
Abstract
Cardiovascular disease is a leading cause of death worldwide, and with the dramatically increasing numbers of heart failure patients in the next 10 years, mortality will only increase [1]. For patients with end-stage heart failure, heart transplantation is the sole option. Regrettably, the number of available donor hearts is drastically lower than the number of patients waiting for heart transplantation. Despite evidence of cardiomyocyte renewal in adult human hearts, regeneration of functional myocardium after injury can be neglected. The limited regenerative capacity due to inadequate proliferation of existing cardiomyocytes is insufficient to repopulate areas of lost myocardium [2]. As a solution, the hypothesis that adult stem cells could be employed to generate functional cardiomyocytes was proposed. One of the early studies that supported this hypothesis involved direct injection of hematopoietic c-kit-positive cells derived from bone marrow into the infarcted heart [3]. However, in sharp contrast, more recent evidence emerged demonstrating that these hematopoietic stem cells only differentiate into cells down the hematopoietic lineage rather than into cardiomyocytes [4, 5], and the focus shifted towards stem cells residing in the heart, called cardiac progenitor cells. These CPCs were extracted and injected into the myocardium to regenerate the heart [6]. In recent years, over 80 pre-clinical studies employing cardiac stem cells in vivo in large and small animals to evaluate the effect on functional parameters were systematically reviewed, identifying differences between large and small animals [7]. Despite the positive outcome of these stem cell therapies on functional parameters, c-kit-positive cardiac progenitor cells were shown to contribute minimally to the generation of functional cardiomyocytes [8, 9]. This heavily debated topic is summarized concisely by van Berlo and Molkentin [10]. Recently, single-cell sequencing and genetic lineage tracing of proliferative cells in the murine heart in both homeostatic and regenerating conditions did not yield a quiescent cardiac stem cell population or other cell types that support transdifferentiation into cardiomyocytes, nor did it support proliferation of cardiac myocytes [11, 12]. Now, the focus is shifting towards exploiting the limited regenerative capacity of the cardiomyocytes themselves, by re-activating proliferation of existing cardiomyocytes through dedifferentiation, reentry into the cell cycle, and cytokinesis. This process is the new focus of research to promote cardiac regeneration, and can be controlled on multiple levels, including cell-cycle manipulation, reprogramming, small molecules, extra-cellular matrix (ECM), proteins, and RNA regulation [13].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
Cardiovascular disease is a leading cause of death worldwide, and with the dramatically increasing numbers of heart failure patients in the next 10 years, mortality will only increase [1]. For patients with end-stage heart failure, heart transplantation is the sole option. Regrettably, the number of available donor hearts is drastically lower than the number of patients waiting for heart transplantation. Despite evidence of cardiomyocyte renewal in adult human hearts, regeneration of functional myocardium after injury can be neglected. The limited regenerative capacity due to inadequate proliferation of existing cardiomyocytes is insufficient to repopulate areas of lost myocardium [2]. As a solution, the hypothesis that adult stem cells could be employed to generate functional cardiomyocytes was proposed. One of the early studies that supported this hypothesis involved direct injection of hematopoietic c-kit-positive cells derived from bone marrow into the infarcted heart [3]. However, in sharp contrast, more recent evidence emerged demonstrating that these hematopoietic stem cells only differentiate into cells down the hematopoietic lineage rather than into cardiomyocytes [4, 5], and the focus shifted towards stem cells residing in the heart, called cardiac progenitor cells. These CPCs were extracted and injected into the myocardium to regenerate the heart [6]. In recent years, over 80 pre-clinical studies employing cardiac stem cells in vivo in large and small animals to evaluate the effect on functional parameters were systematically reviewed, identifying differences between large and small animals [7]. Despite the positive outcome of these stem cell therapies on functional parameters, c-kit-positive cardiac progenitor cells were shown to contribute minimally to the generation of functional cardiomyocytes [8, 9]. This heavily debated topic is summarized concisely by van Berlo and Molkentin [10]. Recently, single-cell sequencing and genetic lineage tracing of proliferative cells in the murine heart in both homeostatic and regenerating conditions did not yield a quiescent cardiac stem cell population or other cell types that support transdifferentiation into cardiomyocytes, nor did it support proliferation of cardiac myocytes [11, 12]. Now, the focus is shifting towards exploiting the limited regenerative capacity of the cardiomyocytes themselves, by re-activating proliferation of existing cardiomyocytes through dedifferentiation, reentry into the cell cycle, and cytokinesis. This process is the new focus of research to promote cardiac regeneration, and can be controlled on multiple levels, including cell-cycle manipulation, reprogramming, small molecules, extra-cellular matrix (ECM), proteins, and RNA regulation [13].
Cardiac neovascularization, the new blood vessels formation in the heart, is an essential part of cardiac regeneration. Neovascularization is distinct from vasculogenesis, which is considered de novo primitive vascular network formation, and occurs when angioblast precursors differentiate into endothelial cells in the developmental stages [14]. Angiogenesis is the formation of a new blood vessel from the existing blood vessels. In the past the focus has been on mechanisms influencing the inhibition of angiogenesis, relating to its role in the spread of tumors [15]. However, there has been a growing interest in tackling ischemic disorders by promoting neovascularization in an attempt to regenerate tissues [16].
This chapter will focus on findings regarding the role of non-coding RNAs in cardiac regeneration. Cardiac regeneration is defined as the repair of cardiac tissue, which in turn enhances or restores the functional capabilities of the heart. Studying the role of these non-coding RNAs in species with inherent cardiac regenerative capacity uncovers and helps to understand the mechanisms that drive cardiac regeneration, such as cardiomyocyte proliferation and neovascularization. First, we elaborate on the regenerative capacity in lower vertebrates and rodents and their role as scientific models, then we elucidate the role of non-coding RNAs in cardiomyocyte proliferation and neovascularization.
2 Cardiac Regeneration in Various Scientific Model Species
Regenerative capacity varies between species. Lower vertebrate species have a high regenerative capacity throughout life, whereas in higher vertebrate species this regenerative capacity decreases after birth. Teleost fish like zebrafish and urodeles like newts or axolotls display robust cardiac regeneration, making them excellent model systems to study the underlying processes for cardiac regeneration. In zebrafish, after apical resection, bleeding is halted by blood clotting in the wound. Then fibrin is deposited, and where mammalian hearts become fibrotic through collagen deposition and scarring, the zebrafish heart replaces lost myocardium by proliferation of cardiomyocytes [17, 18]. Cardiomyocyte proliferation is highest 2 weeks after injury, and 2 months after injury the majority of the lost myocardium has been renewed and cardiac output restored [18, 19]. By employing Cre-based genetic fate mapping it was shown that resident cardiomyocytes dedifferentiate, proliferate, and mature similar to the developmental program to replace the lost myocardium, indicating that stem cells are not the source of regenerated myocardium [20, 21]. Similarly, in urodeles complete regeneration without scarring was observed two to 3 months after the injury [22], and it was demonstrated that sarcomeric gene expression is downregulated during regeneration which supports the notion that adult cardiomyocytes can generate more cardiomyocytes via dedifferentiation, proliferation, and redifferentiation. This concept is known as the dedifferentiation hypothesis [23, 24]. A concise recapitulation of the evolution of the scientific view on cardiomyocyte proliferation was published by Yutzey [25].
Rodent models like mice and rats display full growth and regeneration before and shortly after birth, yet have a reduced cardiac regenerative capacity as adult mammals [19]. Thus, neonatal rodents are excellent models for studying the mechanisms, and adult rodents are a suitable model to test stimulation of regeneration. To study the regenerative capacity of the fetal mice heart, an X-linked mutation, deadly to cardiomyocytes was introduced in female embryos. Due to random X inactivation half of the cardiomyocytes were lost, though at birth the hearts were fully functional. The fetal hearts compensated for the effective loss of 50% of cardiomyocytes by increased healthy cardiac cells proliferation [26]. High regenerative capacity in neonatal mice hearts has been demonstrated in multiple cardiac damage models: myocardial infarction [27], ventricular resection [28], cryoinfarction [29, 30], and clamping [31]. The human heart can also fully recover from injury as demonstrated in a case study of myocardial infarction in neonatal humans as a result of coronary artery occlusion. The infants fully recovered from the ischemic injury [32].
3 Non-coding RNA in Cardiomyocyte Proliferation
In contrast to the aforementioned lower vertebrate species, cardiomyocyte proliferation in adult mammals is, though present [2], insufficient to replenish lost cardiomyocytes due to injury. The proliferative state of cardiomyocytes as observed in embryonic stages of development quickly diminishes as the cells differentiate into the mature phenotype characterized by binucleation and hypertrophy. This development and maturation is regulated by many different factors. The capability of mature cardiomyocytes to again become proliferative is small [33,34,35]. Consequently, after the significant loss of cardiomyocytes as seen in ischemic injury, the heart cannot replace the lost cardiomyocytes and regenerate the myocardium. To increase the regenerative capacity of the heart, revealing the mechanisms underlying the development and proliferation of cardiomyocytes is essential. Recently, non-coding RNAs have been found playing important roles in regulating cardiomyocyte proliferation. These non-coding RNAs and their effect on cardiomyocyte proliferation are listed in Table 9.1. Here, we aim to highlight the discoveries of these non-coding RNAs and their role, organized per non-coding RNA class. At the time of writing the knowledge on the role of non-coding RNA in cardiac regeneration is limited to microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). Other non-coding RNAs such as circular RNA, PIWI-interacting RNAs, or small inhibitory RNAs are not covered, as their functions in cardiac regeneration are still to be further studied.
3.1 MicroRNA
With their function as post-transcriptional regulators and their broad spectrum of targets due to partially complementary binding, miRNAs are potential candidates to regulate cardiomyocyte proliferation. Both the aforementioned models, fish and rodent, are widely used to study the involvement of miRNAs in cardiac regeneration, and cardiomyocyte proliferation specifically. A common approach to characterize miRNAs that could potentially regulate cardiomyocyte regeneration is to compare miRNA expression levels in different stages of development in prenatal and postnatal rodent hearts. Candidate miRNAs are then validated in one or multiple in vivo models of cardiac injury in either or both neonatal and adult rodents. Oftentimes, a model of cardiac injury is employed to replicate the disease as observed in humans. These models are based on either surgical induction, via ischemia or ischemia/reperfusion, or on genetic induction of heart failure (HF). Here, the role of the different miRNAs in cardiomyocyte proliferation is illustrated per disease model.
3.1.1 Surgical Cardiac Injury Models
Through ligation of the left anterior descending coronary artery (LAD), ischemia is induced in the left ventricle, inducing a myocardial infarction (MI), thereby resulting in a loss of cardiomyocytes [36]. The following miRNAs were found to play a role in increasing the regenerative capacity of the heart using this approach.
Initially miR-99/100 and Let-7a/c were identified as key players in cardiomyocyte dedifferentiation in a cardiac apical resection model in zebrafish that naturally regenerate. They were both validated in mice subjected to LAD ligation causing MI. Blocking these miRNAs increased expression of their target proteins farnesyl transferase-beta (FNTß) and SWI/SNF-related matrix associated actin-dependent regulator of chromatin-subfamily A, number 5 (SMARCA5). Upon blocking, increased left ventricular ejection fraction and fractional shortening indicated functional improvements. The resulting cardiac regeneration, induced via dedifferentiation and proliferation of cardiomyocytes, and improved functional effects in both species proved that this mechanism is conserved between species [37].
Over-expression of miR-128 in cardiomyocytes in a neonatal apical resection model, suppressed cardiomyocyte proliferation and hampering cardiac function, thereby inhibiting cardiac regeneration, which can be observed after apical resection in untreated neonatal mice. Additionally, the role of miR-128 in cardiac regeneration was validated by deletion in cardiomyocytes in adult mice demonstrating improved cardiomyocyte proliferation after myocardial infarction induced by permanent LAD ligation. By deleting miR-128 the expression of SUZ12, a chromatin modifier, was enhanced. This in turn suppressed cyclin-dependent kinase inhibitor p27 and activated cell cycle regulators Cyclin E and cyclin dependent kinase 2 (CDK2), promoting cell cycle re-entry in adult cardiomyocytes. Additionally, increased levels of GATA4 were observed, indicative of dedifferentiated cardiomyocytes [38].
In neonatal mice, miR-34a levels were found to be low and in adult mice, miR-34a levels were high, even after cardiac injury. Increasing miR-34a expression levels in neonatal mice resulted in a decreased regenerative capacity and hampered recovery. Inhibiting miR-34a in adult mice hearts after inducing MI increased the regenerative capacity and improved cardiac function, reduced adverse remodeling, decreased fibrosis, and increased cell cycle activity. Further investigation demonstrated that this miR-43a inhibition resulted in higher protein levels of Sirt1, Cyclin D1, and Bcl2. These proteins have been implicated in cell cycle activity, cellular aging, and cell survival. The former was merely protective against cell death, whereas the latter two maintained proliferative and cell cycle capacities [39].
Similarly, the miRNA cluster miR-302-367 was found to be elevated in pre-natal stages compared to post-natal stages. Reactivating the cluster in adult hearts led to cardiomyocyte proliferation, though persistent, prolonged expression resulted in cardiomegaly and ultimately in heart failure. The positive effect of reactivation of the cluster on cardiomyocyte proliferation was confirmed in mice with MI induced by LAD ligation. Transient transfection with the miRNA cluster however prevented the adverse effects of persistent over-expression without compromising the positive effect of increased cardiomyocyte proliferation on the regenerative capacity of the heart. This cluster was found to target components of the Hippo pathway. Specifically, proliferation-associated gene Ccnd1, and consequently Cyclin G1, was elevated, and kinases Mst1 and Mob1b were decreased [40]. In a more recent study, miR-302 was injected intramyocardially using a hydrogel delivery system for local and sustained delivery in adult Confetti mice with a MI. Due to the lineage labeling it was possible to distinguish newly generated cardiomyocytes as a result of clonal expansion. In addition, infarcted mice treated with the miR-302 showed comparable cardiac function to non-infarcted mice, as measured by ejection fraction and fractional shortening. Mice treated with miR-302 mimic demonstrated knock-down of Lats2, Mob1, and Mst1, all components of the Hippo signaling pathway, which controls cell proliferation mediated by YAP [41].
Additionally, miR-199a and miR-590 were identified by using a high-throughput functional screening with a human whole-genome miRNA library, and validated in vivo. Neonatal rats received injections of one of the two miRNAs together with a lipid transfection agent directly in the heart. Four days after treatment the ventricular wall thickness had increased, proliferating cardiomyocytes were found, and no fibrosis was observed. Next, adult mice were treated with AAV9 vectors expressing the two miRNAs after induction of a MI through permanent LAD ligation. Infarct size was significantly decreased in mice treated with miRNAs and cardiac function measured by left ventricular ejection fraction, fractional shortening, and end-systolic anterior wall thickness was preserved [42]. Expanding on these results, miR-199a was overexpressed using AAVs in pigs subjected to MI and reperfusion. Functional parameters improved, such as overall and local contractility. Muscle mass increased while scar tissue decreased, demonstrating that it is possible to regenerate the myocardium in larger mammals by stimulating endogenous repair via cardiomyocyte proliferation [43]. In all, expression of miR-199a and miR-590 after MI reduced infarct size and improved cardiac function by actively stimulating cardiomyocyte proliferation. Both miRs suppressed Homer1 and Hopx, genes involved in calcium signaling and in regulating proliferation, respectively [42, 43]. After inducing MI on day 1 after birth through LAD ligation, the mouse heart recovered fully within 3 weeks, through proliferation of existing cardiomyocytes. This regenerative response was impaired by over-expression of miR-195, one of the members of the miR-15 family, leading to adverse remodeling as observed in adult mice. Next, in an ischemia-reperfusion model of MI the miR-15 family was inhibited during postnatal development into adulthood. This led to an increase in cardiomyocyte proliferation and improved systolic function. Thus, inhibition of the miR-15 family increases the regenerative capacity [44]. In an earlier study by the same group, miR-195 was shown to directly affect cell cycle genes Chek1, Cdc2a, Birc5, Nusap1, and Spag5, supposedly increasing mitotic and cell cycle entry, and cell cycle progression. However, of these, Chek1 is the only gene with a miR-15 binding site that is conserved between mice and humans [45].
3.1.2 Genetic Cardiac Injury Models
When Dicer, which is required for pre-miR processing to mature miRNAs, was deleted in the hearts of embryonic mice using Cre recombinase and under the control of the Nkx2.5 promotor this led to death as a result of heart failure. Thus proper miRNA functioning is required in cardiac development. In the Dicer mutant hearts, miR-1 was dysregulated. Using homologous recombination, miR-1-2 was deleted in mouse embryonic stem cells. Heterozygous animals were intercrossed to create offspring lacking miR-1-2. These mice died early, mostly due to ventricular septum defects, and those that survived suffered from cardiac arrhythmias and hyperplasia due to abnormalities in the cardiomyocyte cell cycle. Loss of miR-1-2 resulted in a loss of lrx5, leading to abnormal repolarization of cardiomyocytes and consequently cardiac arrhythmias. Furthermore, miR-1-2 mutants were hyperplastic as a result of increased mitotic activity in cardiomyocytes. This indicates a potential role for miR-1-2 in stimulating the regenerative capacity, though the observation in this study may be a result of increased proliferation in the early stages of development rather than in adult animals, for cytokinesis was not observed in adult animals [46].
Another example of complete knockout of a miRNA resulting in abnormal cardiomyocyte proliferation is the double knockout of miRNA-133a-1 and miRNA-133a-2 in mice, targeting Cyclin D2 and serum response factor (SRF). Mice lacking genes for one of the variants were normal, but deletion of both genes led to ventricular septal defects and consequently death in embryonic and neonatal animals, as a result of dysregulated cardiomyocyte proliferation, apoptosis, and abnormal expression of smooth muscle genes in the heart. Mice surviving into adulthood perished from heart failure and sudden death. Thus, miR-133a-1 and − 2 are essential for normal cardiac growth and function [47].
Similarly, complete knockout of the miR-17-92 cluster in embryonic, postnatal, and adult mice resulted in smaller hearts and lower proliferation rates in postnatal animals, a reduced number of cardiomyocytes in adult hearts, and decreased cardiac function, demonstrating that this cluster is essential for cardiomyocyte proliferation in embryonic and postnatal hearts. Overexpression of the cluster in a transgenic mouse model demonstrated enlargement of the hearts and thickening of the ventricle walls due to proliferation rather than hypertrophy. Overexpression of miR-17-92 using tamoxifen-inducible Cre recombinase in mice, subjected to MI, attenuated the effects of MI-induced damage and adverse remodeling. miR-17-92 was found to affect phosphatase and tensin homolog (PTEN), and overexpression of PTEN diminished miR-19 (a member of the miR-17-92 cluster) promoted cardiomyocyte proliferation. These results confirmed that the miR-17-92 cluster, and miR-19 specifically, can induce proliferation by suppressing PTEN in cardiomyocytes [48].
Some miRNAs have been validated in vivo without cardiac validation. For example, miRNA array on post-natal day 0 and day 10 rat cardiomyocytes revealed upregulated miR-31a levels on day 10. Inhibition of miR-31a on days 0, 1, and 2 resulted in reduced cardiomyocyte proliferation through RhoBTB1, a subfamily of the Rho small GTPases, suggesting that upregulating miR-31 might increase the generative capacity of the heart through stimulating cardiomyocyte proliferation [49].
Overall, evidence that miRNAs can influence cardiomyocyte proliferation is accumulating. Multiple miRNAs have been identified and validated in vivo. However, it is not yet fully understood how these miRNAs in turn are regulated, nor is it evident that miRNA are the sole regulatory RNAs in cardiac regeneration. Long non-coding RNAs are emerging as regulators of RNAs (mRNA, miRNA, circRNA) as well as DNA and proteins. Their broad complex roles in gene regulation is a popular current topic, and there is evidence for involvement of several lncRNAs in cardiomyocyte proliferation.
3.2 Long Non-coding RNA
Long non-coding RNAs are a class of RNA molecules consisting of over 200 nucleotides that can regulate gene expression, both at transcriptional and post-transcriptional level, in a range of cellular processes, including (de)differentiation and proliferation. The research field of lncRNAs is relatively young and only a limited number of lncRNAs have been explored in the context of cardiovascular regeneration. How the lncRNAs listed in Table 9.1 was identified and how they affect cardiomyocyte proliferation is elaborated on in the following paragraphs.
Three lncRNAs have been identified that influence cardiomyocyte proliferation through affecting cell cycle genes. Endogenous cardiac regeneration-associated regulator (ECRAR) [50], LINCM3 (Gas5), and LINCM9 (Sghrt) [51] exert their function indirectly on one or more Cyclin proteins.ECRAR binds to extracellular signal-regulated kinases 1 and 2 (ERK1/2) activating cyclin D1 and cyclin E1, which both activate E2F transcription factor 1 (E2F1). E2F1 can upregulate ECRAR, creating a positive feedback loop that stimulates cell cycle progression, promoting proliferation in cardiomyocytes. Over-expression of ECRAR stimulated cardiomyocyte proliferation in vivo in the adult rat heart. To assess the effect of ECRAR over-expression in a disease environment, rats with MI from LAD ligation were injected with Adenovirus-mediated ECRAR. Over-expression of ECRAR led to increased proliferation resulting in cardiomyogenesis. Furthermore, infarct size was significantly smaller in rats that were treated with ECRAR, scar formation measured by fibrotic area was less, and functional parameters were improved, suggesting ECRAR enhances the regenerative capacity of the myocardium. This was confirmed by knockdown of ECRAR in naturally regenerative neonatal rat hearts, preventing recovery after MI. ECRAR showed a 12-fold increased expression from the analysis of four datasets of RNA-sequencing in fetal compared to adult human cardiac tissues. This finding was confirmed in rat fetal hearts, where ECRAR expression is high on embryonic day 12 and decreased after birth [50].
Similarly, nuclear RNA-sequencing of single cardiomyocytes from failing and healthy human heart tissue identified heterogeneity in the transcriptomic stress-response [51]. Key nodal lncRNA surfaced that regulate dedifferentiation and cell cycle genes in certain subsets of cardiomyocytes that can potentially regulate cardiac repair. In the diseased cells of a trans-aortic constriction (TAC) mice model, LINCM3 (Gas5) and LINCM9 (Sghrt) were upregulated, and LINCM5 was down-regulated compared to sham operated mice. Both Gas5 and Sghrt are part of signaling pathways related to translation, precursor metabolites generation, oxidative stress response, oxidative phosphorylation, cell proliferation, and cardiac muscle tissue development. This indicated that both lncRNAs could be the main effectors regulating other genes within the same gene regulatory network. This hypothesis was tested by knocking down either of these lncRNAs in adult cardiomyocytes from TAC operated mice. Knockdown of Gas5 down-regulated the expression of Nppa (fetal reprogramming), Dstn (dedifferentiation marker), Ccng1 (cell cycle gene, coding for Cyclin G1), and Ccnd2 (cell cycle gene, coding for Cyclin D2). Gas5 has previously been shown to accumulate in the heart [52] and regulate apoptosis [53] and proliferation [54] in other cell types. Sghrt, at the time of writing, has no previously described function. Suppression of Sghrt did not have any significant effects on either Nppa or Dstn, increased Ccng1, and decreased Ccnd2. Thus, these experiments demonstrated that both lncRNAs can regulate genes in the same regulatory network at a transcriptional level [51].
Three other lncRNAs have been identified to be involved in cardiomyocyte regeneration, though not through affecting cell cycle genes. CAREL [55], MALAT1 [56], and Sirt1 anti-sense lncRNA [57] influence cardiomyocyte proliferation through anti-proliferative and pro-apoptotic pathways, through inhibiting transcription factors in developmental pathways, and through stabilization of mRNA, respectively.
Microarray analysis revealed that lncRNA CAREL was upregulated in postnatal mouse hearts. Over-expression of CAREL in cardiomyocytes of mice diminished their division and proliferation, and the regenerative capacity of neonatal hearts was lost. In contrast, silencing CAREL stimulated cardiac regeneration and promoted cardiac function after injury in both neonatal and adult mice. CAREL binds competitively to the targets of miR-296, Trip53inp1 and Itm2a. In line with previous results, over-expression of miR-296 induced cardiomyocyte proliferation and increased the regenerative capacity. In CAREL transgenic mice, the regenerative capacity was decreased, and could be restored by over-expressing miR-296 [55].
LncRNA MALAT-1 is expressed in adult zebrafish hearts [56]. MALAT-1 knock-out zebrafish showed an enlarged pericardium and other cardiac developmental abnormalities. Cardiac progenitor cell genes nkx2.5 and gata4 were upregulated, hinting at a regulatory role for MALAT-1 [56].
Expression patterns of Silent information regulator factor 2 related enzyme 1 (Sirt1) antisense lncRNA are higher in embryonic and neonatal compared to adult mouse hearts [57]. Sirt1 antisense lncRNA can bind Sirt1 messengerRNA, stabilizing it, and enhancing its translation into Sirt1 protein. Isolated neonatal cardiomyocytes were transfected with Sirt1 antisense lncRNA to examine its role in proliferation. Over-expression resulted in higher cell cycle activity, increased mitosis, and a higher cell number, indicating a positive influence on proliferation. In contrast, knocking down Sirt1 antisense lncRNA decreased the number of cardiomyocytes, cell cycle activity, and mitosis, and increased apoptosis. Next, intra-myocardial injections of LNA targeting Sirt1 antisense lncRNA resulted in decreased levels of proliferation in neonatal mice. Following these experiments, Sirt1 antisense lncRNA was over-expressed in both healthy and LAD-MI adult mice, leading to increased cardiomyocyte proliferation compared to their respective controls and a higher survival rate in the treated LAD-MI mice compared to untreated mice. In the latter group, cardiac output parameters, left ventricular ejection fraction and fractional shortening, improved, and the infarct size was smaller compared to untreated LAD-MI animals. These outcomes indicate that Sirt1 antisense lncRNA positively affects the regenerative capacity in ischemic adult hearts [57].
In addition to affecting gene expression through the aforementioned mechanisms, lncRNAs can act as sponges to miRNAs. Two lncRNAs have been identified that function thusly in affecting cardiomyocyte proliferation.
Recently, lncRNA NR_045363 was discovered to be mainly expressed in cardiomyocytes compared to non-cardiomyocytes, and more in embryonic mouse hearts than in adult mouse hearts [58]. Over-expression of this lncRNA in neonatal mice cardiomyocytes significantly increased proliferation in vitro and in vivo. Knockdown in primary embryonic cardiomyocytes led to decreased proliferation. Furthermore, over-expression in mice subjected to MI resulted in significantly ameliorated left ventricular ejection fraction and fractional shortening. Additionally, using EdU staining cardiomyocyte proliferation was shown to have increased in the animals over-expressing NR_045363. In silico target prediction showed miR-216a as a potential targets of NR_045363. Mir-216a is also a target of LOC101927497, the human ortholog of NR_045363. These predictions were validated in vitro and it showed that knockdown of NR_045363 led to increased miR-216a expression, and over-expression of NR_045363 resulted in decreased miR-216a expression levels. The researchers concluded that NR_045363 may function as a miRNA sponge for miR-216a, thereby preventing down-regulation of the targets of miR-216a, and consequently promoting proliferation [58].
Interestingly, one lncRNA has been identified that affects one of the cardiomyocyte proliferation-associated miRNAs. Cardiomyocyte regeneration related lncRNA (CRRL) has been identified from RNA-sequencing data of human fetal and adult heart tissues. CRRL promotes the expression of Hopx, the gene coding for Homeodomain-only protein, by directly binding miR-199a-3p, thereby removing the inhibition of miR-199a-3p on Hopx mRNA expression [59]. Loss of CRRL in adult rats preserved cardiac function and diminished adverse remodeling post infarct. Knockdown of CRRL in neonatal rat cardiomyocytes promoted proliferation in vitro and in vivo [59]. Thus, downregulation of the Hopx gene by removing the miR-199a-3p sponge CRRL via knockdown has a comparable effect to adding miR-199a as described previously [42].
4 Non-coding RNAs in Neovascularization
The growth of new blood vessels requires proangiogenic stimuli, including growth factors such as vascular endothelial growth factor (VEGF-A) [60]. Certain non-coding RNAs, for example, have the ability to influence these proangiogenic factors. Therefore, non-coding RNAs have the potential to become a therapeutic tool for treating ischemic cardiac tissue [61].
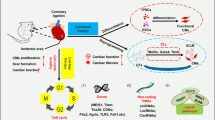
The following part provides an overview of advances in non-coding RNA research on cardiac regeneration through neovascularization. The non-coding RNAs listed in Table 9.2 are also shown in Fig. 9.1, to illustrate the pathways they affect, and their effect on cell cycle progression or apoptosis, respectively.

Schematic representation of non-coding ribonucleic acids (RNA) and their respective targets that affect microvascular development and function through either stimulation (green) or repression (red) of cell cycle progression or apoptosis
4.1 MiRNA
The first example of involvement of miRNA in the regulation of neovascularization was shown by Yang [39]. Their discovery demonstrated that knocking out the miRNA processing enzyme Dicer in mice would result in early death during embryonic development, due to impaired angiogenesis [39, 62]. Following up on that discovery, numerous studies have shown the critical roles of miRNAs in neovascularization.
Over-expression of miR-210 on MI-injured cardiac tissue, showed an increase in neovascularization and angiogenic processes [63]. MiR-210 is known as a hypoxamiR, a label given to miRNAs that have an important role in hypoxic conditions [64]. Improved contractility in mammalian acute cardiac ischemia (ACI) models was observed when exposed to over-expression of miR-210, through the stimulation of hepatocyte growth factor (HGF) expression as well as the effect of miR-210 on left ventricular (LV) remodeling. This study concluded that through administration of miR-210 agonists, increased micro-vessel density was observed, indicating a potential therapeutic tool for patients with ACI [65].
In mice, miR-92a has been observed to control recovery of ischemic tissue and has been noticed to play a role in angiogenesis. Both in vitro and in vivo, over-expression of miR-92a in endothelial cells blocked neovascularization. In MI models, miR-92a antagonists demonstrated enhanced angiogenesis and resulted into recovery of the injured tissue. MiR-92a affects numerous proteins that promote angiogenesis, including integrin subunit α5. An abundance of miR-92a in endothelial cell caused a decline in endothelial cell migration, decreased vascular network formation, blocking of sprouting in a 3D neovascularization model, as well as reduced adhesion of the endothelial cell to fibronectin. MiR-92a influences MAP kinase kinase 4 (MKK4) and Kruppel-like factors-4 (KLF4) by targeting integrin subunit alpha-5, thereby inhibiting cell cycle progression in endothelial cells, and consequently the formation of new blood vessels. MiR-92a can therefore be categorized as an anti-angiogenic factor [62, 66]. Most of these studies were solely conducted in small animal models, and Hinkel et al. [67] took the next step and tested the efficacy of using the therapeutic potential of miR-92a inhibition in a pre-clinical porcine model of ischemia and reperfusion (I/R). The study showed that by using LNA-modified antisense miR-92a (miR-92a inhibitor), when applied regionally with the use of a catheter, the infarct size could be reduced significantly. This consequence resulted in enhanced cardiac function, as measured by LV ejection fraction and LV end-diastolic pressure. Histochemistry in the models confirmed an increased density of capillaries in the post-ischemic MI porcine hearts [67]. This study indicates that anti-angiogenic non-coding RNAs have the potential to be used as therapeutic targets in regenerative blood vessel formation.
MiR-24 is an additional anti-angiogenic miRNA, validated in a MI model in mice. MiR-24 has multiple effects on cardiac vascularization was shown to be upregulated as a consequence of cardiac ischemia. By acting on transcription factor GATA2, usually enriched in EC, as well as affecting PAK4, a p21-activated kinase, miR-24 promotes apoptosis of EC, limits cell sprouting (branching of vessels), and inhibits capillary network formation. Silencing of miR-24-targets as well as over-expression of miR-24, significantly restricted and halted angiogenesis in zebrafish embryos. Complete block of miR-24 decreased the damaged myocardial infarct size in mice, through increased vascularization and reduced apoptosis of the endothelial tissue. This resulted in improved cardiac function and thus miR-24 could be a potential therapeutic candidate for the regeneration of damaged tissue [62, 68]. In a mouse LAD-MI model, miR-24 expression showed increased expression levels in ECs. By blocking miR-24 specifically through local delivery of an adenovirus-mediated decoy, angiogenesis and blood perfusion of the myocardial tissue surrounding the infarcted area increased. In addition, there was a reduction in the infarct size, miR-24 induced fibroblast apoptosis, and overall cardiac function improved. Despite these potentially regenerative measures and effects, miR-24 decoy also increased cardiomyocyte apoptosis. In vitro, miR-24 inhibition supported endothelial cell survival, proliferation, and blood-vessel forming capabilities. Additionally, it led to fibroblast apoptosis, which could result in a reduction in scar formation in vivo, and CM apoptosis. These results were confirmed in vivo 14 days post-MI [69]. Both inhibition [68] and over-expression [70] of miR-24 have yielded positive results in mice with acute MI. Meloni et al. [69] set out to test the results of inhibiting miR-24 on cardiac function in a MI model and observed, 14 days after MI, that cardiac function had improved. These results indicated that the initial positive effect on endothelial cells is stronger than the apoptotic effect in CMs, resulting in a pro-angiogenic response and improved cardiac function 2 weeks after MI. It must be noted that that extended inhibition of miR-24 could lead to increased apoptosis in CMs and have a destructive effect on cardiac function and infarct size.
Wang et al. [71] discovered the importance of miR-126 in ensuring vascular integrity and function. The observation of mutant mice lacking the gene encoding for miR-126 resulted in dead embryos, or embryos suffering from ruptured blood vessels, hemorrhages and systemic edema. These abnormalities can be linked to reduced pro-angiogenic growth factor signaling, through e.g. VEGF and fibroblast growth factor (FGF). A lack of these angiogenic factors can lead to decreased endothelial cell growth, sprouting, and adhesion. MiR-126 as a pro-angiogenic stimulator is linked to the inhibition of Spred-1, which is an inhibiting regulator of MAP kinase signaling. If Spred-1 is over-expressed, it decreases pro-angiogenic signals by VEGF and FGF. In the absence of miR-126, there is no regulation of Spred-1. The group of mutant animals that survived showed malfunctioning cardiac neovascularization following MI induced by permanent LAD ligation, indicating the essential function of miR-126 [71].
In mice that suffer from ACI and in humans with acute coronary syndromes, increased levels of miR-26a has been observed [72]. Expression of miR-26a resulted in endothelial cell cycle arrest, inhibition of endothelial cell migration, sprouting angiogenesis, and blood vessel network formation in Matrigel. Blocking of miR-26a has the opposite effects. Over-expression of miR-26a in vivo in mice inhibited endothelial cell SMAD1 expression. It also resulted into a decrease in exercise-induced angiogenesis. Additionally, miR-26a inhibitor given intravenously resulted in increased levels of SMAD1 expression and readily induced significant levels of angiogenesis within 2 days. The pathway of miR-26a consists of the inhibition of the bone morphogenic protein/SMAD1 signaling pathway in ECs through directly targeting SMAD1. Through this blockage, Id1 expression is decreased, resulting in increased levels of p21 and p27 (regulators of the cell-cycle). leading to reduced infarct size and damage [72].
MiR-377 was found to play a role in paracrine-mediated angiogenesis [73]. In vivo evidence proved that, by knockdown of miR-377, mesenchymal stem cell (MSC) mediated angiogenesis increased, as well as the recovery of cardiac function after MI. Through the transplantation of these MSCs in MI rat hearts, the genetic over-expression of miR-377, its knockdown and a control were compared. Anti-MiR-377 treated hearts showed most myocardial angiogenesis post-MI. Through computational miRNA prediction analysis, VEGF was determined to be a potential target affected by miR-377. Verification of this assumption was performed through Western Blotting as well as through dual luciferase reporter assay. Wen et al. [73] determined that miR-377 can bind to the VEGF untranslated-region (UTR), resulting in its negative regulation on expression [73]. STK35, also known as CLP36-interacting kinase 1, was found to be one of the top targets for miR-377. Indeed knockdown of STK35 resulted into a decreased angiogenic potential of ECs [74]. Goyal et al. concluded that VEGF stimulation in ECs increased STK35 expression, so targeting STK35 would have an antagonistic effect to VEGF. Subsequent studies have demonstrated that myocardial tissue, derived via human cardiac biopsies from patients that suffered from heart failure (HF), showed a significant increase in miR-377 expression compared to non-failing control hearts [74]. The transplantation of miR-377 knockdown hCD34+ cells into ischemic myocardium enhanced the proangiogenic capabilities of the tissue, stimulating LV remodeling and reducing cardiac fibrosis [75].
Not all miRNAs relevant to angiogenesis have been validated in cardiac disease models. Other models have been employed to demonstrate the role of miRNAs in angiogenesis in other tissues such as the eye, muscle tissue, or cerebral tissue. These miRs could potentially be used to play a therapeutic role in cardiac tissue with regards to cardiac regeneration.
For example, miR-132/212 was knocked out in mice subjected to hind-limb ischemia. These animals displayed slower recovery compared to wild-type animals. These results were validated in vitro in a human umbilical vein endothelial cell (HUVEC)/pericyte co-culture by transfection of miR-132 and miR-212i. Addition of these miRs resulted in improved tubule formation, additional junctions, and longer tubule length, whereas inhibiting these miRs resulted in the opposite. By directly inhibiting SPRED1 and RASA1, miR 132/212 modulates the Ras-MAP-kinase pathway and promotes arteriogenesis [76]. MiR-214 has been proven to have an influence on developmental angiogenesis in vivo and in vitro, as well as in adult angiogenesis of mice. Specifically, van Mil et al. [77] demonstrated that miR-214 directly targets Quaking (QKI), a protein instrumental for vascular development. QKI transcript levels were increased in various tissues of mice transfected with antagomiR-214, and QKI knockdown by siRNA as well as miR-214 over-expression demonstrated abnormal vascular sprouting, confirming its importance on vascular formation. Additionally, the role of miR-214 on developmental angiogenesis was shown as antagomir-mediated miR-214 knockdown enhanced mouse retinal developmental angiogenesis. Mechanistic studies indicated that by silencing miR-214, more potent pro-angiogenic growth factors, such as VEGF-A, were secreted and the pro-angiogenic activity of EC-derived conditioned medium was increased, introducing a new pathway to possibly improve therapeutic vascular growth [77].
Established in the study above as well, miRNA influence on VEGF is a determining factor towards the neovascularization of respective tissues. A study by He et al. [78] demonstrated the regulatory function miR-150 has on post-stroke cerebral ischemia in rats via its interaction with VEGF. Through the upregulation of miR-150, the study resulted into decreased levels of vascular density of near-infarcted zones of the brain after middle cerebral artery occlusion. Additionally, miR-150 was seen to counter-effect tube formation, proliferation and migration of brain microvascular endothelial cells. All these results could be linked to the interaction of miR-150 and VEGF, leading to reduced expression. Using a dual-luciferase assay, VEGF was determined to be a direct target of miR-150 [78].
The aforementioned miRNAs play crucial roles in neovascularization and are consequently potential opportunities for therapy. Naturally, expression of miRNAs can be controlled by lncRNAs as illustrated in the subchapter on cardiomyocyte proliferation. Several lncRNAs have been identified to play a role in regulating neovascularization. These are highlighted in the next section.
4.2 LncRNA
LncRNAs have been established as regulators of various mechanisms involving neovascularization. This group of lncRNAs is also known as “Angio-lncRs” [14]. Due to the relative novelty of lncRNAs, lncRNAs that have not been directly validated in cardiac disease models are included in this part if the mechanism plays a role in neovascularization and has the potential to be relevant for cardiac regeneration.
The interplay between previously introduced miR-150 and the lncRNA MIAT was demonstrated with regards to their role in cardiac neovascularization [79]. In cardiac as well as in retinal cells, MIAT functions as a reliever to miR-150-5p repression of the pro-angiogenic growth factor VEGF. By functioning as a sponge to miR-150, it has proven to enhance cardiac hypertrophy in rat derived heart H9c2 cells. MIAT can therefore be labelled as a competitive endogenous RNA. Knockdown of MIAT results in depleted levels of vascular forming networks that result from reduced TNF-α and VEGF. Taken together, this study showed that MIAT functions as an inducer of in pathological angiogenesis [14, 79].
In vivo genetic deletion of MALAT1 as well as pharmacological inhibition of MALAT1 reduced vascular growth, indicating its significance in neovascularization. Using a genetic ablation mouse model, Michalik et al. [80] determined that the lack of MALAT1 resulted in lower neonatal retina vascularization and delayed vessel extension, as opposed to wild-type mice from the same litter. Furthermore, pharmacologically inhibiting MALAT1 with GapmeRs in a hind limb ischemia mouse model hampered blood flow recovery and reduced capillary density. MALAT1 controls the transition between proliferative and migratory phenotypes of ECs, and its silencing through small interfering RNA (siRNA) resulted in the reduction of the number of proliferating ECs. Zhang et al. [81] additionally conducted RNA-immunoprecipitation experiments. These tests demonstrated how MALAT1 has an immediate effect on vascular endothelial growth factor receptor 2 (VEGFR2) to facilitate angiogenesis, indicating that MALAT1 controls intrinsic angiogenesis through direct regulating VEGFR2. The silencing of MALAT1 reduced tube formation, proliferation as well as cell migration in skeletal muscle microvascular endothelial cell [80, 81]. Even though the aforementioned experiments employed the hind limb ischemia model, the results are relevant for cardiac endothelial cells that are in similar pathological remodeling conditions [62].
The lncRNA named MANTIS also affects angiogenesis [82, 83]. In particular HUVECs were used in a Matrigel angiogenesis assay in mice. CRISPR/Cas9-facilitated knockout of MANTIS, or silencing through siRNAs or GapmeRs, decreased angiogenic sprouting and tube formation. MANTIS was discovered to target the endothelial genes SMAD6, SOX18, and COUP-TFII, all important pro-angiogenic genes. Silencing of MANTIS using GapmeRs and siRNAs resulted in decreased protein expression of SMAD6, SOX18, and COUP-TFII in human aortic smooth muscle cells, human coronary artery smooth muscle cells, and in human aortic ECs. Furthermore, depression of any of these three proteins in a spheroid outgrowth assay resulted in poor endothelial sprouting. MANTIS was found to interact with BRG1, part of an ATP-dependent transcription activator family of proteins [82, 83]. MANTIS increases ATP-ase activity of BRG1 by acting as a promoter for BAF155, a subunit of the complex. The BRG1 protein family regulates the alteration and remodeling of the chromatin structure of the genes it acts on, namely SOX18, SMAD6, and COUP-TFII in this particular example. MANTIS can therefore be seen as a promoter of angiogenesis by stimulating transcription of these genes. Leisegang et al. concluded from their data that a decrease in MANTIS levels impaired the endothelial angiogenic function ex vivo as well as in vivo, opening more doors for the relatively unknown domain of lncRNA influence on angiogenic functions [82].
An antisense lncRNA named PUNISHER was found to have a large effect on neovascularization, as its inhibition led to severe vascular defects with problematic branching and decreased vessel formation [83]. These observations were made with the help of human pluripotent stem cell differentiation models. In zebrafish, PUNISHER supports and maintains EC function, yet its particular mechanism is still unknown.
In addition, knockdown of the lncRNA ALIEN resulted into observed down-regulation of 503 genes that contributed to angiogenesis and blood vessel development [83, 84].
5 Discussion
This chapter outlines various non-coding RNAs, their targets, and their effects on cardiac regeneration. By focusing on cardiomyocyte proliferation and cardiac neovascularization as hallmarks of regeneration, research involving the inhibition or enhanced expression of certain non-coding RNAs that resulted in significant alterations in the regenerative abilities of the heart was used to highlight the role non-coding RNAs can play in cardiac regeneration. These results open doors for new research into potential therapeutics based on the influence of non-coding RNAs on cellular pathways.
For non-coding RNAs to be used clinically several challenges have to be overcome and several requirements have to be met. The non-coding RNAs need to be stable, specific, and with high binding affinity, and they need to be delivered efficiently to the target tissue. Modification of non-coding RNAs to improve stability, specificity, and uptake, as well as delivery strategies are reviewed elsewhere [86,87,88,89]. Briefly, non-coding RNAs are modified with chemical modifications, such as 2′ sugar modifications, locked nucleic acids, or phosphodiester and phosphorothioate linkages, to improve stability, specificity, and uptake. Delivery strategies include (biodegradable) biomaterials, lipid-based vehicles, viruses, exosomes, nanoparticles, microbubbles, and (cationic) polymeric drug delivery devices [90, 91]. Their advantages include small size, stability, reduced degradation, improved uptake, and specificity. Local delivery may be facilitated by targeting ligands or localized injections. These strategies need to be investigated in models that allow for inclusion of delivery and surgical practices akin to surgery in humans, and that adequately resemble human pathophysiology. Additional to delivery, bio-distribution and pharmacokinetics/pharmacodynamics of these delivery vehicles need to be characterized. Furthermore, modulation of expression by non-coding RNAs can have profound effects in both the short and the long term. Their targets not only have to be identified and validated in short-term studies, the effect of non-coding RNA modulation also has to be assessed in long-term in vivo studies. As illustrated by Tian et al. and Gabisonia et al. [40, 43], persistent and uncontrolled expression can result in death. Besides these short and long-term effects, off-target effects have to be identified and investigated, for non-coding RNAs can have multiple targets. Only after these rigorous tests have been performed to a satisfactory level, the safety and efficacy of ncRNA therapeutics can be assessed in humans [92, 43]. Therefore, to bring non-coding RNA therapeutics one step closer to clinical applications, research needs to move forward into more representative models of human disease that encompass all aspects of treatment.
References
Benjamin, E.J. Muntner, P. Alonso, A. Bittencourt, M.S. Callaway, C.W. Carson, A.P. Chamberlain, A.M. Chang, A.R. Cheng, S. Das, S.R. Delling, F.N. Djousse, L. Elkind, M.S.V. Ferguson, J.F. Fornage, M. Jordan, L.C. Khan, S.S. Kissela, B.M. Knutson, K.L. Kwan, T.W. Lackland, D.T. Lewis, T.T. Lichtman, J.H. Longenecker, C.T. Loop, M.S. Lutsey, P.L. Martin, S.S. Matsushita, K. Moran, A.E. Mussolino, M.E. O'Flaherty, M. Pandey, A. Perak, A.M. Rosamond, W.D. Roth, G.A. Sampson, U.K.A. Satou, G.M. Schroeder, E.B. Shah, S.H. Spartano, N.L. Stokes, A. Tirschwell, D.L. Tsao, C.W. Turakhia, M.P. VanWagner, L.B. Wilkins, J.T. Wong, S.S. Virani, S. S (2019) American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation 139 (10): e56-e528.
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324(5923):98–102.
Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410(6829):701–5.
Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature. 2004;428(6983):668–73.
Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag JI, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004;428:664–8.
Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114(6):763–76.
Zwetsloot PP, Végh AM, Jansen of Lorkeers SJ, van Hout GP, Currie GL, Sena ES, Gremmels H, Buikema JW, Goumans MJ, Macleod MR, Doevendans PA, Chamuleau SA, Sluijter JP. Cardiac stem cell treatment in myocardial infarction: a systematic review and meta-analysis of preclinical studies. Circ Res. 2016;118(8):1223–32.
van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marbán E, Molkentin JD. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509(7500):337–41.
Sultana N, Zhang L, Yan J, Chen J, Cai W, Razzaque S, Jeong D, Sheng W, Bu L, Xu M, Huang GY, Hajjar RJ, Zhou B, Moon A, Cai CL. Resident c-kit(+) cells in the heart are not cardiac stem cells. Nat Commun. 2015;6:8701.
van Berlo JH, Molkentin JD. Most of the dust has settled: cKit+ progenitor cells are an irrelevant source of cardiac myocytes in vivo. Circ Res. 2016;118(1):17–9.
Kretzschmar K, Post Y, Bannier-Hélaouët M, Mattiotti A, Drost J, Basak O, Li VSW, van den Born M, Gunst QD, Versteeg D, Kooijman L, van der Elst S, van Es JH, van Rooij E, van den Hoff MJB, Clevers H. Profiling proliferative cells and their progeny in damaged murine hearts. Proc Natl Acad Sci. 2018;115(52):E12245–54.
Li Y, He L, Huang X, Bhaloo SI, Zhao H, Zhang S, Pu W, Tian X, Li Y, Liu Q, Yu W, Zhang L, Liu X, Liu K, Tang J, Zhang H, Cai D, Ralf AH, Xu Q, Lui KO, Zhou B. Genetic lineage tracing of nonmyocyte population by dual recombinases. Circulation. 2018;1389(8):793–805.
Bergmann O. Clearing up the mist: cardiomyocyte renewal in human hearts. Eur Heart J. 2019;40(13):1037–8.
Yu B, Wang S. Angio-LncRs: LncRNAs that regulate angiogenesis and vascular disease. Theranostics. 2018;8(13):3654–75.
Folkman J. Tumor angiogenesis. Adv Cancer Res. 1985;43:175–203.
Tabibiazar R, Rockson SG. Angiogenesis and the ischaemic heart. Eur Heart J. 2001;22(11):903–18.
Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, Poss KDA. Dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 2006;127(3):607–19.
Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298(5601):2188–90.
Kikuchi K, Poss KD. Cardiac regenerative capacity and mechanisms. Annu Rev Cell Dev Biol. 2012;28(1):719–41.
Jopling C, Sleep E, Raya M, Martí M, Raya A, Izpisúa Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464(7288):606–9.
Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, MacRae CA, Stainier DYR, Poss KD. Primary contribution to zebrafish heart regeneration by gata4+ cardiomyocytes. Nature. 2010;464(7288):601–5.
Witman N, Murtuza B, Davis B, Arner A, Morrison JI. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol. 2011;354(1):67–76.
Laube F, Heister M, Scholz C, Borchardt T, Braun T. Re-programming of newt cardiomyocytes is induced by tissue regeneration. J Cell Sci. 2006;119(22):4719–29.
Wang WE, Li L, Xia X, Fu W, Liao Q, Lan C, Yang D, Chen H, Yue R, Zeng C, Zhou L, Zhou B, Duan DD, Chen X, Houser SR, Zeng C. Dedifferentiation, proliferation, and redifferentiation of adult mammalian cardiomyocytes after ischemic injury. Circulation. 2017;136(9):834–48.
Yutzey K. Cardiomyocyte proliferation: teaching an old dogma new tricks. Circ Res. 2017;120:627–9.
Drenckhahn J-D, Schwarz QP, Gray S, Laskowski A, Kiriazis H, Ming Z, Harvey RP, Du X-J, Thorburn DR, Cox TC. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev Cell. 2008;15(4):521–33.
Haubner BJ, Adamowicz-Brice M, Khadayate S, Tiefenthaler V, Metzler B, Aitman T, Penninger JM. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging. 2012;4(12):966–77.
Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–80.
Jesty SA, Steffey MA, Lee FK, Breitbach M, Hesse M, Reining S, Lee JC, Doran RM, Nikitin AY, Fleischmann BL, Kotlikoff MI. c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc Natl Acad Sci. 2012;109(33):13380–5.
Strungs, E.G. Ongstad, E.L. O'Quinn, M.P. Palatinus, J.A. Jourdan, L.J. Gourdie, R.G (2013) Cryoinjury models of the adult and neonatal mouse heart for studies of scarring and regeneration. Methods Mol Biol 1037:343–353.
Bryant, D.M. O'Meara, C.C. Ho, N.N. Gannon, J. Cai, L. Lee, R.T (2015) A systematic analysis of neonatal mouse heart regeneration after apical resection. J Mol Cell Cardiol 79:315–318.
Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C, Stein JI, Penninger JM. Functional recovery of a human neonatal heart after severe myocardial infarction. Circ Res. 2016;118(2):16–221.
Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87(2):521–44.
van Amerongen MJ, Engel FB. Features of CM proliferation and its potential for cardiac regeneration. J Cell Mol Med. 2008;12:2233–44.
Bicknell KA, Coxon CH, Brooks G. Can the CM cell cycle be reprogrammed? J Mol Cell Cardiol. 2007;42(4):706–21.
Muthuramu I, Lox M, Jacobs F, De Geest B. Permanent ligation of the left anterior descending coronary artery in mice: a model of post-myocardial infarction remodeling and heart failure. J Vis Exp. 2014;94
Aguirre A, Montserrat N, Zacchigna S, Nivet E, Hishida T, Krause MN, Kurian L, Ocampo A, Vazquez-Ferrer E, Rodriquez-Esteban C, Kumar S, Moresco JJ, Yates JR III, Campistol JM, Sancho-Martinez I, Giacca M, Izpisua Belmonte JC. In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell. 2014;15(5):589–604.
Huang W, Feng Y, Liang J, Yu H, Wang C, Wang B, Wang M, Jiang L, Meng W, Cai W, Medvedovic M, Chen J, Paul C, Davidson WS, Sadayappan S, Stambrook P, Yu X, Wang Y. Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat Commun. 2018;9(1):700.
Yang Y, Cheng HW, Qiu Y, Dupee D, Noonan M, Lin YD, Fisch S, Unno K, Sereti KI, Liao R. MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ Res. 2015;117(5):450–9.
Tian Y, Liu Y, Wang T, Zhou N, Kong J, Chen L, Snitow M, Morley M, Li D, Petrenko N, Zhou S, Lu M, Gao E, Koch WJ, Stewart KM, Morrisey EE. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci Transl Med. 2015;7(279):279–89.
Wang LL, Liu Y, Chung JJ, Wang T, Gaffey AC, Lu M, Cavanough CA, Zhou S, Kanade R, Atluri P, Morrisey EE, Burdick J. Sustained miRNA delivery from an injectable hydrogel promotes cardiomyocyte proliferation and functional regeneration after ischaemic injury. Nat Biomed Eng. 2017;1(12):983–92.
Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492(7429):376–81.
Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, Burchielli S, Collesi C, Zandonà L, Sinagra G, Piacenti M, Zacchigna S, Bussani R, Recchia FA, Giacca M. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature. 2019;569(7756):418.
Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci. 2013;110(1):187–92.
Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, Dorn GW 2nd, van Rooij E, Olson EN. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res. 2011;109(6):670–9.
Zhao Y, Ransom J, Li A, von Vedantham C, Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129(2):303–17.
Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22(23):3242–54.
Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, Pu WT, Liao R, Wang DZ. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res. 2013;112(12):1557–66.
Xiao J, Liu H, Cretoiu D, Toader DO, Suciu N, Shi J, Shen S, Bei Y, Sluijter JP, Das S, Kong X, Li X. miR-31a-5p promotes postnatal cardiomyocyte proliferation by targeting RhoBTB1. Exp Mol Med. 2017;49(10):e386.
Chen Y, Li X, Li B, Wang H, Li M, Huang S, Sun Y, Chen G, Si X, Huang C, Liao W, Liao Y, Bin J. Long non-coding RNA ECRAR triggers post-natal myocardial regeneration by activating ERK1/2 signaling. Mol Ther. 2019;27(1):29–45.
See K, Tan WLW, Lim EH, Tiang Z, Lee LT, Li PYQ, Luu TDA, Ackers-Johnson M, Foo RS. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat Commun. 2017;8(1):225.
Coccia EM, et al. Regulation and expression of a growth arrest-specific gene (gas5) during growth, differentiation, and development. Mol Cell Biol. 1992;12(8):3514–21.
Mourtada-Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009;28(2):195–208.
Yin D. Long noncoding RNA GAS5 affects cell proliferation and predicts a poor prognosis in patients with colorectal cancer. Med Oncol. 2014;31(11):253.
Cai B, Ma W, Ding F, Zhang L, Huang Q, Wang X, Hua B, Xu J, Li J, Bi C, Guo S, Yang F, Han Z, Li Y, Yan G, Yu Y, Bao Z, Yu M, Li F, Tian Y, Pan Z, Yang B. The long noncoding RNA CAREL controls cardiac regeneration. J Am Coll Cardiol. 2018;72(5):534–50.
Wu M, Zhang S, Chen X, Xu H, Li X. Expression and function of lncRNA MALAT-1 in the embryonic development of zebrafish. Gene. 2019;680:65–71.
Li B, Hu Y, Li X, Jin G, Chen X, Chen G, Chen Y, Huang S, Liao W, Liao Y, Teng Z, Bin J. Sirt1 antisense long noncoding RNA promotes cardiomyocyte proliferation by enhancing the stability of Sirt1. J Am Heart Assoc. 2018;7(21)
Wang J, Chen X, Shen D, Ge D, Chen J, Pei J, Li Y, Yue Z, Feng J, Chu M, Nie Y. A long non-coding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J Mol Cell Cardiol. 2019;127:105–14.
Chen G, Li H, Li X, Li B, Zhong L, Huang S, Zheng H, Li M, Jin G, Liao W, Liao Y, Chen Y, Bin J. Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J Mol Cell Cardiol. 2018;122:152–64.
Adair TH, Montani JP. Angiogenesis. San Rafael (CA): Morgan & Claypool Life Sciences Chapter 1, overview of angiogenesis; 2010.
Choong OK, Lee DS, Chen CY, Hsieh P. The roles of non-coding RNAs in cardiac regenerative medicine. Non-coding RNA Research. 2017;2(2):100–10.
Juni R, Abreu R, da Costa Martins P. Regulation of microvascularization in heart failure – an endothelial cell, non-coding RNAs and exosome liaison. Non-coding RNA Research. 2017;2(1):45–55.
Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, Wu JC. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122(11 Suppl):S124–31.
Arif M, Pandey R, Alam P, Jiang S, Sadayappan S, Paul A, Ahmed R. MicroRNA-210-mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. J Mol Med (Berl). 2017;95(12):1369–85.
Fan ZG, Qu XL, Chu P, Gao YL, Gao XF, Chen SL, Tian NL. MicroRNA-210 promotes angiogenesis in acute myocardial infarction. Mol Med Rep. 2018;17(4):5658–65.
Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324(5935):1710–3.
Hinkel R, Penzkofer D, Zahlke S, Fischer A, Husada W, Xu QF, Baloch E, Van Rooij E, Zeiher AM, Kupatt C, et al. Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation. 2013;128(10):1066–75.
Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JTG, Sohn-Lee C. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124(6):720–30.
Meloni M, Marchetti M, Garner K, Littlejohns B, Sala-Newby G, Xenophontos N, Floris I, Suleiman MS, Madeddu P, Caporali A, Emanueli C. Local inhibition of microRNA-24 improves reparative angiogenesis and left ventricle remodeling and function in mice with myocardial infarction. Mol Ther. 2013;21(7):1390–402.
Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208(3):549–60.
Wang S, Aurora AB, Johnson BA, Qi X, Mcanally J, Hill JA, Richardson JA, Bassel-duby R, Olson EN, Hill A, et al. The endothelial-specific MicroRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15(2):261–71.
Icli B, Wara AKM, Moslehi J, Sun X, Plovie E, Cahill M, Marchin JF, Schissler A, Padera RF, Shi J, et al. MicroRNA-26a regulates pathological and physiological angiogenesis by targeting BMP/SMAD1 signaling. Circ Res. 2013;113(11):1231–41.
Wen Z, Huang W, Feng Y, et al. MicroRNA-377 regulates mesenchymal stem cell-induced angiogenesis in ischemic hearts by targeting VEGF. PLoS One. 2014;9(9):e104666.
Goyal A, Behring A, Kumar A, Siess W. STK35L1 associates with nuclear actin and regulates cell cycle and migration of endothelial cells. PLoS One. 2011;6(1):E16249.
Joladarashi D, Garikipati VNS, Thandavarayan RA, et al. Enhanced cardiac regenerative ability of stem cells after ischemia-reperfusion injury: role of human CD34+ cells deficient in microRNA-377. J Am Coll Cardiol. 2015;66(20):2214–26.
Lei Z, van Mil A, Brandt MM, Grundmann S, Hoefer I, Smits M, El Azzouzi H, Fukao T, Cheng C, Doevendans PA, Sluijter JP. MicroRNA-132/212 family enhances arteriogenesis after hindlimb ischemia through modulation of the Ras-MAPK pathway. J Cell Mol Med. 2015;19(8):1994–2005.
van Mil A, Grundmann S, Goumans M, Lei Z, Oerlemans M, Jaksani S, Doevendans P, Sluijter J. MicroRNA-214 inhibits angiogenesis by targeting quaking and reducing angiogenic growth factor release. Cardiovasc Res. 2012;93(4):655–65.
He Q, Li Q, Jin H, Zhi F, Suraj B, Zhu Y, Xia Y, Mao L, Chen X, Hu B. MiR-150 regulates poststroke cerebral angiogenesis via vascular endothelial growth factor in rats. CNS Neurosci Ther. 2016;22(6):507–17.
Yan B, Yao J, Liu JY. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116(7):1143–56.
Michalik KM, You X, Manavski Y. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114(9):1389–97.
Zhang X, Tang X, Hamblin MH, Yin KJ. Lcong non-coding RNA Malat1 regulates angiogenesis in hindlimb ischemia. Int J Mol Sci. 2018;19(6):1723.
Leisegang MS, Fork C, Josipovic I, Richter FM, Preussner J, Hu J, Miller MJ, Epah J, Hofmann P, Günther S, Moll F, Valasarajan C, Heidler J, Ponomareva Y, Freiman TM, Maegdefessel L, Plate KH, Mittelbronn M, Uchida S, Künne C, Stellos K, Schermuly RT, Weissmann N, Devraj K, Wittig I, Boon RA, Dimmeler S, Pullamsetti SS, Looso M, Miller FJ, Brandes RP. Long noncoding RNA MANTIS facilitates endothelial angiogenic function. Circulation. 2017;136(1):65–79.
Trotter KW, Archer TK. The BRG1 transcriptional coregulator. Nucl Recept Signal. 2008;6(1):e004.
Kurian L, Aguirre A, Sancho-Martinez I, Benner C, Hishida T, Nguyen TB, Reddy P, Nivet E, Krause MN, Nelles DA, Esteban CR, Campistol JM, Yeo GW, Belmonte J. Identification of novel long noncoding RNAs underlying vertebrate cardiovascular development. Circulation. 2015;131(14):1278–90.
Gomes C, Spencer H, Ford K, Michel L, Baker A, Emanueli C, Balligand J, Devaux Y. The function and therapeutic potential of long non-coding RNAs in cardiovascular development and disease. Mol Ther Nucleic Acids. 2017;8:494–507.
Lei Z, Sluijter JP, van Mil A. MicroRNA therapeutics for cardiac regeneration. Mini Rev Med Chem. 2015;15(6):441–51.
Tibbitt MW, Dahlman JE, Langer R. Emerging frontiers in drug delivery. J Am Chem Soc. 2016;138(3):704–17.
Van der Ven CFT, Wu PJ, Tibbit MW, van Mil A, Sluijter JPG, Langer R, Aikawa E. In vitro 3D model and miRNA drug delivery to target calcific aortic valve disease. Clin Sci. 2017;131(3):181–95.
Fenton OS, Olafson KN, Pillai PS, Mitchell MJ, Langer R. Advances in biomaterials for drug delivery. Adv Mater. 2018;30
Kwekkeboom RF, Lei Z, Bogaards SJ, Aiazian E, Kamp O, Paulus WJ, Sluijter JP, Musters RJ. Ultrasound and microbubble-induced local delivery of MicroRNA-based therapeutics. Ultrasound Med Biol. 2015;41(1):163–76.
Kwekkeboom RF, Sluijter JP, van Middelaar BJ, Metz CH, Brans MA, Kamp O, Paulus WJ, Musters R. Increased local delivery of antagomir therapeutics to the rodent myocardium using ultrasound and microbubbles. J Control Release. 2016;222:18–31.
Kwekkeboom RF, Lei Z, Doevendans PA, Musters RJ, Sluijter JP. Targeted delivery of miRNA therapeutics for cardiovascular diseases: opportunities and challenges. Clin Sci (Lond). 2014;127(6):351–65.
Acknowledgments
This work was funded by the Netherlands CardioVascular Research Initiative (CVON: The Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science) and Vrienden UMC Utrecht (C.V., J.S.); an unrestricted grant from CELLINK to Vrienden UMC Utrecht (C.V., J.S.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
van der Ven, C.F.T., Hogewoning, B.C.R., van Mil, A., Sluijter, J.P.G. (2020). Non-coding RNAs in Cardiac Regeneration. In: Xiao, J. (eds) Non-coding RNAs in Cardiovascular Diseases. Advances in Experimental Medicine and Biology, vol 1229. Springer, Singapore. https://doi.org/10.1007/978-981-15-1671-9_9
Download citation
DOI: https://doi.org/10.1007/978-981-15-1671-9_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-1670-2
Online ISBN: 978-981-15-1671-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)