
Abstract
Cathepsins are a ubiquitously expressed subset of the papain family of 11 distinct cysteine lysosomal proteases‚ including cathepsins B, C, F, H, K, L, O, S, V, W, and X. They regulate diverse biological processes through protein degradation. Impairment of these processes leads to disease conditions and generates considerable research interest. Cathepsins B and H are fascinating in being functionally different but structurally similar and are involved in neuropeptide and hormone processing. Loss of function has been implicated in various diseases, including cancer. They are also involved in the control of cellular differentiation through regulated proteolysis as well as controlled degradation of the extracellular matrix (ECM). The gain of function in the latter leads to disease states like tumor invasion, metastasis, muscular dystrophy, emphysema, and arthritis. Cathepsin H, uniquely, is an aminopeptidase and endopeptidase, while cathepsin B is an endopeptidase and a carboxypeptidase. This diversity in catalysis and specificity is strongly supported by structure elucidation and folding analysis. This chapter emphasizes the important facts related to cathepsins B and H structure starting from amino acid sequence to gene level and structure–activity relationship of these enzymes. It also attempts to enhance our understanding of cathepsins and presents them as drug targets to control ECM degradation and other components of the disease mechanism.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Lysosomal proteases
- Cathepsin B
- Cathepsin H
- Molecular properties
- Biosynthesis
- Specificity
- Structure
- Function
16.1 Introduction
The last few decades are devoted to a group of lysosomal proteases, especially cysteine cathepsins. The reason behind this is the role of thiol cathepsins in various diseases and involvement in intracellular protein degradation. It has been established that these proteases participate in remodeling and degradation of ECM proteins (Buck et al. 1992; Sires et al. 1995; Riese and Chapman 2000; Wolters and Chapman 2000), control of immune response (Riese et al. 1998; Antoniou et al. 2000), tumor metastasis and invasion (Lah and Kos 1998; Coulibaly et al. 1999), and aging alteration (Juhg et al. 1999; Cuervo and dice 2000) in the cell. Intracellular protein degradation occurs in two major sub-cellular systems: lysosomal and non-lysosomal ubiquitin-proteasome system. The lysosomal pathway is the first major site where protein degradation takes place due to action of combined random and limited action of lysosomal proteases containing cysteine cathepsins (Wolters and Chapman 2000). However, non-lysosomal machinery leading the ubiquitin-proteasome system is the other site where most of the endogenous cellular proteins are degraded (Riese and Chapman 2000). Proteolysis is important not only for the renewal of proteins and disposal of defective protein molecules but also for the energetic mobilization of endogenic proteins. Such proteolytic processing can be regulated by the activation of an inactive precursor, accessibility of peptide bond of a substrate, interaction with protease inhibitor, protease specificity, or combination of these factors (Buck et al. 1992). Protein turnover, multiple sclerosis, bacterial and viral diseases, malignancy and muscular dystrophy, among others, are all initiated and/or sustained by well-characterized thiol-dependent cathepsins. These enzymes, particularly cathepsins B and H, thus have no single but multiple functions and have generated massive interest in their properties and structures.
Therefore, the biochemical nature of cysteine cathepsins by which one may be distinguished from the other is briefly focused here. Further, our knowledge of the physiological substrates and inhibitors, structure and mechanism, and function of most of the thiol cathepsins is inadequate as compared to what we know about the regulation, fine structure, and kinetics of other proteases. Structural differences between various cysteine cathepsins result in variations in their substrate specificity and mechanism of inhibition. Although almost all the cysteine cathepsins have been crystallized, information on the amino acid sequences is not available for all and the specificity of these enzymes (Cathepsins B and H) remains controversial. Until now, the role of signal or pro-sequences during the transport/synthesis is not cleared. The present chapter, therefore, emphasizes relatively well-characterized thiol proteases, cathepsins B and H that illustrate the general characteristics in addition to the abovementioned properties and their diversity.
16.2 Lysosomal Cysteine Proteases
The name cathepsin was derived from the Greek word “Kathepsein” meaning to digest. It was first introduced in 1929 by Willstatter and Bamann to describe an acid protease distinct from pepsin. After a decade, Fruton et al. (1941) identified three enzymes in a crude preparation of cathepsin from bovine spleen which were called cathepsins I, II, and III. These enzymes were reclassified in 1952 by Tallan et al., who proposed the name cathepsins A, B, and C that acted on Z-Glu-Tyr, Bz-Arg-NH2, and Z-Gly-Phe, respectively. Since then, there are about 11 human cysteine proteases, i.e., Cathepsins B, C (J, dipeptidyl peptidase I), F, H(I), K(OC2,O2), L, O, S, V(L2), W(lymphopain), and X(P,Y,Z) (Turk et al. 2001; Rawlings et al. 2010) existing at the sequence level. With the advent of novel concepts and availability of genome sequences (Rossi et al. 2004), this number might probably increase, especially since several new mouse cathepsins without apparent human counterparts have been discovered (Guha and Padh 2008; Vidak et al. 2019). These cathepsins are assigned by simply applying letter designations differing from each other in their distribution, molecular properties, substrate specificity, and sensitivity to inhibitors (Agarwal 1990; Turk et al. 2001). Nomenclature and some molecular properties of lysosomal cysteine cathepsins are summarized in Tables 16.1 and 16.2.
Lysosomal cysteine cathepsins are optimally active at acidic pH values but are unstable at neutral or alkaline pH values. However, cathepsin S is the only exception that retains most of its activity at neutral or slightly alkaline pH (Kirschke et al. 1989). Most of these proteases are glycoprotein in nature, and they are active against large protein substrates and a wide range of small peptides. In 1972, three enzymes isolated from rat liver lysosomes with high proteolytic activity at pH 6–7 were shown to be thiol-dependent; these enzymes are cathepsins B, H, and L (Evered and Whelan 1978). However, two components (B1 and B2) are identified in the same preparation of cathepsin B by Otto (1971), and on the basis of their specificities (McDonald and Ellis 1975) cathepsin B2 is renamed as carboxypeptidase B and cathepsin B1 has been known as cathepsin B. Cathepsin H can readily be distinguished from the other cathepsins by its resistance to high temperatures and by the fact that it possesses both endo- and aminopeptidase activities. Similarly, cathepsin L has been recognized by using Z-Phe-Phe-CHN2 as a potent inhibitor of the enzyme. Likewise, on the basis of substrate specificity and sensitivity to inhibitors (Tables 16.3 and 16.4), several other thiol cathepsins (Table 16.1) isolated from various sources are found different from each other and cathepsins B, H, and L. However, many of the thiol-dependent cathepsins isolated in relatively small amounts are not yet well characterized. For example, cathepsin K (earlier known as cathepsin N), sometimes called “collagenolytic cathepsin,” is analogous to cathepsin L except that it shows slight activity against azocasein (Li et al. 2004). Likewise, beef spleen cathepsin S is similar to cathepsin L, but the two may be differentiated on the pattern of inhibition by Z-Phe-Phe-CHN2 and capability to hydrolyze synthetic substrates (Barrett and Kirschke 1981). Part of the problem in the study of these enzymes lies in their lack of activity toward commonly used synthetic substrates, which hinders attempts to distinguish one enzyme from the other. Unlike most, cathepsins J (now known as cathepsin C) and K may be differentiated on the basis of their high molecular weights (Liao and Lenney 1984). An unstable protease, now known as cathepsin F, is present in cartilages but resists inhibitors of major classes of proteases like PMSF, IAA, pepstatin, and leupeptin (Barrett and McDonald 1980). However, most of the molecular properties of cathepsins O and W are still unknown.
Thiol reagents include 2-mercaptoethanol, cysteine, cysteamine, dithiothreitol, reduced glutathione, and thioglycerol.
16.3 Biosynthesis and Transport
It is well recognized that lysosomal cysteine cathepsins (B, H, and L) are synthesized on membrane-bound ribosomes, which is supported by the fact that most of these enzymes are glycoproteins, as large precursors are moved cotranslationally into the Golgi apparatus through the lumen of the endoplasmic reticulum (Walter and Blobel 1982; Mainferme et al. 1985; Von Figura and Hasilik 1986; Diment et al. 1988; Smith and Gottesman 1989). During the transfer, it is thought that the precursors are subjected to a number of revisions in their carbohydrate and protein content. The exact site for these modifications is still an enigma. The large precursor has an extra peptide, known as a signal peptide or leader sequence having 15–30 bulky hydrophobic amino acids, is restricted to the NH2-terminus (Von Figura and Hasilik 1986). At this moment, a complex formed between cytosolic ribonucleoprotein and signal peptide, which is called signal recognition particle (Walter and Blobel 1982), probably regulates the translation of the enzyme. Subsequently, at the surface of the rough endoplasmic reticulum, the complex is attached to a receptor that is known as specific receptor protein (docking protein) (Meyer et al. 1982) and the emerging protein is transported into the lumen of the rough endoplasmic reticulum. The pro-sequences have generally been believed to mediate the localization of the newly synthesized polypeptide chains to their site of action and/or the regulation of their biological activities. Indeed, recovery of enzymatic activity of cathepsin L following the renaturation (Smith and Gottesman 1989) suggests that the propeptide has a crucial role in the folding and/or stability of the enzyme.
Several pieces of evidence suggest that the selective transport of these proteases from the Golgi bodies to the lysosomes is mainly refereed by a receptor situated in the Golgi that identifies mannose-6-phosphate residues/receptors (MPR) which are present in the precursors of lysosomal enzymes (Dingle 1984; Mainferme et al. 1985; Von Figura and Hasilik 1986). Although considerable work has been done on the transport and processing of cathepsin D (Gieselmann et al. 1983; Matha et al. 2006), from many findings observed by various workers, the role of these receptors is presumed to involve in the formation of a complex with newly synthesized cathepsin in Golgi and deliver the enzyme–receptor complex to an intermediate acidified compartment (endosome). After dissociation of the complex in the endosome, MPR is returned to the Golgi apparatus, while newly synthesized lysosomal cathepsins are transferred to the lysosomes. It should be pointed out that before transferring to lysosomes, a small fraction of these proteases (5–15%) is secreted (Kornfeld 1987). How these secreted enzymes are reached to lysosomes, is not clearly understood, but it seems that they are probably recaptured by receptor-mediated endocytosis and redirect to lysosomes via post-Golgi acidified compartment. It is interesting to note that sorting mechanisms are also possible to exist which do not engage MPR (Dingle 1984; Mainferme et al. 1985). Experimental examinations, however, on the sorting mechanisms for lysosomal cathepsins in the cell are still limited.
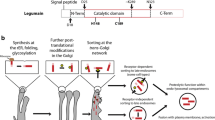
Cathepsins B and H, located in the different lysosomes, bind to the cell membrane. However, cathepsin L is situated in the lysosomes which are dispersed diffusely in liver cells. Thus, an important aspect of the functional share of various cathepsins is their different localizations (Ii et al. 1985; Hara et al. 1988; Kominami et al. 1988). Posttranslational processing and maturation of cathepsins B and H given by Katunuma (2010) are summarized in Fig. 16.1. Cathepsin B is translated as 17, 62, and 252 amino acids of prepart, pro-part, and mature part, respectively (Towatari and Katunuma 1978). After removing prepart cotranslationally, the procathepsins are translocated into Golgi apparatus where 38th-Asn in pro-part and 111th-Asn in the mature part are glycosylated by high mannose-type carbohydrate. Now the mannose-6-phosphate sugars participate as the targeting marker to the lysosomes after cleaving off the pro-part. However, cathepsin H is translated as 21, 114, and 217 amino acids of prepart, pro-part, and mature part, respectively. While the pro-part has two carbohydrate chains at 70th-Asn and 90th-Asn, only one carbohydrate chain is attached at the 99th-Asn in the mature part (Taniguchi et al. 1985; Ishidoh et al. 1987). The commencement of degradation is started from the 47th nicking bond in cathepsin B (Chan et al. 1986) and the 177th nicking bond in cathepsin H (Ishidoh et al. 1987) by some cysteine proteases.

Posttranslational processing and modification of cysteine cathepsins B and H (Kominami et al. 1988)
16.4 Properties of Cathepsins B and H
While cathepsin B has been isolated from various sources such as rat liver (Takio et al. 1983), human liver (Barrett and Kirschke 1981; Musil et al. 1991) and placenta (Swanson et al. 1974), bovine spleen (Otto 1971; McDonald and Ellis 1975) and lymph nodes (Zvonar-Popovic et al. 1979), calf brain (Suhar and Marks 1979), rabbit testis (Scott et al. 1987), buffalo liver (Fazili and Qasim 1986; Salahuddin et al. 1996), spleen (Ahmad et al. 1989), kidney (Lamsal et al. 1997) and lung (Agarwal et al. 2016, 2018), porcine spleen (Takahashi et al. 1984a, b), goat spleen (Agarwal and Khan 1987a; Agarwal et al. 1997), and horse muscle (Yoshida et al. 2015), cathepsin H has been purified from human liver (Schwartz and Barrett 1980), kidney (Popovic et al. 1988), brain and meningioma (Chornaya and Lyannaya 2004), rat spleen (Yamamoto et al. 1984) and liver (Kominami et al. 1985), bovine spleen (Willenbrock and Brocklehurst 1985) and brain (Azaryan and Galoyan 1987), rabbit lung (Singh and Kalnitsky 1978), porcine spleen (Takahashi et al. 1984b), goat liver (Ravish and Raghav 2014), and recently by buffalo lung (Singh et al. 2020). Some of the physicochemical properties of buffalo lung cathepsins B and H studied by us are summarized in Table 16.5. In contrast to cathepsin H, which represents a single-chain enzyme molecule in most of the species, characterization of cathepsin B from porcine spleen (Takahashi et al. 1986a) and goat spleen (Choudhury et al. 1997) shows the presence of two isozymes in these species, suggesting that the cathepsin B isozymes are two separate gene products and/or have a probable tissue/species dependence.
16.4.1 Tissue Distributions
The levels of cathepsins B and H in various rat tissues and peripheral blood cells determined using a sensitive immunoassay (Katunuma and Kominami 1986) are summarized in Table 16.6. Large differences in the concentrations of lysosomal proteases are observed in various tissues. However, it has been verified by immunohistochemical techniques that the activities and concentrations of these proteases are varied within cell type even in one tissue such as brain and liver. The ratio of the levels of cathepsins B and H in tissues also vary: the brain, stomach, esophagus, skeletal muscle, and adrenal gland contain higher levels of cathepsin B, whereas the lung, skin, and liver contain higher levels of cathepsin H (Kominami et al. 1985). Depending on the tissue, several cathepsins represent significant differences in protein expression levels and ratios, implying that each cathepsin(s) may have very specific cellular functions (Zavasnik-Bergant and Turk 2007). While cathepsin H is localized in lysosomes of pancreatic islet A-cells, cathepsin B is contained in those of both A- and B-cells (Watanabe et al. 1988; Uchiyama et al. 1994). This heterogeneity among cysteine proteases in lysosomes may reveal the disparity in metabolic substrates between the two cells.
16.4.2 Storage and Assays
Most of the laboratories are facing a problem to retain the activity of either cathepsin B or H during storage and the optimization of their assay systems. Despite several reports on storage of these enzymes where the activity is lost up to 50% or more (Otto 1971; Barrett and Kirschke 1981), cathepsin B can be stored at 0 °C for a long period in buffer concentration more than 0.1 M, pH 5.0, containing 1 mM EDTA (Khan and Ahmad 1987). Similarly, cathepsin H can be preserved for several months at −20 °C in sodium acetate buffer (0.02 M, pH 4.8) having 1 mM EDTA and 0.02% sodium azide at concentrations more than 1 mg/mL (Singh et al. 2020). As much as 90% activity of these enzymes can be recovered by these methods.
Since the activity of an enzyme depends on both the ionic strength of the buffers and the nature of the buffer components, the maximum catheptic activity can be achieved at low buffer concentrations, preferably in sodium phosphate buffer (0.02 M, pH 6.5), in comparison to other phosphate buffers (Agarwal and Khan 1987b; Singh et al. 2020). These observations also account for the discrepancy in the values of kinetic parameters (Km, Kcat, and Vmax) of cathepsins B and H reported from different laboratories where the buffers of higher ionic strengths were used (Otto 1971; Barrett and Kirschke 1981; Takahashi et al. 1984a, b; Fazili and Qasim 1986). Although the exact mechanism of buffer ions with the amino acids involved in the active site of the enzyme is not known at present, due care should be taken during the choice of buffers for the assay of lysosomal cysteine proteases.
16.4.3 Enzymes’ Nature
Most of the evidence suggests that cathepsins B and H require the integrity of their lone thiol group for the expression of their biological activity. The thiol nature of each enzyme is inferred from its inactivation by stoichiometric amounts of heavy metal ions and thiol-blocking reagents such as IAM, IAA, PHMB, PCMB, and NEM (Otto 1971; Barrett and McDonald 1980; McDonald and Barrett 1986; Agarwal et al. 1997; Singh et al. 2020). Cathepsin B is also inhibited by serum proteins (α2-macroglobulin, IgG, haptoglobin) (Starkey and Barrett 1973; Barrett and Kirschke 1981), endogenous protease inhibitors (cystatins, stefins, kininogens) (Lenney et al. 1979; Katunuma and Kominami 1986; Turk et al. 2012), low-molecular-weight substances (leupeptin, chymostatin, elastatinal, antipain, E-64) (Takahashi et al. 1984b; Yamamoto et al. 1984; Agarwal et al. 1997; Lamsal et al. 1997), and C-Ha-ras gene products (Hiwasa et al. 1987). Likewise, cathepsin H is also inhibited by α2-macroglobulin (Mason 1989) and by the three groups (stefins, cystatins, and kininogens) of intracellular and extracellular protein inhibitors (Machleidt et al. 1986; Abrahamson et al. 1991; Lenarčič et al. 1996). The enzyme binds with these inhibitors more strongly in comparison to cathepsin B (Guncar et al. 1998). In contrast to cathepsin B and other cysteine cathepsins (L, P, S, and K), cathepsin H is only poorly inhibited by antipain, chymostatin, IAA, mercuric chloride, and by irreversible epoxysuccinyl-based inhibitors derived from E-64 (Barrett et al. 1982; Guncar et al. 1998). Since the selectivity and potency of inhibitors are due to their affinity for the specificity sites of enzyme, the discrepancies in inhibitory effects of these compounds on cathepsins B and H indicate the structural difference of these two enzymes. Further cathepsin H is not or less inhibited by leupeptin (Schwartz and Barrett 1980; Singh et al. 2020), and it is, however, powerfully inhibited by substrate analogues composed of a single amino acid residue bound to a diazomethane or fluoromethane group, which react with the active-site cysteine (Angliker et al. 1989). Although the mechanism of inhibition for a number of thiol protease inhibitors is known, it may, however, be possible that some of these inhibitors (whose mechanism is not known) act by mechanisms analogous to those proposed for the action of serine protease inhibitors.
16.4.4 Specificity
Cysteine cathepsins display broad specificity, splitting their substrates preferentially after basic or hydrophobic amino acid residues. This is factual not only for synthetic substrates but also for protein substrates and steady with their roles in intracellular protein degradation (Turk et al. 2000). These thiol-dependent cathepsins show mainly endopeptidase activity except cathepsins C and X, which are exopeptidases only. While cathepsin C is an aminodipeptidase (Turk et al. 2001), cathepsin X is a carboxymonopeptidase (Gunčar et al. 2000). However, cathepsin B shows an unusual property with its specificity or action on various peptide and protein substrates. The protease is generally assumed to be an endopeptidase because it hydrolyzes amide substrates in which the COOH termini are substituted (Barrett 1977; Bond and Butler 1987). It also possesses the activity of a carboxypeptidase which releases dipeptides sequentially (McDonald and Ellis 1975; Barrett 1977; McKay et al. 1983). However, there is no evidence of specificity for basic amino acid residues (Arg-Arg) like synthetic substrates, if insulin B chain, STI, glucagon, or fructose 1,6-bisphosphate aldolase and many other protein substrates are digested with cathepsin B. Although all the cysteine proteases studied to date exhibit endopeptidase activity toward protein and polypeptide substrates, the display of both endo- and exopeptidase activities, depending on the substrate, by cathepsin B seems to be unusual but not unique; cathepsin H, for example, shows endo- and aminopeptidase activity on polypeptide substrates (Barrett and Kirschke 1981; Koga et al. 1992). The dual activities of the enzyme appear compatible with the well-known specificity of cathepsin H hydrolysis of small synthetic substrates. The enzyme can hydrolyze amide bonds of a substrate with a free α-amino group, for instance Arg-NNap. It can also hydrolyze similar substrates with a blocked α-amino group like Bz-Arg-NNap (Barrett and Kirschke 1981). However, the studies by Takahashi et al. (1988) on porcine spleen cathepsin H show that peptide substrates are cleaved exclusively by aminopeptidase activity; it does not hydrolyze large polypeptides or proteins and thus possesses no detectable endopeptidase activity. Similarly, cathepsin B isolated from either porcine spleen (Takahashi et al. 1986b) or goat spleen (Agarwal 1988) is exclusively a dipeptidyl carboxypeptidase with peptide and protein substrates and has no significant endopeptidase activity.
The assumption that cathepsins B and H are endopeptidases is based on their hydrolyzing activity against synthetic substrates which are hydrolyzed by other endopeptidases such as trypsin and papain. In earlier studies (Barrett 1977; Evered and Whelan 1978; Bond and Butler 1987; Brocklehurst et al. 1987), it was believed that the degradation of proteins by cathepsin B or H came from its endopeptidase activity, though this had never been demonstrated directly. Besides, other thiol cathepsins like L, S, and P in mammalian tissue are similar to cathepsin B or H in many physical properties, and some have overlapping enzymatic activities. Thus, they are not easy to eliminate in a purified cathepsins B and H preparation, and this may account for the apparent endopeptidase activity in some enzyme preparations. Hence, the action of proteases on native protein substrates cannot always be expected on the basis of their action on synthetic substrates. However, the observation that cathepsins B and H have “no endopeptidase activity” may be a consequence of the substrates tested or species/tissue disparity because the endopeptidase activity of human, bovine, and rat liver cathepsin B is well recognized (Barrett 1977; Bond and Butler 1987; McKay et al. 1983). But the substrate, oxidized insulin B chain, cleaved endoproteolytically by human liver cathepsin B (McKay et al. 1983) cannot be hydrolyzed to any extent by porcine spleen cathepsins B (Takahashi et al. 1986b) or H (Takahashi et al. 1988). Such drastic differences raise the question of whether they are indeed different enzymes. The species dependence of cathepsin B, however, has been confirmed after observing the differences in catalytic and molecular properties of buffalo, bovine, and goat versions of cathepsin B (Agarwal 1991; Lamsal et al. 1997).
Cezari et al. (2002) have inspected the specificity of subsites S1, S2, S1′, and S2′ for the carboxydipeptidase activity of cathepsin B with internally quenched fluorescent peptides. Subsite S1 preferentially accepts basic amino acids for hydrolysis, though substrates with Phe or amino acids bearing an aliphatic side chain at P1. Despite the existence of Glu245 at S2, the subsite has a clear choice for aromatic amino acid residues, and a substrate with Lys residue at P2 is hydrolyzed better in comparison to one having an Arg residue. S1′ is a hydrophobic subsite and S2′ exhibits a preference for Phe or Trp residues. In the case of cathepsin H, Takahashi et al. (1988) examined aminopeptidase activity with oligopeptide substrates and suggested that the specificity of the enzyme depends primarily on S1-side-chain identification. Preferentially released NH2-terminal residues have large basic (Arg and Lys) or hydrophobic (Phe, Trp, Leu, and Tyr) side chains, whereas the presence of a free α-amino group of substrates is projected to be of secondary significance (Takahashi et al. 1988).
Among the synthetic substrates, the low-molecular-weight peptides containing arginine in the P1 position (Bz-Arg-NNap, Bz-Arg-NPhNO2) are hydrolyzed by cathepsins B and H very efficiently. McDonald and Ellis (1975), however, have shown that substrates containing paired arginine residues are hydrolyzed most effectively only by cathepsin B. If the N-terminus is unsubstituted, as in Arg-Arg-NNap, then the rate is reduced to about 1% of that with the blocked substrate (Z-Arg-Arg-NNap). The most sensitive substrates discovered for cathepsin H are Arg-NNap and Leu-NNap; Leu-NNap is somewhat less susceptible than Arg-NNap (Barrett and Kirschke 1981). However, a number of other synthetic substrates having different leaving groups such as NNapOMe and NMec have also been accounted for (MacGregor et al. 1979; Barrett and McDonald 1980). The NMec derivatives provide a sensitive fluorimetric assay and are often preferred as they constitute less of a health hazard. The more convenient chromogenic and fluorogenic substrates containing the leaving group 7-amino-4-trifluoromethyl coumarin have been worked out for cathepsins B and H (Tchoupé et al. 1991), but there is still no specific substrate for cathepsin L.
In the absence of the mini-chain, substrate specificity in human cathepsin H shifts from aminopeptidase to endopeptidase (Dodt and Reichwein 2003). This is shown by a genetically engineered mutant of human cathepsin H missing the mini-chain, des[Glu(−18)-Thr(−11)]-cathepsin H, which displays endopeptidase activity toward the synthetic substrates; it is not cleaved by wild-type recombinant cathepsin H. Nevertheless, the mutant enzyme does not show significant aminopeptidase activity against H-Arg-NH-Mec, a well-known substrate for native human cathepsin H (Dodt and Reichwein 2003). This has been confirmed earlier for cathepsin B in which kinetic data on the interaction of substrates and inhibitors with recombinant variants support the functional activity of the occluding loop of cathepsin B as the main structural part for determining the exopeptidase activity of the protease (Hasnain et al. 1992; Illy et al. 1997; Nagler et al. 1997). Thus, the kinetic studies on substrate hydrolysis and enzyme inhibition reveal the importance of the mini-chain/occluding loop not only as a structural barrier for endopeptidase-like substrate cleavage but also as a structural framework for transition state stabilization of substrates.
16.5 Structure of Cathepsins B and H
From the amino acid sequences of rat liver (Takio et al. 1983), human liver (Ritonja et al. 1985), bovine spleen (Meloun et al. 1988), and porcine spleen (Takahashi et al. 1984b), cathepsin B represents that the enzyme together with cathepsins H, S, K, and L belong to the papain “superfamily.” According to the nucleotide sequences, cathepsin B from man (Chan et al. 1986) or rat (Fong et al. 1986) synthesized as a polypeptide chain containing 339 amino acid residues is manufactured to the mature single-chain molecule of 254 amino acids (Nishimura and Kato 1987). Likewise, the amino acid sequences of rat liver (Takio et al. 1983), mouse macrophages (Lafuse et al. 1995), and human kidney (Ritonja et al. 1988) cathepsin H agree with those deduced from the rat (Ishidoh et al. 1989) and human (Fuchs et al. 1988) cDNA sequences. Active human kidney cathepsin H is composed of 230 amino acid residues, 222 of which form a single chain and 8 residue long mini-chain, which is disulfide-linked to the rest of the enzyme (Ritonja et al. 1988). From these sequences, it is evident that the mini-chain brought into existence from cathepsin H propeptide is established between propeptide residues Thr83P and Glu76P (Guncar et al. 1998). However, in some of the mammalian tissues, active cathepsin B or H is present as a two-chain molecule comprising of the light (4–5 kDa) and heavy (20–22 kDa) polypeptide chains cross-linked covalently through a disulfide bridge (Machleidt et al. 1986). This suggests that like other proteolytic enzymes, cathepsins B and H are also synthesized in a precursor form from which the mature enzyme is produced by the removal of the pro-sequence through several proteolytic cleavages.
16.5.1 Sequence Homology
One of the important features of the amino acid sequences of cathepsins B, H, and L, and plant protease papain is the presence of somewhat high level of identity (31–56%) in the amino-(active-site cysteinyl) and (28–44%) in the carboxy-terminal (active-site histidinyl) regions but quite less (13–30%) in the middle region (Takio et al. 1983; Dufour 1988; Ritonja et al. 1988). As can be seen in Table 16.7, about 24% of the residues are identical in the N-terminal region within all four enzymes. Low degree of identity in the central (residues 78–152 in cathepsin B) region, a sector in which a large number of deletions/insertions are found, is perhaps due to a single large insertion of 28–30 residues during the long process of divergent evolution (Dufour 1988). However, the functional importance of this part is not understood, still in plant protease papain. In the C-terminal regions where the active site histidinyl is present, a low level of identity (28–44%) in comparison to the cysteinyl regions (31–56%) proposes that the amino acid sequences in the surrounding area of active site histidinyl residue may reflect the diverse peptidase specificities of cathepsins B and H (Takio et al. 1983). Further, the sequences of cathepsins H and L are very much close to those of the plant enzyme papain than to that of cathepsin B, indicating that cathepsin B must have deviated from the common ancestral gene long before cathepsins H and L.
16.5.2 Active Site
The amino acid residues, Cys25 and, as well as Gln19, Asn175, and Trp177, involved in enzyme catalysis are preserved at/around the active site in all cysteine proteinases. Indeed, cathepsins B, H, and L share the amino acid sequence Asn-Ser-Trp (papain residues 175–177) with actinidin and papain. According to X-ray studies on plant thiol proteases, a hydrogen bond formed between Asn175 and the active-site His159 is shielded by Trp177 from the solvent (Dufour 1988). Taking into consideration, a large amount of resemblance in papain and actinidin active-site residues and the sequence homologies among plant and animal cysteine proteinases, all these five enzymes including cathepsins B, H, and L, certainly have same catalytic mechanisms. Any alteration in the active site may happen in the hydrophobic specificity “pocket,” the S2-subsite. For example, the substitution of Ser205 in papain by Glu in cathepsin B results in dissimilar surface characteristics of the S2-subsite binding site (Baker 1980); the side chain of Glu205 may combine with one arginine side chain of the extremely specific substrate Z-Arg-Arg-NMec (Polgar and Csoma 1987).
Whereas six disulfide bridges are restricted in the NH2-terminal half of the molecule, only two cysteine residues (Cys29 and Cys240) of human cathepsin B are unpaired. Topologically Cys29 is the same as the reactive (active-site) cysteine (Cys25 in papain) of all other thiol proteases; Cys240 situated close to the COOH-terminal half of the polypeptide chain surface is unique to cathepsin B (Musil et al. 1991). However, in addition to a disulfide bond produced between Cys205 and Cys80P of the mini-chain, porcine or bovine cathepsin H has three disulfide bridges (Cys22–Cys63, Cys56–Cys95, and Cys154–Cys200) which are topologically equivalent to the disulfide bridges in actinidin (Baker 1980). The active-site cleft of porcine (Guncar et al. 1998) or bovine (Baudys et al. 1991) cathepsin H runs the top of the molecule transversely; the wide ends but thin in the middle of active-site cleft contains catalytic (active-site) residues Cys25, His159, and Gln19. In contrast to all other known structures of cysteine proteases, the imidazole ring of His159 in cathepsin H does not make a thiolate-imidazolium ion pair with Cys25 (Guncar et al. 1998).
16.5.3 Carbohydrate Moieties
While cathepsin B exists in 5–7 molecular forms having pIs in the range of 4.5–5.5 (Table 16.2), cathepsin H presents only in three molecular forms (pIs in the range 6.0–7.1) differing both in carbohydrate contents (Barrett 1977; Barrett and McDonald 1980; McDonald and Barrett 1986). The oligosaccharide structures of lysosomal cathepsins are asparagine-linked and predominantly high mannose type (Kornfeld and Kornfeld 1980). A single N-acetylglucosamine and the fucosylated pentasaccharide are present in a molar ratio of 73:27 in porcine spleen cathepsin B (Takahashi et al. 1984a). In contrast, cathepsin H isolated from the same source shows four high mannose-type oligosaccharides having 6–8 mannose residues (Takahashi et al. 1984b). However, a linear tetrasaccharide and a branched pentasaccharide without fucose (absent in porcine spleen cathepsin B) are reported in rat liver cathepsin B (Taniguchi et al. 1985). Similarly, two (high mannose type) oligosaccharides having 9 and 5 mannose residues are found in rat liver cathepsin H beside the three oligosaccharides present in porcine spleen cathepsin H. In both the cases, although the carbohydrates are linked to Asn 111, the structural differences of asparagine-linked sugar chains reflect species and/or organ specificity of glycoproteins among the rat liver and porcine spleen cathepsins.
16.5.4 Structural Transition
An exclusive feature shared by animal thiol proteases, cathepsins B, H, and L, is that they are freely inactivated at neutral pH (Zvonar-Popovic et al. 1980; Ohtani et al. 1982; Khan et al. 1986; Agarwal and Khan 1987a). In contrast to cathepsin L, cathepsin B is very sensitive toward pH, urea, and guanidine hydrochloride (Agarwal and Khan 1987a; Ahmad et al. 1989; Khan et al. 1992). Buffalo spleen cathepsin B loses its structure as well as the activity irreversibly at alkaline pH; the inactivation of the enzyme is, however, found reversible at acidic pH (Khan et al. 1992). The activity of goat/buffalo enzyme is lost reversibly at denaturant concentrations which does not cause a major change in its secondary structure, and suggest that the inactivation may be ascribed due to slight perturbation in the surroundings of the amino acid residue(s) at and/or around the active site of the enzyme (Agarwal and Khan 1988; Khan et al. 1992). The inactivation process becomes irreversible at high urea/guanidine hydrochloride concentrations leading to the structural changes in the enzyme. Nevertheless, the existence of a multidomain structure in mammalian cathepsin B is first reported after performing a series of denaturation and renaturation experiments by Agarwal and Khan (1988). An important feature of the unfolding–refolding transition of the goat spleen enzyme is that it is not completely reversible and appears to start at extremely low urea concentration. Surprisingly, cathepsin H purified recently from buffalo lung (Singh et al. 2020) unfolds reversibly in two main stages, having a stable intermediate between its native and fully denatured states (unpublished results). The equilibrium and kinetic intermediates have also been confirmed by in vitro studies of cathepsins B, H, and D (Lah et al. 1984; Agarwal and Khan 1988). Although similar data on the precursor “pro-forms” of these enzymes are not available, the non-reversibility of the unfolding transition of the mature enzymes do suggest a role for the pro-sequences in the folding of lysosomal cathepsins.
16.5.5 Secondary Structure
Explorations on the secondary structure of cysteine cathepsins particularly B, H, and L have remained inconclusive so far. According to Garnier et al. (1978), the method predicts the helical structure in cathepsins B, H, and L, respectively, 14, 24, and 16%, whereas the procedure of Chou and Fasman (1974) shows 27, 35, and 30% helical content, respectively. The interpretations of the circular dichroic spectrum (the most commonly used method for determining protein secondary structure) of cathepsin B have varied from one laboratory to the other (Zvonar-Popovic et al. 1980; Bansal et al. 1981; Dufour 1988; Khan et al. 1992). CD spectrum of bovine enzyme conforms to about 12% α-helix and 31% β-sheet (Zvonar-Popovic et al. 1980), whereas buffalo cathepsin B complies with 26% α-helix and 23% β-structure (Khan et al. 1992). This difference in the secondary structure probably seems to be pH-dependent because 34, 65, and 51% α-helix have been reported, respectively, at pHs 5.6, 7.4, and 10.2 in rabbit cathepsin B (Bansal et al. 1981). Moreover, the same pattern is found in cathepsin L where the inactivation at neutral pH is connected to the loss of helical content (40% at pH 5.8 and 17% at pH 7.0) in the enzyme (Dufour et al. 1988). The reason for such an effect may be due to change in the ionization state of histidine side chains which are chiefly situated in the predicted α-helix regions. Further, the ordered structures in cysteine proteases are well preserved in the NH2-terminal and COOH-terminal parts (Dufour 1988). Since the maximum insertions/deletions and substitutions in these proteases emerge in the central region, the most important changes in the secondary structures would have occurred in this part. However, the insertion of 28–30 residues in this region of cathepsin B does not change the overall molecular conformation and conformational organization of active site residues in the enzyme (Dufour 1988).
16.5.6 Gene Structure
Whereas the gene structure of cathepsin H has been worked out from rat (Ishidoh et al. 1989) and murine (Buhling et al. 2011), the structure of cathepsin B has been characterized by a genomic DNA segment encoding mouse (Ferrara et al. 1990) and carp (Tan et al. 2006). The isolated clone (λ32) has all the exons matching to the cDNA sequence except for the leader region. The genomic insert spans 15 kbp consisting nine exons encoding 339 amino acids of mouse preprocathepsin B. However, the gene structure of rat cathepsin H comprises at least 12 exons spanning in total more than 21.5 kbp; cathepsin L gene spans 8.5 kbp and comprises eight exons (Ishidoh et al. 1991). A common characteristic for the gene structure of all examined cysteine cathepsins and aleurain (a thiol protease from aleurone cells) is that intron break points are not established at the joints of the pre-peptide, pro-peptide, and mature enzyme regions. Thus, there is no proof that the gene structure of cathepsin B or H communicates to functional parts.
The number and positions of the introns, however, vary between these cathepsins. For instance, the gene encoding rat cathepsins H and L contain 12 and 8 introns, respectively, whereas the gene structure of mouse cathepsin B represents a minimum of nine introns (Ferrara et al. 1990; Ishidoh et al. 1991). Similarly, in cathepsins B and H genes, five introns break off the two active-site cysteine and histidine residues, instead of two in cathepsin L. Like other cysteine protease genes, the region around the active-site (Cys29) residue (the most conserved region) in cathepsin B is cracked by an intron, but on the contrary with cathepsins H and L the intron break point is positioned immediately following the active site. The differences in both the number and position of introns between thiol protease genes suggest that the relation between the genes is not direct. Since cathepsin H gene is formed of four rather than two ancestral gene parts found in aleurain, and the GC content of dissimilar exons are more uniform for cathepsin B gene than for cathepsins H and L (Ferrara et al. 1990; Ishidoh et al. 1991), the earlier notion that four enzymes (cathepsins B, H, L, and papain) derived from a common ancestral gene seems not to be true. However, the preserved sequence around the cysteinyl active site which has probably evolved in numerous ways in these enzymes proves an important function of this region for hydrolyzing activity of thiol cathepsins.
16.5.7 Crystal Structure
Among the investigated 11 cysteine human cathepsins, cathepsin B is the first one whose crystal structure is determined in the 1990s (Musil et al. 1991) which is followed by cathepsins K (McGrath et al. 1997), L (Fujishima et al. 1997), H (Guncar et al. 1998), V (Somoza et al. 2000), X (Gunčar et al. 2000), C (Turk et al. 2001), F (Somoza et al. 2002), and S (Turkenburg et al. 2002). Although three-dimensional (3D) structure of two more human thiol cathepsins, O and W, is not yet known, a 3D-based sequence arrangement of the mature structure of the nine cysteine cathepsins with identified 3D structure reveals conservation of the active-site residues (Cys25 and His163, cathepsin L numbering), the N-terminus Pro2, the residues interact with the main chain of the bound substrate (Gln19, Gly68, and Trp183) and certain cysteine residues (Turk et al. 2012). The plant thiol proteases for which 3D structures deduced earlier by X-ray diffraction data are papain (Drenth et al. 1968; Kamphuis et al. 1984) and actinidin (Baker and Dodson 1980). These enzymes exhibit the same conservation pattern as present in lysosomal cysteine cathepsins.
The crystal structures of porcine cathepsin H (Guncar et al. 1998) and human cathepsin B (Musil et al. 1991) have been deduced by X-ray crystallography at the resolution of 2.1 Ǻ and 2.15 Ǻ, respectively. In each case, the enzyme consists of a single polypeptide chain folded to form two domains (left-hand “L” and right-hand “R”) with a deep cleft between them. The L-domain is mainly α-helical having the longest central helix and the R-domain is based on a type of β-barrel facing strand(s) which form a coiled structure; the barrel is encircled by α-helix at the bottom. The two domains interrelate via an extended amphipathic interface stabilized by several hydrogen bonds plus hydrophobic interactions. The interface unlocks at the top into a V-shaped active-site cleft where two catalytic residues, cysteine and histidine, are situated. The reactive site cysteine is located at the N-terminus of the central helix of the L-domain, whereas the histidine is situated within the β-barrel residues of the R-domain. A thiolate-imidazolium ion pair formed between two catalytic residues is essential for the proteolytic activity of cathepsin B and other cysteine cathepsins except for cathepsin H (Guncar et al. 1998; Turk et al. 2012).
Human cathepsin B is roughly disk-shaped having a thickness of 30 Ǻ and a diameter of 50 Ǻ (Musil et al. 1991). Out of 248 distinct cathepsin B amino acid residues, 166 α-carbon atoms are topologically equivalent with α-carbon atoms of papain. But several big insertion loops which modify its properties are present on the molecular face. The occluding loop containing 108–119 amino acid residues of the enzyme takes up the back of the active-site cleft resulting in cathepsin B, a carboxypeptidase. Seven disulfide connectivities are in full agreement with those determined for bovine cathepsin B by chemical methods (Baudyš et al. 1990). However, porcine cathepsin H is an ellipsoidal molecule having dimensions 32 × 26.5 × 24 Ǻ (Guncar et al. 1998). Superposition of the α-carbon atoms of cathepsin H on the α-carbon atoms of actinidin, papain, and cathepsin B exhibits that 180 amino acids of cathepsin H and actinidin (Baker 1980) are topologically equal, 173 with papain (Kamphuis et al. 1984) and 156 with human cathepsin B (Musil et al. 1991). However, the mini-chain (an octapeptide linked to the R-domain) attached through a disulfide bridge to Cys205 of the body of cathepsin H is implicated in the steric regulation of the accessibility of the active-site cleft (Baudys et al. 1991) specifying aminopeptidase activity of the enzyme (Guncar et al. 1998). Moreover, the structure strengthens the outline of disulfide bridges in bovine cathepsin H (Baudys et al. 1991); the three disulfide bonds in the enzyme are topologically equivalent to the disulfide bridges in actinidin (Baker 1980). The stability of the mini-chain in cathepsin H and procathepsin H is studied recently by Hao et al. (2018), and the results indicate that the mini-chain is indeed more dynamic in procathepsin H, whereas it reorients to the more stable conformation in cathepsin H during the process of activation.
16.6 Overall Structure–Activity Relationship
One of the significant aspects, i.e., specificity diversity, has now been cleared from the crystal structures of cathepsins B and H. The 3D structures displayed that exopeptidase exhibits extra structural features that alter the active-site cleft (Musil et al. 1991; Guncar et al. 1998). While the active-site cleft expands along the whole length of the two-domain interface in endopeptidases, additional features reduce the number of binding sites in exopeptidases (Turk et al. 2003).
In cathepsin B, substrate binding is governed by a novel insertion loop that packs the active-site cleft on the primed subsites and seems to favor binding of peptide substrates with two residues carboxy-terminal to the scissile peptide bond; the occluding loop of the enzyme uses two histidine residues (His110 and His111) to port the C-terminal carboxylic group of the peptidyl substrate, suggesting an explanation for the well-known dipeptidyl carboxypeptidase activity of the enzyme (Musil et al. 1991). The other subsites neighboring to the reactive site Cys29 are quite similar to papain; Glu245 in the S2 subsite supports the basic P2 side chain. Besides the histidine residues, the hidden Glu171 might stand for a group with pKa of ~5.5 close to the active site, which controls exo- and endopeptidase activity of the enzyme. Since the exact role of these residues remains speculative, it may be further clarified by recombinant methods.
However, cathepsin H utilizes a region from its propeptide part to represent features that fill up the active-site cleft on the non-primed subsites S2 and S3; the enzyme uses a carboxylic group of the main and/or side-chain residues to port the positively charged NH2-terminus of the peptidyl substrate. This carboxylic group is situated at the COOH-terminus of an octapeptide element (originate from the propeptide), termed the mini-chain which remains linked to the active-site cleft through the side chains of Gln78P, Cys80P, and Thr83P of cathepsin H after its activation, providing an essential responsibility of mini-chain in the aminopeptidase activity of the enzyme (Guncar et al. 1998). This has, however, been confirmed after production and characterization of cathepsin H lacking mini-chain, resulting in a switch of its substrate specificity to endopeptidase (Dodt and Reichwein 2003; Vasiljeva et al. 2003). Fascinatingly, in aminopeptidases, glycosylation not only plays a crucial role in stabilizing the structure of the added characteristics but also fills up the active-site cleft tighten the substrate-binding (Turk et al. 2012). Unlike other cysteine proteases, cathepsin H is not inhibited by its own free propeptides (Horn et al. 2005).
16.7 Physiological and Pathological Implications
Lysosomes are the only cellular compartment having a series of hydrolases including cathepsins for the complete degradation of all classes of macromolecules by different modes (Wolters and Chapman 2000; Reiser et al. 2010; Vidak et al. 2019). Although it has not yet been possible to assign a precise function to a particular enzyme, various roles have been proposed for lysosomal thiol-dependent cathepsins. However, pieces of evidence obtained using thiol-specific inhibitors indicate that cysteine cathepsins particularly B, H, and L contribute a significant role in protein turnover (Bohley et al. 1974; Barrett 1977; Evered and Whelan 1978; Brocklehurst et al. 1987; Bromme and Wilson 2011; Verma et al. 2016). For example, the digestion of liver cytosolic proteins by rat liver lysosomal enzymes is entirely due to cathepsin B (Dean 1976) and at pH 6.0 short-lived cytosolic proteins are hydrolyzed in preference to long-lived proteins by cathepsin L (Bohley et al. 1974). In pulmonary emphysema, cathepsin B not only digests lung structural proteins but also inactivates enzymatically α1-proteinase inhibitor and reduces its protective concentration in and around lung tissues (Gairola et al. 1989). Moreover, cysteine cathepsins like H and K found in the lung are associated with inflammatory lung diseases (Chilosi et al. 2009; Faiz et al. 2013).
Apart from the fact in general protein turnover, the cysteine cathepsins could also have a role in the specific processing of proteins and thus in the regulation of enzymatic activity. Cathepsins B and L inactivate aldolase when tested with fructose-1,6-bisphosphate as a substrate (Bond and Butler 1987). The enzyme-treated aldolase showed no detectable change in molecular weight, suggesting that the modification may be significant for the regulation of aldolase activity. Certain other enzymes like glucokinase, pyruvate kinase, tyrosine- and alanine aminotransferases, asparaginase, and glyceraldehyde 3-phosphate dehydrogenase are also inactivated by cathepsin B (Evered and Whelan 1978; Brocklehurst et al. 1987; Barrett et al. 1998). Likewise, cathepsins B and H can also activate peptide hormones and various proteins by cleavage of their precursor forms, e.g., the conversion of proinsulin to insulin (Docherty et al. 1982), proalbumin to albumin (Quinn and Judah 1978), and trypsinogen to trypsin (Otto and Reisenkonig 1975). Furthermore, these enzymes participate in various other physiological processes such as protein synthesis, growth and aging, fertilization, memory, tissue resorption, and modeling (Bond and Butler 1987; Barrett et al. 1998). The exact mechanism of various inactivations or conversions of different enzymes/proteins by cathepsins B and H or other cysteine cathepsins is, however, still speculative.
It has been widely accepted that the extracellular matrix (ECM) is a reservoir for endogenous growth factors. In the body, endogenous proteases such as matrix metalloproteinases and their inhibitors are involved in routine ECM turnover for the maintenance of healthy tissue (Docherty et al. 1992; Burgess et al. 2009; Faiz et al. 2013). Among the papain family of proteases, cathepsins are capable of degrading ECM components with unique collagenolytic activity; cysteine cathepsins B, H, F, K, L, and S have the potential to participate in wound healing (Wolters and Chapman 2000) and ECM remodeling (Lutgens et al. 2007). Besides their role in ECM degradation, they are implicated in major histocompatibility complex (MHC) class II molecules which are expressed on the surface of antigen-presenting cells where after binding with exogenous proteins, MHCs present them to CD4+ T cells. Further, these cathepsins are also involved in the development and progression of cardiovascular diseases such as atherosclerosis, aneurysm, cardiac repair, and cardiomyopathy (Cheng et al. 2012). Nonetheless, imbalance in expression between cysteine cathepsins (S, K, L, and B) and their endogenous inhibitors (cystatin C) may favor proteolysis of ECM in the pathogenesis of such cardiovascular diseases (Wu et al. 2018). There is evidence for cathepsin B as a vital drug target for traumatic brain injury in which the enzyme gets away from its usual subcellular location (lysosome) to ECM (cytoplasm) where the unleashed proteolytic control causes devastation via autophagic, necrotic, apoptotic, and activated glia-induced cell death simultaneously with inflammation and ECM breakdown (Hook et al. 2015).
The activity of cathepsins (B, H, and L) is altered in several disease states such as muscular dystrophy, malignancy, ischemia, hypervitaminosis, multiple sclerosis, diabetes, arthritis, and various forms of cancer cells including the invasion of host tissue and metastasis (Evered and Whelan 1978; Brocklehurst et al. 1987; Turk et al. 2001; Vasiljeva et al. 2006; Victor et al. 2011; Verma et al. 2016). The mechanism for the precise regulation of these proteolytic enzymes however remains to be established. A report published in 1981 that cathepsin B activity is significantly elevated in a variant of B16 melanoma with high metastatic potential (Sloane et al. 1981), and it seems probable that the enzyme release from tumor cells may facilitate invasion and extravasation of tumors. This has been further confirmed by Weiss et al. (1990), who observed that invasive tumor cells enhance the level of cathepsin B in their plasma membranes, which may be used to degrade basement membrane components such as laminin and thereby facilitate tumor invasion. In human melanoma (primary and metastatic) cell lines, cathepsin B is highly expressed at the surface of metastatic but not of primary melanoma cell lines; chemical (CA-074 and CA-074Me) and biological (specific antibodies) inhibitors exert a powerful anti-invasive activity by a mechanism that brings into play the impairment of metastatic cell dissemination. However, in vivo studies (in murine xenografts), human melanoma growth, and artificial lung metastases are significantly reduced by CA-074, suggesting a role for cysteine protease in tumor growth and metastatic potential of human melanoma (Matarrese et al. 2010). Literally, what is the role of protease inhibitors (endogenous/chemical/biological) in tumor invasion and how they regulate the invasive potential of tumor cells is still unclear. Doxorubicin is an effective cytotoxic anticancer drug used for the treatment of malignancies and a broad range of solid tumors, and it shows severe dose-dependent toxicities (Gianni et al. 2003). However, a number of studies on cancer cells in vitro and tumor xenograft in vivo have revealed that cathepsin B-cleavable doxorubicin prodrugs are less toxic in vitro and more effective in vivo, suggesting the role of this enzyme-cleavable prodrugs in cancer therapy (Zhong et al. 2013).
Although fewer studies have been done on cathepsin H in cancer, the activity of the enzyme is elevated in breast cancer, melanoma, tumor invasion, colorectal and prostate carcinomas, and tumor vasculature (Gabrijelcic et al. 1992; Kos et al. 1997; del Re et al. 2000; Waghray et al. 2002; Gocheva et al. 2010). However, reduced cathepsin H expression has also been reported in squamous cell carcinomas of the head and neck and mixed expression patterns in pancreatic cancer cells (Kos et al. 1995; Paciucci et al. 1996). The differences in the expression pattern of cathepsin H in various cancers may indicate highly specific functions for the enzyme in different tissues at various stages of cancer. Moreover, the probable role of cathepsin H in tumor progression is its capability to degrade fibrinogen and fibronectin, suggesting that the enzyme may be occupied in the destruction of ECM components leading to cancer proliferation, migration, and metastasis (Tsushima et al. 1991; Turk et al. 2012). Nevertheless, a study on T3-mediated upregulation of cathepsin H involved not only in extracellular signal-regulated kinase activation but also in increased cell migration reveals that overexpression of cathepsin H in a subset hepatoma is thyroid-hormone-receptor-dependent having a significant role in hepatoma progression (Wu et al. 2011). Apart from cystatins (stefins A, B, and cystatin C), cathepsins B and H have been reported as significant prognostic markers in sera of patients with melanoma and colorectal cancer (Kos and Schweiger 2002).
16.8 Conclusions and Future Perspectives
While cysteine cathepsins like B, H, L, and S show the similarity in terms of physical properties, enzymatic activities, and homology with each other in amino acid sequences including the essential catalytic site region, it is not easy to distinguish these enzymes with respect to their biological functions, and it remains difficult to establish what role they take part in pathophysiological protein degradation. Similarly, a large number of different proteins can act as substrates for cysteine cathepsins in vitro studies, but there is little evidence to confirm that such reactions occur in vivo. Hence the substrate specificity and specific cleavage sites of these cathepsins are smart areas of study for accepting their role in life events and for designing drugs against these enzymes. However, the wide variations in tissue levels of cathepsins B and H are compatible with specific functions of these proteases in distinct tissues. It is, therefore, generally accepted that these enzymes participate in the breakdown of both intra- and extracellular proteins. Despite their several roles in protein catabolism, the exact mechanism of action of each cathepsin is still unknown. How pro-sequences help in the correct folding of a cathepsin molecule is yet to be explained. Moreover, the involvement of cathepsins B and H in inflammatory reactions has been proposed on evidence from inhibition studies and the detection of significant catheptic activities at inflammation sites. However, it is not yet certain that which cysteine cathepsin is directly responsible for the tissue breakdown at inflammatory sites. Gene structure and/or antibodies may be useful to provide a clue for understanding the molecular evolution and functional diversity among cysteine cathepsins.
Cysteine cathepsins are also emerging as major players in tumor progression, making them potential drug targets for a wide range of human cancers. Although cathepsins B and H have been used as drug targets to control ECM degradation and various metabolic activities involved during disease progression, the use of cysteine protease inhibitors (either endogenous, chemical, or biological) may be taken as a pioneering approach in the management of metastatic melanoma as well as on other carcinomas before making therapeutic strategies. Endogenous intracellular inhibitors are likely important in the control of these proteases, and the fluctuations in enzyme activities in cells are due to changes in inhibitor, rather than protease concentrations. Future studies in both clinical samples and preclinical models should now allow us to find out whether these cathepsins have similar or unique roles in different tumor microenvironments. Furthermore, these enzymes have been targeted by pharmacological drugs and inhibitors. Nonetheless, until now, no data are available on the effect of these inhibitors in various pathological events like atherosclerotic cardiovascular disease, neovascularization, polycystic kidney disease, and coronary artery disease.
References
Abrahamson M, Mason RW, Hansson H, Buttle DJ, Grubb A, Ohlsson K (1991) Human cystatin C. role of the N-terminal segment in the inhibition of human cysteine proteinases and in its inactivation by leucocyte elastase. Biochem J 273(3):621–626
Agarwal SK (1988) Specificity of goat spleen cathepsin B. Med Sci Res 16:505–506
Agarwal SK (1990) Proteases cathepsins-A view. Biochem Edu 18(2):67–72
Agarwal SK (1991) Species dependence of mammalian cathepsin B. Med Sci Res 19:485–486
Agarwal SK, Khan MY (1987a) Does cathepsin B play a role in intracellular protein degradation? Biochem Int 15(4):785–792
Agarwal SK, Khan MY (1987b) Influence of buffer constituents on the activity of goat spleen cathepsin B. Med Sci Res 15:387–388
Agarwal SK, Khan MY (1988) A probable mechanism of inactivation by urea of goat spleen cathepsin B. Biochem J 256:609–613
Agarwal SK, Choudhury SD, Lamsal M, Khan MY (1997) Catalytic and physico-chemical characteristics of goat spleen cathepsin B. Biochem Mol Biol Int 42(6):1215–1226
Agarwal SK, Ukil A, Sharma S (2016) Purification and characterization of cathepsin B from buffalo (Bubalus bubalis) lung. J Biochem Technol 7(1):1051–1057
Agarwal SK, Ukil A, Singh S, Sharma S (2018) Progressive study on the physiological role and catalytic properties of buffalo lung cathepsin B. Int J Health Sci Res 8(10):51–57
Ahmad S, Agarwal SK, Khan MY (1989) Purification and some properties of buffalo spleen cathepsin B. J Biosci 14(3):261–268
Angliker H, Wikstrom P, Kirschke H, Shaw E (1989) The inactivation of the cysteinyl exopeptidase cathepsin H and C by affinity-labelling reagents. Biochem J 262:63–68
Antoniou AN, Blockwood SL, Mazzeo D, Watts C (2000) Control of Ag presentation by a single protease cleavage site. Immunity 12(4):391–398
Azaryan A, Galoyan A (1987) Human and bovine brain cathepsin L and cathepsin H: purification, physico-chemical properties, and specificity. Neurochem Res 12:207–213
Baker EN (1980) Structure of actinidin after refinement at 1.7 Ǻ resolution. J Mol Biol 141:441–424
Baker EN, Dodson EJ (1980) Crystallographic refinement of the structure of actinidin at 1.7 Ǻ resolution by fast fourier least-squares methods. Acta Crystallogr 36:559–572
Bansal R, Singh S, Kidwai JR (1981) Conformational studies of cathepsin B: interaction with specific antibodies. Indian J Biochem Biophys 18:110–113
Barrett AJ (1977) In: Barrett AJ (ed) Proteinases in mammalian cells and tissues. Elsevier Scientific Publishing, Amsterdam, pp 181–208
Barrett AJ, Kirschke H (1981) Cathepsin B, cathepsin H and cathepsin L. Methods Enzymol 80:535–561
Barrett AJ, McDonald JK (1980) Mammalian proteases: a glossary and bibliography-endopeptidase, vol 1. Academic, London
Barrett AJ, Kembhavi AA, Brown MA, Kirschke H, Knight CG, Tamai M, Hanada K (1982) L-trans-epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem J 201:189–198
Barrett AJ, Rawlings ND, Woessner JF (1998) Handbook of proteolytic enzymes. Academic, London
Baudyš M, Meloun B, Gan-Erdene T, Pohl J, Kostka V (1990) Disulfide bridges of bovine spleen cathepsin B. Biol Chem 371:485–491
Baudys M, Meloun B, Gan-Erdene M, Fusek M, Mers M, Kostka V, Pohl J, Blake CCF (1991) S-S bridges of cathepsin B and H from bovine spleen: a basis for cathepsin B model building and possible functional implications for discrimination between exo- and endopeptidase activities among cathepsin B, H and L. Biomed Biochim Acta 50:569–577
Bohley P, Kirschke H, Langner J, Wiederanders B, Ansorge S, Hanson H (1974) In: Hanson H, Bohley P (eds) Intracellular protein catabolism. Barth, Leipzig, pp 201–209
Bond JS, Butler PE (1987) Intracellular proteases. Annu Rev Biochem 56:333–364
Bossard MJ, Tomaszek TA, Thompson SK, Amegadzie BY, Hanning CR, Jones C, Kurdyla JT, McNulty DE, Drake FH, Gowen M, Levy MA (1996) Proteolytic activity of human osteoclast cathepsin K. J Biol Chem 271(21):12517–12524
Brocklehurst K, Willenbrock F, Salih E (1987) In: Neuberger A, Brocklehurst K (eds) Hydrolytic enzymes. Elsevier, Amsterdam, pp 39–158
Bromme D, Wilson S (2011) Role of cysteine cathepsins in extracellular proteolysis. In: Parks WC, Mecham RP (eds) Extracellular matrix degradation. Springer, Berlin, pp 23–51
Brömme D, Okamoto K, Want BB, Biroc S (1996) Human cathepsin O2, a matrix protein-degrading cysteine protease expressed in osteoclasts. J Biol Chem 271:2126–2132
Bromme F, Li Z, Barnes M, Mehler E (1999) Human cathepsin V functional expression, tissue distribution, electrostatic surface potential, enzymatic characterization and chromosomal localization. Biochemistry 38:2377–2385
Buck MR, Karustis DG, Day NA, Honn KV, Sloane BF (1992) Degradation of extracellular matrix proteins by human cathepsin B from normal and tumor tissues. Biochem J 28:273–278
Bühling F, Gerber A, Häckel C, Krüger S, Köhnlein T, Brömme D, Reinhold D, Ansorge S, Welte T (1999) Expression of cathepsin K in lung epithelial cells. Am J Respir Cell Mol Biol 20(4):612–619
Buhling F, Kouadio M, Chwieralski CE, Kern U, Hohlfeld JM, Klemm N, Friedrichs N, Roth W, Deussing JM, Peters C, Reinheckel T (2011) Gene targeting of the cysteine peptidase cathepsin H impairs lung surfactant in mice. PLoS One 6(10):1–10
Burgess JK, Boustany S, Moir LM, Weckmann M, Lau JY, Grafton K, Baraket M, Hansbro PM, Hansbro NG, Foster PS, Black JL, Oliver BG (2009) Reduction of tumstatin in asthmatic airways contributes to angiogenesis, inflammation, and hyperresponsiveness. Am J Respir Crit Care Med 181(2):106–115
Cezari MHS, Maria L, Juliano A, Carmona AK, Juliano L (2002) Cathepsin B carboxydipeptidase specificity analysis using internally quenched fluorescent peptides. Biochem J 368(1):365–369
Chan SJ, Segundo SB, McCormick MB, Steiner DF (1986) Nucleotide and predicted amino acid sequences of cloned human and mouse preprocathepsin B cDNAs. Proc Natl Acad Sci U S A 83(11):7721–7725
Cheng XW, Shi GP, Kuzuya M, Sasaki T, Okumura K, Murohara T (2012) Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation 125:1551–1562
Chilosi M, Pea M, Martignoni G, Brunelli M, Gobbo S, Poletti V, Bonetti F (2009) Cathepsin K expression in pulmonary lymphangioleiomyomatosis. Mod Pathol 22(2):161–166
Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, Craik CS (2006) Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem 281(18):12824–12832
Chornaya V, Lyannaya O (2004) Some physicochemical properties of cathepsin H from human meningioma. Exp Oncol 26:278–281
Chou PY, Fasman GD (1974) Prediction of protein conformation. Biochemistry 13(2):222–245
Choudhury SD, Lamsal M, Agarwal SK, Sharma R, Khan MY (1997) On the tissue/species dependence of cathepsin B isozymes. Mol Cell Biochem 177:89–95
Coulibaly S, Sehwihla H, Abrahamson H, Albini A, Cerni C, Clark JL, Ng KM, Katunuma N, Schlappack O, Glossl J, Mach L (1999) Modulation of invasive properties of murine squamous carcinoma cell by heterologous expression of cathepsin B and cystatin C. Int J Cancer 83:526–531
Cuervo AM, Dice JE (2000) When lysosomes get old. Exp Gerontol 35:119–131
Dean RT (1976) The roles of cathepsin B1 and D in the digestion of cytoplasmic proteins in vitro by lysosomal extracts. Biochem Biophys Res Commun 68:518–523
del Re EC, Shuja S, Cai J, Murnane MJ (2000) Alterations in cathepsin H activity and protein patterns in human colorectal carcinomas. Br J Cancer 82:1317–1326
Diment S, Leech MS, Stahl PD (1988) Cathepsin D is membrane-associated in macrophage endosomes. J Biol Chem 263(14):6901–6907
Dingle H (1984) Behaviour, genes, and life histories: complex adaptations in uncertain environments. In: Price PW, Slobodchikoff CH, Gaud WS (eds) A new ecology: novel approaches to interactive systems. Wiley, New York, pp 169–194
Docherty K, Carroll RJ, Steiner DF (1982) Conversion of proinsulin to insulin: involvement of a 31,500 molecular weight thiol protease. Proc Natl Acad Sci U S A 79(15):4613–4617
Docherty AJP, O’Connell J, Crabbe T, Angal S, Murphy G (1992) The matrix metalloproteinases and their natural inhibitors: prospects for treating degenerative tissue disease. Trends Biotechnol 10(6):200–207
Dodt J, Reichwein J (2003) Human cathepsin H: deletion of the mini-chain switches substrate specificity from aminopeptidase to endopeptidase. Biol Chem 384:1327–1332
Drake FH, Dodds RA, James IE, Connor JR, Debouck C, Richardson S, Lee-Rykaczewski E, Rieman D, Barthlow R, Hastings G, Gowen M (1996) Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J Biol Chem 271:12511–12516
Drenth J, Jansonius JN, Koekoek R, Swen HM, Wolthers BG (1968) Structure of papain. Nature 218:929–932
Dufour E (1988) Sequence homologies, hydrophobic profiles and secondary structures of cathepsin B, H and L: comparison with papain and actinidin. Biochimie 70:1335–1342
Dufour E, Dive V, Toma F (1988) Delineation of chicken cathepsin L secondary structure: relationship between pH dependence activity and helix content. Biochim Biophys Acta 955(1):58–64
Evered D, Whelan J (1978) Protein degradation in health and disease. Springer, Berlin
Faiz A, Tjin G, Harkness L, Weckmann M, Bao S, Black JL, Oliver BGG, Burgess JK (2013) The expression and activity of cathepsins D, H and K in asthmatic airways. PLoS One 8(3):e57245
Fazili KM, Qasim MA (1986) Purification and some properties of buffalo liver cathepsin B. J Biochem 100:293–299
Ferrara M, Wojcik F, Rhaissi H, Mordier S, Roux MP, Bechet D (1990) Gene structure of mouse cathepsin B. FEBS Lett 273(1–2):195–199
Fong D, Calhoun DH, Hsieh WT, Lee B, Wells RD (1986) Isolation of a cDNA clone for the human lysosomal proteinase cathepsin B. Proc Natl Acad Sci U S A 83(9):2909–2913
Fonovic M, Brömme D, Turk V, Turk B (2004) Human cathepsin F: expression in baculovirus system, characterization and inhibition by protein inhibitors. Biol Chem 385(6):505–509
Fruton JS, Irving GW, Bergmann M (1941) On the proteolytic enzymes of animal tissues. II. The composite nature of beef spleen cathepsin. J Biol Chem 138:249–262
Fuchs R, Machleidt W, Gassen HG (1988) Molecular cloning and sequencing of a cDNA coding for mature human kidney cathepsin H. Biol Chem 369:469–435
Fujishima A, Imai Y, Nomura T, Fujisawa Y, Yamamoto Y, Sugawara T (1997) The crystal structure of human cathepsin L complexed with E-64. FEBS Lett 407:47–50
Gabrijelcic D, Svetic B, Spaić D, Skrk J, Budihna M, Dolenc I, Popovic T, Cotic V, Turk V (1992) Cathepsins B, H and L in human breast carcinoma. Eur J Clin Chem Clin Biochem 30:69–74
Gairola CG, Galicki NI, Cardozo C, Lai YL, Lesser M (1989) Cigarette smoke stimulates cathepsin B activity in alveolar macrophages of rats. J Lab Clin Med 114:419–425
Garnier J, Osguthorpe DJ, Robson B (1978) Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol 120:97–120
Gianni L, Grasselli G, Cresta S, Locatelli A, Vigano L, Minotti G (2003) Anthracyclines. Cancer Chemother Biol Response Modif 21:29–40
Gieselmann V, Pohlmann R, Hasilik A, von Figura K (1983) Biosynthesis and transport of cathepsin D in cultured human fibroblasts. J Cell Biol 97:1–5
Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, Joyce JA (2010) IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev 24:241–255
Godat E, Lecaille F, Desmazes C, Duchene S, Weidauer E, Saftig P, Bromme D, Vandier C, Lalmanach G (2004) Cathepsin K: a cysteine protease with unique kinin degrading properties. Biochem J 383:501–506
Guha S, Padh H (2008) Cathepsins: fundamental effectors of endolysosomal proteolysis. Indian J Biochem Biophys 45(2):75–90
Guncar G, Podobnik M, Pungercar J, Strukelj B, Turk V, Turk D (1998) Crystal structure of porcine cathepsin H determined at 2.1 Ǻ resolution: location of the mini-chain C-terminal carboxyl group defines cathepsin H aminopeptidase function. Structure 6(1):51–61
Gunčar G, Klemenčič I, Turk B, Turk V, Karaoglanovic-Carmona A, Juliano L, Turk D (2000) Crystal structure of cathepsin X: a flip-flop of the ring of His23 allows carboxy-monopeptidase and carboxy-dipeptidase activity of the protease. Structure 80:305–313
Hao Y, Purtha W, Cortesio C, Rui H, Gu Y, Chen H, Sickmier EA, Manzanillo P, Huang X (2018) Crystal structures of human procathepsin H. PLoS One 13(7):1–14
Hara K, Kominami E, Katunuma K (1988) Effect of proteinase inhibitors on intracellular processing of cathepsin B, H and L in rat macrophages. FEBS Lett 231(1):229–231
Haraguchi CM, Ishido K, Kominami E, Yokota S (2003) Expression of cathepsin H in differentiating rat spermatids: immunoelectron microscopic study. Histochem Cell Biol 120:63–71
Hasnain S, Hirama T, Tam A, Mort JS (1992) Characterization of recombinant rat cathepsin B and nonglycosylated mutants expressed in yeast. J Biol Chem 267:4713–4721
Hiwasa T, Yokoyama S, Ha JM, Noguchi S, Sakiyama S (1987) c-Ha-ras gene products are potent inhibitors of cathepsins B and L. FEBS Lett 211(1):23–26
Hook G, Jacobsen JS, Grabstein K, Kindy M, Hook V (2015) Cathepsin B is a new drug target for traumatic brain injury therapeutics: evidence for E64d as a promising lead drug candidate. Front Neurol 6:178
Horn M, Maresova M, Rulisek L, Masa M, Vasiljeva O, Turk B, Erdene TG, Baudys M, Mares M (2005) Activation processing of cathepsin H impairs recognition by its propeptide. Biol Chem 386:941–947
Ii K, Hizawa K, Kominami E (1985) Different immunolocalizations of cathepsins B, H and L in the liver. J Histochem Cytochem 33:1173–1175
Illy C, Quraishi O, Wang J, Purisima E, Vernet T, Mort JS (1997) Role of the occluding loop in cathepsin B activity. J Biol Chem 272(2):1197–1202
Ishidoh K, Imajoh S, Emori Y (1987) Molecular cloning and sequencing of cDNA for rat cathepsin H: homology in propeptide regions of cysteine proteinases. FEBS Lett 226(1):33–37
Ishidoh K, Kominami E, Katunuma N, Suzuki K (1989) Gene structure of rat cathepsin H. FEBS Lett 253(1–2):103–107
Ishidoh K, Suzuki K, Katunuma N, Kominami E (1991) Gene structures of rat cathepsins H and L. Biomed Biochim Acta 50(4–6):541–547
Juhg H, Lee EY, Lee SI (1999) Age related changes in ultrastructural feature at cathepsin B and D containing neuron in rat cerebral cortex. Brain Res 844(1–2):43–54
Kalnitsky G, Chatterjee R, Singh H, Lones M, Paszkowski A (1983) Bifunctional activities and possible modes of regulation of some lysosomal cysteinyl proteases. In: Katunuma N, Umezawa H, Holzer H (eds) Proteinase inhibitors: medical and biological aspects. Springer, Berlin, pp 263–273
Kamphuis IG, Kalk KH, Swarte MB, Drenth J (1984) Structure of papain refined at 1.65 Ǻ resolution. J Mol Biol 179:233–286
Katunuma N (2010) Posttranslational processing and modification of cathepsin and cystatins. J Signal Transduction 2010:1–8
Katunuma N, Kominami E (1986) In: Turk V (ed) Cysteine proteinases and their inhibitors. Walter de Gruyter and Co, Berlin, pp 219–227
Katunuma N, Murata E, Kakegawa H, Matsui A, Tsuzuki H, Tsuge H, Turk D, Turk V, Fukushima M, Tada Y, Asao T (1999) Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett 458:6–10
Khan MY, Ahmad S (1987) Anomalous behavior of cathepsin B: dependence of activity and stability on salt concentration. Biochem Int 15(1):111–115
Khan MY, Ahmad S, Agarwal SK (1986) On the physiological role of mammalian cathepsin B. IRCS Med Sci 14:1141–1142
Khan MY, Agarwal SK, Ahmad S (1992) Structure-activity relationship in buffalo spleen cathepsin B. J Biochem 111:732–735
Kirschke H, Wiederanders B, Bromme D, Rinne A (1989) Cathepsin S from bovine spleen: purification, distribution, intracellular localization and action on proteins. Biochem J 264:467–473
Klemencic I, Carmona AK, Cezari MH, Juliano MA, Juliano L, Guncar G, Turk D, Krizaj I, Turk V, Turk B (2000) Biochemical characterization of human cathepsin X revealed that the enzyme is an exopeptidase, acting as carboxymonopeptidase or carboxydipeptidase. Eur J Biochem 267:5404–5412
Koga H, Mori N, Yamada H, Nishimura Y, Tokuda K, Kato K, Imoto T (1992) Endo-and aminopeptidase activities of rat cathepsin H. Chem Pharm Bull 40(4):965–970
Kominami E, Tsukahara T, Bando Y, Katunuma N (1985) Distribution of cathepsins B and H in rat tissues and peripheral blood cells. J Biochem 98:87–93
Kominami E, Tsukahara T, Hara K, Katunuma N (1988) Biosyntheses and processing of lysosomal cysteine proteinases in rat macrophages. FEBS Lett 231(1):225–228
Kornfeld S (1987) Trafficking of lysosomal enzymes. FASEB J 1(6):462–468
Kornfeld R, Kornfeld S (1980) In: Lennarz WJ (ed) The biochemistry of glycoproteins and proteoglycans. Plenum Press, New York, pp 1–34
Kos J, Schweiger A (2002) Cathepsins and cystatins in extracellular fluids useful biological markers in cancer. Radiol Oncol 36(2):176–179
Kos J, Smid A, Krasovec M, Svetic B, Lenarcic B, Vrhovec I, Skrk J, Turk V (1995) Lysosomal proteases cathepsins D, B, H, L and their inhibitors stefins A and B in head and neck cancer. Biol Chem 376:401–405
Kos J, Stabuc B, Schweiger A, Krasovec M, Cimerman N, Kopitar-Jerala N, Vrhovec I (1997) Cathepsins B, H, and L and their inhibitors stefin A and cystatin C in sera of melanoma patients. Clin Cancer Res 3:1815–1822
Lafuse WP, Brown D, Castle L, Zwilling BS (1995) IFN-gamma increases cathepsin H mRNA levels in mouse macrophages. J Leukoc Biol 57:663–609
Lah TT, Kos J (1998) Cysteine proteinases in cancer progression and their clinical relevance for prognosis. Biol Chem 379(2):125–130
Lah T, Kosorok MD, Turk V, Pain RH (1984) Conformation, structure and activation of bovine cathepsin D. Biochem J 218:601–608
Lamsal M, Agarwal SK, Choudhury SD, Khan MY (1997) Purification and tissue/species dependence of the specificity of buffalo kidney cathepsin B. Indian J Biochem Biophys 34:461–469
Lautwein A, Burster T, Lennon-Dumenil AM, Overkleeft HS, Weber E, Kalbacher H, Driessen C (2002) Inflammatory stimuli recruit cathepsin activity to late endosomal compartments in human dendritic cells. Eur J Immunol 32:3348–3357
Lecaille F, Chowdhury S, Purisima E, Brömme D, Lalmanach G (2007) The S2 subsites of cathepsins K and L and their contribution to collagen degradation. Protein Sci 16(4):662–670
Lenarčič B, Križaj I, Žunec P, Turk V (1996) Differences in specificity for the interactions of stefins A, B and D with cysteine proteinases. FEBS Lett 395(2–3):113–118
Lenney JF, Tolan JR, Sugai WJ, Lee AG (1979) Thermostable endogenous inhibitors of cathepsins B and H. Eur J Biochem 101:153–161
Li Z, Yasuda Y, Li W, Bogyo M, Katz N, Gordon RE, Fields GB, Bromme D (2004) Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J Biol Chem 279:5470–5479
Liao CR, Lenney FJ (1984) Cathepsins J and K: high molecular weight cysteine proteinases from human tissues. Biochem Biophys Res Commun 124(3):909–916
Lutgens SPM, Cleutjens KBJM, Daemen MJAP, Heeneman S (2007) Cathepsin cysteine proteases in cardiovascular disease. FASEB J 21:3029–3041
Lützner N, Kalbacher H (2008) Quantifying cathepsin S activity in antigen presenting cells using a novel specific substrate. J Biol Chem 283(52):36185–36194
MacGregor RR, Hamilton JW, Kent GN, Shofstal RE, Cohn DV (1979) The degradation of proparathormone and parathormone by parathyroid and liver cathepsin B. J Biol Chem 254:4428–4433
Machleidt W, Ritonja A, PopoviE T, Kotnik M, Brzin J, Turk V, Machleidt I, Mueller-Esterl W (1986) Human cathepsins B, H and L: characterization by amino acid sequences and some kinetics of inhibition by the kininogens. In: Turk V (ed) Cysteine proteinases and their inhibitors. Walter de Gruyter and Co, Berlin/New York, pp 3–18
Mainferme F, Wattiaux R, von Figura K (1985) Synthesis, transport and processing of cathepsin D in Morris hepatoma 7777 cells and rat hepatocytes. Eur J Biochem 153:211–216
Mason RW (1989) Interaction of lysosomal cysteine proteinases with α2-macroglobulin: conclusive evidence for the endopeptidase activities of cathepsins B and H. Arch Biochem Biophys 273(2):367–374
Matarrese P, Ascione B, Ciarlo L, Vona R, Leonetti C, Scarsella M, Mileo AM, Catricalà C, Paggi MG, Malorni W (2010) Cathepsin B inhibition interferes with metastatic potential of human melanoma: an in vitro and in vivo study. Mol Cancer 9:207–220
Matha VL, Derocq D, Prebois C, Katunuma N, Liaudet-Coopman E (2006) Processing of human cathepsin D is independent of its catalytic function and auto-activation: involvement of cathepsins L and B. J Biochem 139:363–371
McDonald JK, Barrett AJ (1986) Mammalian proteases: a glossary and bibliography-exopeptidase, vol 2. Academic Press, London
McDonald JK, Ellis S (1975) On the substrate specificity of cathepsin B1 and B2 including a new fluorogenic substrate for cathepsin B1. Life Sci 17:1269–1276
McGrath ME, Klaus JL, Barnes MG, Bromme D (1997) Crystal structure of human cathepsin K complexed with a potent inhibitor. Nat Struct Biol 4:105–109
McGuire MJ, Lipsky PE, Thiele DL (1997) Cloning and characterization of the cDNA encoding mouse dipeptidyl peptidase I (cathepsin C). Biochim Biophys Acta 1351:267–273
McKay MJ, Offermann MK, Barrett AJ, Bond JS (1983) Action of human liver cathepsin B on the oxidized insulin B chain. Biochem J 213:467–471
Meloun B, Baudys M, Pohl J, Pavlik M, Kostka V (1988) Amino acid sequence of bovine spleen cathepsin B. J Biol Chem 263:9087–9093
Meyer DI, Krause E, Dobberstein B (1982) Secretory protein translocation across membranes-the role of the docking proteins. Nature 297:647–650
Mort JS, Buttle DJ (1997) Cathepsin B. Int J Biochem Cell Biol 29(5):715–720
Musil D, Zucic D, Turk D, Engh RA, Mayr I, Huber R, Popovic T, Turk V, Towatari T, Katunuma N, Bode W (1991) The refined 2.15 Ǻ X-ray crystal structure of human liver cathepsin B: the structural basis for its specificity. EMBO J 10(9):2321–2330
Nagler DK, Storer AC, Portaro FC, Carmona E, Juliano L, Menard R (1997) Major increase in endopeptidase activity of human cathepsin B upon removal of occluding loop contacts. Biochemistry 36(41):12608–12615
Nägler DK, Zhang R, Tam W, Sulea T, Purisima EO, Ménard R (1999) Human cathepsin X: a cysteine protease with unique carboxypeptidase activity. Biochemistry 38:12648–12654
Nishimura Y, Kato K (1987) Intracellular transport and processing of lysosomal cathepsin B. Biochem Biophys Res Commun 148(1):254–259
Ohtani O, Fukuyama K, Epstein WL (1982) Further characterization of cysteine proteinase inhibitors purified from rat and human epidermis. Biochim Biophys Acta 707(1):21–27
Ondr JK, Pham CT (2004) Characterization of murine cathepsin W and its role in cell-mediated cytotoxicity. J Biol Chem 279:27525–27533
Otto K (1971) In: Barrett AJ, Dingle JT (eds) Tissue proteinases. North Holland Publishing Co, Amsterdam, pp 181–207
Otto K, Reisenkonig H (1975) Improved purification of cathepsin Bl and B2. Biochim Biophys Acta 379:462–475
Paciucci R, Berrozpe G, Torà M, Navarro E, García de Herreros A, Real FX (1996) Isolation of tissue-type plasminogen activator, cathepsin H and non-specific cross-reacting antigen from SK-PC-1 pancreas cancer cells using subtractive hybridization. FEBS Lett 385(1–2):72–76
Polgar L, Csoma C (1987) Dissociation of ionizing groups in the binding cleft inversely controls the endo- and exopeptidase activities of cathepsin B. J Biol Chem 262(30):14448–14453
Popovic TZ, Brzin J, Kos J, Lenarcic B, Machleidt W, Ritonja A, Hanada K, Turk V (1988) A new purification procedure of human kidney cathepsin H, its properties and kinetic data. Biol Chem 369:175–183
Quinn PS, Judah JD (1978) Calcium-dependent Golgi-vesicle fusion and cathepsin B in the conversion of proalbumin into albumin in rat liver. Biochem J 172(2):301–309
Ravish I, Raghav N (2014) Curcumin as inhibitor of mammalian cathepsin B, cathepsin H, acid phosphatase and alkaline phosphatase: a correlation with pharmacological activities. Med Chem Res 23:2847–2855
Rawlings ND, Salvesen G (2013) Handbook of proteolytic enzymes, 3rd edn. Academic, London
Rawlings ND, Barrett AJ, Bateman A (2010) MEROPS: the peptidase database. Nucleic Acids Res 38:227–233
Reiser J, Adair B, Reinheckel T (2010) Specialized roles for cysteine cathepsins in health and disease. J Clin Invest 120:3421–3431
Riese RJ, Chapman HA (2000) Cathepsin and compartmentalization in antigen presentation. Curr Opin Immunol 12:107–113
Riese RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER, De Sanctis GT, Ploegh HL, Chapman HA (1998) Cathepsin S activity regulates Ag presentation and immunity. J Clin Invest 101:2351–2363
Ritonja A, Popovic T, Turk V, Wiedermann K, Machleidt W (1985) Amino acid sequence of human liver cathepsin B. FEBS Lett 181:169–172
Ritonja A, Popovic T, Kotnik M, Machleidt W, Turk V (1988) Amino acid sequences of human kidney cathepsins H and L. FEBS Lett 228(2):341–345
Rossi A, Deveraux Q, Turk B, Sali A (2004) Comprehensive search for cysteine cathepsins in the human genome. Biol Chem 385(5):363–372
Ryvnyak VV, Ryvnyak EI, Tudos RV (2004) Electron histochemical localization of cathepsin L in the liver. Bull Exp Biol Med 137:90–91
Salahuddin A, Siddiqui FA, Salahuddin P (1996) Isolation, purification and properties of cathepsin B from buffalo liver. Indian J Biochem Biophys 33:292–297
Santamaria I, Pendas AM, Velasco G, Lopez-Otin C (1998) Genomic structure and chromosomal localization of the human cathepsin O gene (CTSO). Genomics 53:231–234
Santamaria I, Velasco G, Pendas AM, Paz A, Lopez-Otin C (1999) Molecular cloning and structural and functional characterization of human cathepsin F: a new cysteine proteinase of the papain family with a long propeptide domain. J Biol Chem 274:13800–13809
Schwartz WN, Barrett AJ (1980) Human cathepsin H. Biochem J 191:487–497
Scott RP, Ninjoor V, Srivastava PN (1987) Isolation and characterization of cathepsin B from rabbit testis. J Reprod Fertil 79(1):67–74
Siklos M, BenAissa M, Thatcher GR (2015) Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm Sin B 5(6):506–519
Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P (2005) Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A 102:11876–11881
Singh H, Kalnitsky G (1978) Separation of a new α-N-benzoylarginine-β-naphthylamide hydrolase from cathepsin B1. J Biol Chem 253:4319–4326
Singh S, Sharma S, Agarwal SK (2020) A simple purification procedure of buffalo lung cathepsin H, its properties and influence of buffer constituents on the enzyme activity. Biochem Biophys Rep 22:1–8
Sires UI, Schmid TM, Fliszar CJ, Wang ZQ, Gluck SL, Welgus HG (1995) Complete degradation of type X collagen requires the combined action of interstitial collagenase and osteoclast-derived cathepsin B. J Clin Invest 95:2089–2095
Sloane BF, Dunn JR, Honn KV (1981) Lysosomal cathepsin B: correlation with metastatic potential. Science 212(4499):1151–1153
Smith SM, Gottesman MM (1989) Activity and deletion analysis of recombinant human cathepsin L expressed in Escherichia coli. J Biol Chem 264(34):20487–20495
Somoza JR, Zhan H, Bowman KK, Yu L, Mortara KD, Palmer JT, Clark JM, McGrath ME (2000) Crystal structure of human cathepsin V. Biochemistry 39:12543–12551
Somoza JR, Palmer JT, Ho JD (2002) The crystal structure of human cathepsin F and its implications for the development of novel immunomodulators. J Mol Biol 322:559–568
Stachowiak K, Tokmina M, Karpinska A, Sosnowska R, Wiczk W (2004) Fluorogenic peptide substrates for carboxydipeptidase activity of cathepsin B. Acta Biochim Pol 51:81–92
Starkey PM, Barrett AJ (1973) Human cathepsin B1:inhibition by α2-macroglobulin and other serum proteins. Biochem J 131(4):823–831
Suhar A, Marks N (1979) Purification and properties of brain cathepsin B: evidence for cleavage of pituitary lipotropins. Eur J Biochem 101:23–30
Swanson AA, Martin BJ, Spicer SS (1974) Human placental cathepsin B1: isolation and some physical properties. Biochem J 137(2):223–228
Takahashi T, Schmidt PG, Tang J (1984a) Novel carbohydrate structures of cathepsin B from porcine spleen. J Biol Chem 259(10):6059–6062
Takahashi T, Dehdarani AH, Schmidt PG, Tang J (1984b) Cathepsins B and H from porcine spleen. J Biol Chem 259(15):9874–9882
Takahashi T, Yonezawa S, Dehdarani AH, Tang J (1986a) Comparative studies of two cathepsin B isozymes from porcine spleen. J Biol Chem 261(20):9368–9374
Takahashi T, Dehdarani AH, Yonezawa S, Tang J (1986b) Porcine spleen cathepsin B is an exopeptidase. J Biol Chem 261(20):9375–9381
Takahashi T, Dehdarani AH, Tang J (1988) Porcine spleen cathepsin H hydrolyzes oligopeptides solely by aminopeptidase activity. J Biol Chem 263(22):10952–10957
Takio K, Towatari T, Katunuma N, Teller DC, Titani K (1983) Homology of amino acid sequences of rat liver cathepsins B and H with that of papain. Proc Natl Acad Sci U S A 80:3666–3670
Tallan HH, Jones EJ, Fruton JS (1952) On the proteolytic enzymes of animal tissues: beef spleen cathepsin C. J Biol Chem 194:793–705
Tan Y, Osatomi K, Hara K (2006) Gene structure of carp Cyprinus carpio cathepsin B. Fish Sci 72:673–678
Taniguchi T, Mizuochi T, Towatari T (1985) Structural studies on the carbohydrate moieties of rat liver cathepsins B and H. J Biochem 97(3):973–976
Tchoupé JR, Moreau T, Gauthier F, Bieth JG (1991) Photometric or fluorometric assay of cathepsin B, L and H and papain using substrates with an aminotrifluoromethylcoumarin leaving group. Biochim Biophys Acta Protein Struct Mol Enzymol 1076(1):149–151
Tezuka K, Tezuka Y, Maejima A, Sato T, Nemoto K, Kamioka H, Hakeda Y, Kumegawa M (1994) Molecular cloning of a possible cysteine proteinase predominantly expressed in osteoclasts. J Biol Chem 269(2):1106–1109
Tolosa E, Li W, Yasuda Y, Wienhold W, Denzin LK, Lautwein A, Driessen C, Schnorrer P, Weber E, Stevanovic S, Kurek R, Melms A, Bromme D (2003) Cathepsin V is involved in the degradation of invariant chain in human thymus and is overexpressed in myasthenia gravis. J Clin Invest 112:517–526
Towatari T, Katunuma N (1978) Crystallization and amino acid composition of cathepsin B from rat liver lysosomes. Biochem Biophys Res Commun 83(2):513–520
Tsuge H, Nishimura T, Tada Y, Asao T, Turk D, Turk V, Katunuma N (1999) Inhibition mechanism of cathepsin L-specific inhibitors based on the crystal structure of papain–CLIK148 complex. Biochem Biophys Res Commun 266(2):411–416
Tsushima H, Ueki A, Matsuoka Y, Mihara H, Hopsu-Havu VK (1991) Characterization of a cathepsin-H-like enzyme from a human melanoma cell line. Int J Cancer 48:726–732
Turk B, Turk D, Turk V (2000) Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta 1147:98–111
Turk V, Turk B, Turk D (2001) Lysosomal cysteine proteases: facts and opportunities. EMBO J 20(17):4629–4633
Turk D, Turk B, Turk V (2003) Papain-like lysosomal cysteine proteases and their inhibitors: drug discovery targets? Biochem Soc Symp 70:15–30
Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824:68–88
Turkenburg JP, Lamers MBAC, Brzozowski AM, Wright LM, Hubbard RE, Sturt SL, Williams DH (2002) Structure of a Cys25 → Ser mutant of human cathepsin S. Acta Crystallogr D Biol Crystallogr 58:451–445
Uchiyama Y, Wagmi S, Sato N, Watanabe T, Ishido K, Kominami E (1994) Cell and tissue distribution of lysosomal cysteine proteinases, cathepsins B, H, and L, and their biological roles. Acta Histochem Cytochem 27:351–372
Vasiljeva O, Dolinar M, Turk V, Turk B (2003) Recombinant human cathepsin H lacking mini chain is an endopeptidase. Biochemistry 42:13522–13528
Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen BS, Almholt K, Bogyo M, Peters C, Reinheckel T (2006) Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res 66:5242–5250
Verma S, Dixit R, Pandey KC (2016) Cysteine proteases: modes of activation and future prospects as pharmacological targets. Front Pharmacol 7:107
Victor BC, Anbalagan A, Mohamed MM, Sloane BF, Cavallo-Medved D (2011) Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res 13(6):R115
Vidak E, Javoršek U, Vizovišek M, Turk B (2019) Cysteine cathepsins and their extracellular roles: shaping the microenvironment. Cells 8:264–288
Von Figura K, Hasilik A (1986) Lysosomal enzymes and their receptors. Annu Rev Biochem 55:167–193
Waghray A, Keppler D, Sloane BF, Schuger L, Chen YQ (2002) Analysis of a truncated form of cathepsin H in human prostate tumor cells. J Biol Chem 277:11533–11538
Walter P, Blobel G (1982) Signal recognition particle contains a 7S RNA essential for protein translocation across the endoplasmic reticulum. Nature 299:691–698
Wang B, Shi GP, Yao PM, Li Z, Chapman HA, Bromme D (1998) Human cathepsin F: molecular cloning, functional expression, tissue localization, and enzymatic characterization. J Biol Chem 273:32000–32008
Watanabe M, Watanabe T, Ishii Y, Matsuba H, Kimuna S, Fujita T, Kominami E, Katunuma N, Uchiyama Y (1988) Immunocytochemical localization of cathepsins B, H and their endogenous inhibitor, cystatin β in islet endocrine cells of the rat pancreas. J Histochem Cytochem 36:783–791
Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL, Baker JR Jr, Van Echo DA, Von Hoff DD, Leyland-Jones B (1990) Hypersensitivity reactions from taxol. J Clin Oncol 8(7):1263–1268
Wex T, Levy B, Smeekens SP, Ansorge S, Desnick RJ, Bromme D (1998) Genomic structure, chromosomal localization, and expression of human cathepsin W. Biochem Biophys Res Commun 248:255–261
Willenbrock F, Brocklehurst K (1985) Preparation of cathepsin B and H by covalent chromatography and characterization of their catalytic sites by reaction with a thiol- specific two-protonic-state reactivity probe. Biochem J 227:511–519
Willstätter R, Bamann E (1929) Über die proteasen der magenschleimhaut. Erste abhandlung über die enzyme der leukocyten. Hoppe Seylers Z Physiol Chem 180:127–143
Wolters PJ, Chapman HA (2000) Importance of lysosomal cysteine proteases in lung disease. Respir Res 1:170–177
Wu SM, Huang YH, Yeh CT, Tsai MM, Liao CH, Cheng WL, Chen WJ, Lin KH (2011) Cathepsin H regulated by the thyroid hormone receptors associate with tumor invasion in human hepatoma cells. Oncogene 30:2057–2069
Wu H, Du Q, Dai Q, Ge J, Cheng X (2018) Cysteine protease Cathepsins in atherosclerotic cardiovascular diseases. J Atheroscler Thromb 25(2):111–123
Yamamoto K, Kamata O, Kato Y (1984) Separation and properties of three forms of cathepsin H-like cysteine proteinase from rat spleen. J Biochem 95:477–484
Yasuda Y, Li Z, Greenbaum D, Bogyo M, Weber E, Bromme D (2004) Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J Biol Chem 279(35):36761–36770
Yoshida A, Ohta M, Kuwahara K, Cao MJ, Hara K, Osatomi K (2015) Purification and characterization of cathepsin B from the muscle of horse Mackerel Trachurus japonicas. Mar Drugs 13(11):6550–6565
Zavasnik-Bergant T, Turk B (2007) Cysteine proteases: destruction ability versus immunomodulation capacity in immune cells. Biol Chem 388(11):1141–1149
Zhong YJ, Shao LH, Li Y (2013) Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy. Int J Oncol 42(2):373–383
Zvonar-Popovic T, Kregar I, Turk V (1979) Isolation of cathepsin B and α-N-benzoylarginine-β-naphtylamide hydrolase by covalent chromatography on activated thiol Sepharose. Croat Chem Acta 52:411–416
Zvonar-Popovic T, Lah T, Kregar I, Turk V (1980) Some characteristics of cathepsin B and α-N-Benzoylarginine-β-Naphthylamide hydrolase from bovine lymph nodes. Croat Chem Acta 53(3):509–517
Acknowledgments
This work was supported by the Department of Higher Education, Government of Uttar Pradesh, for Centre of Excellence in Biochemistry and Indian Council of Medical Research, New Delhi. The infrastructural facilities provided by the University of Lucknow, Lucknow, are also gratefully acknowledged.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Agarwal, S.K., Singh, S., Sharma, S. (2020). Structural and Functional Dynamics of Lysosomal Cysteine Proteases with Particular Reference to Cathepsin B and Cathepsin H. In: Singh, D., Tripathi, T. (eds) Frontiers in Protein Structure, Function, and Dynamics. Springer, Singapore. https://doi.org/10.1007/978-981-15-5530-5_16
Download citation
DOI: https://doi.org/10.1007/978-981-15-5530-5_16
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-5529-9
Online ISBN: 978-981-15-5530-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)