
Abstract
Autophagy in cancer acts as a double-edged sword whose functional discrepancies precisely depend on cancerization, progression, and type. During stress, they promote cancer cell survival, induce carcinogenesis due to their accumulated genetic mutations or abnormal cell signaling, initiating fast replication capacity, promoting more aggressiveness, and resistant to programmed cell death. Consequently, the study has drawn focus on autophagy in cancer. However, convincing preclinical and clinical evidence on the cytoprotective in addition to the lethal roles of autophagy for cancer stem cells (CSCs) are missing. There are quite a lot of clinical trials ongoing to manipulate autophagy and in this manner decide the result of disease therapy. The clinical relevance of this work encompasses autophagy modifiers, such as rapamycin and chloroquine that control autophagy in anticancer therapy, since autophagy plays roles in both tumor suppression and promotion. Further detailed examination of autophagy in cancer is required to understand how an increased function of autophagy in the tumor microenvironment, stemness, migration and invasion, dormancy, and drug resistance could be tweaked for enhanced therapeutic benefit by eradicating minimal residual disease and preventing metastasis. Here, we recapitulate how autophagy modulates the therapeutic potential to exterminate CSCs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Introduction
Pluripotent cancer stem cells (CSCs) are subset of cancer cells that accentuate their ability to self-renew and (Aponte and Caicedo 2017) differentiate into all somatic cell lineages by indefinite cell division giving rise to the heterogeneous tumor populations and maintain their undifferentiated state (Liu et al. 2013). When a very small population of CSCs was introduced into an immunocompromised mice, it initiated the formation of the original tumor (Ghiaur et al. 2012). They are phenotypically slow cycling and their self-renewing capacity is accountable for tumor growth, resistance to therapy, and recurrence after treatment.
Autophagy is a double-edged sword in the progression of neoplasia and has further produced immense hurdles for researchers to explore its impression on carcinogenesis and tumor development. It has labeled tumor-suppressive and tumor-promoting functions (White and DiPaola 2009). Cytoprotective role of autophagy prevents malignant transformation through the ability to empower the premalignant cells by efficiently meeting up with the increased energy requirements by recycling cellular components that are important in maintaining the physiological tissue homeostasis. This attribute propagates their accommodation within the stress (metabolic, genotoxic, and inflammatory) occurring after the malignant transformation induced in response to anticancer (chemo/targeted/radiotherapy) treatment. Stresses including nutrient and energy stress, ER stress, danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), hypoxia, redox stress, and mitochondrial damage induce autophagy, alongside EMT and stemness. The cytoprotective role of autophagy can turn into a cell-suicidal weapon causing cell death in cancer cells. Defective autophagy has been linked with increased oncogenesis. For instance, low expression of Beclin-1 (Atg6) in some types of cancers of the prostate, breast, and ovary because of monoallelic mutations (Qu et al. 2003). However, the presence of heterozygosity in mice for the beclin-1 gene makes it cancer prone (Qu et al. 2003; Yue et al. 2003) due to absence of functional of Beclin-1.
Cancer progression shows a degree of dependency on the existence of CSCs. The role of autophagy in cancer is multifaceted and has been studied extensively. High levels of autophagy contribute to pluripotency of CSCs in other cancer types, including colorectal cancer (Kantara et al. 2014), pancreatic cancer (Rausch et al. 2012; Viale et al. 2014), glioblastoma (Galavotti et al. 2013), chronic myeloid leukemia (Bellodi et al. 2009), and bladder cancer (Ojha et al. 2016). Despite recent advancement in research, the underlying molecular mechanism inducing autophagy in CSCs remains to be determined. It is difficult to explain how autophagy promotes stemness, have been preserved across different cancer. Mitophagy is a selective autophagy that unambiguously plays an important role in the quality control and homeostasis of mitochondria. Mitochondrial functional pathways play a crucial role in a vital interaction between cancer cells and stromal cells for cancer cell initiation, progression, and treatment response. They emanate a profound role in sustaining CSCs in adverse conditions and initiating their metabolic reprogramming to support the increased bioenergetic demand of the tumor. Transcription factors like SMAD (Nazio et al. 2019), NF-Κb (Zhang et al. 2016), MITF (Moller et al. 2019), STAT3 (Marcucci et al. 2017; Zhang et al. 2016), FOXO (Naka et al. 2010), ATF4 (Pallmann et al. 2019), NANOG (Liu et al. 2017), regulate autophagy and mitophagy in the induction of EMT and maintenance of CSCs. Like autophagy, mitophagy acts in cancer as bimodal processes. Unfortunately, there are unanswered roles of canonical autophagy in cancer (Gewirtz 2014). Therefore, does mitophagy has a role in cancer? CSCs play an unbiased role in promoting therapy resistance leading to tumor recurrence (Shibue and Weinberg 2017), and autophagy deliberately endorses disseminated tumor cells (DTCs) which further lead to the metastatic expansion of tumors (Sosa et al. 2014). To understand how autophagy and mitophagy can inhibit to repress both the above phenotypes are challenging task for translational cancer. Recent studies have linked CSCs with chemoresistance and cancer relapse, autophagy, mitophagy, and CSCs showcase novel perspectives on potential therapeutic targets for enhancing anticancer drug sensitivity. The study of autophagy in cancer has been therapeutically manipulated by many investigators and various clinical trials that are already ongoing to regulate the result of disease therapy.
10.2 Autophagy/Mitophagy Drives Cancer Stem Cells Fate
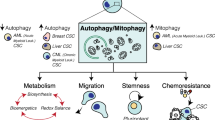
CSCs are a heterogeneous population; they escalate tumor growth and progression by accelerating the proliferative potential and constitute a source for recurrence of cancer. Functional properties of cancer cells are influenced by epigenetic, genetic, and microenvironmental factors. To proliferate in its microenvironment, CSCs have a functional correlation with autophagy and mitophagy. Autophagy, a catabolic pathway enables CSCs to show autophagy dependence and may act as an onco-suppressive depending on tumor stage and type. They exploit the pro-survival attribute of autophagy at the later stage of oncogenesis to meet up with high-energy demands by a supply of metabolites. ATG-encoded gene products play a significant role in CSCs of numerous cancers. Beclin 1/Atg6 modulates CSC plasticity and tumorigenesis in vivo. However, in different cancers, Beclin 1 acts as a tumor suppressor, like human prostate, breast, and ovarian tumors (Liang et al. 1999; Qu et al. 2003; Shen et al. 2008). Improved survival in patients is observed having high Beclin 1 levels affected by large B-cell lymphoma, high-grade gliomas, or hepatocellular carcinoma (Ding et al. 2008; Huang et al. 2011; Pirtoli et al. 2009). The stemness was augmented by the transformation of CD133− to CD133+ cells due to the inhibition of mTOR affecting the liver tumor cells by interrupting the differentiation and stimulating the tumor development in vivo (Yang et al. 2011). Suppression of autophagy by knockdown of autophagic proteins Atg5 and Atg7, curtails stemness markers, such as Sox2, Nanog, and Oct4, resulting colorectal CSCs to undergo suppressed cell proliferation and improved cell senescence (Sharif et al. 2017). In colorectal cancers, mutations in Atg5, Atg12 have been described (Kang et al. 2009) while deletion of Atg5 or Atg7 is supporting the advancement of liver hepatomas (Takamura et al. 2011). Autophagy induction by overexpressing Atg4A protein promotes mammosphere formation and hence increases CSC numbers and in vivo tumorigenesis (Wolf et al. 2013). The conditional knockout of Atg3 affected the continued existence of CML cells and leukemogenesis (Altman et al. 2011). Inhibition of Atg4B resulted in its increased phosphorylation followed by arresting the tumor growth in animal models in a subset of glioblastoma cancer (Huang et al. 2017). The depletion of ATG4B impaired the survivability of CML stem/progenitor cells (Rothe et al. 2014). Knockout of Atg4C in mice increased the propensity to develop fibrosarcomas induced by methylcholanthrene, hence play a tumor-suppressor role. Contrastingly, its tumorigenic role in breast cancer was delineated (Antonelli et al. 2017). Tumor suppressive role of Atg4D expression was observed in colorectal carcinogenesis (Gil et al. 2018). Moreover, its tumor-promoting role was highlighted when cancer cells were sensitized to chemotherapeutic drugs on ATG4D silencing (Betin and Lane 2009) (Fig. 10.1).

The basal level autophagy and mitophagy are important for cell metabolism. When there is stress due to anticancer therapy, autophagy, and mitophagy get impaired, while they are activated due to internal and external factors leading to either suppression or progression of cancer
EMT (epithelial to mesenchymal transition) signaling is an important characteristic of CSCs (Shibue and Weinberg 2017). Autophagy signaling is strongly correlated to EMT in enhancing the metastatic potential of CSCs to migrate by maintaining their mesenchymal signature in the later stages of metastasis. Interestingly, during early metastasis autophagy decreases the invasion and migration of tumor cells in situ. In glioblastoma cells, blocked cell migration and invasion were caused by nutrient deprivations and mTOR inhibition (Catalano et al. 2015). Using specific siRNAs directed against the autophagy-related factors DRAM1 and p62 proteins, autophagy-controlled bioenergetic metabolism, migration/invasion of glioblastoma CSCs was thwarted while the mesenchymal phenotype was restored on autophagy upregulation (Galavotti et al. 2013). Furthermore, in glioblastoma cells, enhanced migration and invasion with EMT regulators continued with knockdown of Beclin 1, Atg5, and Atg7 (Catalano et al. 2015). EMT promotes stemness and can give rise to CSCs through the core stemness factors POU5F1, Sox2, and Nanog, including Slug and Twist that maintains the pluripotency of CSCs and tumor-propagating properties (Mani et al. 2008). Hypoxia and TGF-β through MITF (Caramel et al. 2013), Sox2, and Nanog (Sharif et al. 2017) promote EMT via activating autophagy. Autophagy may promote tumor cell dormancy, lipid metabolism, mitochondrial function, and CSCs existence in muscle stem cells and HSCs (Ho et al. 2017; Warr et al. 2013). It ensures a reversible dormant pool of CSCs potentially making a contribution to tumor repopulation and preventing irreversible senescence (Ho et al. 2017). Autophagy plays a decisive role in the survival of disseminated tumor cells (DTCs) at secondary location to establish drug resistance, minimum residual disease, and metastatic dormancy (Sosa et al. 2014). Interestingly, these DTCs are CSCs that are relatively quiescent and motile state expressing upregulated CSC markers in the bone marrow of breast cancer patients (Balic et al. 2006). Furthermore, a selective form of autophagy known as mitophagy promotes stemness. It abrogates senescence by disrupting the ROS-induced DNA damage and has a principal role in maintaining the stem cell population renewal and homeostasis. It has been reported to maintain hepatic CSCs by regulating p53 localization. Therefore, inhibition of mitophagy phosphorylates p53 by PINK1 leading to its translocation to the nucleus where Oct4 and Sox2 induction of Nanog get alienated. Mitophagy evokes CSCs dependence more on glycolysis for energy needs and hence contributes to its quiescent state. Recent evidence suggests that mitochondrial dysfunction also encourages oncogenesis (Boya et al. 2018). Mitochondrial ROS due to BNIP3 loss subsequently resulted from defects in mitophagy followed by mammary neoplastic progression to metastasis (Chourasia et al. 2015) (Table 10.1).
10.3 Targeting Autophagy/Mitophagy: New Therapeutic Strategies
CSC generation, differentiation, plasticity, migration/invasion, and immune resistance are very much dependent on the variation of autophagy/mitophagy. During anticancer therapy, CSCs remain at the dormant stage to cope with intracellular and environmental stress, involving oxidative stress triggered by overproduction of reactive oxygen species (ROS). These dormant cells arise from EMT tumor cells and become non-cycling autophagic CSC which are later maneuvered on the release of paracrine factors (like MET, TGF-β receptor, IL-6 receptor, PDGFR, EGFR, FGFR, Hedgehog/Smoothened, WNT/Frizzled, Gas6/AXL, and Notch ligands) to cycling CSC with low autophagy. Thus, autophagy and mitophagy enable CSCs to colonize, migrate and metastasize, defy apoptosis and antitumor drugs and hence become therapy-resistant by its self-renewal property and replace the pool of differentiated tumor cells (Marcucci et al. 2017) (Table 10.2).
Autophagy/mitophagy has an inevitable role in cancer cell survival, metastasis, and therapy resistance. The potentially new targeted therapeutic strategy is to use double or triple combinatorial doses of drugs or antibodies and/or radiation to modulate autophagic machinery to efficiently eradicate CSCs. Chemotherapy is a widespread treatment strategy for cancer therapy that engulfs dividing cells and disrupts cancer–cell division. However, several studies have revealed that the overall success rate of chemotherapy is often restricted via the upregulation of cytoprotective activation of autophagy in CSCs which protects cancer cells subjected to anticancer therapy. Cancer chemotherapeutic drugs 5-Fluorouracil (5FU) and cisplatin used in various solid cancers, like, gallbladder and colorectal cancers show autophagy-regulated chemoresistance (Ferreira et al. 2016; Liang et al. 2014; Park et al. 2013). Additionally, the CXCL12/CXCR4 axis is prompted in colorectal cancer and is linked with potential progression of cancer, such as invasion, metastasis, and chemoresistance. Subsequently, grants 5-fluorouracil (5-FU) resistance by increasing autophagy both in vitro and in vivo (Yu et al. 2017).
Suppression of autophagy preferentially stimulated in multiple molecular pathways that govern CSCs growth and differentiation, includes Notch (Li et al. 2018), Sonic Hedgehog (Fan et al. 2019), Wnt/β-catenin (Pai et al. 2017), NF-kβ (Trocoli and Djavaheri-Mergny 2011), transforming growth factor-β (Kiyono et al. 2009), and fibroblast growth factor (Chen et al. 2018) signaling cascades lead to sensitization of cancer cells to anticancer therapy. The appreciating effect of the Wnt/β-catenin pathway is inhibited by FH535 and its derivative (FH535-N) alone and in combination with sorafenib through nullification of the autophagic flux in hepatocellular carcinoma (Turcios et al. 2019). Hyperactivation of PI3K/Akt/mTOR pathway in GBM and its inhibition exerts antineoplastic activity by targeting CSCs, supporting differentiation, and inhibiting cell migration and invasion prospective of GSCs (Li et al. 2016). Balance is the key between Beclin1 and Bcl2/Bcl-xL that supports the concept of the presence of a complex relationship between autophagy and apoptosis, which seems important in the context of cancer and cancer therapy (Kim et al. 2014). JNK-mediated protective autophagy increased Bcl2 expression followed by an increased autophagic flux and conferred chemoresistance in colon cancer (Sui et al. 2014).
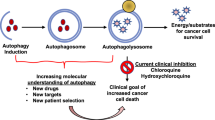
Evolving clinical and experimental evidence indicates that CSCs have clinical significance as they are bestowed with intrinsic resistance to radio- and chemotherapy owing to the indulgence of autophagy (Chen et al. 2012; Vitale et al. 2015). Targeting components of the autophagic machinery can be recruited as the hopeful target to selectively eliminate CSCs facilitating cancer cell growth/progression/metastasis and enhancing the effectiveness of radio- and chemotherapy (Nazio et al. 2019; Ojha et al. 2015; Perez-Hernandez et al. 2019). Henceforth, these findings completely indicate that autophagy suppression and its activation, both, can be deemed to be promising approaches for sensitizing CSCs to anticancer therapy, evaluated by the reduction of the number of CSCs. So, the development of new anticancer drugs focuses on CSCs which is key to the problem required to be resolved in drug clinical trials (Fig. 10.2).

The conventional anticancer therapy is incapable to target the CSCs that shoot to cancer relapse. Autophagy plays a Janus role in cancer cell modulation, acts protective during tumor relapse, and lethal via programmed cell death. Newly discovered combinatorial treatments target both the bulk tumor and cancer stem cells leading to elimination of persistent CSCs and tumor regression
10.4 Conclusion
Development of autophagy inhibitors, specific mitophagy inhibitors have been proven beneficial, given the fears about global autophagy suppression for tissue homeostasis and that mitophagy has a crucial functional role earlier credited to general autophagy. Focusing on selective inhibitors will pave an unexplored path of how autophagy is responsible for determining stemness, dormancy-whether DTCs are autophagy-dependent CSCs, and which autophagy functions will be significant in promoting drug resistance and cancer recurrence. Further research is requisite before CSCs can be treated by regulating autophagy and mitophagy. Desirable therapeutic impacts of anticancer reagents have not been achieved by only targeting autophagy using autophagy modulators; to the contrary, it has enacted as a pro-survival response by supplying nutrients to cancer cells. Consequently, clinical trials that aim autophagy by a combination of autophagy alterations and anticancer components are appropriate to consider autophagy as a possible effectual therapeutic approach in anticancer therapy. The conjunction of these techniques hopefully deciphers the vital mechanisms necessary for maintaining cancer stemness and will play an important role in designing more efficient and effective personalized therapeutic strategies.
References
Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, Ilkayeva O, Jia W, He YW, Rathmell JC (2011) Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 30:1855–1867
Antonelli M, Strappazzon F, Arisi I, Brandi R, D’Onofrio M, Sambucci M, Manic G, Vitale I, Barila D, Stagni V (2017) ATM kinase sustains breast cancer stem-like cells by promoting ATG4C expression and autophagy. Oncotarget 8:21692–21709
Aponte PM, Caicedo A (2017) Stemness in cancer: stem cells, cancer stem cells, and their microenvironment. Stem Cells Int 2017:5619472
Aryal P, Kim K, Park PH, Ham S, Cho J, Song K (2014) Baicalein induces autophagic cell death through AMPK/ULK1 activation and downregulation of mTORC1 complex components in human cancer cells. FEBS J 281:4644–4658
Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, Datar RH, Cote RJ (2006) Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res 12:5615–5621
Balic A, Sorensen MD, Trabulo SM, Sainz B Jr, Cioffi M, Vieira CR, Miranda-Lorenzo I, Hidalgo M, Kleeff J, Erkan M, Heeschen C (2014) Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther 13:1758–1771
Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R, Van Etten RA, Donato N, Hunter A, Dinsdale D, Tirro E, Vigneri P, Nicotera P, Dyer MJ, Holyoake T, Salomoni P, Calabretta B (2009) Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 119:1109–1123
Betin VM, Lane JD (2009) Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci 122:2554–2566
Billington RA, Genazzani AA, Travelli C, Condorelli F (2008) NAD depletion by FK866 induces autophagy. Autophagy 4:385–387
Boya P, Codogno P, Rodriguez-Muela N (2018) Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development 145:dev146506
Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J, Hutchinson P, Tse G, Lachuer J, Puisieux A, Pringle JH, Ansieau S, Tulchinsky E (2013) A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 24:466–480
Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V, Limatola C, Cecconi F, Di Bartolomeo S (2015) Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol 9:1612–1625
Cea M, Cagnetta A, Fulciniti M, Tai YT, Hideshima T, Chauhan D, Roccaro A, Sacco A, Calimeri T, Cottini F, Jakubikova J, Kong SY, Patrone F, Nencioni A, Gobbi M, Richardson P, Munshi N, Anderson KC (2012) Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 120:3519–3529
Chaterjee M, van Golen KL (2011) Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res 2011:362938
Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488:522–526
Chen CH, Changou CA, Hsieh TH, Lee YC, Chu CY, Hsu KC, Wang HC, Lin YC, Lo YN, Liu YR, Liou JP, Yen Y (2018) Dual inhibition of PIK3C3 and FGFR as a new therapeutic approach to treat bladder cancer. Clin Cancer Res 24:1176–1189
Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, Macleod KF (2015) Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16:1145–1163
Dany M, Gencer S, Nganga R, Thomas RJ, Oleinik N, Baron KD, Szulc ZM, Ruvolo P, Kornblau S, Andreeff M, Ogretmen B (2016) Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 128:1944–1958
Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, Shi GM, Wang XY, Ke AW, Wu B, Fan J (2008) Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res 68:9167–9175
Fan J, Zhang X, Wang S, Chen W, Li Y, Zeng X, Wang Y, Luan J, Li L, Wang Z, Sun X, Shen B, Ju D (2019) Regulating autophagy facilitated therapeutic efficacy of the sonic Hedgehog pathway inhibition on lung adenocarcinoma through GLI2 suppression and ROS production. Cell Death Dis 10:626
Ferreira JA, Peixoto A, Neves M, Gaiteiro C, Reis CA, Assaraf YG, Santos LL (2016) Mechanisms of cisplatin resistance and targeting of cancer stem cells: adding glycosylation to the equation. Drug Resist Updat 24:34–54
Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G (2012) Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. PLoS One 7:e47357
Francipane MG, Lagasse E (2013) Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget 4:1948–1962
Frieboes HB, Huang JS, Yin WC, McNally LR (2014) Chloroquine-mediated cell death in metastatic pancreatic adenocarcinoma through inhibition of autophagy. JOP 15:189–197
Fu Y, Chang H, Peng X, Bai Q, Yi L, Zhou Y, Zhu J, Mi M (2014) Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS One 9:e102535
Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, Dinsdale D, Condorelli F, Brandner S, Campanella M, Grose R, Jones C, Salomoni P (2013) The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 32:699–712
Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G (2017) Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov 16:487–511
Gewirtz DA (2014) The four faces of autophagy: implications for cancer therapy. Cancer Res 74:647–651
Ghiaur G, Gerber JM, Matsui W, Jones RJ (2012) Cancer stem cells: relevance to clinical transplantation. Curr Opin Oncol 24:170–175
Ghosh N, Matsui W (2009) Cancer stem cells in multiple myeloma. Cancer Lett 277:1–7
Gil J, Ramsey D, Pawlowski P, Szmida E, Leszczynski P, Bebenek M, Sasiadek MM (2018) The influence of tumor microenvironment on ATG4D gene expression in colorectal cancer patients. Med Oncol 35:159
Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, Passegue E (2017) Autophagy maintains the metabolism and function of young and old stem cells. Nature 543:205–210
Huang JJ, Zhu YJ, Lin TY, Jiang WQ, Huang HQ, Li ZM (2011) Beclin 1 expression predicts favorable clinical outcome in patients with diffuse large B-cell lymphoma treated with R-CHOP. Hum Pathol 42:1459–1466
Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, Song X, Shi T, Yang Y, Sastry N, Horbinski CM, Lu S, Stupp R, Kessler JA, Nishikawa R, Nakano I, Sulman EP, Lu X, James CD, Yin XM, Hu B, Cheng SY (2017) MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 32(840–855):e848
Huang H, Song J, Liu Z, Pan L, Xu G (2018) Autophagy activation promotes bevacizumab resistance in glioblastoma by suppressing Akt/mTOR signaling pathway. Oncol Lett 15:1487–1494
Jangamreddy JR, Ghavami S, Grabarek J, Kratz G, Wiechec E, Fredriksson BA, Rao Pariti RK, Cieslar-Pobuda A, Panigrahi S, Los MJ (2013) Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: differences between primary and cancer cells. Biochim Biophys Acta 1833:2057–2069
Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, Xu J, Kondo Y, Bekele BN, Colman H, Lang FF, Fueyo J (2007) Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst 99:1410–1414
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, Ahn CH, Yoo NJ, Lee SH (2009) Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol 217:702–706
Kantara C, O’Connell M, Sarkar S, Moya S, Ullrich R, Singh P (2014) Curcumin promotes autophagic survival of a subset of colon cancer stem cells, which are ablated by DCLK1-siRNA. Cancer Res 74:2487–2498
Katayama R, Koike S, Sato S, Sugimoto Y, Tsuruo T, Fujita N (2009) Dofequidar fumarate sensitizes cancer stem-like side population cells to chemotherapeutic drugs by inhibiting ABCG2/BCRP-mediated drug export. Cancer Sci 100:2060–2068
Kim SY, Song X, Zhang L, Bartlett DL, Lee YJ (2014) Role of Bcl-xL/Beclin-1 in interplay between apoptosis and autophagy in oxaliplatin and bortezomib-induced cell death. Biochem Pharmacol 88:178–188
Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, Sugimoto K, Miyazono K (2009) Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 69:8844–8852
Kumar D, Shankar S, Srivastava RK (2013) Rottlerin-induced autophagy leads to the apoptosis in breast cancer stem cells: molecular mechanisms. Mol Cancer 12:171
Kumar D, Shankar S, Srivastava RK (2014) Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett 343:179–189
Kwitkowski VE, Prowell TM, Ibrahim A, Farrell AT, Justice R, Mitchell SS, Sridhara R, Pazdur R (2010) FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 15:428–435
Li X, Wu C, Chen N, Gu H, Yen A, Cao L, Wang E, Wang L (2016) PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 7:33440–33450
Li LQ, Pan D, Zhang SW, Xie D-Y, Zheng XL, Chen H (2018) Autophagy regulates chemoresistance of gastric cancer stem cells via the Notch signaling pathway. Eur Rev Med Pharmacol Sci 22:3402–3407
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676
Liang X, Tang J, Liang Y, Jin R, Cai X (2014) Suppression of autophagy by chloroquine sensitizes 5-fluorouracil-mediated cell death in gallbladder carcinoma cells. Cell Biosci 4:10
Lin YC, Lin JF, Wen SI, Yang SC, Tsai TF, Chen HE, Chou KY, Hwang TI (2017) Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J Med Sci 33:215–223
Liu A, Yu X, Liu S (2013) Pluripotency transcription factors and cancer stem cells: small genes make a big difference. Chin J Cancer 32:483–487
Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, Cho C, Machida K, Chen D, Ou JJ (2017) Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell 68(281–292):e285
Lu J, Sun D, Gao S, Gao Y, Ye J, Liu P (2014) Cyclovirobuxine D induces autophagy-associated cell death via the Akt/mTOR pathway in MCF-7 human breast cancer cells. J Pharmacol Sci 125:74–82
Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, Lazova R, Klump V, Pawelek JM, Xu X, Xu W, Schuchter LM, Davies MA, Herlyn M, Winkler J, Koumenis C, Amaravadi RK (2014) Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest 124:1406–1417
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715
Marcucci F, Ghezzi P, Rumio C (2017) The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol Cancer 16:3
Mohammed A, Janakiram NB, Brewer M, Ritchie RL, Marya A, Lightfoot S, Steele VE, Rao CV (2013) Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl Oncol 6:649–659
Moller K, Sigurbjornsdottir S, Arnthorsson AO, Pogenberg V, Dilshat R, Fock V, Brynjolfsdottir SH, Bindesboll C, Bessadottir M, Ogmundsdottir HM, Simonsen A, Larue L, Wilmanns M, Thorsson V, Steingrimsson E, Ogmundsdottir MH (2019) MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci Rep 9:1055
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, Hirao A (2010) TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463:676–680
Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F (2019) Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ 26:690–702
Ojha R, Jha V, Singh SK, Bhattacharyya S (2014) Autophagy inhibition suppresses the tumorigenic potential of cancer stem cell enriched side population in bladder cancer. Biochim Biophys Acta 1842:2073–2086
Ojha R, Bhattacharyya S, Singh SK (2015) Autophagy in cancer stem cells: a potential link between chemoresistance, recurrence, and metastasis. Biores Open Access 4:97–108
Ojha R, Singh SK, Bhattacharyya S (2016) JAK-mediated autophagy regulates stemness and cell survival in cisplatin resistant bladder cancer cells. Biochim Biophys Acta 1860:2484–2497
Pai SG, Carneiro BA, Mota JM, Costa R, Leite CA, Barroso-Sousa R, Kaplan JB, Chae YK, Giles FJ (2017) Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol 10:101
Pallmann N, Livgard M, Tesikova M, Zeynep Nenseth H, Akkus E, Sikkeland J, Jin Y, Koc D, Kuzu OF, Pradhan M, Danielsen HE, Kahraman N, Mokhlis HM, Ozpolat B, Banerjee PP, Uren A, Fazli L, Rennie PS, Jin Y, Saatcioglu F (2019) Regulation of the unfolded protein response through ATF4 and FAM129A in prostate cancer. Oncogene 38:6301–6318
Park JM, Huang S, Wu TT, Foster NR, Sinicrope FA (2013) Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol Ther 14:100–107
Perez-Hernandez M, Arias A, Martinez-Garcia D, Perez-Tomas R, Quesada R, Soto-Cerrato V (2019) Targeting autophagy for cancer treatment and tumor chemosensitization. Cancers (Basel) 11:1599
Pirtoli L, Cevenini G, Tini P, Vannini M, Oliveri G, Marsili S, Mourmouras V, Rubino G, Miracco C (2009) The prognostic role of Beclin 1 protein expression in high-grade gliomas. Autophagy 5:930–936
Previs RA, Coleman RL, Harris AL, Sood AK (2015) Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res 21:955–961
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112:1809–1820
Radogna F, Cerella C, Gaigneaux A, Christov C, Dicato M, Diederich M (2016) Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene 35:3839–3853
Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, Tan KS, Heitjan DF, DeMichele AM, Vaughn DJ, Redlinger M, Alavi A, Kaiser J, Pontiggia L, Davis LE, O’Dwyer PJ, Amaravadi RK (2014) Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 10:1391–1402
Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S, Kallifatidis G, Kaczorowski A, Groth A, Gross W, Gebhard MM, Schemmer P, Werner J, Salnikov AV, Zentgraf H, Buchler MW, Herr I (2012) Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol 227:325–335
Rebecca VW, Nicastri MC, Fennelly C, Chude CI, Barber-Rotenberg JS, Ronghe A, McAfee Q, McLaughlin NP, Zhang G, Goldman AR, Ojha R, Piao S, Noguera-Ortega E, Martorella A, Alicea GM, Lee JJ, Schuchter LM, Xu X, Herlyn M, Marmorstein R, Gimotty PA, Speicher DW, Winkler JD, Amaravadi RK (2019) PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov 9:220–229
Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot MF, Lamberton A, Mathieu M, Bertrand T, Marquette JP, El-Ahmad Y, Filoche-Romme B, Schio L, Garcia-Echeverria C, Goulaouic H, Pasquier B (2014) A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol 10:1013–1019
Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, McAfee Q, Fisher J, Troxel AB, Piao S, Heitjan DF, Tan KS, Pontiggia L, O’Dwyer PJ, Davis LE, Amaravadi RK (2014) A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10:1359–1368
Rothe K, Lin H, Lin KB, Leung A, Wang HM, Malekesmaeili M, Brinkman RR, Forrest DL, Gorski SM, Jiang X (2014) The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 123:3622–3634
Sanchez CG, Penfornis P, Oskowitz AZ, Boonjindasup AG, Cai DZ, Dhule SS, Rowan BG, Kelekar A, Krause DS, Pochampally RR (2011) Activation of autophagy in mesenchymal stem cells provides tumor stromal support. Carcinogenesis 32:964–972
Schneider L, Giordano S, Zelickson BR, Johnson M, Benavides G, Ouyang X, Fineberg N, Darley-Usmar VM, Zhang J (2011) Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic Biol Med 51:2007–2017
Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, Bielawski J, Ogretmen B (2012) Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol 8:831–838
Shao S, Li S, Qin Y, Wang X, Yang Y, Bai H, Zhou L, Zhao C, Wang C (2014) Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int J Oncol 44:1661–1668
Sharif T, Martell E, Dai C, Kennedy BE, Murphy P, Clements DR, Kim Y, Lee PW, Gujar SA (2017) Autophagic homeostasis is required for the pluripotency of cancer stem cells. Autophagy 13:264–284
Shen Y, Li DD, Wang LL, Deng R, Zhu XF (2008) Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 4:1067–1068
Shibue T, Weinberg RA (2017) EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 14:611–629
Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14:611–622
Stein EM, Tallman MS (2016) Emerging therapeutic drugs for AML. Blood 127:71–78
Sui X, Kong N, Wang X, Fang Y, Hu X, Xu Y, Chen W, Wang K, Li D, Jin W, Lou F, Zheng Y, Hu H, Gong L, Zhou X, Pan H, Han W (2014) JNK confers 5-fluorouracil resistance in p53-deficient and mutant p53-expressing colon cancer cells by inducing survival autophagy. Sci Rep 4:4694
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25:795–800
Takeda M, Koseki J, Takahashi H, Miyoshi N, Nishida N, Nishimura J, Hata T, Matsuda C, Mizushima T, Yamamoto H, Ishii H, Doki Y, Mori M, Haraguchi N (2019) Disruption of endolysosomal RAB5/7 efficiently eliminates colorectal cancer stem cells. Cancer Res 79:1426–1437
Trocoli A, Djavaheri-Mergny M (2011) The complex interplay between autophagy and NF-kappaB signaling pathways in cancer cells. Am J Cancer Res 1:629–649
Turcios L, Chacon E, Garcia C, Eman P, Cornea V, Jiang J, Spear B, Liu C, Watt DS, Marti F, Gedaly R (2019) Autophagic flux modulation by Wnt/beta-catenin pathway inhibition in hepatocellular carcinoma. PLoS One 14:e0212538
Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF (2014) Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514:628–632
Vitale I, Manic G, Dandrea V, De Maria R (2015) Role of autophagy in the maintenance and function of cancer stem cells. Int J Dev Biol 59:95–108
Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegue E (2013) FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494:323–327
White E, DiPaola RS (2009) The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 15:5308–5316
Wolf J, Dewi DL, Fredebohm J, Muller-Decker K, Flechtenmacher C, Hoheisel JD, Boettcher M (2013) A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res 15:R109
Xiong L, Liu Z, Ouyang G, Lin L, Huang H, Kang H, Chen W, Miao X, Wen Y (2017) Autophagy inhibition enhances photocytotoxicity of Photosan-II in human colorectal cancer cells. Oncotarget 8:6419–6432
Yan C, Luo L, Guo CY, Goto S, Urata Y, Shao JH, Li TS (2017) Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett 388:34–42
Yang Z, Zhang L, Ma A, Liu L, Li J, Gu J, Liu Y (2011) Transient mTOR inhibition facilitates continuous growth of liver tumors by modulating the maintenance of CD133+ cell populations. PLoS One 6:e28405
Yao JC, Phan AT, Jehl V, Shah G, Meric-Bernstam F (2013) Everolimus in advanced pancreatic neuroendocrine tumors: the clinical experience. Cancer Res 73:1449–1453
Yu X, Shi W, Zhang Y, Wang X, Sun S, Song Z, Liu M, Zeng Q, Cui S, Qu X (2017) CXCL12/CXCR4 axis induced miR-125b promotes invasion and confers 5-fluorouracil resistance through enhancing autophagy in colorectal cancer. Sci Rep 7:42226
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100:15077–15082
Zhang Z, Duan Q, Zhao H, Liu T, Wu H, Shen Q, Wang C, Yin T (2016) Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-kappaB/STAT3 signaling cascade. Cancer Lett 382:53–63
Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL, Huang C, Li P, Gao N (2015) A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 11:1259–1279
Zhu H, Wang D, Liu Y, Su Z, Zhang L, Chen F, Zhou Y, Wu Y, Yu M, Zhang Z, Shao G (2013) Role of the hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int 13:119
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sinha, N. (2020). Relevance of Autophagy in Cancer Stem Cell and Therapeutic. In: Bhutia, S.K. (eds) Autophagy in tumor and tumor microenvironment . Springer, Singapore. https://doi.org/10.1007/978-981-15-6930-2_10
Download citation
DOI: https://doi.org/10.1007/978-981-15-6930-2_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-6929-6
Online ISBN: 978-981-15-6930-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)