
Abstract
Sarcopenia, the age-related loss of skeletal muscle mass, is characterized by a deterioration of muscle quantity and quality, leading to a gradual slowing of movement, a decline in strength and power, increased risk of fall-related injury, and often frailty. Chronic kidney disease (CKD) is characterized by the gradual loss of renal function over a period of months to years. CKD is a catabolic state, leading to renal sarcopenia. This chapter focuses on the recent advances of pharmacological approaches for attenuating normal and CKD-induced sarcopenia. A myostatin-inhibiting antibody is the most important candidate to prevent normal sarcopenia in humans, but is needed for time to determine the effect for CKD-induced sarcopenia. Although treatment with ghrelin seems to be applicable for both types of sarcopenia in humans, further validation of this trial is necessary by increasing the sample size, varying the range of doses during treatment, and observing other outcomes. Supplementation with ursolic acid is also an intriguing candidate to combat normal and CKD-induced sarcopenia, although further systematic and fundamental research is needed for this treatment on humans.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
11.1 Introduction
Sarcopenia is widely accepted as causing an age-related decline of muscle mass, quality, and strength. In addition, it is often used to describe both the cellular processes (denervation, mitochondrial dysfunction, and inflammatory and hormonal changes) and the outcomes such as decreased muscle strength, mobility, and function; a greater risk of falls; and reduced energy needs. Sarcopenia can be considered “primary” (or age related) when no other cause is evident but aging itself. Primary sarcopenia is especially associated with overall reduction of the physical exercise. Secondary sarcopenia usually occurs when one or more identifiable causes coexist. This condition is a proxy of chronic or acute diseases prevalent in older persons such as diabetes, several types of cachexia (e.g., chronic obstructive pulmonary disease [COPD], cancer cachexia, chronic heart failure, and CKD), stroke, and hip fracture. Von Haeling et al. [1] estimated its prevalence at 5–13% for elderly people aged 60–70 years and 11–50% for those aged 80 years or older.
CKD is characterized by the gradual loss of renal function over a period of months to years. In addition to renal deficiency, CKD is a major risk multiplier in patients with diabetes, hypertension, heart disease, and stroke [2]. The prevalence of CKD varies between 7 and 12% in the general population worldwide and increases with age, affecting more than 30% of people over 65 years of age [3]. CKD is a catabolic state leading to renal sarcopenia, which is characterized by the loss of skeletal muscle strength and physical function [4]. To explain molecular mechanism of CKD-induced sarcopenia, there are many candidates such as the ubiquitin-proteasome system (UPS), caspase-3-mediated apoptosis, autophagy, imbalance between the anabolic insulin/IGF-I and catabolic myostatin signaling pathways, and IL-6 and TNF-a-mediated inflammatory pathways [5]. In contrast, in normal sarcopenic mammalian muscles, there is no apparent evidence for enhancement in several negative regulators such as UPS, calpain, and inflammatory pathway’s downstream mediator, NF-κB (nuclear factor-kappaB) [6, 7]. In addition, many recent studies indicated an apparent functional defect of autophagy-dependent signaling in sarcopenic muscle [8,9,10] different to the activation of this in CKD-induced sarcopenia.
The progression of normal and CKD-induced sarcopenia is effectively prevented by the exercise, in particular, resistance training [11, 12]. Inhibiting myostatin is an important option of attenuating muscle wasting, such as cachexia, and sarcopenia [7]. More recent studies indicated the possible application of new pharmacologic agents such as ursolic acid and fibroblast growth factor 19] to prevent muscle atrophy including CKD model mice [13, 14]. In addition, new intriguing candidate for attenuating CKD has recently emerged [15, 16]. This chapter summarizes the recent strategies for inhibiting normal and CKD-induced sarcopenia.
11.2 Pharmacological Approach
11.2.1 Myostatin Inhibition
Myostatin, a novel member of the transforming growth factor-β superfamily, regulates negative muscle growth [17]. Mutations of myostatin in mice can lead to marked hypertrophy and/or hyperplasia in developmental stage. In addition, mouse skeletal muscles engineered to overexpress the naturally occurring myostatin inhibitor follistatin, or a dominant negative form of main myostatin receptor (ActRIIB) all display similar, if not greater, increases in size [17].
The increased levels of myostatin are widely accepted to lead to muscle wasting including muscle atrophy due to unloading in mice and humans [6] and with severe atrophy in HIV patients. Whether myostatin expression levels increase in sarcopenic muscles have yielded conflicting results [6, 18, 19]. Sarcopenic muscles of mice seem to exhibit the abundant Smad3 (possible myostatin-downstream regulator) but not myostatin [18]. In addition, muscle loading induced more abundant myostatin in satellite cells in type II fibers in older males than in younger males, although myostatin in satellite cells of each muscle fiber is equally expressed regardless of age at baseline [19]. Therefore, it seems that myostatin-dependent signaling is locally activated in sarcopenic mammalian muscles.
Phase 2 humanized mAb against myostatin LY2495655 showed dose-dependent and significant increases in appendicular lean mass at weeks 8 and 16 as compared with placebo after total hip arthroplasty randomized clinical trial of 400 patients [20]. Many researchers have conducted experiments to inhibit myostatin in models of muscle disorders such as cancer cachexia, amyotrophic lateral sclerosis, and Duchenne muscular dystrophy (DMD) [21, 22]. Although several researchers actively try to determine the effect of pharmacological inhibition of myostatin for DMD patients, it is much difficult for obtaining positive effects and few possibilities of clinical application. Indeed, a randomized clinical trial that used the same compound for DMD patients had a trend toward improved muscle mass and performance (6-min walk test) but was stopped early due to nonmuscle side effects (i.e., epistaxis and telangiectasias) [23]. In contrast, several investigators basically examined the effect of inhibiting myostatin to counteract sarcopenia using animals. Lebrasseur et al. [24] showed significantly greater muscle mass and increased performance such as distance to exhaustion and treadmill time in aged mice after treatment with a myostatin inhibitor (PF-354, 24 mg/kg) for 4 weeks. They also observed the decreased levels of phosphorylated Smad3 and Muscle ring-finger protein 1 (MuRF1) in aged muscle after pharmacological myostatin inhibition. In addition, Murphy et al. [22] showed that a lower dose of PF-354 (10 mg/kg) significantly increased the fiber size and in situ force of hindlimb muscle of 21-month-old senescent mice. More recently, Becker et al. [25] conducted a randomized, Phase 2 trial of a myostatin antibody (LY2495655: LY, 315 mg) by subcutaneous injection using worldwide frail elderly aged 75 years or older (the USA, France, Australia, etc.). At 24 weeks, the LY group significantly increased the least-squares mean change in appendicular lean body mass (LBM) than the placebo group. In addition, treatment with LY for elderly subjects for 24 weeks improved functional characteristics such as stair climbing time and chair rise with arms from the baseline [25]. These lines of evidence clearly highlight the therapeutic potential of the antibody-directed inhibition of myostatin for treating sarcopenia.
In mice and rats with CKD, there is apparent increase in myostatin expression in muscle [26, 27]. There is some report for pharmacological inhibition of myostatin for CKD mice but not those of humans. Subcutaneous injection of an anti-myostatin peptibody into CKD mice reversed the loss of body weight (5–7% increase) and mass of hindlimb muscles such as tibialis anterior, gastrocnemius, extensor digitorum longus, and soleus (~10% increase) [26]. In addition, myostatin inhibition suppressed circulating inflammatory cytokines (interferon-γ [IFNγ], interleukin [IL]-6, tumor necrosis factor-α [TNF-α], macrophage colony-stimulating factor, etc.) and mRNA expression of these cytokines (TNF-α and IL-6) in CKD mice. Furthermore, this treatment enhances the rate of protein synthesis, satellite cell function, and improves IGF-I intracellular signaling [26]. Although the effect of pharmacological myostatin inhibition for CKD was clearly obtained using model mice, there is no clear evidence for CKD human trial by myostatin inhibition. A pilot randomized controlled trial (RCT) study of PINTA 745, an anti-myostatin peptibody, in patients undergoing hemodialysis has completed recruiting, but results are not yet published (www.clinicaltrials.gov NCT01958970). The primary outcome is percentage change in lean mass using Dual-energy X-ray absorptiometry (DXA) after 4 weeks of treatment. To the best of our knowledge, this is the only study in patients with kidney disease. Hopefully, myostatin antagonism will be tested in patients with predialysis CKD in the future.
11.2.2 Anabolic Steroids
In males, levels of testosterone decrease by 1% per year, and those of bioavailable testosterone by 2% per year from age 30 [28]. In women, testosterone levels drop rapidly between the ages of 20 and 45. There is a significant decrease in testosterone level with renal impairment (progressing CKD stage) [29]. The decreased testosterone level in CKD patients had a significant correlation with increased level of sperm cytoplasmic droplets and for total neutral glucosidase activity [29]. Testosterone increases muscle protein synthesis, and its effects on muscle are modulated by several factors including the genetic background, nutrition, and exercise. Systemic reviews [30] have indicated that supplementation with testosterone attenuates some sarcopenic characteristics such as decreases in the grip strength [31] and muscle mass [32]. A study of long-term (6 months) supraphysiological treatment with testosterone in a placebo-controlled study showed increased leg lean body mass and leg and arm strength [33]. Although there are significant increases in strength among elderly males given high doses of testosterone, the potential risks may outweigh the benefits. Thrombotic complication, sleep apnea, and an increased risk of prostate cancer are associated risks with testosterone therapy in older men.
Novel, nonsteroidal compounds, called selective androgen receptor modulators (SARMs), have shown tissue-selective activity and improved pharmacokinetic properties but lower systemic side effects. SARMs have been tested in the healthy elderly patients and in cancer-associated sarcopenia [34, 35]. In fact, improvements in physical function (stair climb speed) and LBM were shown in a Phase 2, double-blind study of enobosarm, a nonsteroidal SARM, in healthy postmenopausal and elderly men [35]. This study showed that only 1 mg of enobosarm increased LBM in advanced cancer patients. In addition, another type of SARM, GSK2881078, was recently investigated in healthy older men and women [36]. Repeated DXA and magnetic resonance imaging cross-sectional thigh scan revealed that treatment with GSK2881078 once daily for 50 days resulted in significant greater lean mass with no serious adverse effects in spite of transient elevations of alanine aminotransferase. Whether these drugs are effective in treating normal and CKD-induced sarcopenia should be determined by further studies with more strict clinical trials.
Using 43 hemodialysis patients, Supasyndh et al. [37] tested a supplementation of oxymetholone, a more mild anabolic steroid, under RCT for a 24-week duration. They observed a significant increase in fat-free mass, subjective scores of physical function, and a grip strength for this treatment. During 24 weeks, these hemodialysis patients exhibited the atrophy of both types I and II muscle fibers and decreased the IGF-I receptor mRNA level under placebo treatment, but these were blocked by treatment with oxymetholone and surprisingly induce hypertrophy of type I fibers. More recently, I found very unique study for this field. Kim et al. [38] determined the effect of oxymetholone (50 mg/kg) for old mice subjected to chronic forced exercise. Chronic annual exercise for 28 days elicited the abundant expression of reactive oxygen species, partially due to downregulation of glutathione, superoxide dismutase, and catalase activity, but it restored these levels at baseline after supplementation with oxymetholone. This treatment elicited the fiber hypertrophy of the gastrocnemius and soleus muscles due to attenuation of the expression of myostatin, sirtuin1, and muscle-atrophy genes [38]. However, there is little report of oxymetholone treatment for sarcopenia or CKD in mice model or humans. Collectively, these facts should be elucidated by many studies to assess benefits versus risks for normal and CKD-induced sarcopenia by treatment with anabolic steroids, androgens, or similar substances.
11.2.3 Ghrelin
Ghrelin, a 28-amino acid peptide, is mainly produced by cells in the intestines, stomach, and hypothalamus. Ghrelin is a natural ligand for the growth hormone (GH)-secretagogue receptor that possesses a unique fatty acid modification. Ghrelin can stimulate GH secretion and modulate energy homeostasis by promoting adiposity and enhancing food intake. In contrast, ghrelin suppresses the production of anorectic proinflammatory cytokines (IL-1β, IL-6, and TNF-α) on T lymphocytes and monocytes [39]. Ghrelin and low-molecular-weight agonists of the ghrelin receptor are considered attractive candidates for the treatment of cachexia [40]. Nagaya et al. [41] demonstrated that intravenous human ghrelin (2 μg/kg, twice daily, 3 weeks) significantly increased in the lean body mass, hand grip strength, and Karnofsky performance score in patients with chronic obstructive pulmonary disease. In addition, Nagaya et al. [42] demonstrated that similar intravenous treatment with human ghrelin significantly improved several parameters (e.g., lean body mass measured by Dual-energy X-ray Absorption and left ventricular ejection fraction) in patients with chronic heart failure. Pietra et al. [43] demonstrated that oral treatment with anamorelin HCl (ANAM) to rats significantly and dose dependently increased food intake and body weight at all dose levels (3, 10, and 30 mg/kg) compared with the control and significantly increased GH levels at 10 or 30 mg/kg doses. In addition, Phase 3 trials of two types of anamorelin (ROMANA1 and ROMANA2) were conducted in patients with non-small cell lung cancer and cachexia [44]. Twelve weeks of both anamorelin treatments induced significant increases in the LBM but not handgrip strength in these cachectic patients with very low levels of adverse effects. However, a more recent review pointed out that heterogeneity existed in the clinical effects of anamorelin [45]. Intriguingly, such a treatment with ghrelin was tried in elderly people. In a long-term (1 year) placebo-controlled study, an oral ghrelin mimetic (MK-677) was observed to increase an appetite in healthy older adults (over 60 years old), although this study did not show a significant increase in strength or function by the ghrelin mimetic treatment [46]. Anamorelin HCl (ONO-7643) is a potent and selective novel ghrelin receptor agonist that mimics the N-terminal active core of ghrelin [47].
It is possible that unacylated ghrelin (UnAG) lowers several defective symptoms (e.g., oxidative stress), resulting in muscle mass loss. Cappellari et al. [48] determined the effect of treatment with UnAG for 5/6 nephrectomy (Nx) CKD rat model. They demonstrated that UnAG administration (200 μg twice a day) normalizes CKD-induced loss of gastrocnemius muscle mass and a cluster of high tissue mitochondrial reactive oxygen species generation, high proinflammatory cytokines, and low insulin signaling activation. More specifically, UnAG administration ameliorates the downregulation of the amount of activated (phosphorylated) Akt, mammalian target of rapamycin (mTOR), p70 ribosomal protein S6 kinase (p70S6K), GSK3β, and PRAS40 in the muscle of CKD model rats. In addition, 8 weeks’ treatment with UnAG (0.1 nmol/g BW) to similar CKD model mice improved all of the declined parameters such as exercise endurance, muscle mass, mitochondrial amount, and the expression of peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1α) [49]. Surprisingly, treatment with ghrelin increased exercise endurance in CKD mice more markedly than those of more popular and general treatment with IGF-I [49]. Very intriguingly, some trial of ghrelin mimetic was conducted using hemodialysis patients. Campbell et al. [50] performed a randomized crossover double-blind study in assessing the effect of MK-0677 versus placebo on IGF-I levels, the primary outcome, in hemodialysis patients (22 subjects, 3-month crossover). Although there is no significant change in many blood parameters such as IL-6, TNF-α, adiponectin, GH, and insulin by treatment with MK-0677, the geometric mean IGF-I blood levels were 1.76-fold greater following the treatment with ghrelin. Therefore, further validation of this trial is necessary by increasing the sample size, varying the range of doses during treatment, and observing other outcomes.
11.2.4 Ursolic Acid
A pentacyclic triterpenoid, ursolic acid, is the major waxy component in apple peel, and it is also found in many other edible plants. Ursolic acid exerts beneficial effects in animal models of diabetes and hyperlipidemia [51]. Kunkel et al. [52] showed that ursolic acid reduced two skeletal atrophy-inducing models, such as muscle denervation and fasting, by predictive analysis, using connectivity mapping. Under fasting, ursolic acid might increase muscle mass by inhibiting atrophy-promoting muscle gene (atrogin-1 and MuRF1) expression [52]. Chronic administration of ursolic acid for unstressed normal mice with appears to elicit muscle hypertrophy, probably due to the decrease in mRNA expression of atrogin-1 and MuRF1. Ursolic acid may increase skeletal muscle Akt phosphorylation in vivo. Intriguingly, ursolic acid alone was not sufficient to activate the receptor of insulin and IGF-I but done rapidly only by combining with insulin and IGF-I [52]. Thus, ursolic acid’s hypertrophy-promoting effect seems to be indirect. Some reports have determined whether the treatment with ursolic acid attenuates sarcopenia in mice. Using 22-month-old mice, Ebert et al. [53] investigated the effect of 0.27% addition of ursolic acid for standard diet to the weight of the body, muscle, or several organs. Although treatment with ursolic acid did not modulate the weight of the body, heart, liver, or epididymal and retroperitoneal fat, this improved the weight of quadriceps, the fiber size of type IIB but not type IIX, grip strength, and specific force. Intriguingly, a subset of the mRNAs repressesd by ursolic acid in aged muscle are positively regulated by activating transcription factor 4 (ATF4) [53]. Thus, it is possible that the attenuating effect of ursolic acid for aged muscle atrophy is modulated via ATF4-dependent signaling. In contrast, using a mouse model of CKD, a long-term (21 days) effect with ursolic acid was verified (oral gavage, 100 mg/kg) [14]. Treatment with ursolic acid has been demonstrated to markedly attenuate CKD-induced muscle atrophy by inhibiting the expression of inflammatory cytokines, such as IL-6 and TNF-α and myostatin, which elicit several muscle-specific ubiquitin ligases (MUSA1, MuRF1, and atrogin-1). In addition, they demonstrated that ursolic acid significantly attenuated the levels of phosphorylation of p38, signal transducer and activator of transcription 3, and NF-κB (p65) in the muscle affected by CKD. However, its hypertrophic effects in humans are less clear. Supplementation with ursolic acid (50 mg/day for loquat leaf extract) did not improve physical performance (balance, 4 m gait speed, and chair-rise ability), muscle mass, and strength (knee extension and handgrip strength) in healthy adults (aged between 19 and 65 years and body mass index ranging from 18.5 to 30.0 kg/m2) than those who took placebo [54]. In addition, supplementation with ursolic acid (3 g) for resistance-trained men did not modify serum level of insulin and IGF-I and muscle IGF-I-dependent pathways (IGF-I receptor, Akt, mTOR, and p70S6K) than those adapted to a single bout of resistance training [55]. Further research is needed to elucidate the effect of supplementation with ursolic acid on human skeletal muscle and the attenuation of sarcopenia and CKD.
11.2.5 The Other Candidates
AST-120, a charcoal absorbent, is used in indoxyl sulfate (IS)-targeted therapeutics because it absorbs indole, a precursor of IS, in the gut flora, resulting in a reduced level of IS synthesis in the liver [56]. In CKD mice, the weights of body and several skeletal muscles (tibialis anterior, soleus, and gastrocnemius) were markedly decreased. Compared with sham mice, IL-6 and atrophy-accelerating factors (myostatin and atrogin-1) were significantly enhanced in the skeletal muscle of CKD mice, whereas muscular Akt phosphorylation was decreased [15]. In addition, a reduced exercise capacity was observed for the CKD mice, which was accompanied by an increased muscular autophagy and the decreased expression of muscular PGC-1α and mitochondrial oxidative capacity. An AST-120 treatment (charcoal oral absorbent, 8 w/w% in powder diet) significantly restored these degraded symptoms, including muscle atrophy observed in CKD mice to the sham levels as well as a decrease in IS levels. An L-carnitine (560 mg/kg) or teneligliptin (60 mg/kg) treatment by drinking water also restored them to the sham levels without changing IS levels [15]. There are other reports of AST-120 amelioration for chronic kidney disease in mice model [57, 58]. Intriguingly, treatment with AST-120 for mice subjected to renal failure (RF) significantly downregulated urinary albumin excretion and mRNA expression levels of plasminogen activator inhibitor 1. IS binds to aryl hydrocarbon receptor (AhR), activates cytochrome P450 family 1 subfamily A member 1 (Cyp1a1), and induces vascular inflammation [59]. The skeletal muscle of RF mice, which contained elevated levels of IS, displayed significantly higher Cyp1a1 expression than that of control mice, suggesting that accumulated IS induced Cyp1a1 expression. AST-120 restored Cyp1a1 expression levels in the RF mice to control levels [58].
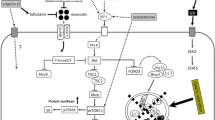
5-aminolevulinic acid (ALA) is a mitochondria-activating substance, which is synthesized from glycine and succinyl-CoA by the action of ALA synthase in mitochondria. ALA supplementation with sodium ferrous citrate (SFC) has been shown to promote mitochondrial electron transport and increased ATP production [60]. Fujii et al. [16] investigated the effect of supplementation with low- and normal-dose ALA for normal and CKD-induced sarcopenia in mice. Normal dose of ALA (0.03%) significantly increased the grip power and muscle weight in both aged (100 weeks of age) and CKD model 5/6 Nx mice. Surprisingly, low dose of ALA (0.003%) also significantly improved the extent of decrease in running distance in both symptoms probably due to modulation of mitochondrial amount and those of normal dose [16]. The efficacy of ALA on physical performance of elderly women has also been recently reported by Masuki et al. [61]. Since low dose of ALA supplementation is effective for muscle endurance in mice, ALA would be an intriguing candidate for improving normal and CKD-induced sarcopenia in humans. Figure 11.1 represents an overview of pharmacological interventions for sarcopenia in CKD.

Pharmacological intervention affects different mediators in CKD-induced sarcopenic muscle. Recent findings suggest that the myostatin-Smad pathway inhibits protein synthesis probably due to blocking the functional role of Akt. In animal studies, ursolic acid supplementation blocks the myostatin- and TNF-α-dependent signaling, resulting in the attenuation of muscle atrophy. Treatment with an ursolic acid and ghrelin upregulates the amount of IGF-I and then stimulates protein synthesis by activating the Akt/mTOR/p70S6K pathway. CKD-induced sarcopenic muscle exhibits a marked upregulation of myostatin and several atrogenes, which are effectively ameliorated by anabolic steroids. ALK activin receptor-like kinase, ActRIIB activin receptor IIB, IGF-I insulin-like growth factor I, TSC tuberous sclerosis complex, TORC1 component of TOR signaling complex 1, Rheb Ras homolog enriched in brain, mTORC1 mammalian target of rapamycin complex 1, eIF4E eukaryotic initiation factor 4E, p70S6K p70 ribosomal protein S6 kinase, FOXO forkhead box O, TNF-α tumor necrosis factor-α, IKK inhibitor of kB kinase, NF-κB nuclear factor-kappaB, MuRF1 muscle ring-finger protein 1
11.3 Conclusion
The recent advances in muscle biology have led to new hopes for hormonal, pharmacological, and nutritional treatment of muscle wasting. In the case of CKD-induced sarcopenia, there are many candidates such as UPS, apoptosis, autophagy, imbalance between anabolic and catabolic pathway, or inflammatory signaling [5]. In normal sarcopenic mammalian muscles, there is no demonstration for enhancement in UPS, calpain, and inflammatory pathway [6, 7], in spite of an apparent functional defect of autophagy-dependent signaling [8,9,10]. Although there are several differences in the molecular mechanism between normal and CKD-induced sarcopenia, several candidates (myostatin inhibitor, ghrelin, and ursolic acid) seem to exhibit similar positive effect for both symptoms. Further research is needed to elucidate the effect of these supplementations on these two sarcopenias in humans.
References
von Haehling S, Morley JE, Anker SD. An overview of sarcopenia: facts and numbers on prevalence and clinical impact. J Cachexia Sarcopenia Muscle. 2010;1:129–33.
Couser WG, Remuzzi G, Mendis S, Tonelli M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011;80:1258–70.
Romagmnami P, Remuzzi G, Glassock R, Levin A, Jager KJ, Tonelli M, Massy Z, Wanner C, Anders HJ. Chronic kidney disease. Nat Rev Dis Primers. 2017;3:17088.
Moorthi RN, Avin KG. Clinical relevance of sarcopenia in chronic kidney disease. Curr Opin Nephrol Hypertens. 2017;26:219–28.
Yoshida T, Delafontaine P. Mechanisms of cachexia in chronic disease states. Am J Med Sci. 2015;350:250–6.
Sakuma K, Aoi W, Yamaguchi A. Current understanding of sarcopenia: possible candidates modulating muscle mass. Pflügers Arch. 2015;467:213–29.
Sakuma K, Aoi W, Yamaguchi A. Molecular mechanism of sarcopenia and cachexia: recent research advances. Pflügers Arch. 2017;469:573–91.
Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R, Sandri M. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014;8:1509–21.
Jiao J, Demontis F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr Opin Pharmacol. 2017;34:1–6.
Sakuma K, Kinoshita M, Ito Y, Aizawa M, Aoi W, Yamaguchi A. p62/SQSTM1 but not LC3 is accumulated in sarcopenic muscle of mice. J Cachexia Sarcopenia Muscle. 2016;7:204–12.
Roshanravan B, Gamboa J, Wilund K. Exercise and CKD: skeletal muscle dysfunction and practical application of exercise to prevent and treat physical impairments in CKD. Am J Kidney Dis. 2017;69:837–52.
Sakuma K, Yamaguchi A. Molecular mechanisms in aging and current strategies to counteract sarcopenia. Curr Aging Sci. 2010;3:90–101.
Benoit B, Meugnier E, Castelli M, Chanon S, Vieille-Marchiset A, Durand C, Bendridi N, Pesenti S, Monternier PA, Durieux AC, Freyssenet D, Rieusset J, Lefai E, Vidal H, Ruzzin J. Fibroblast growth factor 19 regulates skeletall muscle mass and ameliorates muscle wasting in mice. Nat Med. 2017;23:990–6.
Yu R, Chen JA, Xu J, Cao J, Wang Y, Thomas SS, Hu Z. Suppression of muscle wasting by the plant-derived compound ursolic acid in a model of chronic kidney disease. J Cachexia Sarcopenia Muscle. 2017;8:327–41.
Enoki Y, Watanabe H, Arake R, Fujimura R, Ishiodori K, Imafuku T, Nishida K, Sugimoto R, Nagao S, Miyamura S, Ishima Y, Tanaka M, Matsushita K, Komaba H, Fukagawa M, Otagiri M, Maruyama T. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J Cachexia Sarcopenia Muscle. 2017;8:735–47.
Fujii C, Miyashita K, Mitsuishi M, Sato M, Fujii K, Inoue H, Hagiwara A, Endo S, Uto A, Ryuzaki M, Nakajima M, Tanaka T, Tamaki M, Muraki A, Kawai T, Itoh H. Treatment of sarcopenia and glucose intolerance through mitochondrial activation by 5-aminolevulinic acid. Sci Rep. 2017;7:4013.
Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86.
Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and notch induces CDK inhibitors is old muscle stem cells. Nature. 2008;454:528–32.
McKay BR, Ogborn DI, Bellamy LM, Tarnopolsky MA, Parise G. Myostatin is associated with age-related human muscle stem cell dysfunction. FASEB J. 2012;26:2509–21.
Woodhouse L, Gandhi R, Warden SJ, Poiraudeau S, Myers SL, Benson CT, Hu L, Ahmad QI, Linnemeier P, Gomez EV, Benichou O. A phase 2 randomized study investigating the efficacy and safety of myostatin antibody LY2495655 versus placebo in patients undergoing elective total hip arthroplasty. J Frailty Aging. 2016;5:62–70.
Murphy KT, Ryall JG, Snell SM, Nair L, Koopman R, Krasney PA, Ibebunjo C, Holden KS, Loria PM, Salatto CT, Lynch GS. Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am J Pathol. 2010;176:2425–34.
Murphy KT, Koopman R, Naim T, Léger B, Trieu J, Ikebunjo C, Lynch GS. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010;24:4433–42.
Campbell C, McMillan HJ, Mah JK, Tarnopolsky M, Selby K, McClure T, Wilson DM, Sherman ML, Escolar D, Attie KM. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: results of a randomized, placebo-controlled clinical trial. Muscle Nerve. 2017;55:458–64.
Lebrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects on performance and metabolic outcomes in aged mice. J Gerontol Series A Biol Sci Med Sci. 2009;64:940–8.
Becker C, Lord SR, Studenski SA, Warden SJ, Dielding RA, Recknor CP, Hochberg MC, Ferrari SL, Blain H, Binder EF, Rolland Y, Poiraudeau S, Benson CT, Myers SL, Hu L, Ahmad QI, Pacuch KR, Gomez EV, Benichou O, STEADY Group. Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015;3:948–57.
Wang D-T, Yang Y-J, Huang R-H, Zhang Z-H, Lin X. Myostatin activates the ubiquitin-proteasome and autophagy-lysosome systems contributing to muscle wasting in chronic kidney disease. Oxidative Med Cell Longev. 2015;2915:684965.
Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, Song Y, Min H, Wang X, Du J, Mitch WE. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011;25:1653–63.
Feldman HA, Longcope C, Derby CA, Johannes CB, Araujo AB, Coviello AD, Bremner WJ, McKinlay JB. Age trends in the level of serum testosterone and other hormones in middle-aged men: longituidinal results from the Massachusetts male aging study. J Clin Endocrinol Metab. 2002;87:589–98.
Lehtihet M, Hylander B. Semen quality in men with chronic kidney disease and its correlation with chronic kidney disease stages. Andrologia. 2015;47:1103–8.
Bhasin S, Calof O, Storer TW, Lee ML, Mazer NA, Jasuja R, Montori VM, Gao W, Dalton JT. Drug insight: testosterone and selective androgen receptor modulators as anabolic therapies for physical dysfunction in chronic illness and ageing. Nat Clin Pract Endocrinol Metab. 2006;2:146–59.
Bakhshi V, Elliott M, Gentili A, Godschalk M, Mulligan T. Testosterone improves rehabilitation outcomes in ill older men. J Am Geriatr Soc. 2000;48:550–3.
Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, Lieberman SA, Tipton K, Wolfe RR, Urban RJ. Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am J Physiol Endocrinol Metab. 2002;282:E601–7.
Sinha-Hikim I, Cornford M, Gaytan H, Lee ML, Bhasin S. Effects of testosterone supplementation on skeletal muscle fiber hypertrophy and satellite cells in community-dwelling older men. J Clin Endocrinol Metab. 2006;91:3024–33.
Crawford J, Prado CMM, Ann Johnston M, Gralla RJ, Taylor RP, Hancock ML, Dalton JT. Study design and rationale for the phase 3 clinical development program of enobosarm, a selective androgen receptor modulator, for the prevention and treatment of muscle wasting in cancer patients (POWER trials). Curr Oncol Rep. 2016;18:37.
Dalton JT, Barnette KG, Bohl CE, Hancock ML, Rodriguez D, Dodson ST, Morton RA, Steiner MS. The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial. J Cachexia Sarcopenia Muscle. 2011;2:153–61.
Neil D, Clark RV, Magee M, Billiard J, Chan A, Xue Z, Russell A. GSK2881078, a SARM, produces dose-dependent increases in lean mass in healthy older men and women. J Clin Endocrinol Metab. 2018;103:3215–24.
Supasyndh O, Satirapoj B, Aramwit P, Viroonudomphol D, Chaiprasert A, Thanachatwej V, Vanichakarn S, Kopple JD. Clin J Am Soc Nephrol. 2013;8:271–9.
Kim K-Y, Ku S-K, Lee K-W, Song C-H, An WG. Muscle-protective effects of Schisandrae Fructus extracts in old mice after chronic forced exercise. J Ethnopharmacol. 2018;212:175–87.
Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan SR, Lillard JW Jr, Taub DD. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest. 2004;114:57–66.
Akamizu T, Kangawa K. Ghrelin for cachexia. J Cachexia Sarcopenia Muscle. 2010;1:169–76.
Nagaya N, Itoh T, Murakami S, Oya H, Uematsu M, Miyatake K, Kangawa K. Treatment of cachexia with ghrelin in patients with COPD. Chest. 2005;128:1187–93.
Nagaya N, Moriya J, Yasumura Y, Uematsu M, Ono F, Shimizu W, Ueno K, Kitakaze M, Miyatake K, Kangawa K. Effects of ghrelin administration on left ventricular function, exercise capacity, and muscle wasting in patients with chronic heart failure. Circulation. 2004;110:3674–9.
Pietra C, Takeda Y, Tazawa-Ogata N, Minami M, Yuanfeng X, Duus EM, Northrup R. Anamorelin HCl (ONO-7643), a novel ghrelin receptor agonist, for the treatment of cancer anorexia-cachexia syndrome: preclinical profile. J Cachexia Sarcopenia Muscle. 2014;5:329–37.
Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, Fearon KC. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomized, double-blind, phase 3 trials. Lancet Oncol. 2016;17:519–31.
Bai Y, Hu Y, Zhao Y, Yu X, Xu J, Hua Z, Zho Z. Anamorelin for cancer anorexia-cachexia syndrome: a systematic review and meta-analysis. Support Care Cancer. 2017;25:1651–9.
Bach MA, Rockwood K, Zetterberg C, Thamsborg G, Hébert R, Devogelaer JP, Christiansen JS, Rizzoli R, Ochsner JL, Beisaw N, Gluck O, Yu L, Schwab T, Farrington J, Taylor AM, Ng J, Fuh V, MK 0677 Hip Fracture Study Group. The effects of MK-0677, an oral growth hormone secretagogue, in patients with hip fracture. J Am Geriatr Soc. 2004;52:516–23.
Nass R, Gaylinn BD, Thorner MO. The ghrelin axis in disease: potential therapeutic indications. Mol Cell Endocrinol. 2011;340:106–10.
Cappellari GG, Semolic A, Ruozi G, Vinci P, Guarnieri G, Bortolotti F, Barbetta D, Zanetti M, Giacca M, Barazzoni R. Unacylated ghrelin normalizes skeletall muscle oxidative stresss and prevents muscle catablism by enhancing tissue mitophagy in experimental chronic kidney disease. FASEB J. 2017;31:5159–71.
Tamaki M, Hagiwara A, Miyashita K, Wakino S, Inoue H, Fujii K, Sato M, Mitsuishi M, Muraki A, Hayashi K, Doi T, Itoh H. Improvement of physical decline through combined effects of muscle enhancement and mitochondrial activation by a gastric hormone ghrelin in male 5/6Nx model mice. Endocrinology. 2015;156:3638–48.
Campbell GA, Patrie JT, Gaylinn BD, Thorner MO, Bolton WK. Oral ghrelin receptor agonist MK-0677 increases serum insulin-like growth factor 1 in hemodialysis patients: a randomized blinded study. Nephrol Dial Transplant. 2018;33:523–30.
Wang ZH, Hsu CC, Huang CN, Yin MC. Anti-glycative effects of oleanolic acid and ursolic acid in kidney of diabetic mice. Eur J Pharmacol. 2009;628:255–60.
Kunkel SD, Suneja M, Ebert SM, Bongers KS, Fox DK, Malmberg SE, Alipour F, Shields RK, Adams CM. mRNA expression signatures of human skeletal muscle atrophy identify a natural compound that increases muscle mass. Cell Metab. 2011;13:627–38.
Ebert SM, Dyle MC, Bullard SA, Dierdorff JM, Murry DJ, Fox DK, Bongers KS, Lira VA, Meyerholz DK, Talley JJ, Adams CM. Identification and small molecule inhibition of an activating transcription factor 4 (ATF4)-dependent pathway to age-related skeletal muscle weakness and atrophy. J Biol Chem. 2015;42:25497–511.
Cho YH, Lee SY, Kim CM, Kim ND, Cho S, Lee C-H, Shin J-H. Effect of loquat leaf extract on muscle strength, muscle mass, and muscle function in healthy adults: a randomized double-blinded, and placebo-controlled trial. Evid Based Complement Alternat Med. 2016;2016:4301621.
Church DD, Schwarz NA, Spillane MB, McKinley-Barnard SK, Andre TL, Ramirez AJ, Willoughby DS. l-Leucine increases skeletal muscle IGF-I but does not differentially increases Akt/mTORC1 signaling and serum IGF-I compared to ursolic acid in response to resistance exercise in resistance-trained men. J Am Coll Nutr. 2016;35:627–38.
Niwa T, Ise M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J Lab Clin Med. 1994;124:96–104.
Nishikawa M, Ishimori N, Takada S, Saito A, Kadoguchi T, Furihara T, Fukushima A, Matsushima S, Yokota T, Kinugawa S, Tsutsui H. AST-120 ameliorates lowered exercise capacity and mitochondrial biogenesis in the skeletal muscle from mice with chronic kidney disease via reducing oxidative stress. Nephrol Dial Transplant. 2015;30:934–42.
Sato E, Saigusa D, Mishima E, Uchida T, Miura D, Morikawa-Ichinose T, Kisu K, Sekimoto A, Saito R, Oe Y, Matsumoto Y, Tomioka Y, Mori T, Takahashi M, Sato H, Abe T, Niwa T, Ito S. Impact of the oral adsorbent AST-120 on organ-specific accumulation of uremic toxins: LC-MS/MS and MS imaging techniques. Toxins. 2018;10:19.
Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18:207–50.
Ogura S, Maruyama K, Hagiya Y, Sugiyama Y, Tsuchiya K, Takahashi K, Abe F, Tabata K, Okura I, Nakajima M, Tanaka T. The effect of 5-aminolevulinic acid on cytochrome c oxidase activity in mouse liver. BMC Res Notes. 2011;4:66.
Masuki S, Morita A, Kamijo Y, Ikegawa S, Kataoka Y, Ogawa Y, Sumiyoshi E, Takahashi K, Tanaka T, Nakajima M, Nose H. Impact of 5-aminolevulinic acid with iron supplementation on exercise efficiency and home-based walking training achievement in older women. J Appl Physiol. 2016;120:87–96.
Acknowledgements
This work was supported by a research Grant-in-Aid for Scientific Research C (No. 17 K01755) from the Ministry of Education, Culture, Sports, and Science and Technology of Japan.
Conflict of Interest
Kunihiro Sakuma and Akihiko Yamaguchi declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sakuma, K., Yamaguchi, A. (2020). Pharmacological Intervention for Sarcopenia in Chronic Kidney Disease. In: Kato, A., Kanda, E., Kanno, Y. (eds) Recent Advances of Sarcopenia and Frailty in CKD. Springer, Singapore. https://doi.org/10.1007/978-981-15-2365-6_11
Download citation
DOI: https://doi.org/10.1007/978-981-15-2365-6_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-2364-9
Online ISBN: 978-981-15-2365-6
eBook Packages: MedicineMedicine (R0)