT protein. WNT signaling is required for several developmental and post-developmental neuroscientific processes, including synaptogenesis and CNS regionalization (Tang 2014; Wada and Okamoto 2009; Wodarz and Nusse 1998; Rosso and Inestrosa 2013; Bielen and Houart 2014; Abu-Khalil et al. 2004; Bengoa-Vergniory and Kypta 2015; Burden 2000; Inestrosa and Varela-Nallar 2015; Onishi et al. 2014). Thus, any disturbance in WNT signaling has the potential to cause the development of CNS-related diseases (Mulligan and Cheyette 2017; Okerlund and Cheyette 2011).
Investigations of both genetically engineered animal models and Human Induced Pluripotent Stem Cell (hiPSC) models have shown the critical role of spatiotemporal WNT signaling throughout animal development (Mulligan and Cheyette 2017). It has also been shown that irregularities in WNT may cause several types of mental illnesses, such as autism, schizophrenia, bipolar disorder, and developmental problems (Kalkman 2012; Mulligan and Cheyette 2017; Okerlund and Cheyette 2011; Oron and Elliott 2017; Kwan et al. 2016a; Martin et al. 2013). Many of the genes and epigenetic factors involving ASD have been identified as affecting common biological processes such as epigenetic modification, WNT, and synaptic transmission (Oron and Elliott 2017; Hormozdiari et al. 2015; Krumm et al. 2014).
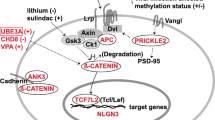
The WNT pathway is classified into two major pathways: (1) β-catenin-dependent (canonical pathways) and (2) β-catenin-independent (noncanonical pathways), both of which are key players in neural development and related neurodevelopmental disorders (Grainger and Willert 2018; Komiya and Habas 2008). Several genetic variations associated with and/or documented in ASD cases include either fundamental elements of the β-catenin-dependent pathway, such as CTNNB1 (β-catenin) (Kalkman 2012; Mulligan and Cheyette 2017; Krumm et al. 2014; O’Roak et al. 2012a) and adenomatous polyposis coli (APC) (Mohn et al. 2014), or β-catenin-independent pathway, such as PRICKLE2 (Sowers et al. 2013), implying that these two pathways are key players in ASD etiology. A list of selected ASD-associated genes that the authors have found to influence ASD-related pathways is presented in Table 1.1. Possible interactions between the ASD-associated genes and WNT pathway are illustrated in Fig. 1.1.

Plausible interactions between the ASD-related genes and WNT signaling. The majority of molecules encoded by ASD-related genes either play a key role in WNT signaling pathways or are modulators. A plus sign signifies upregulation and a negative sign implies downregulation. The figure was created using BioRender (https://biorender.com/)
1.3 Genetic Etiologies
Core elements of the canonical WNT signaling
In humans, the components of the β-catenin-dependent route includes many WNT ligands, frizzled receptors, LDL receptor family co-receptors, and intracellular and extracellular modulators (Klaus and Birchmeier 2008). WNT ligands are modified cysteine-rich proteins, and to function, they need to be glycosylated and palmitoylated (Komiya and Habas 2008). WNT1, WNT2, WNT3, and WNT9B are known to be involved with ASD. ASD individuals usually harbor a unique missense genetic variant in WNT1 (S88R, rs61758378) that has demonstrated higher activation of the WNT/β-catenin pathway compared to WNT1 wild type i.e. without this variant (Martin et al. 2013). Beyond that, ASD Individuals have been found to have uncommon pathogenic variants in WNT2, WNT3, and WNT9B (Wassink et al. 2001; Marui et al. 2010; Lin et al. 2012; Levy et al. 2011). Therefore, it is surprising that in people with ASD, WNT3 expression in the prefrontal cortex is more pronounced (Chow et al. 2012). The above finding suggests that WNT signaling is overactive and that overactivation could play a key role in ASD etiology.
Several studies have revealed that WNT1 is crucial for cerebellar and midbrain development in animal models (Thomas and Capecchi 1990; McMahon and Bradley 1990; McMahon et al. 1992). Furthermore, it has been shown that WNT2 is necessary for the development of cortical dendrites as well as the production of dendritic spines. Moreover, it was shown that WNT2 expression could be regulated by a protein referred to as brain-derived neurotrophic factor (BDNF), which is a key player in neuron survival and growth (Hiester et al. 2013). It has also been shown that altered dendritic spines were associated with neurodegenerative and neurodevelopmental problems (Hiester et al. 2013). In addition, WNT3 has been shown to be required for gastrulation and for the regulation of hippocampus neurogenesis (Lie et al. 2005; Liu et al. 1999). Increased inhibitory synaptic density and decreased excitatory synapse counts have been seen in cortical layer 6 neurons during late mouse gestation when the function of T-brain-1, a T-box transcription factor and one of the high-confidence ASD-associated genes, is lost (Fazel Darbandi et al. 2018). WNT9b is known to promote lip/palate formation and fusion (Jin et al. 2012; Juriloff et al. 2006), although it is not known what function it plays in neurodevelopment.
The functions of WNT receptors, in addition to the other WNT ligands (such as FZD1-FZD10 protein family), in ASD etiology remain poorly understood (Onishi et al. 2014; MacDonald and He 2012). Research shows that the duplication or deletion in FZD9 receptors might potentially impair brain development and thus cause ASD (Kalkman 2012). In addition, the administration of WNT2 has been found to stimulate the overexpansion of neurons in dopaminergic pathways in the midbrain of mice, resulting in the mice engaging in repetitive behavior (Sousa et al. 2010).
β-Catenin, an adherent junction component linked to E-cadherin, is an endogenous protein encoded by the gene CTNNB1, a key regulator of the WNT signaling pathway. β-Catenin is a major intracellular molecule in the classic WNT signaling pathway that plays essential roles in development and illness (Clevers and Nusse 2012; Grigoryan et al. 2008). High levels of the β-catenin signaling pathway have been reported to contribute to aberrant brain development in people with ASD. De novo CTNNB1 mutations have been implicated, not only in ASD, but also in intellectual disabilities, microcephaly, speech impairment, and motor delay (Krumm et al. 2014; O’Roak et al. 2012a, b; Kuechler et al. 2015; Sanders et al. 2012). In the Simons Foundation Autism Research Initiative (SFARI) database, CTNNB1 has a genetic score of “1,” defined as high confidence, indicating that there are at least three de novo likely gene-disrupting genetic variants being reported in association with ASD (Table 1.1). De novo pathogenic variants in this gene were discovered in two different studies utilizing ASD probands from the Simons Simplex Collection (Sanders et al. 2012). In both humans and animals, CTNNB1 haploinsufficiency has been linked to neuronal loss, craniofacial deformities, and hair follicle abnormalities (Dubruc et al. 2014). Conditional β-catenin ablation in mouse embryo dorsal neural folds represses PAX3 and CDX2 expression at the dorsal posterior neuropore. Leading to reduced expression of the WNT/β-catenin signaling target genes T, TBX6, and FGF8 at the tailbud, resulting in spina bifida aperta, caudal axis bending, and tail truncation (Zhao et al. 2014). Conditional ablation of catenin in parvalbumin interneurons in mice resulted in poor object recognition and social interactions, and increased repetitive behaviors, both of which are essential features of ASD individuals, but they also showed improvement in spatial memory (Dong et al. 2016). The mice exhibited reduced c-Fos activity in the cortex but not in the dentate gyrus or the amygdala, suggesting that β-catenin has a cell type-specific role in the regulation of cognitive and autistic-like behaviors (Dong et al. 2016).
There are a number of ASD-linked genes, besides CTNNB1, that are believed to affect β-catenin functions; among them is adenomatous polyposis coli (APC) (Zhou et al. 2007). APC is a tumor suppressor that functions as a negative regulator of β-catenin and plays a significant role in the β-catenin-destruction complex (MacDonald et al. 2009). APC-inactivating gene variations in humans have been linked to ASD (Zhou et al. 2007; Barber et al. 1994). APC protein deletion mutation in mice was found to lead to autism-like disabilities, as well as learning and memory deficits in APC conditional knockout compared to wild-type littermates (Mohn et al. 2014). In addition, APC knockout forebrain neurons had higher levels of β-catenin and increased transcript levels of the canonical WNT target genes Dkk1, Sp5, Neurog1, and SYN2 (Mohn et al. 2014). Furthermore, APC knockout mouse lysates from the hippocampus, cortical, and striatal areas were shown to have greater β-catenin levels than control mice (Mohn et al. 2014), along with seizures, behavioral traits, and cognitive deficits (Pirone et al. 2017). These findings imply that WNT/β-catenin signaling activity might be associated with ASD. Besides being an important regulator of the β-catenin level, APC has other roles that are critical for neurogenesis and function, including its role in regulating microtubule and actin cytoskeleton dynamics (Zumbrunn et al. 2001; Akiyama and Kawasaki 2006) and as an mRNA-binding protein with several of its targets involved in brain development (Preitner et al. 2014).
Another gene that plays an important role in the WNT pathway is the transcription factor 7-like 2 (TCF7L2), also known as TCF4. TCF7L2 is one of the TCF/LEF1 transcription factors in the WNT/β-catenin signaling pathway that aid in the initiation of gene transcriptional responses when WNT ligands engage their receptors on the membrane and the signal is transduced to the nucleus (Grant et al. 2006) (Fig. 1.1). TCF7L2 has been implicated in developmental delays (DDs), intellectual disabilities (IDs), neurodevelopmental disorders (NDDs), and attention-deficit hyperactivity disorder (ADHD) (De Rubeis et al. 2014; Iossifov et al. 2014; Dias et al. 2021).
Recently, two de novo loss-of-function mutations in the TCF7L2 gene have been identified in ASD cases (De Rubeis et al. 2014; Iossifov et al. 2014). In addition, 11 people with de novo TCF7L2 mutations who had a syndromic neurodevelopmental condition were identified, four of which had ASD (Dias et al. 2021). It has been shown that TCF7L2, like the key WNT co-receptor Lrp6, is crucial for the development of thalamocortical axonal projections in mice (Zhou et al. 2004; Lee et al. 2017) implying that abnormal thalamocortical axonal inputs might be playing a vital role in the development of the disorder. Nevertheless, there is still a paucity of knowledge about the connection of other transcription factors from the TCF/Lef1 family with ASD. Furthermore, the significance of TCF4 in brain development remains unknown and will need to be investigated further in future research.
Core elements of the noncanonical WNT signaling
Whole-exome sequencing (WES) conducted on ASD-affected families has identified genetic variations of the WNT/PCP pathway genes PRICKLE1 and PRICKLE2. PRICKLE1 acts as a nuclear receptor that negatively regulates the WNT/β-catenin signaling pathway. Moreover, it is involved in the planar cell polarity route, which has several roles, such as convergent extension during gastrulation and neural tube closure, whereas the function of PRICKLE2 remains unclear. Pathogenic variants in PRICKLE1 and PRICKLE2 have previously been linked to epilepsy (EP) (Bosoi et al. 2011; Tao et al. 2011), a disease that is often associated with ASD (Buckley and Holmes 2016). According to an in vitro study, PRICKLE1 and PRICKLE2 promote neurite outgrowth through a dishevelled-dependent mechanism (Fujimura et al. 2009). PRICKLE1 +/− mice were reported to have ASD-like characteristics, such as abnormal social interactions and disturbed circadian rhythms. In addition, PRICKLE1 is required for synapsin1 (Syn1) function in the pre-synapse (Paemka et al. 2013), while PRICKLE2 interacts with postsynaptic density protein-95 (PSD95) and NMDA receptors in the postsynapse (Hida et al. 2011). Mice with disrupted PRICKLE2 were reported exhibiting impaired social behavior, learning disabilities, and behavioral rigidity. Moreover, mouse models with ASD were shown to exhibit behavioral and physiological abnormalities that are similar to those of PRICKLE2 mice (Sowers et al. 2013). Furthermore, rare on-synonymous PRICKLE2 genetic variants (p.E8Q and p.V153I) have been identified in individuals with ASD (Sowers et al. 2013). Dendrite branching, synapse number, and PSD size were all reduced in PRICKLE2-deficient mice’s hippocampal neurons (Sowers et al. 2013). The discovery of a PRICKLE2-containing 3p interstitial deletion in identical twins with ASD adds to the evidence that PRICKLE2 plays a role in ASD (Okumura et al. 2014). PRICKLE2’s interaction with PSD-95 is improved by Vangl2, a key component in the noncanonical WNT/PCP pathway (Nagaoka et al. 2015). The role of other PCP genes in ASD etiology, and the signaling interaction between the PCP and WNT/β-catenin pathways, should be investigated in the future.
1.4 Modulators and Effectors of WNT Signaling in ASD Etiology
Several genes have been implicated in ASD, such as chromodomain helicase DNA-binding protein 8 (CHD8) (Willsey et al. 2013; Cotney et al. 2015), ankyrin-G (ANK3) (Shi et al. 2013; Iqbal et al. 2013; Bi et al. 2012), DIX domain-containing 1 (DIXDC1) (Kwan et al. 2016b), prostaglandin E2 (PGE2) (Wong et al. 2016), and HECT domain E3 ubiquitin ligase (UBE3A) (Yi et al. 2017) (Fig. 1.1). In addition, recent research finds that neuroligin 3 (NLGN3), an ASD-associated gene, is a direct downstream target of WNT/β-catenin signaling during synaptogenesis (Medina et al. 2018) (Fig. 1.1). Furthermore, due to the identification of multiple people with genetic variants in phosphatase and tensin homolog (PTEN), it has been described as a high-risk candidate ASD-associated gene that plays a role in WNT signaling (O’Roak et al. 2012b; Spinelli et al. 2015; Frazier et al. 2015; McBride et al. 2010).
Canonical WNT signaling is one of the key pathways controlled by CHD8 (Thompson et al. 2008; Nishiyama et al. 2012). CHD8 acts as a transcription repressor by altering the structure of chromatin (Kwan et al. 2016a). It binds β-catenin and inhibits the WNT signaling pathway, which is vital in vertebrates’ early development and morphogenesis. Alternatively, spliced transcript variants encoding various isoforms have been discovered in CHD8 (Thompson et al. 2008). Due to its presence at active transcription sites with H3K4me3 or H3K27ac histone modifications, it is believed that CHD8 directly activates genes by binding to the transcriptional start site and boosting transcription factor activity or recruitment, according to the theory. It might also have an indirect effect on transcription by interacting with altered histone sites and other co-regulators to make chromatin more accessible to transcription factors (Barnard et al. 2015; Sugathan et al. 2014; Wilkinson et al. 2015; Cotney et al. 2015). CHD8 binding to p53 causes the development of a trimeric complex on chromatin with histone H1, which reduces p53-dependent transactivation and death during early embryogenesis (Nishiyama et al. 2009). CHD8 is also necessary for the expression of E2 adenovirus promoter-binding factor target genes during the cell cycle’s G1/S transition (Subtil-Rodríguez et al. 2014). In mice, the CHD8 gene deletion causes embryonic lethality (Nishiyama et al. 2009), whereas its heterozygous loss-of-function variants are associated with macrocephaly, craniofacial deformities, and behavioral impairments (Platt et al. 2017). Its knockdown in human neural progenitor cells changes the expression of neuronal development genes (Wilkinson et al. 2015). In the nucleus accumbens (NAc) region of the brain in CHD8+/− mice, WNT signaling is upregulated, suggesting the crucial function CHD8 plays in WNT signaling regulation in the NAc (Platt et al. 2017).
CHD8 is considered the most potential single candidate gene for non-syndromic ASDs (O’Roak et al. 2012a, b; Barnard et al. 2015; Krumm et al. 2014, 2015; Sanders 2015; Bernier et al. 2014). Multiple de novo, truncating, or missense mutations in CHD8 have been found in people with ASDs (O’Roak et al. 2012a, b; Neale et al. 2012; Sugathan et al. 2014; Bernier et al. 2014; Talkowski et al. 2012; McCarthy et al. 2014). Novel risk loci as Balanced chromosomal abnormalities (BCAs) in the CHD8 was found in people with ASD or other neurodevelopmental problems (Talkowski et al. 2012). An analysis of rare coding variation in 3871 ASD cases and 9937 ancestry-matched or paternal controls identified CHD8 as a gene with high statistical significance with an FDR of 0.01, indicating that this gene had a 99% chance of being a true autism gene (De Rubeis et al. 2014). The gene CHD8 has been identified as an antagonist to the signaling pathway that regulates canonical WNT signaling. This finding is consistent with the notion that increased canonical WNT signaling activity causes excessive proliferation of embryonic neural progenitor cells in the brain, which might help to explain in part the macrocephaly (or “big brain”) phenotype observed in cases (Bernier et al. 2014). In Bernier and colleagues study (loss of funcation variants in CHD8 was identified in children with developmental delay and ASD; a phenotypic comparison of patients with CHD8 variants in this report revealed recurrent phenotypes and dysmorphic facial features suggestive of a syndromic form of ASD (Bernier et al. 2014). According to in vivo research, CHD8 loss-of-function pathogenic variant might activate more canonical WNT signals, resulting in macrocephaly, and ASD-like symptoms (Platt et al. 2017). In addition, recent research found that multiple CHD8-controlled genes were implicated in abnormal head size (Sugathan et al. 2014; Wang et al. 2015; Merner et al. 2016). It has been revealed that CHD8 is a positive regulator of WNT/β-catenin signaling neural progenitor cells while also negatively regulating the pathway in non-neuronal cells (Durak et al. 2016). This finding suggests that CHD8 modulates WNT signaling in a cell-specific manner and that some CHD8 mutations might not be as straightforward as WNT signaling loss-of-function mutations. Further studies are needed to understand how CHD8 modulates WNT signaling in diverse brain cell types, and how patients with CHD8 pathogenic variants develop macrocephaly. It is also worth noting that WNT signaling is only one of several neurodevelopmental pathways regulated by CHD8, and recent research has revealed several other mechanisms, such as chromatin remodeling. Thus, more studies are required to explore the involvement of CHD8 in WNT signaling at various stages in the development of brain. This is critical to gain deeper insights into how CHD8 mutations might impair embryonic brain development.
Another gene that has been implicated in ASD is the ANK3 gene, which encodes a scaffolding protein referred to as ankyrin-G (Shi et al. 2013; Iqbal et al. 2013; Bi et al. 2012). The protein was first discovered in the axonal initial segment and nodes of Ranvier of neurons, where it plays a role in axonal initial segment assembly and neuronal polarity (Hedstrom et al. 2008; Kordeli et al. 1995). In general, the ankyrin family of proteins are thought to aid in anchoring integral membrane proteins to the cytoskeleton. Furthermore, they are involved in a wide range of activities, including cell motility, activation, proliferation, contact, and maintenance of specialized membrane domains, among other things. Ankyrin-G promotes cell-cell contact by binding to E-cadherin at a conserved location separate from that of β-catenin and transporting it to the cell adhesion site with 2-spectrin in early embryos and cultured epithelial cells (Kizhatil et al. 2007). Ankyrin-G is abundant in the embryonic brain’s ventricular zone, where it governs neural progenitor cell growth (Durak et al. 2015). Because of alternative splicing and different beginning exons, there are multiple protein isoforms of ankyrin-G13,17. The isoforms have distinct roles and tissue distributions, with some being expressed exclusively in the brain. Rare polymorphisms identified in ASD patients are mostly found in the brain-specific exons 371, 2, 3, 4, 5, 6, 7, and 8 (Bi et al. 2012; Ferreira et al. 2008; Schulze et al. 2009; Psychiatric GWAS Consortium Bipolar Disorder Working Group 2011; Tesli et al. 2011; Baum et al. 2008; Iqbal et al. 2013; Kosmicki et al. 2017). In ASD cases, whole-genome and whole-exome sequencing investigations have documented several genetic variants in ANK3 (Shi et al. 2013; Iqbal et al. 2013; Bi et al. 2012). Loss of function in ANK3 promotes neural progenitor cell proliferation and nuclear β-catenin expression, most likely by disrupting the β-catenin/cadherin connection (Durak et al. 2015).
Several missense mutations in DIXDC1 have been identified in individuals with ASD (Kwan et al. 2016b). These mutations hinder DIXDC1 isoform 1 phosphorylation, resulting in dendritic and spine development defects (Kwan et al. 2016b). DIXDC1 encodes for a protein that acts as a positive regulator of the WNT signaling pathway that modulates excitatory neuron dendrite formation and synapse function in the mouse cortex (Kwan et al. 2016b). MARK1, which has also been associated with ASD, phosphorylates DIXDC1 to regulate dendritic and spine formation via isoform-specific cytoskeletal network regulation (Kwan et al. 2016b). DIXDC1-deficient animals were found to exhibit behavioral abnormalities, including decreased social interaction, which can be relieved by pharmacological inhibition of glycogen synthase kinase 3 (GSK3) to increase WNT/β-catenin signaling (Martin et al. 2018; Kivimäe et al. 2011). These findings point to a possible approach to ASD treatment, including modification of WNT/β-catenin signaling activity. Exome sequencing of the DIXDC1 gene has shown a higher burden of rare sequencing-disrupting SNVs among ASD cases in comparison to controls (Martin et al. 2018). In DIXDC1 knockout neurons, it has been reported that rare DIXDC1 missense variants in ASD cases failed to rescue deficits in glutamatergic synapse density and spine density, with a subset of DIXDC1 missense variants displaying hyperactivity in WNT/β-catenin signaling activity as opposed to dominant-negative effects on spine density and glutamatergic synapse density in wild-type neurons. The existence of functionally relevant ASD missense pathogenic variants in controls, on the other hand, complicates the genetic evidence connecting DIXDC1 to ASD, with reports of high functionally relevant ASD missense variants in controls. Lack of information regarding the mode of inheritance and segregation of variants in ASD cases, along with the presence of sequence-disrupting DIXDC1 variants in controls, all make it very challenging to understand the link between DIXDC1 and ASD.
PGE2, an endogenous lipid molecule, has been shown to alter the expression of downstream WNT pathway genes previously linked to neurodevelopmental problems (Wong et al. 2016). The primary regulator of PGE2 synthesis is cyclooxygenase-2 (COX2). COX2/PGE2 signaling abnormalities have been linked to ASD (Wong et al. 2016). In addition, a growing body of evidence shows that a variety of environmental risk factors, such as inadequate dietary supplementation, infections, misoprostol use during pregnancy, air pollutants, or chemicals, can have a negative effect on PGE2 levels and can be indirectly linked to ASD (Tamiji and Crawford 2010; Wong et al. 2015; Bandim et al. 2003; Landrigan 2010). Furthermore, PGE2 has been shown to downregulate PTGS2 while upregulating MMP9 and CCND1 in undifferentiated stem cells. On the other hand, in differentiating neuronal cells, it upregulates WNT3, TCF4, and CCND1 expression (Wong et al. 2016).
Another gene that has been implicated in ASD etiology is UBE3A which encodes an E3 ubiquitin-protein ligase, which is part of the ubiquitin protein degradation mechanism. This imprinted gene is expressed maternally in the brain and biallelically in other organs. Several studies have shown genetic associations and rare polymorphisms in the UBE3A gene that are linked to ASD. A link was discovered in the collaborative linkage study of autism families and rare variations were discovered in instances of European ancestry (Nurmi et al. 2001). Another polymorphism identified in UBE3A was T485A which is a de novo autism-linked UBE3A pathogenic variants that was reported to be involved in ubiquitinating numerous proteasome subunits, decreasing their number and activity, stabilizing nuclear β-catenin, and activating the canonical WNT pathway more efficiently in comparison to wild-type UBE3A (Yi et al. 2017). Dysfunction of UBE3A has been associated with cancer, Angelman syndrome, ADHD, DD/NDD, extrapyramidal symptoms (EPS), ID, and EP (Yi et al. 2017).
Pathogenic variants in the neurexin and neuroligin families have also been implicated in the development of autism spectrum disorders (Südhof 2008). Neurexins and neuroligins interact cooperatively to regulate the synaptic activity, both excitatory and inhibitory, in the brain (Knight et al. 2011). NRXN1, NLGN1, NLGN3, CNTN4, CNTN6, and CNTNAP2 are examples of superfamily genes that have a function in autism spectrum disorder (Berg and Geschwind 2012). Knockout ASD mouse models of genes such as NRXN1, SHANK3, FMR1, and CNTNAP2 were found to have imbalances in excitation and inhibition in brain regions, and these knockout models exhibited social interaction deficits and reduced ultrasonic vocalizations, which overlapped with behavioral endophenotypes relevant to ASD (Rosti et al. 2014). Imbalances in excitation and inhibition in brain regions were discovered in knockout ASD mouse models of genes such as NRXN1, CNTNAP2, FMR1, and SHANK3. It has been shown that ASD-affected individuals have variants in the neuroligins NLGN3 and NLGN4 (Jamain et al. 2003). These type I transmembrane proteins serve as adhesion molecules for brain cells and are required to establish and develop synaptic connections in the brain (Zhang et al. 2017). According to chromatin immunoprecipitation and promoter luciferase experiments, WNT/β-catenin signaling directly controls the production of NLGN3, a transcription factor (Medina et al. 2018). It would however be essential to establish whether or not WNT/β-catenin signaling influences the expression of additional ASD-associated genes. It is noteworthy that neuronal activity is regulated by genes such as GRIN2B, SCN1A, and SCN2A, and these genes are thought to be involved in the mediation of synaptic plasticity. In addition, they encode for ion channels (O’Roak et al. 2012a). It is noteworthy to mention that UBE3A, PCDH10, DIA1, and NHE9/SLC9A9 are similarly affected by neuronal activity that regulates transcription factors (Morrow et al. 2008; Flavell et al. 2008).
Given the finding of several individuals with phosphatase and tensin homolog (PTEN) mutations, PTEN is considered a high-risk autism candidate gene that plays a role in WNT signaling (O’Roak et al. 2012b; Spinelli et al. 2015; Frazier et al. 2015; McBride et al. 2010). Furthermore, individuals with heterozygous PTEN pathogenic variants are also at risk of developing macrocephaly, implying that PTEN regulates brain size, which is thought to affect specific ASD instances (Page et al. 2009; Kwon et al. 2006; Chen et al. 2015; Vogt et al. 2015; Clipperton-Allen and Page 2014, 2015; Takeuchi et al. 2013; Tilot et al. 2015; Zhou and Parada 2012).
1.5 Altered Sonic Hedgehog (SHH) Signaling in ASD
SHH signaling is one such pathway that regulates neurogenesis and neuronal processes during CNS development (Choy and Cheng 2012). SMO-SHH signaling is implicated in various neurological activities, including neuronal cell proliferation and survival (Álvarez-Buylla and Ihrie 2014). While the function of primary neural cilia in CNS patterning during embryonic development is well understood, their relevance in adult CNS plasticity has only recently been discovered (Kirschen and Xiong 2017). SHH signaling at the main cilium has been investigated and defined, as shown in Fig. 1.2 (Seppala et al. 2017). In the absence of SHH activity, PTCH suppresses SMO effectively. This causes Gli (glioma-associated oncogene/transcription factor) proteins to be phosphorylated, after which they are proteolytically truncated into repressor forms that inhibit transcriptional activity. SHH binding to PTCH, on the other hand, induces internalization followed by breakdown, resulting in SMO buildup and protein phosphorylation. As a consequence, Gli is carried to the cytoplasm and enters the nucleus in its complete form, boosting target transcription even more. In children with ASD, pathological functions for SHH, Indian hedgehog (IHH), and BDNF have been proposed (Halepoto et al. 2015). SHH signaling influences both neurogenesis and neuronal patterning during the development of the central nervous system during pregnancy. Neurological diseases such as ASD are caused by an imbalance in SHH signaling in the brain (Patel et al. 2017). In the context of ASD, SHH has also been linked to oxidative stress (Ghanizadeh 2012). Autistic children exhibited substantially greater levels of oxygen free radicals (OFR) and serum SHH protein, suggesting that oxidative stress and SHH play a role in the development of ASD (Al-Ayadhi 2012). As shown in Fig. 1.2, the connection between ASD-associated genes and SHH signaling is illustrated.

Possible associations between the ASD-linked genes and SHH signaling. PTCHD1, EN2, and DHCR7 genes are all potential ASD-associated genes. A negative sign denotes downregulating activity. The figure was created using BioRender (https://biorender.com/)
The SHH pathway has also been implicated in ASD, and several pathogenic variants in patched domain-containing 1 (PTCHD1) have been identified. A pathogenic variant in this gene has been found to lead to problems with synaptic connections and irregularities in neuronal transmissions in male mice, resulting in hyperactivity and cognitive deficits (Tora et al. 2017; Ung et al. 2018) and behavioral abnormalities (Tora et al. 2017). However, while the PTCHD1 protein does not seem to play a role in SHH-dependent signaling when tested using a loss-of-function approach, it does appear to affect synaptic transmission in the mouse dentate gyrus (Tora et al. 2017). In addition it has been shown that PTCHD1 interacts with PSD95 and SAP102, two postsynaptic proteins (Ung et al. 2018). PTCHD1 deficiency (PTCHD1-/y) in male mice causes widespread changes in synaptic gene expression, including changes in the expression of the immediate-early expression genes EGR1 and NPAS4, and disruptions in excitatory synaptic structure and neuronal excitatory activity in the hippocampus, which results in cognitive dysfunction, motor impairments, and hyperactivity (Ung et al. 2018).
Pathogenic variants in the 7-dehydrocholesterol reductase (DHCR7) gene have also been linked to ASD. Increased cholesterol production was found to have a negative effect on the activation of the transmembrane protein smoothened (SMO), which is responsible for transmitting SHH signals, and its localization to the major cilium (Blassberg et al. 2016). The transcription factor engrailed 2 (EN2) has also been implicated in the development of ASD (Brune et al. 2008; Sen et al. 2010; Wang et al. 2008; Yang et al. 2008, 2010). The higher levels of EN2 seen in individuals with the EN2 ASD-associated haplotype (rs1861972-rs1861973 A-C) indicated that EN2 is associated with ASD susceptibility (Choi et al. 2014; Bi et al. 2012; Gharani et al. 2004). According to the evidence from postmortem samples, rising EN2 levels have been associated with increased SHH expression (Choi et al. 2014). During brain development, SHH is one of the genes that coexpress EN2 and other genes (Wechsler-Reya and Scott 1999; Simon et al. 2005). Furthermore, both mice with EN2 pathogenic variants and autistic humans had cerebellar structural abnormalities that were similar (Gharani et al. 2004).
1.6 Altered Retinoic Acid (RA) Signaling in ASD
RA, a functional metabolite of vitamin A, is a necessary morphogen during vertebrate development (Niederreither and Dollé 2008; Duester 2008). RA can influence several developmental genes that include RA response elements (RARE) and their regulatory areas. By binding to nuclear receptors known as retinoic acid receptors (RARs) and retinoid X receptors (RXRs), RA mediates both genomic transcriptional effects and non-genomic effects such as retinoylation (RA acylation), a posttranslational modification of proteins (Rhinn and Dollé 2012; Das et al. 2014). A number of co-activators and co-repressors have been identified as modulators of RA signaling activity (Das et al. 2014). RA is necessary for neural patterning, differentiation, proliferation, and establishment of neurotransmitter systems in the developing CNS (Zieger and Schubert 2017). Cortical neuron generation is regulated by RA from the meninges (Siegenthaler et al. 2009). During embryonic development, RA aids in the regulation of a group of HOX (homeobox) genes that form the upper-body pattern, both anteriorly and posteriorly, and are implicated in brain patterning. Dopaminergic and GABAergic neurons are involved in brain cell development in RA. It is also a critical inducing factor for the development of motor neurons from pluripotent stem cells (Faravelli et al. 2014).
Furthermore, RA is necessary for the appropriate functioning of motor neurons. RA has also been linked to neuronal migration and neurogenesis in the granular zone of the hippocampus, the sub-ventricular zone, and the olfactory bulb (Ghyselinck and Duester 2019). The above mentioned functions of RA indicate that neurodevelopmental diseases and RA signaling pathways are linked.
A sufficient quantity of retinol is required for the regular functioning of the RA signaling system, assuming that all of the enzymes involved in the RA pathway and nuclear factors operate properly. One of the key reasons for reduced intracellular RA signaling is retinol insufficiency. In China, some autistic people had lower levels of retinol than the normal control group, which might be a synergistic component in the development of ASD symptoms (Cheng et al. 2021). Retinol supplements have been shown to increase RAR expression and reduce ASD symptoms. Some investigations have shown that retinol deficiency in rats during pregnancy reduces RA receptor expression (RAR, beta isoform) in the hypothalamus, resulting in autistic-like symptoms in the neonates (Lai et al. 2018). In rats, a lack of vitamin A has been linked to ASD-like behavior (Lai et al. 2018). It has also been claimed that ASD is caused by an abnormality in the interaction of RA and sex hormones (Niculae and Pavăl 2016).
The nuclear receptors for RA have also been implicated in the pathophysiology of ASD. RORs (retinoic acid-related orphan receptors) operate as transcriptional regulators upon RA binding, triggering transcription of numerous genes. Autistic people were shown to have reduced protein expression of the RA-related orphan receptor alpha (RORA) due to hypermethylation (Nguyen et al. 2010), and RORA mutations have been linked to ASD (Sayad et al. 2017). RORA has been shown to transcriptionally control numerous ASD-relevant genes, including NLGN1 (Sarachana and Hu 2013). P89L missense variant of NLGN1, identified in ASD cases, has been linked to alterations in cellular localization, protein degradation, and spine formation impairment. Mice with heterozygous P89L in the NLGN1 have also been found to exhibit aberrant social behavior, which is a crucial hallmark of ASD (Nakanishi et al. 2017).
Furthermore, an immunohistochemical examination of the postmortem brains of autistic people revealed a reduced quantity of ROR alpha protein (Nguyen et al. 2010). Disruption of the retinoic acid enzymatic production pathway was found to be associated with ASD phenotypes and retinoic acid nuclear receptors, which have also been implicated in the pathophysiology of ASD. Therefore, more research is needed to establish a link between ASD pathogenesis and involvement of RAR and ROR agonists in autism treatment.
Another approach for detecting ASD signals is quantitative electroencephalography (EEG) analysis. Details of individual characterization of EEG fluctuations in ASD subjects could aid in examining brain issues, which would be helpful in observing automatic groupings and random draws of the patient population when analyzing sensory-processing issues of the brain and peripheral system (Ryu et al. 2021).
The conversion of retinol to RA with the aid of the enzyme (ALDH) retinaldehyde dehydrogenase, which assures the concentration of RA in the cell, is an essential metabolic step in the RA pathway. In vitro, over-ubiquitinoylation by UBE3A has been shown to increase the breakdown rate of this enzyme’s isoform aldehyde dehydrogenase 1 family member A2 (ALDH 1A2). Autistic characteristics were detected in mice with UBE3A overexpression (Xu et al. 2017). In addition, loss-of-function pathogenic variants in the UBE3A have been linked to Angelman syndrome, demonstrating the role of UBE3A in brain development (Khatri and Man 2019). Surprisingly, UBE3A overexpression suppresses ALDH1A2 and inhibits RA-mediated synaptic plasticity in ASD, which might be improved with RA supplementation (Xu et al. 2018). All-trans-RA can increase CD38 expression in ASD lymphoblastoid cell lines, but CD38-deficient animals display ASD-like behavior (Riebold et al. 2011; Kim et al. 2016). In BTBR mice, beta-carotene, a precursor of vitamin A, is a viable therapy for autistic-like behavior (Avraham et al. 2019). In a mouse model, a synthetic RORA/G agonist was investigated for its ability to ameliorate autistic symptoms (Wang et al. 2016). A South American cohort’s WES has revealed a link between RA signaling genes, including a RA-synthesizing gene ALDH1A3 and the RORA-regulated FOXN1, and ASD (Moreno-Ramos et al. 2015). A subpopulation of autistic people were shown to have low levels of ALDH1A1 (Pavăl et al. 2017). ASD and other abnormalities linked with proximal 1p36 deletions might be caused by de novo variants in arginine-glutamic acid dipeptide repeats (RERE) that encode a nuclear receptor coregulator for RA signaling (Fregeau et al. 2016). These findings point to potential pharmacological methods for ASD treatment that include targeting RA and associated signaling pathways. Figure 1.3 depicts the potential connections between ASD-associated genes and RA signaling.

Possible interactions between ASD-associated genes and RA signaling. UBE3A influences ALDH1A expression, which in turn affects the RA signaling pathway. RORA is linked to ASD, which impacts NLGN1. ASD is also linked to the RA signaling coregulator RERE. The figure was created using BioRender (https://biorender.com/)
1.7 Altered Fibroblast Growth Factor (FGF) Signaling in ASD
FGF signaling is critical in brain patterning, and its disruption might result in a variety of neurological disorders (Turner et al. 2016). In the mammalian FGF family, there are 18 secreted FGFs and 4 tyrosine kinase FGF receptors (FGFRs) whose interaction is controlled by cofactors and external binding proteins (Ornitz and Itoh 2015). When FGFRs are activated, tyrosine residues are phosphorylated, resulting in interactions between cytosolic adaptor proteins and RAS-MAPK, PI3K-AKT, PLC, and STAT intracellular signaling pathways (Ornitz and Itoh 2015). It has been proposed that dysregulation of FGF signaling plays a role in the etiology of ASD (Iwata and Hevner 2009). Cortical abnormalities in autistic brains, for example, have been linked to faulty FGF signaling (Turner et al. 2016; Amaral et al. 2008). ASD is thought to be caused by a disruption in the number of excitatory and inhibitory synapses (Terauchi et al. 2010). Mutant mice FGF22 or FGF7 knockout (KO) showed defective synapse formation in hippocampal CA3 pyramidal neurons (Terauchi et al. 2010), indicating the pathogenic significance of dysregulated FGF signaling in ASD. The metabotropic glutamate receptor 5 (mGluR5) loss-of-function pathogenic variant causes abnormal dendritogenesis in cortical neurons, which is one of the traits identified in autistic brains, by raising nerve growth factor (NGF) and FGF10 mRNA levels (Huang and Lu 2017). Although several members of FGF gene subfamilies have been linked to CNS development and/or function, many of their roles remain largely unknown, and no direct evidence of altered FGF signaling in ASD has been reported. Nevertheless, it has been well documented that epigenetic mechanisms (Zhu et al. 2007), and posttranslational modifications of FGFs and FGFRs (Sarabipour and Hristova 2016; Triantis et al. 2010; Wheeler and Clinkenbeard 2019; Kucinska et al. 2019; Porebska et al. 2018), are involved in the regulation of the expressions of FGF/FGFR signaling (Xie et al. 2020). Figure 1.4 summarizes the plausible mechanisms by which FGF signaling modulates cell fate.

Possible mechanisms of FGF signaling. A simplified depiction of the possible mechanisms through which an FGF signal might be modulated to influence cell fate. The figure was created using BioRender (https://biorender.com/)
1.8 Altered TGF-β/BMP Signaling in ASD
TGF-β/activin and bone morphogenetic protein (BMP)/growth and differentiation factor (GDF) are the two subcategories of the TGF-β superfamily (Zi et al. 2012). BMPs are the most abundant in the TGF-β superfamily (Lory and Rosen 2018), and they play an essential role in nervous system development (Bond et al. 2012). Their signaling has been demonstrated to be dysregulated in ASD. BMPs influence gene expression via both canonical (Smad-dependent) and noncanonical pathways (such as the MAPK cascade) (Wang et al. 2014). The binding of BMPs to type I or type II serine/threonine kinase receptors results in a heterotetrameric complex in the canonical pathway. The type II receptor then transphosphorylates the type I receptor, following which the type I receptor phosphorylates the R-Smads (Smad1/5/8). Phosphorylated Smad1/5/8 and the co-Smad (Smad4) translocate to the nucleus and regulate gene expression. BMP signaling is modulated by various factors, including plasma membrane co-receptors and external and intracellular factors (Wang et al. 2014). The BTBR T + Itpr3tf/J (BTBR) mice are commonly utilized in ASD studies (Ansari et al. 2017). TGF-β levels have been found to be lower in BTBR mice than in B6 mice (Ansari et al. 2017). TGF-β expression levels in the spleen and brain tissues of BTBR mice were found to be significantly higher than in adenosine A2A receptor (A2AR) agonist CGS 21680 (CGS)-treated mice (Ansari et al. 2017). Components of the TGF-β pathway were identified as novel hyperserotonemia-related ASD genes in a network-based gene set enrichment analysis (NGSEA) based on loss-of-function and missense de novo variants (Chen et al. 2017). Figure 1.5 summarizes the interactions between ASD-associated genes and TGF-β/BMP signaling.

Possible interactions between ASD-associated genes and TGF-β/BMP signaling. ASD-associated gene-encoded proteins, such as NLGN3/4, FMR1, DLX, and UBE3A, interact with BMP signaling. Plus sign indicates upregulation; minus sign indicates downregulation. The figure was created using BioRender (https://biorender.com/)
Fragile X syndrome (FXS) is the most frequent heritable form of ASD and intellectual impairment caused by FMR1 silencing (Kashima et al. 2016). Depletion of the FMR1 protein (FMRP) leads to increased bone morphogenetic protein type II receptor (BMPR2) and activation of a noncanonical BMP signaling component, LIM domain kinase 1 (LIMK1), which stimulates actin rearrangement to promote neurite outgrowth and synapse formation (Kashima et al. 2016). Increased BMPR2 and LIMK1 activity has been observed in the prefrontal cortex of FXS patients compared to healthy participants (Kashima et al. 2016).
UBE3A has been associated with the regulation of synapse growth and endocytosis by inhibiting BMP signaling (Li et al. 2016). The ubiquitin-proteasome pathway degrades the BMP receptor Tkv, a direct substrate of UBE3A (Li et al. 2016). Through the BMP signaling pathway, Drosophila UBE3A is known to modulate neuromuscular junction (NMJ) development in presynaptic neurons (Li et al. 2016). Drosophila UBE3A mutants have been found to be viable and productive. Nevertheless, they have been demonstrated to have impaired endocytosis in the NMJs and upregulated BMP signaling in the nervous system, owing to an increase in Tkv (Li et al. 2016).
The DLX genes, which encode homeodomain transcription factors, have also been linked to ASD (Liu et al. 2009; Hamilton et al. 2005; Rubenstein and Merzenich 2003). These genes regulate craniofacial patterning, differentiation, and survival of forebrain inhibitory neurons (Hamilton et al. 2005). The BMP-binding endothelial regulator (Bmper) is upregulated in a cell line overexpressing DLX5 (Sajan et al. 2011), indicating that dysregulated DLX activity in individuals with ASD might lead to aberrant BMP signaling.
1.9 Signaling Crosstalk in ASD
While it is known that crosstalk occurs across signaling pathways in many developmental stages and illnesses, availability of evidence on autistic models is limited. Research has shown that the WNT signaling proteins might affect both developmental and inflammatory processes. It was shown that WNT signaling and SHH signaling are connected and influence each other at the transcriptional level (Chatterjee and Sil 2019). Cell-line studies have shown that Gli stimulates β-catenin nuclear translocation by E-cadherin and Snail (Li et al. 2007). Gli regulates WNT5a and WNT2b to elevate WNT signaling (Katoh and Katoh 2009). Casein kinase 1 (CK1), p53, PTEN, and GSK3 are protein modulators in these signaling pathways, and each serves a different purpose. They collaborate to boost β-catenin and Gli signaling (Song et al. 2014). Due to evidence of cross talk between WNT and SHH/Gli signaling in colon cancer, this signaling pathway is now a possible candidate for colon cancer therapy (Song et al. 2015). CK1 and GSK3 are known to have antagonistic functions in regulating β-catenin and Gli1 (Wang and Li 2006; Tempé et al. 2006; Hart et al. 1999), and a detrimental impact on TCF and downstream genes of metastatic colon cancer. Also, they were shown to have opposing roles (Varnat et al. 2010). Another problem is that the gene for a kinase inhibitor (Sufu), which represses Gli1 activity, has been shown to change the distribution of β-catenin between the nucleus and cytoplasm (Meng et al. 2001; Dunaeva et al. 2003; Tukachinsky et al. 2010). Loss of p53 or PTEN causes Gli1 and β-catenin to activate in colon cancer (Varnat et al. 2010; Rychahou et al. 2008). The authors have seen that the use of SMO inhibitors keeps Gli1, an upstream effector, from becoming active, which prevents the amount of active β-catenin from rising and increases the nuclear exclusion of β-catenin (Arimura et al. 2009). Furthermore, Gli1 prevents Gli3R, whereas Gli3R stops Gli1 (Varnat et al. 2010). Additionally, β-catenin activity has been shown to reduce with Gli3R (Ulloa et al. 2007). There is evidence that Gli1 boosts the expression of WNT4, WNT2b, and WNT7b (Li et al. 2007).
Research on the embryonic development of WNT signaling, TGF-β, RA signaling, and BMP revealed that interactions exist among them (Hayward et al. 2008; Boyle et al. 2011; Pelullo et al. 2019). There are a number of ways that WNT signaling might also interact with cytokine signaling and NF-B to support inflammatory responses (Koopmans et al. 2017).
The development of the zebrafish tailbud (Stulberg et al. 2012) and the mouse craniofacial area are thought to be influenced by the crosstalk between the FGF and WNT pathways (Wang et al. 2011). Researchers have shown that increased FGF and WNT signaling is positive (Stulberg et al. 2012). The gene Fgf8 was shown to play a key role in the induction of neural tissues in the face. It is particularly active in the anterior neural ridge and face ectoderm, with WNT/β-catenin signaling enhancing its role and β-catenin mutation leading to aberrant levels of Fgf8 expression in the face ectoderm (Glinka et al. 2011). WNT boosts FGF signaling by boosting Erk phosphorylation in the MAPK branch (Stulberg et al. 2012). FGF blocks WNT antagonists dkk1 and notum1a, which results in WNT signaling being raised (Stulberg et al. 2012). A mutated UBE3A gene, linked to ASD, affects both the WNT and BMP signaling pathways, indicating their possible connection (Li et al. 2016; Yi et al. 2017). Xu and her colleagues observed UBE3A as negatively regulating ALDH1A2, which means that this gene makes RA formation more difficult (Xu et al. 2018). The ASD-associated gene might additionally influence BMP signaling, even though it has been suggested that it was likely to be a direct target of WNT/β-catenin signaling (Medina et al. 2018). The above results indicate that ASD-connected genes enhance the signaling of cross talk via morphogenetic pathways, emphasizing the necessity for further study into the interconnections between signals in normal physiological and pathological circumstances. WNT and BMP signaling cross talk seems to be the result of a tissue-specific process (Itasaki and Hoppler 2010). Signaling interactions occur contextually in the eye, because WNT signaling positively controls RA signaling in the dorsal optic cup but suppresses RA signaling during orofacial development (Itasaki and Hoppler 2010).
Cyclosporine-enhanced cell proliferation has also been seen in human gingival fibroblasts (Chung and Fu 2013). In addition, interactions between TGF-β and SHH pathways have been found in cancer (Javelaud et al. 2012). TGF-β, a growth factor, binds to receptors found on cancer cell membranes, including the neuropilin-1 (NRP1). NRP1 has been demonstrated to promote the canonical SMAD2/3 signaling in response to TGF-β (Glinka et al. 2011). Also, NRP1 transduction is strengthened by HH signaling, and NRP1 has been shown to increase HH target gene activation by assisting the SMO/SUFU interconnection (Hillman et al. 2011; Hochman et al. 2006). While it is essential in the growth of SMO-associated cancers (Fan et al. 2010), it has been shown that TGF-β might increase GLI2 and GLI1 expression by blocking PKA activity (Pierrat et al. 2012). According to the hierarchical structure of cross talk, TGF-β upregulates SHH, resulting in increased SHH synthesis and cell proliferation in gingival fibroblasts. This occurs through cyclosporine, which enhances SHH production (Chung and Fu 2013).
1.10 Nongenetic ASD Etiologies
With ASD susceptibility estimated to be 40–80% hereditary, the risk is probably driven by environmental variables, which are most likely involved in ASD etiology via epigenetic regulation as the primary mechanism.
Hundreds of plausible environmental risk variables have been identified, including increased parental age, viral infections, and prenatal exposure to anticonvulsants, such as valproic acid (VPA). All the proposed risk factors can cause various neurodevelopmental diseases with disrupted WNT signaling (O’Roak et al. 2012a; Rasalam et al. 2005; Kong et al. 2012; Ohkawara et al. 2015).
1.11 Advancing Paternal Age
Since the 1970s, there has been speculation about a connection between paternal age and ASD (Allen et al. 1971; Treffert 1970). A study has reported that increasing paternal age was significantly associated with the susceptibility to ASD, even after adjusting for confounding factors, such as maternal age (Reichenberg et al. 2006). De novo germline pathogenic variants or changes in genomic imprinting might have a role, or at least in part, in the observed association. Nevertheless, further research is needed to validate these findings.
1.12 Viral Infection
New research suggests a link between viral infections and neurodegenerative and neurobehavioral disorders such as autism. Infection with a virus at key stages of early in utero neurodevelopment might increase the chance of autism in the child. Clinical and epidemiological research has revealed associations between congenital CMV infection and autistic symptoms (Stubbs 1978; Stubbs et al. 1984; Markowitz 1983). An antibody response to the virus, positive viral culture from the urine, decreased hearing, and retinal inflammation revealed evidence of congenital CMV infection. During pregnancy or at birth, CMV infection is a significant cause of sensorineural hearing loss (Grosse et al. 2008; Fowler et al. 1999) and other neurological impairments (Dollard et al. 2007; Townsend et al. 2013). Earlier case studies found that children with congenital CMV infection had typical autistic traits such as inability to build strong interpersonal relationships, poor eye contact, delayed use of language, and nonthematic usage of items.
In 1990, there were numerous reports of severely impaired children with autism who also had congenital CMV infection (Ivarsson et al. 1990). Yamashita and colleagues (Sarabipour and Hristova 2016) demonstrated positive serum CMV-specific IgM antibodies and CMV-DNA in the urine of children with typical autistic disorders. The brain magnetic resonance imaging (MRI) findings indicated an unusually intense region in the periventricular white matter, indicating disrupted myelination. Seten et al. (Seten et al. 2004) discussed the potential function of congenital and perinatal CMV infection in inducing an altered immune response or autoimmune process. The infection or the resulting immune response might impair the development of brain regions or structures, leading to autism. Engman et al. (Engman et al. 2015) also examined the frequency of congenital CMV infection in a representative sample of children with ASD. Congenital CMV infection was found in 1 of the 33 infants with autistic disorder and intellectual deficits, corresponding to a 0.2% prevalence in the general Swedish newborn population. Sakamoto et al. (Sakamoto et al. 2015) hypothesized that congenital CMV infection was involved in a subset of children with ASD since the rate of CMV infection was greater than the incidence of congenital CMV infection in Nagasaki, Japan.
There has been growing evidence describing a connection between rubella infection in early pregnancy and ASD (Chess 1971). Recent studies have demonstrated that in utero viral immune activation results in persistent hyper- and hypomethylated CpGs at WNT signaling genomic regions (WNT3, WNT7B, WNT8A) (Richetto et al. 2017), leading to the disruption of the transcription of downstream target genes, which is thought to have a role in the development of ASD. Other viral infections that have been implicated in ASD include Zika (Nielsen-Saines et al. 2019; Abtibol-Bernardino et al. 2020), cytomegalovirus, and seasonal and pandemic influenza infections (Atladóttir et al. 2012; Deykin and Macmahon 1979).
1.13 Valproic Acid (VPA)
VPA is a medication used to treat EP and bipolar disorder. VPA usage during early pregnancy has been associated with ASD and autistic features in children (Moore et al. 2000). Prenatal VPA exposure in rats has been reported to be related to autism-like phenotypes (Zhang et al. 2012). Prenatally VPA treatment in rats has been shown to cause an imbalance in oxidative homeostasis, which increases vulnerability to autism (Zhang et al. 2012; Qin and Dai 2015). In addition, VPA-administered rats demonstrated reduced social contact, among other autism-like behavioral features. Sulindac, a small-molecule inhibitor of the WNT/β-catenin signaling pathway (Zhang et al. 2012; Qin and Dai 2015), has been reported to be capable of reversal of the VPA-induced autistic-like behaviors. It also caused a decrease in p-GSK3β expression and an increase in β-catenin expression in the hippocampus, prefrontal lobe, and cerebellum (Qin and Dai 2015).
In addition, GATA-3 is a transcription factor that is required for brain development (Tsarovina et al. 2010). It is implicated in the WNT (Notani et al. 2010) and TGF-β/BMP (Kim et al. 2018; Forsman et al. 2013) signaling pathways. It has been shown to be associated with ASD, after exposure to valproate, thalidomide, and alcohol, which might contribute to the development of ASD (Rout and Clausen 2009).
1.14 Precision Medicine Approaches in ASD
Various pathophysiological processes and etiologies characterize ASD and therefore precision medicine seems to be the most promising way of finding treatment options for ASD (Uddin et al. 2019; Al-Dewik and Alsharshani 2020; Styles et al. 2020). The field of precision medicine attempts to mix groundbreaking pathophysiologically based treatments (biomarker stratification) with testing to determine which therapeutic course of action might assist a particular person (Loth et al. 2016). Understanding the molecular and cellular pathways associated with ASD might enable the development of more effective treatment options. The current approach has more significant information about the molecular and cellular processes involved in ASD (Loth et al. 2016). However, following recent therapeutic research failures involving monogenic ASD variants, new challenges have emerged which include conceptual and methodological problems (i.e., the difficulty of translating from animal models to humans and then conducting suitable clinical trials). Other issues are the impact of placebo effects, clinical trial design, and the need to identify mechanistically plausible, measurable outcomes (Loth et al. 2016). Additionally, as many as 70% of individuals with ASD are likely to have a psychiatric or physical co-condition (Simonoff et al. 2008), which might complicate treatment, which could cause an impairment that will last far into adulthood (Shaltout et al. 2020). A transdisciplinary, multidisciplinary, and collaborative approach would be required to deal with the many issues that future research projects must cover.
1.15 Future Perspectives and Conclusions
Research and knowledge of ASD are growing, thanks to many efforts that have been rigorous and comprehensive. Nonetheless, a multi-domain expert collaboration is required to have a cohesive picture of ASD and adequately evaluate its increasing genetic, epidemiological, and environmental components. During embryogenesis, dysregulation of various signaling neurodevelopmental pathways such as WNT, BMP/TGF, SHH, FGF, or RA seems to induce ASD and alter neurogenesis. The signaling pathways might function upstream or downstream of ASD causative genes. Several studies have revealed that the pathways mentioned above play important roles in developing targeted therapies for ASD. However, comprehensive developmental investigations are needed to determine the time window during which disruption of these signals has the most significant impact on brain structure and function, and the subsequent impairment in behavior.
Furthermore, such research might aid in the understanding of the processes involved in ASD etiology, and the upstream and downstream signaling pathways involved. The interaction between different signaling pathways in neuronal and glial cells for ASD should be explored, as this might aid in devising therapy and addressing the disturbed signaling in a cell-specific manner. Only a few ASD risk genes have been investigated in the context of disrupted signaling pathways so far. The exploration of functions of additional ASD genes in neurodevelopment and the control of other signaling pathways might improve the existing knowledge of the processes underlying ASD etiology. Early identification is necessary for a successful therapy that treats the symptoms while reversing neurons to normal circumstances. In addition to the clinical value of a genetic diagnosis, early diagnosis has also been proven to enhance the understanding, give a feeling of empowerment, and improve the quality of life of the affected child and the parents. As a result, additional investigations are needed to comprehend the mechanistic picture of signaling pathways, communication in ASD, and causative gene interaction with these pathways.
References
Abrahams BS, Geschwind DH (2008) Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet 9:341
Abtibol-Bernardino MR, de Almeida Peixoto LdFA, de Oliveira GA, de Almeida TF, Rodrigues GRI, Otani RH, Soares Chaves BC, de Souza Rodrigues C, de Andrade ABCA, de Fatima Redivo E et al (2020) Neurological findings in children without congenital microcephaly exposed to Zika virus in utero: a case series study. Viruses 12:1335. https://doi.org/10.3390/v12111335
Abu-Khalil A, Fu L, Grove EA, Zecevic N, Geschwind DH (2004) Wnt genes define distinct boundaries in the developing human brain: implications for human forebrain patterning. J Comp Neurol 474:276–288
Akiyama T, Kawasaki Y (2006) Wnt signaling and the actin cytoskeleton. Oncogene 25:7538–7544. https://doi.org/10.1038/sj.onc.1210063
Al-Ayadhi LY (2012) Relationship between sonic hedgehog protein, brain-derived neurotrophic factor and oxidative stress in autism spectrum disorders. Neurochem Res 37:394–400
Al-Dewik N, Alsharshani M (2020) New horizons for molecular genetics diagnostic and research in autism spectrum disorder. In: Essa M, Qoronfleh M (eds) Personalized food intervention and therapy for autism spectrum disorder management. Advances in neurobiology, vol 24. Springer, Cham. https://doi.org/10.1007/978-3-030-30402-7_2
Al-Dewik N et al (2020) Overview and introduction to autism spectrum disorder (ASD). In: Essa M, Qoronfleh M (eds) Personalized food intervention and therapy for autism spectrum disorder management. Advances in neurobiology, vol 24. Springer, Cham. https://doi.org/10.1007/978-3-030-30402-7_1
Allen J, DeMeyer MK, Norton JA, Pontius W, Yang E (1971) Intellectuality in parents of psychotic, subnormal, and normal children. J Autism Child Schizophr 1(3):1311–1326
Álvarez-Buylla A, Ihrie RA (2014) Sonic hedgehog signaling in the postnatal brain. Semin Cell Dev Biol 33:105–111
Amaral DG, Schumann CM, Nordahl CW (2008) Neuroanatomy of autism. Trends Neurosci 31:137–145
Ansari MA, Attia SM, Nadeem A, Bakheet SA, Raish M, Khan TH et al (2017) Activation of adenosine A2A receptor signaling regulates the expression of cytokines associated with immunologic dysfunction in BTBR T(+) Itpr3(tf)/J mice. Mol Cell Neurosci 82:76–87
Arimura S, Matsunaga A, Kitamura T, Aoki K, Aoki M, Taketo MM (2009) Reduced level of smoothened suppresses intestinal tumorigenesis by down-regulation of Wnt signaling. Gastroenterology 137:629–638
Atladóttir H, Henriksen TB, Schendel DE, Parner ET (2012) Autism after infection, febrile episodes, and antibiotic use during pregnancy: an exploratory study. Pediatrics 130:e1447–e1454. https://doi.org/10.1542/peds.2012-1107
Avraham Y, Berry EM, Donskoy M, Ahmad WA, Vorobiev L, Albeck A et al (2019) Beta-carotene as a novel therapy for the treatment of “autistic like behavior” in animal models of autism. Behav Brain Res 364:469–479
Bae SM, Hong JY (2018) The Wnt signaling pathway and related therapeutic drugs in autism spectrum disorder. Clin Psychopharmacol Neurosci 16(2):129–135
Bandim JM, Ventura LO, Miller MT, Almeida HC, Costa AES (2003) Autism and Möbius sequence: an exploratory study of children in northeastern Brazil. Arq Neuropsiquiatr 61:181–185
Barber JC, Ellis KH, Bowles LV, Delhanty JD, Ede RF, Male BM et al (1994) Adenomatous polyposis coli and a cytogenetic deletion of chromosome 5 resulting from a maternal intrachromosomal insertion. J Med Genet 31:312–316
Barnard RA, Pomaville MB, O’Roak BJ (2015) Mutations and modelling of the chromatin remodeler CHD8 define an emerging autism etiology. Front Neurosci 9:477. https://doi.org/10.3389/fnins.2015.00477
Baum AE et al (2008) A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry 13:197–207
Bengoa-Vergniory N, Kypta RM (2015) Canonical and noncanonical Wnt signaling in neural stem/progenitor cells. Cell Mol Life Sci 72:4157–4172
Berg JM, Geschwind DH (2012) Autism genetics: searching for specificity and convergence. Genome Biol 13:247–247. https://doi.org/10.1186/gb-2012-13-7-247
Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT et al (2014) Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158(2):263–276. https://doi.org/10.1016/j.cell.2014.06.017
Bi C, Wu J, Jiang T, Liu Q, Cai W, Yu P et al (2012) Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat 33:1635–1638
Bielen H, Houart C (2014) The Wnt cries many: Wnt regulation of neurogenesis through tissue patterning, proliferation, and asymmetric cell division. Dev Neurobiol 74:772–780
Blassberg R, Macrae JI, Briscoe J, Jacob J (2016) Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome. Hum Mol Genet 25(4):693–705
Bond AM, Bhalala OG, Kessler JA (2012) The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev Neurobiol 72:1068–1084
Bosoi CM, Capra V, Allache R, Trinh VQ, De Marco P, Merello E, Drapeau P, Bassuk AG, Kibar Z (2011) Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Hum Mutat 32:1371–1375
Boyle SC, Kim M, Valerius MT, McMahon AP, Kopan R (2011) Notch pathway activation can replace the requirement for Wnt4 and Wnt9b in mesenchymal-to-epithelial transition of nephron stem cells. Development 138:4245–4254. https://doi.org/10.1242/dev.070433
Brune CW, Korvatska E, Allen-Brady K, Cook EH, Dawson G, Devlin B et al (2008) Heterogeneous association between engrailed-2 and autism in the CPEA network. Am J Med Genet B Neuropsychiatr Genet 147B:187–193
Buckley AW, Holmes GL (2016) Epilepsy and autism. Cold Spring Harb Perspect Med 6:a022749
Burden SJ (2000) Wnts as retrograde signals for axon and growth cone differentiation. Cell 100:495–497
Chatterjee S, Sil PC (2019) Targeting the cross talks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharmacol Res 142:251–261. https://doi.org/10.1016/j.phrs.2019.02.027
Chen Y, Huang WC, Sejourne J, Clipperton-Allen AE, Page DT (2015) Pten mutations alter brain growth trajectory and allocation of cell types through elevated beta-catenin signaling. J Neurosci 35(28):10252–10267. https://doi.org/10.1523/JNEUROSCI.5272-14.2015
Chen R, Davis LK, Guter S, Wei Q, Jacob S, Potter MH et al (2017) Leveraging blood serotonin as an endophenotype to identify de novo and rare variants involved in autism. Mol Autism 8:14
Cheng B, Zhu J, Yang T, Guo M, Lai X, Li Q, Chen J, Li T (2021) Vitamin A deficiency increases the risk of gastrointestinal comorbidity and exacerbates core symptoms in children with autism spectrum disorder. Pediatr Res 89:211–216. https://doi.org/10.1038/s41390-020-0865-y
Chess S (1971) Autism in children with congenital rubella. J Autism Child Schizophr 1:33–47
Choi J, Ababon MR, Soliman M, Lin Y, Brzustowicz LM, Matteson PG et al (2014) Autism associated gene, engrailed2, and flanking gene levels are altered in post-mortem cerebellum. PLoS One 9:e87208
Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L et al (2012) Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet 8:e1002592
Choy SW, Cheng SH (2012) Hedgehog signaling. Vitam Horm 88:1–23
Chung Y, Fu E (2013) Crosstalk between Shh and TGF-β signaling in cyclosporine-enhanced cell proliferation in human gingival fibroblasts. PLoS One 8:e70128
Clevers H, Nusse R (2012) Wnt/β-catenin signaling and disease. Cell 149:1192–1205
Clipperton-Allen AE, Page DT (2014) Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Hum Mol Genet 23(13):3490–3505. https://doi.org/10.1093/hmg/ddu057
Clipperton-Allen AE, Page DT (2015) Decreased aggression and increased repetitive behavior in Pten haploinsufficient mice. Genes Brain Behav 14(2):145–157. https://doi.org/10.1111/gbb.12192
Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J et al (2015) The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun 6:6404. https://doi.org/10.1038/ncomms7404
Das BC, Thapa P, Karki R, Das S, Mahapatra S, Liu TC et al (2014) Retinoic acid signaling pathways in development and diseases. Bioorg Med Chem 22:673–683
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE et al (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–215
Deykin EY, Macmahon B (1979) Viral exposure and autism. Am J Epidemiol 109:628–638. https://doi.org/10.1093/oxfordjournals.aje.a112726
Dias C, Pfundt R, Kleefstra T, Shuurs-Hoeijmakers J, Boon EMJ, van Hagen JM et al (2021) De novo variants in TCF7L2 are associated with a syndromic neurodevelopmental disorder. Am J Med Genet A 185(8):2384–2390
Dollard SC, Grosse SD, Ross DS (2007) New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol 17:355–363. https://doi.org/10.1002/rmv.544
Dong F, Jiang J, McSweeney C, Zou D, Liu L, Mao Y (2016) Deletion of CTNNB1 in inhibitory circuitry contributes to autism-associated behavioral defects. Hum Mol Genet 25:2738–2751
Dubruc E, Putoux A, Labalme A, Rougeot C, Sanlaville D, Edery P (2014) A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am J Med Genet A 164A:1571–1575
Duester G (2008) Retinoic acid synthesis and signaling during early organogenesis. Cell 134:921–931
Dunaeva M, Michelson P, Kogerman P, Toftgard R (2003) Characterization of the physical interaction of Gli proteins with SUFU proteins. J Biol Chem 278:5116–5122
Durak O, de Anda FC, Singh KK, Leussis MP, Petryshen TL, Sklar P et al (2015) Ankyrin-G regulates neurogenesis and Wnt signaling by altering the subcellular localization of β-catenin. Mol Psychiatry 20:388–397
Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, Tsai LH (2016) Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 19(11):1477–1488
Engman ML, Sundin M, Miniscalco C, Westerlund J, Lensohn-Fuchs I, Gillberg C, Fernell E (2015) Prenatal acquired cytomegalovirus infection should be considered in children with autism. Acta Paediatr 104:792–795. https://doi.org/10.1111/apa.13032
Fan Q, He M, Sheng T, Zhang X, Sinha M, Luxon B et al (2010) Requirement of TGFbeta signaling for SMO-mediated carcinogenesis. J Biol Chem 285:36570–36576
Faravelli I, Bucchia M, Rinchetti P, Nizzardo M, Simone C, Frattini E, Corti S (2014) Motor neuron derivation from human embryonic and induced pluripotent stem cells: experimental approaches and clinical perspectives. Stem Cell Res Ther 5:1–13. https://doi.org/10.1186/scrt476
Fazel Darbandi S, Robinson Schwartz SE, Qi Q, Catta-Preta R, Pai EL, Mandell JD et al (2018) Neonatal Tbr1 dosage controls cortical layer 6 connectivity. Neuron 100:831–845.e7
Ferreira MA et al (2008) Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 40:1056–1058
Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME (2008) Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60:1022–1038. https://doi.org/10.1016/j.neuron.2008.11.029
Forsman CL, Ng BC, Heinze RK, Kuo C, Sergi C, Gopalakrishnan R et al (2013) BMP-binding protein twisted gastrulation is required in mammary gland epithelium for normal ductal elongation and myoepithelial compartmentalization. Dev Biol 373:95–106
Fowler KB, Dahle AJ, Boppana SB, Pass RF (1999) Newborn hearing screening: will children with hearing loss caused by congenital cytomegalovirus infection be missed? J Pediatr 135:60–64. https://doi.org/10.1016/S0022-3476(99)70328-8
Frazier TW, Embacher R, Tilot AK, Koenig K, Mester J, Eng C (2015) Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry 20(9):1132–1138. https://doi.org/10.1038/mp.2014.125
Fregeau B, Kim BJ, Hernández-García A, Jordan VK, Cho MT, Schnur RE et al (2016) De novo mutations of RERE cause a genetic syndrome with features that overlap those associated with proximal 1p36 deletions. Am J Hum Genet 98:963–970
Fujimura L, Watanabe-Takano H, Sato Y, Tokuhisa T, Hatano M (2009) Prickle promotes neurite outgrowth via the Dishevelled dependent pathway in C1300 cells. Neurosci Lett 467:6–10
Geschwind DH (2008) Autism: many genes, common pathways? Cell 135:391
Ghanizadeh A (2012) Malondialdehyde, Bcl-2, superoxide dismutase and glutathione peroxidase may mediate the association of sonic hedgehog protein and oxidative stress in autism. Neurochem Res 37:899–901
Gharani N, Benayed R, Mancuso V, Brzustowicz LM, Millonig JH (2004) Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol Psychiatry 9:474–484. https://doi.org/10.1038/sj.mp.4001498
Ghyselinck NB, Duester G (2019) Retinoic acid signaling pathways. Development 146:dev167502. https://doi.org/10.1242/dev.167502
Glinka Y, Stoilova S, Mohammed N, Prud’homme GJ (2011) Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis 32:613–621
Golden CE, Buxbaum JD, De Rubeis S (2017) Disrupted circuits in mouse models of autism spectrum disorder and intellectual disability. Curr Opin Neurobiol 48:106–112
Grainger S, Willert K (2018) Mechanisms of Wnt signaling and control. Wiley Interdiscip Rev Syst Biol Med e1422
Grant SFA, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J et al (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 38:320–323
Grigoryan T, Wend P, Klaus A, Birchmeier W (2008) Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev 22:2308–2341
Grosse SD, Ross DS, Dollard SC (2008) Congenital cytomegalovirus (CMV) infection as a cause of permanent bilateral hearing loss: a quantitative assessment. J Clin Virol 41:57–62. https://doi.org/10.1016/j.jcv.2007.09.004
Halepoto DM, Bashir S, Zeina R, Al-Ayadhi LY (2015) Correlation between hedgehog (Hh) protein family and brain-derived neurotrophic factor (BDNF) in autism spectrum disorder (ASD). J Coll Physicians Surg Pak 25(12):882–885
Hamilton SP, Woo JM, Carlson EJ, Ghanem N, Ekker M, Rubenstein JLR (2005) Analysis of four DLX homeobox genes in autistic probands. BMC Genet 6:52
Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H et al (1999) The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol 9:207–210
Hayward P, Kalmar T, Arias AM (2008) Wnt/Notch signaling and information processing during development. Development 135:411–424. https://doi.org/10.1242/dev.000505
Hedstrom KL, Ogawa Y, Rasband MN (2008) AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol 183:635–640
Hida Y, Fukaya M, Hagiwara A, Deguchi-Tawarada M, Yoshioka T, Kitajima I, Inoue E, Watanabe M, Ohtsuka T (2011) Prickle2 is localized in the postsynaptic density and interacts with PSD-95 and NMDA receptors in the brain. J Biochem 149:693–700
Hiester BG, Galati DF, Salinas PC, Jones KR (2013) Neurotrophin and Wnt signaling cooperatively regulate dendritic spine formation. Mol Cell Neurosci 56:115–127
Hillman RT, Feng BY, Ni J, Woo WM, Milenkovic L, Hayden Gephart MG et al (2011) Neuropilins are positive regulators of hedgehog signal transduction. Genes Dev 25:2333–2346
Hochman E, Castiel A, Jacob-Hirsch J, Amariglio N, Izraeli S (2006) Molecular pathways regulating pro-migratory effects of hedgehog signaling. J Biol Chem 281:33860–33870
Hormozdiari F, Penn O, Borenstein E, Eichler EE (2015) The discovery of integrated gene networks for autism and related disorders. Genome Res 25:142–154
Huang JY, Lu HC (2017) mGluR5 tunes NGF/TrkA signaling to orient spiny stellate neuron dendrites toward thalamocortical axons during whisker-barrel map formation. Cereb Cortex 28(6):1991–2006
Inestrosa NC, Varela-Nallar L (2015) Wnt signaling in neuronal differentiation and development. Cell Tissue Res 359:215–223
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D et al (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515:216–221
Iqbal Z, Vandeyer G, van der Voet M, Waryah AM, Zahoor MY, Besseling JA et al (2013) Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum Mol Genet 22:1960–1970
Itasaki N, Hoppler S (2010) Crosstalk between Wnt and bone morphogenic protein signaling: a turbulent relationship. Dev Dyn 239:16–33
Ivarsson S, Bjerre I, Vegfors P, Ahlfors K (1990) Autism as one of several disabilities in two children with congenital cytomegalovirus infection. Neuropediatrics 21:102–103. https://doi.org/10.1055/s-2008-1071471
Iwata T, Hevner RF (2009) Fibroblast growth factor signaling in development of the cerebral cortex. Develop Growth Differ 51:299–323
Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC et al (2003) Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34:27–29
Javelaud D, Pierrat MJ, Mauviel A (2012) Crosstalk beten TGF-β and hedgehog signaling in cancer. FEBS Lett 586:2016–2025
Jin YR, Han XH, Taketo MM, Yoon JK (2012) Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development 139:1821–1830
Juriloff DM, Harris MJ, McMahon AP, Carroll TJ, Lidral AC (2006) Wnt9b is the mutated gene involved in multifactorial nonsyndromic cleft lip with or without cleft palate in A/WySn mice, as confirmed by a genetic complementation test. Birth Defects Res A Clin Mol Teratol 76:574–579
Kalkman HO (2012) A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol Autism 3:10. https://doi.org/10.1186/2040-2392-3-10
Kashima R, Roy S, Ascano M, Martinez-Cerdeno V, Ariza-Torres J, Kim S et al (2016) Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci Signal 9:ra58
Katoh Y, Katoh M (2009) Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med 9:873–886. https://doi.org/10.2174/156652409789105570
Khatri N, Man H-Y (2019) The autism and Angelman syndrome protein Ube3A/E6AP: the gene, E3 ligase ubiquitination targets and neurobiological functions. Front Mol Neurosci 12:109. https://doi.org/10.3389/fnmol.2019.00109
Kim S, Kim T, Lee HR, Jang EH, Ryu HH, Kang M et al (2016) Impaired learning and memory in CD38 null mutant mice. Mol Brain 9:16
Kim HW, Hong R, Choi EY, Yu K, Kim N, Hyeon JY et al (2018) A probiotic mixture regulates T cell balance and reduces atopic dermatitis symptoms in mice. Front Microbiol 9:2414
Kirschen GW, Xiong Q (2017) Primary cilia as a novel horizon between neuron and environment. Neural Regen Res 12:1225–1230
Kivimäe S, Martin PM, Kapfhamer D, Ruan Y, Heberlein U, Rubenstein JLR et al (2011) Abnormal behavior in mice mutant for the Disc1 binding partner, Dixdc1. Transl Psychiatry 1:e43
Kizhatil K, Davis JQ, Davis L, Hoffman J, Hogan BLM, Bennett V (2007) Ankyrin-G is a molecular partner of E-cadherin in epithelial cells and early embryos. J Biol Chem 282:26552–26561
Klaus A, Birchmeier W (2008) Wnt signaling and its impact on development and cancer. Nat Rev Cancer 8:387–398. https://doi.org/10.1038/nrc2389
Knight D, Xie W, Boulianne GL (2011) Neurexins and neuroligins: recent insights from invertebrates. Mol Neurobiol 44:426–440. https://doi.org/10.1007/s12035-011-8213-1
Komiya Y, Habas R (2008) Wnt signal transduction pathways. Organogenesis 4:68–75. https://doi.org/10.4161/org.4.2.5851
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G et al (2012) Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488:471–475. https://doi.org/10.1038/nature11396
Koopmans T, Eilers R, Menzen M, Halayko A, Gosens R (2017) β-catenin directs nuclear factor-κB p65 output via CREB-binding protein/p300 in human airway smooth muscle. Front Immunol 8:1086. https://doi.org/10.3389/fimmu.2017.01086
Kordeli E, Lambert S, Bennett V (1995) AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270:2352–2359
Kosmicki JA et al (2017) Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet 49:504–510
Krumm N, O’Roak BJ, Shendure J, Eichler EE (2014) A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 37(2):95–105. https://doi.org/10.1016/j.tins.2013.11.005
Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX et al (2015) Excess of rare, inherited truncating mutations in autism. Nat Genet 47(6):582–588. https://doi.org/10.1038/ng.3303
Kucinska M et al (2019) Differential regulation of fibroblast growth factor receptor 1 trafficking and function by extracellular galectins. Cell Commun Signal 17:65
Kuechler A, Willemsen MH, Albrecht B, Bacino CA, Bartholomew DW, van Bokhoven H et al (2015) De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet 134:97–109
Kwan V, Unda BK, Singh KK (2016a) Wnt signaling networks in autism spectrum disorder and intellectual disability. J Neurodev Disord 8:45. https://doi.org/10.1186/s11689-016-9176-3
Kwan V, Meka DP, White SH, Hung CL, Holzapfel NT, Walker S et al (2016b) DIXDC1 phosphorylation and control of dendritic morphology are impaired by rare genetic variants. Cell Rep 17:1892–1904
Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50(3):377–388. https://doi.org/10.1016/j.neuron.2006.03.023
Lai X, Wu X, Hou N, Liu S, Li Q, Yang T, Miao J, Dong Z, Chen J, Li T (2018) Vitamin A deficiency induces autistic-like behaviors in rats by regulating the RARβ-CD38-oxytocin axis in the hypothalamus. Mol Nutr Food Res 62:1700754. https://doi.org/10.1002/mnfr.201700754
Landrigan PJ (2010) What causes autism? Exploring the environmental contribution. Curr Opin Pediatr 22:219–225
Lee M, Yoon J, Song H, Lee B, Lam DT, Yoon J et al (2017) Tcf7l2 plays crucial roles in forebrain development through regulation of thalamic and habenular neuron identity and connectivity. Dev Biol 424:62–76
Levy D, Ronemus M, Yamrom B, Lee Y, Leotta A, Kendall J et al (2011) Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70:886–897
Li X, Deng W, Lobo-Ruppert SM, Ruppert JM (2007) Gli1 acts through Snail and E-cadherin to promote nuclear signaling by beta-catenin. Oncogene 26:4489–4498. https://doi.org/10.1038/sj.onc.1210241
Li W, Yao A, Zhi H, Kaur K, Zhu YC, Jia M et al (2016) Angelman syndrome protein Ube3a regulates synaptic growth and endocytosis by inhibiting BMP signaling in Drosophila. PLoS Genet 12:e1006062
Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A et al (2005) Wnt signaling regulates adult hippocampal neurogenesis. Nature 437:1370–1375
Lin PI, Chien YL, Wu YY, Chen CH, Gau SS, Huang YS et al (2012) The WNT2 gene polymorphism associated with speech delay inherent to autism. Res Dev Disabil 33:1533–1540
Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A (1999) Requirement for Wnt3 in vertebrate axis formation. Nat Genet 22:361–365
Liu X, Novosedlik N, Wang A, Hudson ML, Cohen IL, Chudley AE et al (2009) The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. Eur J Hum Genet 17:228–235
Lory JW, Rosen V (2018) Bone morphogenetic protein-based therapeutic approaches. Cold Spring Harb Perspect Biol 10:a022327
Loth E, Murphy DG, Spooren W (2016) Defining precision medicine approaches to autism spectrum disorders: concepts and challenges. Front Psychiatry 7:188
MacDonald BT, He X (2012) Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb Perspect Biol 4:a007880
MacDonald BT, Tamai K, He X (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17:9–26
Markowitz PI (1983) Autism in a child with congenital cytomegalovirus infection. J Autism Dev Disord 13:249–253. https://doi.org/10.1007/BF01531564
Martin PM, Yang X, Robin N, Lam E, Rabinowitz JS, Erdman CA et al (2013) A rare WNT1 missense variant overrepresented in ASD leads to increased WNT signal pathway activation. Transl Psychiatry 3:e301
Martin PM, Stanley RE, Ross AP, Freitas AE, Moyer CE, Brumback AC, Iafrati J, Stapornwongkul KS, Dominguez S, Kivimäe S, Mulligan KA, Pirooznia M, McCombie WR, Potash JB, Zandi PP, Purcell SM, Sanders SJ, Zuo Y, Sohal VS, Cheyette B (2018) DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/β-catenin signaling. Mol Psychiatry 23(2):467–475. https://doi.org/10.1038/mp.2016.184
Marui T, Funatogawa I, Koishi S, Yamamoto K, Matsumoto H, Hashimoto O et al (2010) Association between autism and variants in the wingless-type MMTV integration site family member 2 (WNT2) gene. Int J Neuropsychopharmacol 13:443–449
Matoba N, Liang D, Sun H, Aygün N, McAfee JC, Davis JE et al (2020) Common genetic risk variants identified in the SPARK cohort support DDHD2 as a candidate risk gene for autism. Transl Psychiatry 10(1):265
McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, Herman GE (2010) Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res 3(3):137–141. https://doi.org/10.1002/aur.132
McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, Mistry M, Pavlidis P, Solomon R, Ghiban E et al (2014) De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry 19(6):652–658. https://doi.org/10.1038/mp.2014.29
McMahon AP, Bradley A (1990) The Wnt-1 (int-1) proto-oncogene is required for the development of a large region of the mouse brain. Cell 62:1073–1085
McMahon AP, Joyner AL, Bradley A, McMahon JA (1992) The midbrain-hindbrain phenotype of Wnt-1-/Wnt-1-mice results from stepwise deletion of engrailed-expressing cells by 9.5 days postcoitum. Cell 69:581–595
Medina MA, Andrade VM, Caracci MO, Avila ME, Verdugo DA, Vargas MF et al (2018) Wnt/β-catenin signaling stimulates the expression and synaptic clustering of the autism-associated Neuroligin 3 gene. Transl Psychiatry 8:45
Meng X, Poon R, Zhang X, Cheah A, Ding Q, Hui CC et al (2001) Suppressor of fused negatively regulates beta-catenin signaling. J Biol Chem 276:40113–40119
Merner N, Forgeot d’Arc B, Bell SC, Maussion G, Peng H, Gauthier J, Crapper L, Hamdan FF, Michaud JL, Mottron L et al (2016) A de novo frameshift mutation in chromodomain helicase DNA-binding domain 8 (CHD8): a case report and literature review. Am J Med Genet A 170(5):1225–1235. https://doi.org/10.1002/ajmg.a.37566
Mohn JL, Alexander J, Pirone A, Palka CD, Lee SY, Mebane L et al (2014) Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol Psychiatry 19:1133–1142
Moore SJ, Turnpenny P, Quinn A, Glover S, Lloyd DJ, Montgomery T et al (2000) A clinical study of 57 children with fetal anticonvulsant syndromes. J Med Genet 37:489–497
Moreno-Ramos OA, Olivares AM, Haider NB, de Autismo LC, Lattig MC (2015) Whole-exome sequencing in a South American cohort links ALDH1A3, FOXN1 and retinoic acid regulation pathways to autism spectrum disorders. PLoS One 10:e0135927
Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM et al (2008) Identifying autism loci and genes by tracing recent shared ancestry. Science 321:218–223. https://doi.org/10.1126/science.1157657
Mulligan KA, Cheyette BN (2017) Neurodevelopmental perspectives on Wnt signaling in psychiatry. Mol Neuropsychiatry 2(4):219–246
Nagaoka T, Tabuchi K, Kishi M (2015) PDZ interaction of Vangl2 links PSD-95 and Prickle2 but plays only a limited role in the synaptic localisation of Vangl2. Sci Rep 5:12916
Nakanishi M, Nomura J, Ji X, Tamada K, Arai T, Takahashi E et al (2017) Functional significance of rare neuroligin 1 variants found in autism. PLoS Genet 13(8):e1006940
Nakashima N, Yamagata T, Mori M, Kuwajima M, Suwa K, Momoi MY (2010) Expression analysis and mutation detection of DLX5 and DLX6 in autism. Brain Dev 32(2):98–104
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V et al (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485(7397):242–245. https://doi.org/10.1038/nature11011
Nguyen A, Rauch TA, Pfeifer GP, Hu VW (2010) Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J 24:3036–3051. https://doi.org/10.1096/f.10-154484
Niculae AS, Pavăl D (2016) From molecules to behavior: an integrative theory of autism spectrum disorder. Med Hypotheses 97:74–84
Niederreither K, Dollé P (2008) Retinoic acid in development: towards an integrated view. Nat Rev Genet 9:541–553
Nielsen-Saines K, Brasil P, Kerin T, Vasconcelos Z, Gabaglia CR, Damasceno L, Pone M, de Carvalho LMA, Pone SM, Zin AA (2019) Delayed childhood neurodevelopment and neurosensory alterations in the second year of life in a prospective cohort of ZIKV-exposed children. Nat Med 25:1213–1217. https://doi.org/10.1038/s41591-019-0496-1
Nishiyama M, Oshikawa K, Tsukada Y, Nakagawa T, Iemura S, Natsume T et al (2009) CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol 11:172–182
Nishiyama M, Skoultchi AI, Nakayama KI (2012) Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-β-catenin signaling pathway. Mol Cell Biol 32(2):501–512. https://doi.org/10.1128/MCB.06409-11
Notani D, Gottimukkala KP, Jayani RS, Limaye AS, Damle MV, Mehta S et al (2010) Global regulator SATB1 recruits beta-catenin and regulates T(H)2 differentiation in Wnt-dependent manner. PLoS Biol 8:e1000296
Nurmi EL, Bradford Y, Chen Y-h, Hall J, Arnone B, Gardiner MB et al (2001) Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics 77(1):105–113
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP et al (2012a) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485:246–250
O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K et al (2012b) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338(6114):1619–1622. https://doi.org/10.1126/science.1227764
Ohkawara T, Katsuyama T, Ida-Eto M, Narita N, Narita M (2015) Maternal viral infection during pregnancy impairs the development of fetal serotonergic neurons. Brain Dev 37:88–93. https://doi.org/10.1016/j.braindev.2014.03.007
Okerlund ND, Cheyette BNR (2011) Synaptic Wnt signaling—a contributor to major psychiatric disorders. J Neurodev Disord 3:162–174
Okumura A, Yamamoto T, Miyajima M, Shimojima K, Kondo S, Abe S et al (2014) 3p interstitial deletion including PRICKLE2 in identical twins with autistic features. Pediatr Neurol 51:730–733
Onishi K, Hollis E, Zou Y (2014) Axon guidance and injury-lessons from Wnts and Wnt signaling. Curr Opin Neurobiol 27:232–240
Ornitz DM, Itoh N (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4:215–266
Oron O, Elliott E (2017) Delineating the common biological pathways perturbed by ASD’s genetic etiology: lessons from network-based studies. Int J Mol Sci 18:828
Paemka L, Mahajan VB, Skeie JM, Sors LP, Ehaideb SN, Gonzalez-Alegre P, Sasaoka T, Tao H, Miyagi A, Ueno N, Takao K, Miyakawa T, Wu S, Darbro BW, Ferguson PJ, Pieper AA, Britt JK, Wemmie JA, Rudd DS, Wassink T, El-Shanti H, Mefford HC, Carvill GL, Manak JR, Bassuk AG (2013) PRICKLE1 interaction with SYNAPSIN I reveals a role in autism spectrum disorders. PLoS One 8:e80737
Page DT, Kuti OJ, Prestia C, Sur M (2009) Haploinsufficiency for Pten and Serotonin transporter cooperatively influences brain size and social behavior. Proc Natl Acad Sci U S A 106(6):1989–1994. https://doi.org/10.1073/pnas.0804428106
Patel SS, Tomar S, Sharma D, Mahindroo N, Udayabanu M (2017) Targeting sonic hedgehog signaling in neurological disorders. Neurosci Biobehav Rev 74:76–97
Pavăl D, Rad F, Rusu R, Niculae A, Colosi HA, Dobrescu I et al (2017) Low retinal dehydrogenase 1 (RALDH1) level in prepubertal boys with autism spectrum disorder: a possible link to dopamine dysfunction? Clin Psychopharmacol Neurosci 15(3):229–236
Pelullo M, Zema S, Nardozza F, Checquolo S, Screpanti I, Bellavia D (2019) Wnt, notch, and TGF-β pathways impinge on hedgehog signaling complexity: an open window on cancer. Front Genet 10:711. https://doi.org/10.3389/fgene.2019.00711
Pierrat MJ, Marsaud V, Mauviel A, Javelaud D (2012) Expression of microphthalmia-associated transcription factor (MITF), which is critical for melanoma progression, is inhibited by both transcription factor GLI2 and transforming growth factor-β. J Biol Chem 287:17996–18004
Pilarowski GO, Vernon HJ, Applegate CD, Boukas L, Cho MT, Gurnett CA et al (2018) Missense variants in the chromatin remodeler CHD1 are associated with neurodevelopmental disability. J Med Genet 55(8):561
Pirone A, Alexander J, Lau LA, Hampton D, Zayachkivsky A, Yee A et al (2017) APC conditional knock-out mouse is a model of infantile spasms with elevated neuronal β-catenin levels, neonatal spasms, and chronic seizures. Neurobiol Dis 98:149–157. https://doi.org/10.1016/j.nbd.2016.11.002
Platt RJ, Zhou Y, Slaymaker IM, Shetty AS, Weisbach NR, Kim JA, Sharma J, Desai M, Sood S, Kempton HR et al (2017) Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep 19:335–350. https://doi.org/10.1016/j.celrep.2017.03.052
Porebska N et al (2018) Targeting cellular trafficking of fibroblast growth factor receptors as a strategy for selective cancer treatment. J Clin Med 8:7
Preitner N, Quan J, Nowakowski DW, Hancock ML, Shi J, Tcherkezian J et al (2014) APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell 158:368–382. https://doi.org/10.1016/j.cell.2014.05.042
Psychiatric GWAS Consortium Bipolar Disorder Working Group (2011) Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 43:977–983
Qin L, Dai X (2015) Effect of sulindac on improving autistic behaviours in rats. Nan Fang Yi Ke Da Xue Xue Bao 35:1162–1165
Rasalam AD, Hailey H, Williams JHG, Moore SJ, Turnpenny PD, Lloyd DJ et al (2005) Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol 47:551–555. https://doi.org/10.1017/s0012162205001076
Reichenberg A, Gross R, Weiser M, Bresnahan M, Silverman J, Harlap S et al (2006) Advancing paternal age and autism. Arch Gen Psychiatry 63(9):1026–1032
Rhinn M, Dollé P (2012) Retinoic acid signaling during development. Development 139:843–858
Richetto J, Massart R, Weber-Stadlbauer U, Szyf M, Riva MA, Meyer U (2017) Genome-wide DNA methylation changes in a mouse model of infection-mediated neurodevelopmental disorders. Biol Psychiatry 81:265–276
Riebold M, Mankuta D, Lerer E, Israel S, Zhong S, Nemanov L et al (2011) All-trans retinoic acid upregulates reduced CD38 transcription in lymphoblastoid cell lines from autism spectrum disorder. Mol Med 17:799–806
Rosso SB, Inestrosa NC (2013) WNT signaling in neuronal maturation and synaptogenesis. Front Cell Neurosci 7:103
Rosti RO, Sadek AA, Vaux KK, Gleeson JG (2014) The genetic landscape of autism spectrum disorders. Dev Med Child Neurol 56:12–18. https://doi.org/10.1111/dmcn.12278
Rout UK, Clausen P (2009) Common increase of GATA-3 level in PC-12 cells by three teratogens causing autism spectrum disorders. Neurosci Res 64:162–169
Rubenstein JLR, Merzenich MM (2003) Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2:255–267
Rychahou PG, Kang J, Gulhati P, Doan HQ, Chen LA, Xiao SY et al (2008) Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc Natl Acad Sci U S A 105:20315–20320
Ryu J, Bar-Shalita T, Granovsky Y, Weissman-Fogel I, Torres E (2021) Personalized biometrics of physical pain agree with psychophysics by participants with sensory over responsivity. J Pers Med 11:93. https://doi.org/10.3390/jpm11020093
Sajan SA, Rubenstein JLR, Warchol ME, Lovett M (2011) Identification of direct downstream targets of Dlx5 during early inner ear development. Hum Mol Genet 20:1262–1273
Sakamoto A, Moriuchi H, Matsuzaki J, Motoyama K, Moriuchi M (2015) Retrospective diagnosis of congenital cytomegalovirus infection in children with autism spectrum disorder but no other major neurologic deficit. Brain Dev 37:200–205. https://doi.org/10.1016/j.braindev.2014.03.016
Sanders SJ (2015) First glimpses of the neurobiology of autism spectrum disorder. Curr Opin Genet Dev 33:80–92. https://doi.org/10.1016/j.gde.2015.10.002
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485:237–241
Sarabipour S, Hristova K (2016) Mechanism of FGF receptor dimerization and activation. Nat Commun 7:10262
Sarachana T, Hu VW (2013) Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism 4(1):14
Sayad A, Noroozi R, Omrani MD, Taheri M, Ghafouri-Fard S (2017) Retinoic acid-related orphan receptor alpha (RORA) variants are associated with autism spectrum disorder. Metab Brain Dis 32:1595–1601
Schulze TG et al (2009) Two variants in Ankyrin 3 (ANK3) are independent genetic risk factors for bipolar disorder. Mol Psychiatry 14:487–491
Sen B, Singh AS, Sinha S, Chatterjee A, Ahmed S, Ghosh S et al (2010) Family-based studies indicate association of Engrailed 2 gene with autism in an Indian population. Genes Brain Behav 9:248–255
Seppala M, Fraser GJ, Birjandi AA, Xavier GM, Cobourne MT (2017) Sonic hedgehog signaling and development of the dentition. J Dev Biol 5:6
Seten TL, Posey DJ, McDougle CJ (2004) Brief report: autistic disorder in three children with cytomegalovirus infection. J Autism Dev Disord 34:583–586. https://doi.org/10.1007/s10803-004-2552-y
Shaltout E, Al-Dewik N, Samara M, Morsi H, Khattab A (2020) Psychological comorbidities in autism spectrum disorder. In: Essa M, Qoronfleh M (eds) Personalized food intervention and therapy for autism spectrum disorder management. Advances in neurobiology, vol 24. Springer, Cham. https://doi.org/10.1007/978-3-030-30402-7_6
Shi L, Zhang X, Golhar R, Otieno FG, He M, Hou C et al (2013) Whole-genome sequencing in an autism multiplex family. Mol Autism 4:8
Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA et al (2009) Retinoic acid from the meninges regulates cortical neuron generation. Cell 139:597–609
Simon HH, Scholz C, O’Leary DDM (2005) Engrailed genes control the developmental fate of serotonergic and noradrenergic neurons in mid-and hindbrain in a gene dose-dependent manner. Mol Cell Neurosci 28:96–105
Simonoff E, Pickles A, Charman T, Chandler S, Loucas T, Baird G (2008) Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J Am Acad Child Adolesc Psychiatry 47(8):921–929. https://doi.org/10.1097/CHI.0b013e318179964f
Song L, Li Z-Y, Liu W-P, Zhao M-R (2014) Crosstalk beten Wnt/β-catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol Ther 16:1–7. https://doi.org/10.4161/15384047.2014.972215
Song L, Li ZY, Liu WP, Zhao MR (2015) Crosstalk beten Wnt/β-catenin and hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol Ther 16:1–7
Sousa KM, Villaescusa JC, Cajanek L, Ondr JK, Castelo-Branco G, Hofstra W, Bryja V, Palmberg C, Bergman T, Wainwright B et al (2010) Wnt2 regulates progenitor proliferation in the developing ventral midbrain. J Biol Chem 285:7246–7253. https://doi.org/10.1074/jbc.M109.079822
Sowers LP, Loo L, Wu Y, Campbell E, Ulrich JD, Wu S et al (2013) Disruption of the non-canonical Wnt gene PRICKLE2 leads to autism-like behaviors with evidence for hippocampal synaptic dysfunction. Mol Psychiatry 18:1077–1089
Spinelli L, Black FM, Berg JN, Eickholt BJ, Leslie NR (2015) Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet 52(2):128–134. https://doi.org/10.1136/jmedgenet-2014-102803
Stubbs EG (1978) Autistic symptoms in a child with congenital cytomegalovirus infection. J Autism Child Schizophr 8:37–43. https://doi.org/10.1007/BF01550276
Stubbs EG, Ash E, Williams CP (1984) Autism and congenital cytomegalovirus. J Autism Dev Disord 14:183–189. https://doi.org/10.1007/BF02409660
Stulberg MJ, Lin A, Zhao H, Holley SA (2012) Crosstalk beten Fgf and Wnt signaling in the zebrafish tailbud. Dev Biol 369:298–307
Styles M, Alsharshani D, Samara M, Alsharshani M, Khattab A, Qoronfleh MW et al (2020) Risk factors, diagnosis, prognosis and treatment of autism. Front Biosci (Landmark Ed) 25:1682–1717
Subtil-Rodríguez A, Vázquez-Chávez E, Ceballos-Chávez M, Rodríguez-Paredes M, Martín-Subero JI, Esteller M et al (2014) The chromatin remodeller CHD8 is required for E2F-dependent transcription activation of S-phase genes. Nucleic Acids Res 42:2185–2196
Sudhof TC (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455:903
Südhof TC (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455:903–911. https://doi.org/10.1038/nature07456
Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J et al (2014) CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A 111(42):E4468–E4477. https://doi.org/10.1073/pnas.1405266111
Takeuchi K, Gertner MJ, Zhou J, Parada LF, Bennett MV, Zukin RS (2013) Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc Natl Acad Sci U S A 110(12):4738–4743. https://doi.org/10.1073/pnas.1222803110
Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM et al (2012) Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149(3):525–537. https://doi.org/10.1016/j.cell.2012.03.028
Tamiji J, Crawford DA (2010) The neurobiology of lipid metabolism in autism spectrum disorders. Neurosignals 18:98–112
Tang SJ (2014) Synaptic activity-regulated Wnt signaling in synaptic plasticity, glial function and chronic pain. CNS Neurol Disord Drug Targets 13:737–744
Tao H, Manak JR, Sors L, Mei X, Kiyonari H, Abe T, Dahdaleh NS, Yang T, Wu S, Chen S, Fox MH, Gurnett C, Montine T, Bird T, Shaffer LG, Rosenfeld JA, McConnell J, Madan-Khetarpal S, Berry-Kravis E, Griesbach H, Saneto RP, Scott MP, Antic D, Reed J, Boland R, Ehaideb SN, El-Shanti H, Mahajan VB, Ferguson PJ, Axelrod JD et al (2011) Mutations in prickle orthologs cause seizures in flies, mice, and humans. Am J Hum Genet 88:138–149
Tempé D, Casas M, Karaz S, Blanchet-Tournier M-F, Concordet JP (2006) Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol 26:4316–4326
Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H (2010) Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465:783–787
Tesli M et al (2011) Association analysis of ANK3 gene variants in Nordic bipolar disorder and schizophrenia case-control samples. Am J Med Genet B Neuropsychiatr Genet 156B:969–974
Thomas KR, Capecchi MR (1990) Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature 346:847–850
Thompson BA, Tremblay V, Lin G, Bochar DA (2008) CHD8 is an ATP-dependent chromatin remodelling factor that regulates beta-catenin target genes. Mol Cell Biol 28:3894–3904
Tilot AK, Frazier TW 2nd, Eng C (2015) Balancing proliferation and connectivity in PTEN-associated autism spectrum disorder. Neurotherapeutics 12(3):609–619. https://doi.org/10.1007/s13311-015-0356-8
Tora D, Gomez AM, Michaud JF, Yam PT, Charron F, Scheiffele P (2017) Cellular functions of the autism risk factor PTCHD1 in mice. J Neurosci 37:11993–12005
Townsend CL, Forsgren M, Ahlfors K, Ivarsson S-A, Tookey PA, Peckham CS (2013) Long-term outcomes of congenital cytomegalovirus infection in Sden and the United Kingdom. Clin Infect Dis 56:1232–1239. https://doi.org/10.1093/cid/cit018
Treffert DA (1970) Epidemiology of infantile autism. Arch Gen Psychiatry 22(5):431–438
Triantis V et al (2010) Glycosylation of fibroblast growth factor receptor 4 is a key regulator of fibroblast growth factor 19-mediated down-regulation of cytochrome P450 7A1. Hepatology 52:656–666
Tsarovina K, Reiff T, Stubbusch J, Kurek D, Grosveld FG, Parlato R et al (2010) The Gata3 transcription factor is required for the survival of embryonic and adult sympathetic neurons. J Neurosci 30:10833–10843
Tukachinsky H, Lopez LV, Salic A (2010) A mechanism for vertebrate hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol 191:415–428
Turner CA, Eren-Koçak E, Inui EG, Watson SJ, Akil H (2016) Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Semin Cell Dev Biol 53:136–143
Uddin M, Wang Y, Woodbury-Smith M (2019) Artificial intelligence for precision medicine in neurodevelopmental disorders. NPJ Digit Med 2(1):112
Ulloa F, Itasaki N, Briscoe J (2007) Inhibitory Gli3 activity negatively regulates Wnt/beta-catenin signaling. Curr Biol 17:545–550
Ung DC, Iacono G, Méziane H, Blanchard E, Papon MA, Selten M et al (2018) Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Mol Psychiatry 23(5):1356–1367
Varnat F, Siegl-Cachedenier I, Malerba M, Gervaz P, Ruiz i Altaba A (2010) Loss of WNT-TCF addiction and enhancement of HH-GLI1 signaling define the metastatic transition of human colon carcinomas. EMBO Mol Med 2:440–457
Vogt D, Cho KK, Lee AT, Sohal VS, Rubenstein JL (2015) The parvalbumin/somatostatin ratio is increased in Pten mutant mice and by human PTEN ASD alleles. Cell Rep 11(6):944–956. https://doi.org/10.1016/j.celrep.2015.04.019
Wada H, Okamoto H (2009) Roles of noncanonical Wnt/PCP pathway genes in neuronal migration and neurulation in zebrafish. Zebrafish 6:3–8
Wang B, Li Y (2006) Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc Natl Acad Sci U S A 103:33–38
Wang L, Jia M, Yue W, Tang F, Qu M, Ruan Y et al (2008) Association of the ENGRAILED 2 (EN2) gene with autism in Chinese Han population. Am J Med Genet B Neuropsychiatr Genet 147B:434–438
Wang Y, Song L, Zhou CJ (2011) The canonical Wnt/β-catenin signaling pathway regulates Fgf signaling for early facial development. Dev Biol 349:250–260
Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M et al (2014) Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Dis 1:87–105
Wang P, Lin M, Pedrosa E, Hrabovsky A, Zhang Z, Guo W, Lachman HM, Zheng D (2015) CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol Autism 6:55. https://doi.org/10.1186/s13229-015-0048-6
Wang Y, Billon C, Walker JK, Burris TP (2016) Therapeutic effect of a synthetic RORα/γ agonist in an animal model of autism. ACS Chem Neurosci 7:143–148
Wassink TH, Piven J, Vieland VJ, Huang J, Swiderski RE, Pietila J et al (2001) Evidence supporting WNT2 as an autism susceptibility gene. Am J Med Genet 105:406–413
Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron 22:103–114
Wheeler JA, Clinkenbeard EL (2019) Regulation of fibroblast growth factor 23 by iron, EPO, and HIF. Curr Mol Biol Rep 5:8–17
Wilkinson B, Grepo N, Thompson BL, Kim J, Wang K, Evgrafov OV, Lu W, Knowles JA, Campbell DB (2015) The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl Psychiatry 5(5):e568. https://doi.org/10.1038/tp.2015.62
Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA et al (2013) Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155:997–1007
Wodarz A, Nusse R (1998) Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol 14:59–88
Wong CT, Wais J, Crawford DA (2015) Prenatal exposure to common environmental factors affects brain lipids and increases the risk of developing autism spectrum disorders. Eur J Neurosci 42:2742–2760
Wong CT, Ussyshkin N, Ahmad E, Rai-Bhogal R, Li H, Crawford DA (2016) Prostaglandin E2 promotes neural proliferation and differentiation and regulates Wnt target gene expression. J Neurosci Res 94:759–775. https://doi.org/10.1002/jnr.23759
Xie Y, Su N, Yang J et al (2020) FGF/FGFR signaling in health and disease. Signal Transduct Target Ther 5:181. https://doi.org/10.1038/s41392-020-00222-7
Xu X, Li C, Gao X, Xia K, Guo H, Li Y, Hao Z, Zhang L, Gao D, Xu C et al (2017) Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res 28:48–68. https://doi.org/10.1038/cr.2017.132
Xu X, Li C, Gao X, Xia K, Guo H, Li Y et al (2018) Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res 28:48–68
Yang P, Lung FW, Jong YJ, Hsieh HY, Liang CL, Juo SH (2008) Association of the homeobox transcription factor gene ENGRAILED 2 with autistic disorder in Chinese children. Neuropsychobiology 57:3–8
Yang P, Shu B-C, Hallmayer JF, Lung FW (2010) Intronic single nucleotide polymorphisms of engrailed homeobox 2 modulate the disease vulnerability of autism in a Han Chinese population. Neuropsychobiology 62:104–115
Yi JJ, Paranjape SR, Walker MP, Choudhury R, Wolter JM, Fragola G et al (2017) The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/β-catenin pathway by inhibiting the proteasome. J Biol Chem 292:12503–12515
Yoo HJ, Cho IH, Park M, Cho E, Cho SC, Kim BN, Kim JW, Kim SA (2008) Association between PTGS2 polymorphism and autism spectrum disorders in Korean trios. Neurosci Res 62(1):66–69. https://doi.org/10.1016/j.neures.2008.05.008
Yuen RK, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B et al (2016) Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med 1:160271–1602710
Yuen RKC, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV et al (2017) Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20(4):602–611
Zhang Y, Sun Y, Wang F, Wang Z, Peng Y, Li R (2012) Downregulating the canonical Wnt/β-catenin signaling pathway attenuates the susceptibility to autism-like phenotypes by decreasing oxidative stress. Neurochem Res 37:1409–1419
Zhang X, Rui M, Gan G, Huang C, Yi J, Lv H et al (2017) Neuroligin 4 regulates synaptic growth via the bone morphogenetic protein (BMP) signaling pathway at the Drosophila neuromuscular junction. J Biol Chem 292:17991–18005
Zhao T, Gan Q, Stokes A, Lassiter RN, Wang Y, Chan J, Han JX, Pleasure DE, Epstein JA, Zhou CJ (2014) β-catenin regulates Pax3 and Cdx2 for caudal neural tube closure and elongation. Development (Cambridge, England) 141(1):148–157. https://doi.org/10.1242/dev.101550
Zhou J, Parada LF (2012) PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol 22(5):873–879. https://doi.org/10.1016/j.conb.2012.05.004
Zhou CJ, Pinson KI, Pleasure SJ (2004) Severe defects in dorsal thalamic development in low-density lipoprotein receptor-related protein-6 mutants. J Neurosci 24:7632–7639
Zhou XL, Giacobini M, Anderlid BM, Anckarsäter H, Omrani D, Gillberg C et al (2007) Association of adenomatous polyposis coli (APC) gene polymorphisms with autism spectrum disorder (ASD). Am J Med Genet B Neuropsychiatr Genet 144B(3):351–354
Zhu X, Lee K, Asa SL, Ezzat S (2007) Epigenetic silencing through DNA and histone methylation of fibroblast growth factor receptor 2 in neoplastic pituitary cells. Am J Pathol 170:1618–1628
Zi Z, Chapnick DA, Liu X (2012) Dynamics of TGF-β/Smad signaling. FEBS Lett 586:1921–1928
Zieger E, Schubert M (2017) New insights into the roles of retinoic acid signaling in nervous system development and the establishment of neurotransmitter systems. Int Rev Cell Mol Biol 330:1–84
Zoghbi HY (2003) Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302:826
Zumbrunn J, Kinoshita K, Hyman AA, Näthke IS (2001) Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 β phosphorylation. Curr Biol 11:44–49. https://doi.org/10.1016/s0960-9822(01)00002-1
Acknowledgement
The authors want to thank their respective institutions for their continued support.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Ethics declarations
Ethics Approval and Consent to Participate: Not applicable.
Consent for Publication: Not applicable.
Availability of Data and Materials: Not applicable.
Competing/Conflict of Interest: The authors declare no conflicts of/or competing interests.
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Younes, S.N. et al. (2022). Principal Molecular Pathways Affected in Autism Spectrum Disorder. In: Qoronfleh, M.W., Essa, M.M., Saravana Babu, C. (eds) Proteins Associated with Neurodevelopmental Disorders. Nutritional Neurosciences. Springer, Singapore. https://doi.org/10.1007/978-981-15-9781-7_1
Download citation
DOI: https://doi.org/10.1007/978-981-15-9781-7_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-9780-0
Online ISBN: 978-981-15-9781-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)