
Abstract
This review article deals with comprehensive information about the evolutionary history of introns with their localization and functions in the gene transcripts of colorectal cancer precisely. In this way, the major breakthrough in the molecular biology discipline was the discovery of introns by Richard Robert and Phil Sharp in 1977. Firstly, noncoding regions are recognized by various assortments of regulatory ncRNA sequences such as circular RNA, telomere-associated RNA, small nuclear RNA, Piwi-interacting RNA, small interfering RNA, small nucleolar RNA, microRNA, and long noncoding RNA. Fortunately, splicing process of mRNA strand deals with the excision of introns via spliceosomal proteins into mature mRNA which is witnessed only in eukaryotic organisms and devoid of the splicing machinery components in the prokaryotic organisms. The major focal point relies on intronic genes mainly involved in the progression of colorectal cancer with preliminary information. An alternative splicing process takes place in mRNA that implicates in intron retention leading to varied gene expression in cells and tissues and their promotion in colorectal cancer. Therefore, colorectal cancer-associated diseases have paved the way to know more about the intronic genes mainly concentrated among them in the progression of the related diseases. Hence, the focus of the researchers is toward the fascinating cellular and molecular biology aspects of the regulatory intronic sequences known to enhance as well as repress particular gene expression in tumor microenvironment of colorectal cancer by analyzing the genome and proteome levels for the betterment of human kind that is intended for various therapeutic purposes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Introns (noncoding sequences)
- mRNA
- Spliceosomal proteins
- Alternate splicing process
- Intron retention
- Colorectal cancer
13.1 Introduction
Eukaryotic genome consists of DNA sequences which are transcribed into pre-mRNA, which is exemplified by the organization of intron-exon structure and endures posttranscriptional and posttranslational process to produce a desired protein. Consequently, intervened introns are excised among the adjoining exons, to form the pre-mRNA [1]. In particular, the coding regions specifically undergo translation, while in the noncoding regions, introns also undergo the translation process but the protein produced is acclaimed to be regulated in various splicing processes which in turn advance varied gene expression and cancer development [2]. Introns appear to influence any phase of mRNA maturation together with transcription process such as mRNA stability, nuclear transport, and polyadenylation. Splicing introns imply an expurgation of spliceosomal introns from the genome where the spliceosome consists of five snRNAs and more than 150 proteins which are coded by intron-bearing genomes itself. The transcription of lengthy introns persists up to several hours as the elongation time is found to be 60 bases for every second facilitated with the help of RNA polymerase [3]. Therefore, the host of cis-regulatory facet assists in the identification of splicing joint by the spliceosome. As a consequence, intrusion of normal splicing pattern paves the way to more than half of the genetic disorders in humans (Table 13.1).
13.1.1 Elucidation of Intron Evolution
The preponderance of the existing intron-rich mammals is elevated in contrast to the preceding eukaryotic ancestors populated with introns. As a result, when the intron is excised from the gene, it turns out to be an element of post-splicing complexes that follow de-branching and destruction. Therefore, the RNA gene is entrenched within the intron; its expression is witnessed on the intron exclusion and outlasts its intronic mass (Fig. 13.1). Functional consequence of an intron might be compatible with the point of conservation of intron position. Excision of intron exon structure indicates sequence construction of orthologous genes which helps in the assessment of intron position (Fig. 13.2). Such constructions reveal the intron position is at times preserved throughout the long evolutionary times in orthologous genes [1].

Intronic position in site of intron insertion along the mRNA

Comparison of intron positions between orthologous genes
The eukaryotic hierarchy reveals an array of intron concentration (Fig. 13.3). An investigation of intron mass and eukaryote phylogeny illustrates that it is not always the instance that early division eukaryotes are intron deprived and that late division eukaryotes are luxuriant intron. Analysis of eukaryotic genome aids in empathizing the intron gain or loss. During evolution, the locations of definite introns are preserved between extremely divergent and vibrant among eukaryotes [2] (Fig. 13.4).

The hierarchy of the origin of introns. (a) Early theory of introns. (b) Late theory of introns (LUCA, last universal common ancestor)

Various types of intronic functions
Consequently, the life expectancy of an intron is of five stages which independently allude to the capacities that are related with each phase. Diverse functional antisense elements arise from intronic regions and are considered as intron-hosted RNA genes that are triggered during transcription process and are also required for alternative splicing (Fig. 13.5) [3].

Dynamic aspects of gain and loss of introns
13.1.2 Different Classes of Introns Residing in Prokaryotes and Eukaryotes
Spliceosomal introns, group I self-splicing intron, group II self-splicing introns, and tRNA introns are categorized as the four main arrays of introns to be inherent in pre-nuclear mRNA. Moreover, the intronic form is discrete due to its construction and phylogenetic dispersal [4, 5] (Table 13.2).
13.1.3 Noncoding RNAs in Eukaryotes
In human beings, approximately 25,000 genes are recognized which effectively transcribe into mature RNA, where it is comprised of 20% coding (exons) and 80% noncoding (introns) sequences. Consequently, the genome contains greater part of noncoding sequences also known as “junk DNA.” The major focal point is on functional noncoding RNAs which are generated in a small percentage which is an assorted regulatory biological mechanism, for instance, like gene expression, propagation, differentiation and senescence of cell, and epigenetic modification along with other cellular processes which lead to numerous diseases by the dysregulation of human genome [6].
13.1.4 Description of Different Types of Regulatory ncRNA
-
MicroRNAs (miRNAs)
MicroRNA is a lavish group of small noncoding RNAs which do not encode for protein synthesis. They have a significant task as a tumor suppressor or an oncogene which is misled by mutations and abnormal gene expression. Thus, the microRNA modification will prompt the development of various types of cancer [7].
-
Circular RNA (ciRNA)
Circular RNA is a sort of contended endogenous RNA in the lineage of long noncoding RNAs which is known to be stable in eukaryotic cell. ciRNA has diverse functionalities such as the capacity of reorganizing the genomic sequences, fortification against exonuclease at the 3′ poly(A) tail, and also an epigenetic regulator [7].
-
Long noncoding RNA (lncRNA)
Resistance of colorectal cancer to chemotherapy is due to the encoding long noncoding RNA which restrains the cell multiplication, differentiation, programmed cell death, and metastasis [8]. Some of the colorectal cancer-associated long noncoding RNAs influence the gene expression by epigenetic alteration that necessitates DNA methylation, histone scaffolding, chromosomal instability, and pseudogenes appropriately (Fig. 13.6).

Classification of noncoding RNAs
13.1.5 Functional Characteristic of Noncoding Sequences in Cancer
Tumor is an unrestrained proliferation of cells residing in a tumor microenvironment of a specific tissue site triggered by the dysregulation of the signaling mechanism by the “oncogene” or “tumor suppressor gene” appropriately. Consequently, it advances in their abnormal gene expression, cell growth, protein profiles, differentiation, and epigenetic modulation, and very few instances may be due to familial inherited gene in their germ line. Genome-correlated analysis in cancer divulges in the fact that nearly 75–85% of cancer-linked single-nucleotide polymorphisms transpire in the regulatory noncoding sequences such as intergenic or intronic regions [9] (Fig. 13.7).

Single-nucleotide polymorphisms (SNPs) existing in the genome dispersal (%) of a range of cancers. It is revealed to be widely encoded by the noncoding sequences (intergenic, intronic) and minority of them by the coding sequences
13.2 Colorectal Cancer and Associated Factors
Colorectal cancer is the third most prevalent disease affecting both the genders worldwide which is mostly predominant in men with >50 years of age in Western countries while found in minor incidence in India. The dynamic power of tumorigenesis is due to the chromosomal mutations and epigenetic modifications, which either activate oncogenes or cease the task of tumor suppressor genes, which subsequently progress in the development of cancer from neoplasia to metastasis. Initial genetic changes start in an early adenoma and accumulate as it transforms to carcinoma and ultimately to invasive and metastatic tumor. So, the molecular pathogenesis of colorectal cancer includes the familial adenomatous polyposis (FAP) and Lynch syndrome and hereditary nonpolyposis colorectal cancer (HNPCC) (Fig. 13.8).

Precariousness in genomic pathways engaged in colorectal cancer
13.3 Genetic Background of the Colorectal Cancer-Associated Diseases
13.3.1 Adenomatous Polyposis Colon Cancer
Adenomatous polyposis coli (APC) is recognized to be a tumor suppressor protein concealed by the APC gene which is endowed as the pioneer gene transformed in sporadic and inherited colon cancer. The APC gene is frequently mutated by either frameshift or nonsense mutation generating a misfolded protein which leads to various syndromes related to colorectal cancer. Normally, the cell cycle checkpoints involve G1/S (start or restriction point) and G2/M checkpoint, and spindle checkpoints are the barricade which is known to regulate cell cycle appropriately without any errors. If the mutational sequences are replicated and synthesized, it will advance in various genetic diseases. In adenomatous polyposis colon cancer, the alteration from G1 to S phase cell cycle is obstructed by the tumor suppressor gene, i.e., APC gene. Subsequently, the Gardner syndrome, familial adenomatous polyposis, Turcot syndrome, and attenuated familial adenomatous polyposis are the APC-related polyposis conditions in colorectal cancer. Wnt signaling and β-catenin pathway take part in the colorectal tumorigenesis of sporadic and familial colorectal cancer (Fig. 13.9).

Pathological interpretation of colon cancer tissue prototype
13.3.2 Lynch Syndrome
Lynch syndrome is also known as “hereditary nonpolyposis colorectal cancer” which is transmitted by germline mutations through microsatellite instability and mismatch repair pathways in colorectal cancer. MLH1, MSH2, MSH6, and PMS2 are the most frequently mutated genes in case of mismatch repair pathway which is known to be the diagnostic marker in colorectal cancer [10]. Therefore, deficit of DNA in mismatch repair activity acts as an indicator of microsatellite instability. Consequently, the preponderance of mismatch repair deficiency in sporadic colorectal cancer is suitable to the epigenetic silencing of MLH1 gene expression that is induced by overmethylation of the promoter [11] (Fig. 13.10).

Properties of introns
13.3.3 Intron Retention
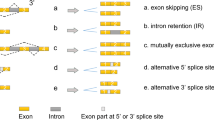
Alternative splicing is described as an arbitrary splicing of the introns from premature mRNA which eventually affects the multiple exon genes in humans. Intron retention, alternative 5′ or 3′ control, and exonic regions are the three major divisions of alternate splicing. Among the three divisions mentioned above, intron retention plays an intense role in causing cancer of various types. Intron retention is characterized as the conservation of introns in the coding vicinity or flanked by the introns in the untranslated region which directs the way to mis-splicing of mature RNA during the transcription process which ultimately end results in the origination of destructive proteins during the translation process [6]. Thus, the generation of detrimental proteins may lead to an assorted gene expression which roots the basis of various genetic diseases. Therefore, the translation process is inhibited by the contrivance of nonsense-intervened decay or exosome deprivation which facilitates in the excision of introns [12]. Transcripts containing introns often contain the premature termination codons which instigate the nonsense-intervened decay (Fig. 13.11). In general, the nonsense-intervened decay disintegrates the transcripts retaining introns with a premature termination codon which is situated in greater than 50–55 nucleotides upstream of an exon to exon junction [13]. Finally, the plausibility of intron retention can be influenced by different factors such as GC content, expression of splicing factor, extent of introns, alteration of chromatin structure or nucleosome packing, and potency of splice site [14].

Role of intron-sustained transcripts. (a) Downgrading the gene expression through eliciting the nonsense-intervened decay. (b) Intron-preserving transcripts might endure deprivation in the nucleus as a result of inhibiting the transport of mRNA which in turn obstructs the translation process. (c) Creation of new isoforms along with precise biological act
13.3.4 Intron Retention Performs a Crucial Role in Gene Expression
Our main focus is on intron retention where the noncoding genes are retained within the coding regions of the gene due to the alternative excision process. Theoretically, an alternative splicing process of exonic genes is known to widely influence the human population. During the transcription process, the pre-mRNA consists of 5′ capping, 3′ poly(A) tail with interspersed exons and introns appropriately (Fig. 13.12) [15].

Schematic representation of alternative splicing process
The captivating information about the major and minor compounds contributing to the excision process of heteronuclear RNA encompassing the dominant complex which adds to their role in nucleus, whereas, the less significant complex acts on the division of introns with lower incidence in the multicellular genomes [16]. Thus, the coding regions undergo the translation process generating a functional protein [15]. However, in alternative splicing process, the exon-exon junction complex with up frameshift proteins such as UP-1, UP-2, and UP-3 are bound to the mRNA which proceeds to the protein synthesis. In the translation process, the premature stop codons which are situated prior the termination site lead to the preservation of introns in the coding sequences which advance in the unexpected production of silencing or harmful detrimental proteins without a specific function. In general, the premature stop codons are situated in upstream of greater than 55 nucleotides in the mRNA transcript which eventually signals the nonsense-intervened decay process to activate and undergo the necessary degradation process. So, to circumvent the initiation of such forms of unknown proteins are conceded to promote the deprivation of the transcripts via nonsense-mediated decay. The nonsense-mediated decay process disintegrates the proteins which are accumulated in the processing bodies organized in the cytoplasm of a cell. Thus, the intron retention in gene expression results in varied roles in cell cycle, cell differentiation, cancer, and even genetic disorders. Consequently, our focal point will be on the intron-retaining genes extensively present in colorectal cancer to assess their diverse properties of noncoding RNAs which may be the long noncoding RNA, small interfering RNA, and various other types of noncoding RNAs. The preservation of intronic transcripts in the mature RNA is identified to cause latent destructive end product, if it undergoes translation. According to the literature analysis, intron retention is known to be associated with progression of tumor such as the instigation of oncogene besides reconciling the tumor suppressor gene. So, the profusion of mature RNA comprising the introns residing in tumor cells is recognized to enhance multiplicity of tumor transcriptomes [17]. Abnormal excision of the noncoding regions from the mRNA leads to limited or complete preservation of the introns. Fortunately, the final intron is prone to restrain the normal as well as cancer tissues and also cause disease by the point mutation in the nucleotide sequence. The molecular level of the chromatin packing of the DNA involves the nucleosome which is compactly packed in two forms such as euchromatin and heterochromatin in which the intron retention has a crucial role in influencing the histone variation, nucleosome compaction, as well as the chromatin modifications at the gene promoter level expressing a range of tumor development with distorted gene expression in their tumor microenvironment. At this point, we will discuss about “mirtron” discerned to arise from the microRNA known to be refined in the introns during the splicing process that creates a loop which is self-regulated and devoid of the microprocessor that is exported to the cytosol of the cell [13]. Frequently, retaining the noncoding sequences within the mRNA constituent and cleaving in the cytoplasm are certain alterations in the transcriptional level advancing in the cellular multiplicity of eukaryotic cells mainly human cells [16].
13.3.5 Noncoding RNA Genes [Human]
-
H19 (nonprotein coding)
H19 gene is found to explicit from the maternally inherited chromosome situated in p-arm (15.5) of the chromosome 11. Therefore, the consequence of the gene is the lengthy noncoding RNA which performs as a tumor repression. So, eventually the mutation in this gene will lead to diverse genetic disorders.
-
MIR137 microRNA 137 (1p21.3), MIR126 microRNA 126 (9q34.3), MIR33A microRNA 33a (22q13.2), MIR335 microRNA 335 (7q32.2), MIR33B microRNA 33b (17p11.2), and MIR21 microRNA 21 (17q23.1)
In multicellular organisms, miRNAs are short about 20–24 nucleotides ncRNAs transcribed by RNA polymerase II which can end result as noncoding or protein coding which affects the transcriptional control. Then, the initial transcript is spliced by the Drosha ribonuclease III enzyme to generate a prototype miRNA, which in turn is further excised by cytoplasmic Dicer ribonuclease to produce mature miRNA and antisense miRNA star products. Translational reticence or destability of the mature mRNA is due to the improper base pairing with miRNA which is eventually conceded by the RNA-provoked silencing complex.
-
CDKN2B-AS1 (antisense RNA 1)
This gene is imprinted in the p-arm (21.3) of chromosome 9 which resides within the gene cluster. Epigenetic silencing of the neighboring genes in the cluster is due to the interaction of polycomb suppressive complex-1 and complex-2 with the functional RNA molecule. Some of the alternatively processed transcript variants have been perceived in the form of circular RNA molecule. This gene seizes the prime locus for various disease abnormalities such as endometriosis, intracranial aneurysm, glaucoma, periodontitis, type-2 diabetes, cancer, and Alzheimer’s disease.
-
KCNQ1OT1 (antisense transcript 1)
KCNQ1OT1 gene is situated in p-arm (15.5) of chromosome 11 which is a nonprotein coding gene specifically expressed in maternally or paternally inherited chromosomes which enclose two clusters of epigenetically controlling genes. This gene is regulated by functionally imprinted control region present in the intron of KCNQ1, and the DNA is known to be unmethylated in maternally derived chromosomes. KCNQ1OT1 transcript is the antisense to the KCNQ1 gene which is an uncleaved lengthy ncRNA found to interrelate with chromatin affected by epigenetic alteration. The transcript plays a key role in colorectal carcinogenesis.
-
CCAT 1 and CCAT 2 (colon cancer-associated transcript 1 and 2)
CCAT gene is situated in the q-arm of chromosome 8 (24.21) which generates a long noncoding RNA gene to facilitate the tumor progression such as cell propagation, differentiation, invasion, and metastasis. It is known to be highly regulated in colon cancer which interrelates with myc oncoprotein and controls metabolism in an allele-specific manner.
Introns play a vital role in the gene expression of proteins which are translated from the intron retention segment of the messenger RNA. The alternative splicing process comes into picture with a varied multiplicity of proteins generated that alters the gene expression in eukaryotes. Thus, the crucial alternative excision process occurs in the nucleus of the eukaryotic cell with post translational variation such as methylation, sumoylation, and phosphorylation which influence the splicing mechanism appropriately. The splicing machinery affects the tumor microenvironment such as proliferation, differentiation, invasion, and metastasis of a cancer by affecting the mutations in the regulatory site. Therefore, the majority of the human genes is affected by the alternative splicing process [18]. MicroRNAs are derived from the intronic regions of the genes which are protein encoded in Homo sapiens, through the analysis of the genome populated with complete intron-coded genes comprised of 22–45% approximately. A phylogenetic conservation of miRNA may provide an additional advantage to the assimilation into transcriptional systems [19]. Currently, the genome-wide analysis studies gives the entire information about the gene loci, location, region, traits, disease probability, and other functional classes such as single-nucleotide polymorphism (SNPs) [20] evaluated through genotyping of wide populace. Unpredictably, the significant genome-wide analysis study of the SNPs and haplotypes is mostly resided in the noncoding regions impeding their functions during the molecular processes [21]. The cleavage of introns from the messenger RNA that in turn modifies the open reading frame (ORF) as well as the protein production. RNA sequencing analysis which utilizes the conserved introns, and predicting their gene structure and the intron prediction algorithms eliminate the introns with <50 nucleotides and STAR aligner that overcomes the RNA sequencing analysis by recognizing very short introns in the mRNA sequences [22]. Various literature reviews have suggested that intron retention mechanism paves an extensive evolutionary conservation of the homologous and heterologous genome diversity. They aid in regulating the genome multiplicity by assisting the species predilection through the epigenetic modification [23]. Therefore, the splicing process is in particular assessed by the kinetics phenomenon, which senses the polymerase activity as well as the length of introns that may be short or long [24]. The current progression of high-throughput transcriptome method emphasizes the persistent disposition of the human genome to transcription process that divulges the nonprotein encoding genes with their functional transcripts with the aim of genome intricacy. Hence, an efficient transcription unit might produce numerous molecules consisting of regulatory noncoding RNA; proteins rely upon the requirement of a cell according to their external factors [25]. Ultimately, the variation in the expression of specific noncoding RNAs engaged in the stemness of colorectal cancer by employing miRNAs serves as a new tool to reverse the cancer stem cell phenotype and overcome the therapy resistance significantly.
13.4 Cancer Stem Cells
Initially, the cancer stem cells are derived from progenitor or differentiated cells which are usually known to reside in the inner recesses of a tumor mass that holds the capability of self-renewal as well as a diverse family of cancer cells. There are specific surface markers that typically distinguish cancer stem cells that are isolated from various solid tumors including the colon. The two hypotheses of cancer stem cells are the mutations of the oncogene that build up within the adult cells or embryonic stem cells leading to an uncontrolled multiplication of cells, and the other one is the cellular dedifferentiation into a stem cell-like state [26]. Cancer stem cells are of two subdivisions, namely, stationary cancer stem cells and mobile cancer stem cells. The stationary cancer stem cells reside in the epithelial tissues which are active in tumor mass proliferation and cannot disseminate to other distant sites, and the mobile cancer stem cells divide indefinitely that leads to the metastasis of cancer to other parts of the body. It has been suggested that the colon cancer stem express CD44 and CD166. CD133 and epithelial-specific antigen surface marker characteristics of CSCs/CSLCs are their ability to invade and metastasize by acquiring epithelial-mesenchymal transition (EMT) phenotype, which can be determined by analyzing the expression of E-cadherin and vimentin representing Wnt effectors and notch signals. Most of the human malignancies emerge from tissues that contain an active population of stem cells. The stem cells are increasingly recognized as the focus of cancer-causing events, since both genetic and epigenetic alterations may lead to carcinogenesis processes [27]. This is primarily due to the tumor bursting through the intestinal wall and spreading through the lymph nodes and systemically through the bloodstream to distant organs. The colon’s luminal surface consists of one single layer of columnar epithelial cells that are folded into the lumen to form finger-like protrusions. The spaces between those folds are known as Lieberkuhn’s crypts, the intestine’s functional network. There are four distinct cell lineages in the colonic epithelium: enterocytes, goblet cells, endocrine cells, and Paneth cells. The small undifferentiated cells such as the crypt base columnar cells are known to hold the intestinal stem cells that are found to upsurge to the epithelial lineage. The stem cells have the potential of asymmetrical division which arises to give similar daughter cells as well as the transit amplifying cells that multiply and single out into goblet cells, endocrine cells, and enterocyte in the course of upward movement through the crypt. Here comes the Paneth cell which maintains the microenvironment of stem cell by the release of mucosal defense barriers that change the intestinal microflora through the growth factors and regulatory molecules.
13.4.1 Exacerbation of Cancer Stem Cells in Conjunction with Intronic Genes During the Development of Colorectal Cancer
Cancer stem cells have a distinct microenvironment encompassing the inclination of oxygen levels, chemokines receptors, cyclooxygenase, cytokines, molecules, and growth factors enhancing in the progression of colorectal cancer. Pro-cancer stem cell cytokines such as hepatocyte growth factor, prostaglandin E2, bone morphogenetic protein, and tumor niche-generating interleukins are found to be intensified in the cancer stem cell assembly. The major organ involved in the metastasis of colorectal cancer is the liver and also the growth factor such as chemokine receptor 4 (CXCR4). Stromal-derived factor 1 is chiefly articulated in the liver assisting in the transit of circulating CXCR4 colorectal cancer cells [28]. Wnt, Sonic hedgehog, bone morphogenetic protein (BMP), β-catenin, tumor growth factor-beta (TGF-β), and notch are the major signaling pathways engaged in the homeostasis of colorectal cancer stem cells precisely. Some of the cellular processes such as proliferation, differentiation, migration, and cell death majorly rely on the homeostatic self-renewal of the intestine which ultimately depends on the evolutionarily conserved signaling pathway [29]. Eventually, the microRNAs control several cancer processes like transformation, tumor cell duplication, epithelial-mesenchymal transition (EMT), invasion, and metastasis which are mainly involved in the inhibition of gene expression in pathways that regulate cell processes, for instance, cell cycle, apoptosis, and miRNA migration. Intronic gene such as mir-21 is found to be overexpressed nearly in all malignancies such as breast cancer, glioblastoma, colorectal cancer, lung cancer, pancreatic cancer, and leukemia. In due course, pluripotency and differentiation are known to be through the alteration of stem cells through microRNA [30].
13.5 Future Direction
This chapter encompasses on the intronic genes mainly involved in the progression of colorectal cancer with preliminary information. Researchers turned their focal point toward the mammalian cell and determined that the noncoding sequences (junk DNAs) are known to perform a key role in the development of cancer. Then, the alternative splicing process takes place in the messenger RNA strand that implicates in the retention of introns leading to a varied gene expression as well as promotion in colorectal cancer. So, the intron retention is known by sensing the premature termination codons in the mRNA strand and triggers the nonsense-mediated decay process appropriately. Therefore, colorectal cancer-associated diseases such as the adenomatous polyposis colorectal cancer and Lynch syndrome have paved the way to know more about the genes and what are all the intronic genes mainly concentrated among them in the development of colorectal cancer. As a result, the concentration of the protein output and gene expression is known to be influenced by the intron retention.

Thus, the noncoding regions in the genome can be predicted by RNA sequencing method and interpreting the obtained results from the normal to the diseased form focusing on colorectal cancer. They can also knock down the associated intronic genes in the tumor microenvironment of colorectal cancer which may be beneficial in the tumor proliferation and differentiation. Recent advancement has hypothesized that conjunction of intronic gene with cancer stem cells is known to be progressed in the colorectal cancer precisely. Ultimately, the current circumstances of research fields are accomplished to work with the intriguing noncoding sequences engaged to play a crucial role at certain neoplastic transformation in the normal microflora of the colorectal by analyzing the domino effect of the intronic genes in the molecular phase precisely.
References
Chorev M, Carmel L (2012) The function of introns. Front Genet 3:55
Niu D-K, Yang Y-F (2011) Why eukaryotic cells use introns to enhance gene expression: splicing reduces transcription associated mutagenesis by inhibiting topoisomerase I cutting activity. Biol Direct 6:24
Jeffares DC, Mourier T, Penny D (2006) The biology of intron gain and loss. Trends Genet 22(1):16–22
Haugen P, Simon DM, Bhattacharya D (2005) The natural history of group I introns. Trends Genet 21(2):111–119
Irimia M, Roy SW (2014) Origin of spliceosomal introns and alternative splicing. Cold Spring Harb Perspect Biol 6(6):a016071
Middleton R, Gao D, Thomas A et al (2017) Irfinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 18(1):51
Esquela-Kerscher A, Slack FJ (2006) Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer 6(4):259–269
Lizarbe MA, Fernández-Lizarbe S, Calle-Espinosa J et al (2017) Colorectal cancer: from the genetic model to posttranscriptional regulation by noncoding RNAs. Hindawi Biomed Res Int 2017:7354260
Cheetham SW, Gruhl F, Mattick JS, Dinger ME (2013) Long noncoding rnas and the genetics of cancer. Br J Cancer 108:2419–2425
Alhopuro P, Sammalkorpi H et al (2011) Candidate driver genes in microsatellite-unstable colorectal cancer. Int J Cancer 130(7):1558–1566
Sameer AS (2013) Colorectal cancer: molecular mutations and polymorphisms. Front Oncol 3:114
Jung H, Lee D, Lee J et al (2015) Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 47(11):1242–1248
Dvinge H, Bradley RK (2015) Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7:45
Wong JJ-L, Au AYM et al (2015) Intron retention in mRNA: no longer nonsense. Bioessays 38:41–49
Fang X et al (2016) SNORD126 promotes HCC and CRC cell growth by activating the PI3K-AKT pathway through FGFR2. J Mol Cell Biol Adv 9(3):243–255
Wong JJ-L et al (2015) Intron retention in mRNA: no longer nonsense. Bioessays 38:41–49
Middleton R et al (2017) IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 18:51
Buckley PT (2014) Cytoplasmic intron retention, function, splicing, and the sentinel RNA hypothesis. WIREs RNA 5:223–230
Martinez-Montiel N et al (2018) Alternative splicing as a target for cancer treatment. Int J Mol Sci 19:545
Steiman-Shimony A et al (2018) Assessing the functional association of intronic mirnas with their host genes. RNA 24:991–1004
Abebrese EL et al (2017) Identification of human short introns. PLoS One 12(5):e0175393
Rohlin A et al (2017) Expanding the genotype–phenotype spectrum in hereditary colorectal cancer by gene panel testing. Fam Cancer 16:195–203
Hube F et al (2017) Short intron-derived ncRNAs. Nucleic Acids Res 45(8):4768–4781
Schmitz U et al (2017) Intron retention enhances gene regulatory complexity in vertebrates. Genome Biol 18:216
Bartonicek N et al (2017) Intergenic disease-associated regions are abundant in novel transcripts. Genome Biol 18:241
Roy S et al (2012) Cancer stem cells in colorectal cancer: genetic and epigenetic changes. J Stem Cell Res Ther 7(Suppl 6):10342
Kim SW et al (2017) Widespread intra-dependencies in the removal of introns from human transcripts. Nucleic Acids Res 45(16):9503–9513
Munro MJ et al (2017) Cancer stem cells in colorectal cancer: a review. J Clin Pathol 71(2):110–116
Huang T et al (2013) Noncoding RNAs in cancer and cancer stem cells. Chin J Cancer 32:582–593
Zhou Y et al (2018) Cancer stem cells in progression of colorectal cancer. Oncotarget 9(70):33403–33415
Acknowledgments
The authors are thankful to Science and Engineering Research Board (SERB) (EMR/20l7/001877) for providing the Core Research Grant to Prof. Surajit Pathak and Chettinad Academy of Research and Education for the research support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Gopi, J., Gopinath, M., Sun, XF., Pathak, S., Banerjee, A. (2020). Functionality of Intron-Specific Genes and Cancer Stem Cells in the Progression of Colorectal Cancer. In: Pathak, S., Banerjee, A. (eds) Cancer Stem Cells: New Horizons in Cancer Therapies. Springer, Singapore. https://doi.org/10.1007/978-981-15-5120-8_13
Download citation
DOI: https://doi.org/10.1007/978-981-15-5120-8_13
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-5119-2
Online ISBN: 978-981-15-5120-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)