
Abstract
Autophagy is an evolutionary conserved self-degradation process that occurs ubiquitously in eukaryotes. It plays an important role in maintenance of cellular homeostasis by balancing the energy resources or through removal of misfolded proteins and damaged organelles. During autophagy, the recycling of the long-lived proteins or organelles is executed through their engulfment into double-membrane autophagosome followed by their lysosomal degradation via formation of autophagolysosome. Interestingly, autophagy is under tight regulation by a group of genes called autophagy-related genes (ATG) in association with various signalling pathways. Literature review suggests that autophagy is implicated in numerous developmental and other physiological processes such as cell differentiation, cell survival, cell death, nutrient starvation response and its dysregulation, often, leads to many pathological conditions including cancer. Generally, under normal physiological conditions, basal autophagy occurs in all cells but it is induced only in response to specific intra- or extra-cellular stimuli. In cancer, depending on the context, autophagy can be paradoxical in nature (i.e. tumour-suppressive or tumour-promoting) and has also been documented to have the remarkable role in development of chemoresistance, thus, justifying the effectiveness of cancer therapeutic intervention through stimulation or inhibition of autophagy. Henceforth, in this chapter, we have summarised the autophagy in a nutshell, with focus on its mechanism, monitoring methods, regulation and context-dependent role in cancer and explored how the manipulation of autophagy could be beneficial towards improved cancer cure, as evident from the numerous in vitro and in vivo studies as well as clinical trials.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
12.1 Introduction
Autophagy is a dynamic process responsible for degradation and turnover of cellular proteins and organelle. It is accomplished through sequestration of target cellular constituents or organelle into double-membrane vesicles called autophagosome, which in turn fuses with lysosome to form autophagolysosome, wherein they are degraded by lysosomal proteases for recycling. It is an evolutionary conserved ubiquitous process occurring in eukaryotes (Klionsky and Emr 2000; Levine and Klionsky 2004). In recent times, autophagy has gained immense attention in clinical research owing to their versatile role in diverse physiological and pathophysiological conditions, amongst which cancer is of particular importance. Autophagy occurs at basal level in most of the cells. However, it can also be induced in response to specific stimuli, wherein autophagy is context-dependent (Mizushima 2007; White 2012; Amaravadi et al. 2016). However, the differences between basal autophagy and stimuli-induced autophagy and their relevance are not yet well-understood.
The Greek term ‘autophagy’ meaning ‘self-eating’ was coined by Christian de Duve in 1963 based on the electron microscopic studies displaying single or double-membrane vesicles containing parts of sequestered cytoplasm with variable degree of disintegrated organelles, especially mitochondria and other intracellular structures. Remarkable progress in understanding autophagy has been reported in the last few decades by decoding its molecular mechanism and significance in various physiological processes (Klionsky 2007; Levy et al. 2017). The breakthrough discovery of the detailed mechanism of regulation and execution of autophagy at molecular level in yeast Saccharomyces cerevisiae by Yoshinori Ohsumi has been awarded the 2016 Nobel prize in Physiology and Medicine.
12.2 Types of Autophagy
Autophagy can be categorised into three different types: macro-autophagy, micro-autophagy and chaperon-mediated autophagy. Macroautophagy involves engulfment of cytoplasmic proteins and organelle into double-membrane bound vesicles called autophagosome, which is trafficked to lysosome to form autolysosome for degradation by lysosomal proteases. In contrast, microautophagy is characterised by the internalization of the substrate through invagination of lysosomal or endosomal membrane followed by their lysosomal degradation (Li et al. 2012). However, in chaperon-mediated autophagy (CMA), the cargo protein contains KFERQ-like pentapeptide motif, which is recognised by cytosolic chaperone protein called heat shock cognate 70 (HSP-70) for their translocation to the lysosomal lumen through interaction with lysosomal-associated membrane protein 2A (LAMP 2A) receptor (Kaushik et al. 2011). Although both micro and macro-autophagy are capable of targeting large structures through selective and non-selective mechanism, CMA is constitutively selective in nature and thereby, restricted to turnover of specific protein with well-defined KFERQ motif. It is important to note that non-selective autophagy involves the direct engulfment of the cytoplasm and its components into the autophagosome (in macroautophagy) or through invagination of the lysosomal membrane (in microautophagy). While, in contrast, selective autophagy is mediated by specific targeting of the cargo, either cellular proteins or organelles, hallmarked with degradation signal (most commonly, ubiquitin in mammals) through interaction with autophagy cargo receptor, which serves as molecular bridge, for their degradation by autophagy (Kaur and Debnath 2015; Levy et al. 2017).
12.3 Mechanism of Autophagy
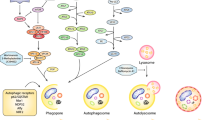
Autophagy is a complex, multi-step process under the intricate control of a set of 30 evolutionary conserved, autophagy-regulated genes (ATG), which were identified in yeast and mostly, have well-recognised mammalian orthologue. It divided into three distinct stages: autophagosome biogenesis, fusion with lysosome and lysosomal degradation of intravesicular constituents (Fig. 12.1). The autophagosome formation is initiated at the phagophore assembly point through the activation of ULK (UNC 51-like kinase) complex comprising of ULK1, ULK2 and ATG13, FIP200 (FAK family kinase interacting protein of 200 kDa) and ATG 101. This is followed by the nucleation stage when the ULK complex targets class III PI3 kinase complex—consisting of Beclin 1 (Atg6 in yeast), VPS34 (vacuolar protein sorting 34; also known as PIK3C3), ATG14, UVRAG (UV radiation resistance-associated gene protein; also known as p63) and AMBRA1 (activating molecule in BECN1-regulated autophagy protein 1)—promotes production of autophagosome-specific phosphatidyl-inositol-3-phosphate. Finally, the ATG5–ATG12–ATG16 complex along with ATG4B–ATG7 complex facilitates the expansion of the autophagosome membrane through lipidation of the microtubule-associated protein light chain 1 (LC3I), which is the mammalian homologue of yeast Atg8, and GABARAP (γ-aminobutyric type A (GABAA)-receptor associated protein) with phosphatidylethanolamine (PE) to form LC3II and GABARAP-II, which in turn co-localise followed by their recruitment to the membrane. Interestingly, LC3BII, the well-known autophagosome marker found on the autophagosomal membrane, has been reported to facilitate the hemifusion of membranes and cargo selection for degradation, possibly through regulation of variable protein–protein interaction (Mizushima 2007; Levy et al. 2017; Meijer and Codogno 2004; Mizushima et al. 2011; Onorati et al. 2018). However, the significance of LCB-related molecules in autophagy needs further investigation (Fig. 12.1).

The mechanism of autophagy. Autophagy is a multistep cellular process comprising of autophagosome initiation, elongation of the autophagosomal membrane, sequestration of the cargo and fusion of the autophagosome with the lysosome for degradation of the constituents. It can be inhibited at particular steps by specific inhibitors (such as 3MA—early phase autophagy inhibitor that inhibits autophagosome formation and chloroquine, hydroxychloroquine, bafilomycin A—late phase autophagy inhibitors that prevents the fusion of autophagosome with lysosome)
12.4 Methods of Monitoring Autophagy
In present-day autophagy research, the detection and quantification of autophagosome along with biochemical validation of the autophagic markers comprises the principal methods of monitoring autophagy. The electron microscopy is the most conventional and oldest method that enables the visualisation of the autophagosome at the ultrastructural level. It is of immense interest to note that in 1950s, the autophagy was first discovered by electron microscopic study of the lysosome (Klionsky 2007). However, advancement of autophagy research called for the formulation of easier and more accessible assays of autophagy detection. The advent of LC3B as the signature of autophagosome has simplified the revelation of autophagy by the light microscopic detection of LC3B or GFP–LC3B puncta. Finally, the conversion of LC3I to LC3 II by immunoblotting with LC3 antibody is the widely employed biochemical assay to confirm autophagy. Furthermore, immunoblot depicting the turnover of p62 is also used to expose autophagy (Mizushima 2004; Mizushima et al. 2010; Yoshii and Mizushima 2017).
Nonetheless, Levine et al. have highlighted the misconception of the direct correlation of the number of autophagosome with the autophagic activity (Mizushima et al. 2010). Owing to the dynamic nature, at any point of time, the number of autophagosome is the function of the balance between their formation rate and fusion rate with the lysosome. Henceforth, the autophagosome accumulation represents either induction of autophagy or suppression of the downstream pathway necessitating the measurement of the autophagic flux, in absence and presence of pharmacological inhibitors and activators, as an essential parameter for uncovering the status of autophagy. The commonly used pharmacological inhibitors include PI3-kinase inhibitors (such as wortmannin, 3-MA and LY294002), microtubule-disrupting agents (e.g. nocodazole), etc. while rapamycin and its analogue, CCI-779, BH3 mimetics (ABT737) and many others are used as autophagy activators. Further, manipulation of the autophagy by knockdown or knockout and over-expression of the ATG genes are also adopted to analyse autophagic flux. The methods used to measure autophagy comprises of LC3 turnover assay, degradation of LC3 and other selective targets, specifically p62 as well as radiolabelled long-lived protein and mRFP-GFP-LC3 assay. The mRFP–GFP–LC3 assay is an interesting test, which exploits the principle of lysosomal stability of RFP versus the quenching of GFP in acidic lysosomal compartment and thus, ascertains the localisation of LC3 depending on their fluorescence properties (Mizushima et al. 2010; Mizushima 2004; Yoshii and Mizushima 2017). Owing to the limitation of each of these assays, combination of the independent experimental methods is usually recommended as the most appropriate technique to estimate autophagy.
12.5 Regulation of Autophagy
Numerous signalling pathways have been involved in up and down-regulation of autophagy. However, the lack of information to understand the detailed molecular mechanism of the autophagy regulation in both cancer and normal cells calls for further investigation (Fig. 12.2).

Regulation of autophagy. The different molecular signalling cascade involved in modulation of autophagy. The green arrows indicate activation and the red indicates inhibition of autophagy
12.5.1 The PI3K–AKT–mTOR Signalling Pathway
According to recent reports, phosphatidylinositol 3-kinase (PI3K)–AKT–mTOR pathway is important for negative regulation of autophagy. AKT, a serine-threonine kinase, activates mTOR, a TOR kinase, which leads to suppression of autophagy. Studies in yeast have demonstrated that TOR kinase, which lies upstream of the autophagy-related genes, serves as the guard in autophagy initiation (Schmelzle and Hall 2000). Moreover, in mammalian cells, mTOR integrates with growth factor signalling cascade, thereby, regulating autophagy. It is interesting to note that class I and III PI3K have opposing role in regulation of autophagy: Class I PI3K, which is activated through growth factor receptor, inhibits autophagy while activation of class III PI3K facilitates autophagy by promoting sequestration of cytoplasmic cargo (Petiot et al. 2000). The tumour suppressor genes, like oncogenic RAS and phosphatase and tensin homologue (PTEN), also regulate autophagy through PI3K–AKT–mTOR pathway. Oncogenic RAS activates class I PI3K while PTEN deactivates class I PI3K, thereby, suppressing and initiating autophagy, respectively, through modulation of AKT (Arico et al. 2001). In addition, mutation of PTEN, located on chromosome 10q23, in various cancers activates AKT and thus, inhibits autophagy. PI3K–AKT–mTOR signalling pathway is dependent on nutrient availability like nitrogen or amino acids, which leads to transcriptional and translational regulation by p70s6 kinase and 4E binding protein 1 (Wang and Klionsky 2003).
12.5.2 Beclin 1 (BECN1) and Other Pathways
Beclin 1 is a coiled coil protein which is a BCL-2 interacting gene product. It is the first reported molecule to directly link tumourigenesis with autophagy. Previous reports have indicated significant role of class III PI3K in regulation of autophagosome formation and also, promotion of transport of the lysosomal enzymes from trans-golgi network (TGN) to the lysosome. Hence, BECN1 binds to class III PI3K to form BECN1–PI3K complex, which localises in TGN and presumably, facilitates sorting of putative autophagosomal components followed by autophagy induction (Liang et al. 1999).
Other molecules implicated in regulation of autophagy in cancer cells include BCL2 and its family members (BNIP3 and HSPIN1), death-associated protein kinase (DAPK), death-associated related protein kinase 1 (DRP1) and mitogen-activated kinases. BNIP3 (BCL2–adenovirus E1B 19-kDa-interacting protein 3) and HSPIN1 (a human homologue of the Drosophila melanogaster spin gene product) are the member of the BCL2 homology 3 (BH3)-only subfamily of the BCL2 family proteins (Vande Velde et al. 2000). They have been reported to induce caspase-independent autophagic cell death in various cancer cell lines. In addition, literature survey has documented that bone marrow-derived cells from BAX and BAK-deficient mice or murine embryonic fibroblast (MEF) are apoptosis resistant but susceptible to autophagy induction upon withdrawal of growth factor or exposure to the chemotherapeutic agent, etoposide (Lum et al. 2005). These, collectively, strengthens the relevance of BNIP3 and HSPIN1 in regulation of autophagy. The DAPK, DRP1 and mitogen-activated protein kinases belong to the family of serine–threonine kinases that regulate a plethora of cellular responses including autophagy. For example, DAPK and DRP1, which are regulated by Ca2+–calmodulin, induce autophagy in MCF7 and HeLa cell (Inbal et al. 2002). While the stimulation of extracellular signal-regulated kinases ERK1 and ERK2, by the RAS–RAF1–mitogen-activated protein kinase (MEK) signalling cascade, induces autophagy in HT-29 colon cancer cell (Ogier-Denis et al. 2000) and buffers the metabolic stress (Degenhardt et al. 2006). For instance, during nutrient starvation, autophagy serves as the alternative energy reservoir whereas it also expedites the adaptation of cancer cells to cellular damage by removing the damaged proteins and organelles (Mizushima 2007).
12.6 Autophagy: The Double-Edged Sword
Autophagy has versatile role in diverse cellular processes and diseases. Basal autophagy occurs constitutively and performs its homeostatic function in conjugation with proteasome degradation pathway to facilitate protein and organelle quality control (Mathew et al. 2007; Mizushima 2007; Ravikumar et al. 2002). It has also been reported to help in elimination of pathogens and apoptotic bodies (Colombo 2007; Qu et al. 2007).
Mounting evidences suggest that autophagy has a pivotal role in cancer, although, its role in sustaining cell survival or inducing cell death is paradoxical (Baehrecke 2005). Autophagy is a well-conserved survival mechanism in several tumour types, which is rendered by protecting the cancer cells from undergoing programmed cell death. It is the most widely used mechanism of the cancer cells to survive. Therefore, inhibition of the autophagy is often exploited as the most feasible approach to sensitise the tumour cells to apoptosis and forms the basis of numerous cancer clinical trials. Nonetheless, in some situations, autophagy can also induce cell death, which is called programmed cell death type II (PCD II) or lethal autophagy. However, the autophagic cell death and apoptosis can be distinguished based on morphological and biochemical features. For instance, in contrast to apoptosis, autophagic cell death is caspase-independent and characterised by degradation of Golgi apparatus, polyribosome and endoplasmic reticulum prior to nuclear destruction (Bursch et al. 2000). Interestingly, during anti-cancer treatment, protective autophagy is initially triggered at the early stage by sequestering the damaged organelle and protein. But once the cellular damage crosses a certain threshold, lethal autophagy or death-inducing is activated to remove the damaged cells from the tissue (Kondo et al. 2005). Although apoptosis and autophagy are interconnected but little is known about the crosstalk between them. Recently, prothymosin-α, inhibitor of apoptosome formation in neuron, has been identified as plausible candidate for modulating the switch between apoptosis and autophagy (Kondo et al. 2005). Intriguingly, autophagy is dependent on multiple factors such as the nature and duration of stimulus, cell type, etc. For example, arsenic oxide (As2O3)-induced autophagy in glioma cells while in leukaemia cells, it triggered apoptotic cell death. Similarly, in contrast to the DNA alkylating agent, cisplatins, temozolomide (TMZ) induced autophagy, instead of apoptosis, in several cancer cell lines (Pelicano et al. 2003; Kanzawa et al. 2004). Moreover, it is interesting to document that while tamoxifen induced apoptosis in some cells, it also induced autophagy in other and both apoptosis and autophagy in the rest of breast cancer cells (Bursch et al. 1996). Henceforth, the modern cancer researchers have focused on investigation of the intricate regulation of autophagy and deciphering the interlink between the apoptosis and autophagy.
12.7 Role of Autophagy in Chemoresistance
A large number of recent studies suggests autophagy plays pivotal role in development of chemoresistance (Datta et al. 2017; Hu et al. 2012); in addition, various articles provide increasing evidences that inhibition of autophagy, in combination with various anticancer drugs can augment cytotoxicity on cancer cells leading to attenuation of chemoresistance development and metastasis process (Datta et al. 2019; Follo et al. 2018; Levy et al. 2014) (Fig. 12.3).

The modulation of chemoresistance and chemosensitivity by autophagy. Schematic diagram depicting the activation of different cellular signalling pathways in cancer cells through autophagy induction or inhibition, leading to chemoresistance or chemosensitivity, respectively
Epirubicin, one of the leading drugs used for breast cancer treatment, has shown evidences of autophagy induction in MCF7 breast cancer cell lines, which leads to cytoprotection of the cells from the chemotherapeutic stress induced by this drug. Similarly, autophagy inhibition has also shown elevated cytotoxic effect of various chemotherapeutic drugs like 5-fluorouracil, irinotecan in colorectal cancer, oesophageal cancer, etc. (Chen et al. 2011; Sasaki et al. 2010). Likewise, in human hepatocarcinoma cell lines, autophagy level gets elevated with oxaliplatin treatment, and suppression of autophagy enhances oxaliplatin-induced cell death (Guo et al. 2013). Some of the leading drugs for treatment of lung cancers, like topetocan and paclitaxel, also shows elevated autophagy levels in lung cancer cells, which may ultimately aid in development of chemoresistance against these drugs and inhibition of autophagy has shown promising role in prevention of chemoresistance development against these drugs in lung cancer cells (Datta et al. 2019; Goldberg et al. 2012).
There are varieties of molecular mechanisms via which autophagy induction may lead to chemoresistance development in various cancers; epidermal growth factor is a key regulatory factor for cell survival. Through its binding to cell surface receptors, EGF can induce the activation of three signalling pathways that aids in cancer development and progression, Ras/MAPK, PI3K/Akt and JAK/STATs (Henson and Gibson 2006). In malignant peripheral nerve sheath tumour (MPNST) PD168393, an EGFR-TKI, may induce autophagy as a cytostatic but not a cytotoxic response in malignant peripheral nerve sheath tumour (MPNST) cells that was accompanied by suppression of Akt and mTOR activation. The aberrant expression of PI3K/AKt pathway may also aid in chemoresistance development and PI3K/Akt inhibitors may also lead to increased cytotoxicity of chemotherapeutic drugs against cancer cells by autophagy blockage. In many pre-clinical models autophagy inhibition has shown increased cytotoxic effect, by elevated p53 activity. Vascular endothelial growth factor-C (VEGF-C) is a secreted growth factor involved in many oncogenic processes, which shows autophagy promoting activities in many cancer cells and VEGF-C inhibitors have been reported to increase cytotoxic effect of anti-cancer drugs by downregulation of cellular autophagy. Activation of MAPK14/p38 also triggers survival-promoting autophagy to protect tumour cells against the cytotoxic effects of chemotherapeutic drugs. In addition, various micro-RNAs may also play key role in chemoresistance development by either inhibition or up-regulation of cellular autophagy, for example inhibition of miR30a (a potent autophagic inhibitor) may lead to chemoresistance development and elevated expression of miR30a may aggravate cytotoxicity of cancer cells by inhibition of autophagy; similarly, miR-199a-5p (an autophagic inducer) may lead to chemoresistance development to cisplatin and vice versa. Moreover, recent reports suggest that some paclitaxel resistant cell lines also show reduced expression of miR16 and 17, which usually exhibits inhibitory effects on beclin-1 expression and elevated expression of these miRNAs may increase sensitivity of these resistant cell lines towards paclitaxel by down-regulation of autophagy (Chatterjee et al. 2015).
However, in spite of its clear prosurvival role, autophagy has also shown to have a prodeath role under certain circumstances, following treatment with a specific set of chemotherapeutic agents, either by enhancing the induction of apoptosis or mediating ‘autophagic cell death’ by K-RAS, ERK pathways.
12.8 Autophagy Inhibitors
The autophagy inhibitors, whose effectiveness in in vivo and safety in clinical trials have been approved by the FDA, are the antimalarial drugs chloroquine (CQ) and its derivative hydroxychloroquine (HCQ); these are lysomotrophic drugs which raise the lysosomal pH, thereby preventing fusion of lysosomes with autophagosomes and thus, preventing autophagosomal degradation (Fox 1993; Mauthe et al. 2018). Both CQ and HCQ have been investigated in preclinical studies or clinical trials. In addition to antimalarial drugs, inhibition of autophagy by either pharmacological approaches or via genetic silencing of autophagy regulatory genes such as Beclin 1, ATG6, ATG5, ATG7 or ATG12 (Table 12.1) also results in sensitisation of cancer cells to a variety of chemotherapeutic drugs. Different autophagy inhibitors block autophagy at different well-defined stages. For example, another antimalarial drug bafilomycin A1 can inhibit autophagosome fusion with lysosomes and autophagosome degradation in the final stage of autophagy. Class III PI3K inhibitors (3-methyladenine (3-MA), LY294002 and Wortmannin) or knockdown of autophagy regulatory genes are involved in the initiation/expansion stage of autophagy (Liu et al. 2013; Zhao et al. 2012) (Table 12.1).
Although some previous articles have linked autophagy with cell death (Acharya et al. 2011; Lin and Baehrecke 2015; Paul et al. 2020), increasing number of recent research articles have also displayed the promising role of autophagy in cancer cell survival, wherein autophagy inhibition enhanced the chemo-sensitivity of cancer cells towards a wide range of chemotherapeutic drugs (Bhattacharya et al. 2016; Cournoyer et al. 2019; Dyczynski et al. 2018; Ganguli et al. 2014; Pagotto et al. 2017). In addition, many reports also suggest that autophagy inhibition may prevent chemo-resistance development in many cancer cell lines (Belounis et al. 2016; Datta et al. 2019). Hence, literature review has established the differential role of autophagy under different conditions. Therefore, finding the exact role of autophagy in a given cancer type, under a given condition is the key factor in determining the clinical approach for apt cancer chemotherapy.
12.9 Clinical Trials
Owing to the opposing, context-dependent role of autophagy in cancer, several studies have proposed that manipulation of autophagy, by stimulation or inhibition, could enhance the efficacy of multiple cancer therapies. However, till date, chloroquine (CQ) and hydroxychloroquine (HCQ) are the only clinically approved and available drugs to inhibit autophagy in clinical models. As tabulated in Table 12.2, mounting preclinical evidences have documented that the inhibition of autophagy with CQ or HCQ alone or in combination with other drugs or radiation caused significant improvement in clinical outcome in cancer patients (Barnard et al. 2014; Briceno et al. 2003; Chude and Amaravadi 2017; Eldredge et al. 2013; Levy et al. 2017; Mahalingam et al. 2014; Rangwala et al. 2014; Rojas-Puentes et al. 2013; Vogl et al. 2014).
12.10 Conclusions
The significance of autophagy in tumourigenesis and cancer treatment makes it an important target for therapeutic intervention. However, till date, autophagy and its role in cancer are poorly understood. Therefore, the attempt to manipulate autophagy should be designed depending on its specific role in that particular scenario of malignancy. The two different and competing approaches of autophagy modulation are generally adopted towards improvement in cancer therapy. Firstly, cancer cells undergoing lethal autophagy could be exposed to mTOR inhibitors such as rapamycin and its derivatives: CCI-779, RAD001 and AP23573 in order to aggravate autophagic cell death culminating in suppression of a broad range of tumours (Chan 2004). Secondly, in contrast to the above strategy, inhibition of protective autophagy, with autophagy inhibitors such as chloroquine, hydroxychloroquine, bafilomycin A, etc. enhances the therapeutic potential of cancer therapeutics through sensitisation of the cancer cells to apoptotic cell death, as supported by mounting number of clinical trials (Barnard et al. 2014; Chude and Amaravadi 2017; Kanzawa et al. 2003; Levy et al. 2017; Mahalingam et al. 2014; Rangwala et al. 2014). However, both of these attempts in modulation of autophagy yield best outcome when combined with conventional cancer therapies.
Presently, numerous research groups throughout the globe have focused on delineating the detailed mechanism and signalling network of autophagy and understanding its intricate role in various types and stages of cancer. Henceforth, these extensive studies could enlighten new strategies of enhancing the efficacy of the currently available therapeutic options towards successful cancer cure.
References
Acharya BR, Bhattacharyya S, Choudhury D, Chakrabarti G (2011) The microtubule depolymerizing agent naphthazarin induces both apoptosis and autophagy in A549 lung cancer cells. Apoptosis 16:924–939
Amaravadi R, Kimmelman AC, White E (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30:1913–1930
Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E (2001) The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem 276:35243–35246
Baehrecke EH (2005) Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 6:505–510
Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH (2014) Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 10:1415–1425
Belounis A, Nyalendo C, Le Gall R, Imbriglio TV, Mahma M, Teira P, Beaunoyer M, Cournoyer S, Haddad E, Vassal G, Sartelet H (2016) Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer 16:891
Bhattacharya S, Das A, Datta S, Ganguli A, Chakrabarti G (2016) Colchicine induces autophagy and senescence in lung cancer cells at clinically admissible concentration: potential use of colchicine in combination with autophagy inhibitor in cancer therapy. Tumour Biol 37:10653–10664
Briceno E, Reyes S, Sotelo J (2003) Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg Focus 14:e3
Bursch W, Ellinger A, Kienzl H, Torok L, Pandey S, Sikorska M, Walker R, Hermann RS (1996) Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis 17:1595–1607
Bursch W, Ellinger A, Gerner C, Frohwein U, Schulte-Hermann R (2000) Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann N Y Acad Sci 926:1–12
Chan S (2004) Targeting the mammalian target of rapamycin (mTOR): a new approach to treating cancer. Br J Cancer 91:1420–1424
Chatterjee A, Chattopadhyay D, Chakrabarti G (2015) MiR-16 targets Bcl-2 in paclitaxel-resistant lung cancer cells and overexpression of miR-16 along with miR-17 causes unprecedented sensitivity by simultaneously modulating autophagy and apoptosis. Cell Signal 27:189–203
Chen YS, Song HX, Lu Y, Li X, Chen T, Zhang Y, Xue JX, Liu H, Kan B, Yang G, Fu T (2011) Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis Esophagus 24:437–443
Chude CI, Amaravadi RK (2017) Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci 18:1279
Colombo MI (2007) Autophagy: a pathogen driven process. IUBMB Life 59:238–242
Cournoyer S, Addioui A, Belounis A, Beaunoyer M, Nyalendo C, Le Gall R, Teira P, Haddad E, Vassal G, Sartelet H (2019) GX15-070 (Obatoclax), a Bcl-2 family proteins inhibitor engenders apoptosis and pro-survival autophagy and increases chemosensitivity in neuroblastoma. BMC Cancer 19:1018
Datta S, Choudhury D, Das A, Das Mukherjee D, Das N, Roy SS, Chakrabarti G (2017) Paclitaxel resistance development is associated with biphasic changes in reactive oxygen species, mitochondrial membrane potential and autophagy with elevated energy production capacity in lung cancer cells: a chronological study. Tumour Biol 39. https://doi.org/10.1177/1010428317694314
Datta S, Choudhury D, Das A, Mukherjee DD, Dasgupta M, Bandopadhyay S, Chakrabarti G (2019) Autophagy inhibition with chloroquine reverts paclitaxel resistance and attenuates metastatic potential in human nonsmall lung adenocarcinoma A549 cells via ROS mediated modulation of beta-catenin pathway. Apoptosis 24:414–433
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10:51–64
Dyczynski M, Yu Y, Otrocka M, Parpal S, Braga T, Henley AB, Zazzi H, Lerner M, Wennerberg K, Viklund J, Martinsson J, Grander D, De Milito A, Pokrovskaja Tamm K (2018) Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett 435:32–43
Eldredge HB, Denittis A, Duhadaway JB, Chernick M, Metz R, Prendergast GC (2013) Concurrent whole brain radiotherapy and short-course chloroquine in patients with brain metastases: a pilot trial. J Radiat Oncol 2:315
Follo C, Cheng Y, Richards WG, Bueno R, Broaddus VC (2018) Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol Carcinog 57:319–332
Fox RI (1993) Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin Arthritis Rheum 23:82–91
Ganguli A, Choudhury D, Datta S, Bhattacharya S, Chakrabarti G (2014) Inhibition of autophagy by chloroquine potentiates synergistically anti-cancer property of artemisinin by promoting ROS dependent apoptosis. Biochimie 107(Pt B):338–349
Goldberg SB, Supko JG, Neal JW, Muzikansky A, Digumarthy S, Fidias P, Temel JS, Heist RS, Shaw AT, McCarthy PO, Lynch TJ, Sharma S, Settleman JE, Sequist LV (2012) A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J Thorac Oncol 7:1602–1608
Guo XL, Li D, Sun K, Wang J, Liu Y, Song JR, Zhao QD, Zhang SS, Deng WJ, Zhao X, Wu MC, Wei LX (2013) Inhibition of autophagy enhances anticancer effects of bevacizumab in hepatocarcinoma. J Mol Med (Berl) 91:473–483
Henson ES, Gibson SB (2006) Surviving cell death through epidermal growth factor (EGF) signal transduction pathways: implications for cancer therapy. Cell Signal 18:2089–2097
Hu YL, Jahangiri A, Delay M, Aghi MK (2012) Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res 72:4294–4299
Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A (2002) DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol 157:455–468
Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I (2003) Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res 63:2103–2108
Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S (2004) Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 11:448–457
Kaur J, Debnath J (2015) Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16:461–472
Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, Orenstein SJ, Wong E, Cuervo AM (2011) Chaperone-mediated autophagy at a glance. J Cell Sci 124:495–499
Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721
Kondo Y, Kanzawa T, Sawaya R, Kondo S (2005) The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 5:726–734
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH, Foreman NK, Thorburn A (2014) Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov 4:773–780
Levy JMM, Towers CG, Thorburn A (2017) Targeting autophagy in cancer. Nat Rev Cancer 17:528–542
Li WW, Li J, Bao JK (2012) Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69:1125–1136
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676
Lin L, Baehrecke EH (2015) Autophagy, cell death, and cancer. Mol Cell Oncol 2:e985913
Liu F, Liu D, Yang Y, Zhao S (2013) Effect of autophagy inhibition on chemotherapy-induced apoptosis in A549 lung cancer cells. Oncol Lett 5:1261–1265
Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120:237–248
Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, Giles FJ, Carew JS (2014) Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 10:1403–1414
Mathew R, Karantza-Wadsworth V, White E (2007) Role of autophagy in cancer. Nat Rev Cancer 7:961–967
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N, Mari M, Reggiori F (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14:1435–1455
Meijer AJ, Codogno P (2004) Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol 36:2445–2462
Mizushima N (2004) Methods for monitoring autophagy. Int J Biochem Cell Biol 36:2491–2502
Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861–2873
Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140:313–326
Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132
Ogier-Denis E, Pattingre S, El Benna J, Codogno P (2000) Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem 275:39090–39095
Onorati AV, Dyczynski M, Ojha R, Amaravadi RK (2018) Targeting autophagy in cancer. Cancer 124:3307–3318
Pagotto A, Pilotto G, Mazzoldi EL, Nicoletto MO, Frezzini S, Pasto A, Amadori A (2017) Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cells. Cell Death Dis 8:e2943
Paul S, Chakrabarty S, Ghosh S, Nag D, Das A, Dastidar DG, Dasgupta M, Dutta N, Kumari M, Pal M, Chakrabarti G (2020) Targeting cellular microtubule by phytochemical apocynin exhibits autophagy-mediated apoptosis to inhibit lung carcinoma progression and tumorigenesis. Phytomedicine 67:153152
Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P (2003) Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem 278:37832–37839
Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P (2000) Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 275:992–998
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B (2007) Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128:931–946
Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, Piao S, Troxel AB, Evans TL, DeMichele AM, Nathanson KL, O’Dwyer PJ, Kaiser J, Pontiggia L, Davis LE, Amaravadi RK (2014) Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 10:1369–1379
Ravikumar B, Duden R, Rubinsztein DC (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11:1107–1117
Rojas-Puentes LL, Gonzalez-Pinedo M, Crismatt A, Ortega-Gomez A, Gamboa-Vignolle C, Nunez-Gomez R, Dorantes-Gallareta Y, Arce-Salinas C, Arrieta O (2013) Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat Oncol 8:209
Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, Kaneko M, Kitayama J, Takahashi K, Nagawa H (2010) Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 10:370
Schmelzle T, Hall MN (2000) TOR, a central controller of cell growth. Cell 103:253–262
Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH (2000) BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol 20:5454–5468
Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, Paul TM, Nichols CW, Porter DL, Kaplan J, Mallon G, Bradner JE, Amaravadi RK (2014) Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 10:1380–1390
Wang CW, Klionsky DJ (2003) The molecular mechanism of autophagy. Mol Med 9:65–76
White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12:401–410
Yoshii SR, Mizushima N (2017) Monitoring and measuring autophagy. Int J Mol Sci 18:1865
Zhao S, Ma CM, Liu CX, Wei W, Sun Y, Yan H, Wu YL (2012) Autophagy inhibition enhances isobavachalcone-induced cell death in multiple myeloma cells. Int J Mol Med 30:939–944
Acknowledgement
D.D.M. is thankful to “DBT-Research Associateship program in Biotechnology and Life Sciences” under Department of Biotechnology (Govt. of India) for the financial support
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Das Mukherjee, D., Datta Choudhury, S., Chakrabarti, G. (2020). Targeting Autophagy in Cancer: Therapeutic Implications. In: Bhutia, S.K. (eds) Autophagy in tumor and tumor microenvironment . Springer, Singapore. https://doi.org/10.1007/978-981-15-6930-2_12
Download citation
DOI: https://doi.org/10.1007/978-981-15-6930-2_12
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-6929-6
Online ISBN: 978-981-15-6930-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)