
Abstract
Non-alcoholic fatty liver disease (NAFLD) will become a dominant cause of hepatocellular carcinoma (HCC) in the coming decade. Whereas the exact molecular mechanisms underlying the progression from simple steatosis, through steatohepatitis, to HCC remains largely unclear, emerging evidence has supported a central role of defective autophagy in the pathogenesis of NAFLD and its complications. Autophagy not only regulates lipid metabolism and insulin resistance, but also protects hepatocytes from injury and cell death. Nevertheless, in inflammation and tumorigenesis, the role of autophagy is more paradoxical. In NAFLD, defective hepatic autophagy occurs at multiple levels through numerous mechanisms and is causally linked to NAFLD-related HCC. In this chapter, we summarize the regulation and function of autophagy in NAFLD and highlight recent identification of potential pharmacological agents for restoring autophagic flux in NAFLD.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Epidemiology and General Mechanism of NAFLD and NAFLD-Related HCC
10.1.1 Epidemiology of NAFLD and NAFLD-Related HCC
The community prevalence of non-alcoholic fatty liver disease (NAFLD) has increased from less than 10% in the 1980s to current rates of 15–30% or higher. NAFLD affects 15–40% of the general population in Asia [1]. The pathological spectrum of NAFLD comprises hepatic steatosis alone, hepatic steatosis with lobular inflammation, and non-alcoholic steatohepatitis (NASH). More than 30% of people with NAFLD may have NASH, which may progress to cirrhosis (in 10–29% of NASH patients) and ultimately hepatocellular carcinoma (HCC) (in 4–27% of NASH-induced cirrhosis patients) [1]. NAFLD is strongly associated with metabolic syndrome (i.e. obesity, diabetes, insulin resistance and dyslipidemia). The relative risk for HCC in obese men with body mass index (BMI) >35 was 4.52 compared to those with BMI between 18.5 and 24.9. Type II diabetes is also found to double the risk of HCC. With the increasing prevalence of obesity in children and adolescents, it is expected that NASH will become a dominant cause of HCC in the future with increasing number of patients presenting at an earlier age [1].
10.1.2 Molecular Basis of NASH and NAFLD-Related HCC
The current paradigm of NASH pathogenesis is that “toxic lipid species”, including free fatty acids or free cholesterol, trigger cell death and inflammatory response. Such changes are accompanied by metabolic alterations, such as insulin resistance, and overproduction of free radicals from the mitochondria, causing lipid peroxidation, cytokine production, and necrosis [1, 2]. Some advances have also been made in understanding the molecular characteristics of NAFLD-related HCC. Hyperinsulinemia associated with type II diabetes could activate IRS-1 and the downstream mitogen-activated protein kinases (MAPK) and phosphoinositide-3-kinase (PI3K)/Akt pathways to promote cell proliferation and survival. Pro-inflammatory cytokines, such as interleukin (IL)-6 and tumor necrosis factor (TNF) α, could also mediate activation of oncogenic transcription factors, including activator protein-1 (AP-1), nuclear factor-κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3). Imbalance of adipokine signaling (i.e. hypoadiponectinemia and hyperlectinemia) associated with increased adiposity is also linked to malignant phenotypes, such as unchecked cell cycle progression, evasion of apoptosis, and enhanced invasiveness and metastasis [1, 2]. However, little is known about how the effect of aberrant fatty accumulation in the liver is directly converted to oncogenic signals. As a consequence, there is minimal intervention in the clinic to prevent HCC development in NASH patients at risk of such progression. Thus, there is a compelling need to elucidate the molecular mechanisms of NAFLD-related HCC and to identify potential therapeutic targets to control this disorder.
10.2 Mechanism and Functions of Autophagy
10.2.1 Cellular Mechanism of Autophagy
Macroautophagy (hereafter referred to as autophagy) is a major process in which the cell digests its own contents. This self-cannibalistic pathway is instigated by the sequestration of cytosolic cargos, such as proteins and damaged organelles, by the phagophore followed by the formation of double-membrane structures known as autophagosomes. In the late phase, autophagosomes merge with lysosomes to produce autolysosomes. The sequestered materials are then degraded by acidic hydrolases to release free amino acids [3]. In this way, autophagy serves as an important pathway for energy production in time of starvation. Autophagy has been shown to have crosstalk with diverse signaling pathways and possess the ability to regulate other cellular and tissue processes, such as cell proliferation, apoptosis, differentiation, and inflammation. In this regard, altered autophagosomal-lysosomal pathway has been connected to many pathological conditions, including such as cancer, infection, autoimmunity, inflammatory diseases, neurodegeneration and aging [4].
10.2.2 Molecular Checkpoints of Autophagy
Although autophagy could be regulated at multiple levels, its execution converges on multiple mediators collectively known as “autophagy-related proteins”, which are involved in the abovementioned multi-step machinery [5]. The initiation of autophagosome formation is regulated by the nutrient-sensing mammalian target of rapamycin (mTOR) through the unc-51-like kinase 1/2 (ULK1/2)-mAtg13-focal adhesion kinase family interacting protein of 200 kDa (FIP200) complex. Under growth-permissive conditions, mTOR binds and represses the ULK1/2-mAtg13-FIP200 complex and thereby inhibiting FIP200 phosphorylation and recruitment of Atg proteins. However, under growth factor- or nutrient-deprived conditions, mTOR dissociates from the ULK1/2-mAtg13-FIP200 complex to unmask the kinase activities of ULK1/2, resulting in assembly of Atg proteins at the autophagosome formation site [6]. Beclin 1, the mammalian orthologue of the yeast Apg6/Vps30, could mediate multiple vesicle-trafficking pathways and plays a central role in autophagy. Beclin1 is a Bcl-2-interacting protein which exists in complexes of at least three different configurations: Beclin 1-hVps34-p150-Atg14, Beclin 1-hVps34-p150-UVRAG-Bif1 and Beclin 1-hVps34-p150-Rubicon-UVRAG [7]. The former functions at the early stage of autophagosome formation whereas the latter two complexes facilitate autophagosomal membrane curvature and the maturation phase, respectively [7]. LC3, an autophagosomal ortholog of yeast Atg8, is another major regulator of autophagy, in which conversion of a cytosolic truncated form of LC3 (LC3-I) to its lipidated, autophagosomal membrane-associated form (LC3-II) is required for autophagosome formation [8].
10.3 Roles of Autophagy in NAFLD-Associated Biological Processes
10.3.1 Lipid Metabolism
Autophagosomal sequestration of triglycerides and cholesterol derived from lipid droplets in liver has been described and termed lipophagy. In autolysosomes, triglycerides are broken down by acidic hydrolases to produce free fatty acid, which are utilized for mitochondrial β-oxidation. In this capacity, lipophagy functions to regulate intracellular lipid stores and energy homeostasis. Accordingly, blockade of autophagy by pharmacological inhibitor or silencing expression autophagy related genes caused the retention of triglycerides and lipid droplets [9], reduced free fatty acid oxidation, and lowered the secretion of very-low-density lipoprotein (VLDL) from hepatocytes. Induction of hepatic autophagy through liver specific overexpression of Atg7 thus has been shown to alleviate the metabolic stress and mitigate hepatic steatosis in ob/ob mice [10]. Two pro-autophagic transcription factors FOXO1 and transcription factor EB (TFEB) also alleviate steatosis [11, 12]. Short-term treatment with pharmacological activators of autophagy, namely carbamazepine and rapamycin could reduce liver steatosis and triglyceride levels in the liver and blood [13]. These findings support a lipolytic role of autophagy in the liver.
10.3.2 Insulin Resistance
Defective autophagy has been linked to the development of insulin resistance. FOXO1-mediated suppression of autophagy conferred insulin resistance in genetically obese mice or mice fed with high-fat diet. Deficient hepatic autophagy of obese mice also promoted ER stress to induce insulin resistance [10, 12]. Concordantly, restoration of autophagy by hepatocyte-specific overexpression of Atg7 in obese mice normalized the insulin sensitivity and improved glucose tolerance [10].
10.3.3 Hepatocellular Injury
Recurrent hepatocellular injury and necroinflammation could lead to progression of simple steatosis to NASH, cirrhosis or even HCC. Autophagy functions to clear damaged organelles and, in this capacity, protects against cell death by removing abnormal mitochondria, which produce oxidative stress or trigger apoptosis through the intrinsic pathway [14,15,16]. Consistently, silencing Atg5 could blunt the cytoprotective function of autophagy and thereby enhancing hepatocyte death induced by menadione, which causes oxidative stress and mitochondrial cytochrome release [14]. Autophagy has also been shown to protect hepatocytes from extrinsic pathway of apoptosis, including death triggered by necrosis factor-related apoptosis-inducing ligand (TRAIL) [17]. Activating autophagy could also attenuate cell death in hepatocytes loaded with palmitic acid [18].
10.3.4 Inflammation
Autophagy could be a potent suppressor of inflammation. Findings from genetic and functional studies have pinpointed defective autophagy as a contributing factor to several autoimmune disorders, particularly in Crohn’s disease of which inflammation plays a key role in its pathogenesis [19]. Mechanistically, autophagy clears damaged mitochondria that release reactive oxygen species and mitochondrial DNA, thereby suppressing the activation of inflammasomes and Toll-like receptor 9 [20, 21]. Autophagy is also required for the degradation of p62/SQSTM1, an activator of NF-κB that promotes the transcription of pro-inflammatory cytokines [22, 23]. Through these mechanisms, autophagy dampens the transcription and/or maturation of pro-inflammatory cytokines to suppress inflammation [24, 25].
However, it is noteworthy that autophagy could in some biological contexts paradoxically promote inflammation. For instance, autophagy mediates the production of pro-inflammatory cytokines induced by avian influenza H5N1 pseudotyped particle via NF-κB and p38 mitogen-activated protein kinase (MAPK) signaling pathways [26]. Autophagy also enhances lipopolysaccharides-induced lung inflammation and neutrophil recruitment [27]. Enforced expression of hepatitis B virus X (HBx) protein, an oncogenic and pro-inflammatory protein, also induces autophagy in normal hepatocytes, in which knockdown of ATG5 and ATG7 mitigated HBx-induced activation of NF-κB and production of pro-inflammatory cytokines IL-6, IL-8, and CXCL2 in cultured hepatocytes [28].
10.4 Autophagic Impairment in NAFLD
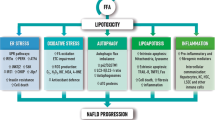
Emerging data suggests that obesity and long-term high-fat diet feeding might impair the autophagosomal-lysosomal system at multiple levels (Fig. 10.1), namely blockade of autophagosome formation, inhibition of autophagosome-lysosome fusion and mitigation of lysosome function [29].

Multi-level inhibition of autophagosome-lysosome system by steatosis in non-alcoholic fatty liver diseases
10.4.1 Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) provides the oxidizing environment for synthesis, folding and posttranslational modification of cellular proteins and is the primary storage organelle for intracellular Ca2+. Disruption of ER homeostasis could have severe cellular consequences. When the ER stress occurs as a result of accumulation of misfolded proteins, cells activate a protective called unfolded protein response (UPR), which is consisted three major molecular circuitries – [1] IRE-1 (inositol requiring enzyme 1)-mediated alternative splicing of XBP1 (X-box binding protein 1); [2] nuclear translocation and activation of ATF6 (activating transcription factor 6); activation of PERK (PKR-like ER kinase)-eIF2α (eukaryotic initiation factor 2 α)-ATF4 cascade. Activation of these pathways presumably helps to reduce the protein load and increase the folding capacity of the ER [30]. González-Rodríguez et al. recently demonstrated that the autophagic flux is impaired in cell-line and animal models of NAFLD as well as in clinical specimens of NAFLD patients. Interestingly, abrogation of endoplasmic reticulum (ER) could alleviate such impairment. In particular, knockdown of C/EBP homologous protein (CHOP), a pro-apoptotic mediator induced by both ATF4 and ATF6 upon ER stress, has been shown to partially alleviate autophagic impairment and hepatocyte apoptosis [18]. A subsequent mechanistic study by Wang and colleagues demonstrated that ER stress-induced asparagine synthetase overexpression contribute to increased generation of asparagine and thereby inhibiting lysosome acidification. The induction of asparagine synthetase was mediated through the PERK (PKR-like ER kinase)-eIF2α-ATF4 cascade. Interestingly, both steatotic- and asparagine-treated hepatocytes showed reduced lysosomal acidity as evidenced by impaired cathepsin D cleavage and reduced number of acidic vesicular organelles. Such deficits were attributed to the retention of lysosomal Ca2+, of which positive charge presumably prevents the transport of protons into lysosomes. Accordingly, knockdown of asparagine synthetase in steatotic hepatocytes restored autophagic flux [31]. These results also reverberated previous findings that cathepsin B, D, and L expression was significantly decreased in the liver from NAFLD patients [32].
10.4.2 Adipokines and Cytokines
Patients with NASH have been shown to exhibit dysregulated cytokine profiles in liver tissues, plasma and peripheral blood monocyte [33]. Reduced levels of adiponectin, an adipose-derived adipokine, are associated with obesity and NASH. Interestingly, adiponectin knockout attenuated high fat diet-induced autophagic defects, such as accumulation of p62/SQSTM1, and liver injury without reversing liver weights and hepatic steatosis, suggesting that reduced levels of adiponectin play an auto-protective role possibly through normalizing autophagy [34]. C-X-C motif chemokine 10 (CXCL10) is a crucial pro-inflammatory factor in chronic hepatitis. In animal models of NASH, ablation of CXCL10 by neutralizing monoclonal antibody or gene knockout has been to protect against hepatocyte injury and steatohepatitis development. These protective effects were accompanied by rectification of autophagic flux impairment. Bafilomycin A1, an inhibitor of lysosomal vacuolar type H+-ATPase and autolysosome formation, abolished the rectifying effect of CXCL10 ablation in cultured hepatocytes, indicating CXCL10 impaired late-stage autophagy in NAFLD [35].
10.4.3 Overactivation of mTOR Signaling
In both genetic and dietary models of obesity, a severe inhibition of autophagosome formation has been demonstrated. The mTOR signaling, which is a major suppressor of autophagosome formation, and FOXO1 downstream of AKT has been shown to be overactivated in steatotic liver, presumably owing to increased amino acid concentration and hyperinsulinemia [12, 36, 37]. High-fat diet rich in saturated fatty acids has been shown to elevate the expression and activity of Sirtuin 3 (Sirt3), which renders hepatocytes susceptible to palmitate-induced cell death. Mechanistically, Sirt3 upregulation results in manganese superoxide dismutase deacetylation and activation, which depleted intracellular superoxide contents, leading to AMP-activated protein kinase (AMPK) inhibition and mTOR complex 1 activation and thereby suppressing autophagy [38].
10.4.4 Altered Membrane Lipid Content
Koga and colleagues established an in vitro fusion assay using different lysosomal/autophagic compartments isolated from mouse liver. They found that altered membrane lipid composition induced by 25 mM methyl-beta-cyclodextrin in vitro or feeding animals with high-fat diet in vivo could reduce autophagosome-lysosome fusion up to 70% [39].
10.4.5 Foxo3a Downregulation
Autophagic impairment might occur in mesenchymal cells of steatotic livers. Palmitate and lipopolysaccharides have been shown to synergistically reduce Foxo3a expression in Kupffer cells, in which downregulation of Foxo3a increased blockage of autophagy flux. The protective effect of Foxo3a was found to be mediated through its transcriptional target Bim, whose overexpression also restored autophagy influx [40].
10.5 The Role of Autophagy in Tumorigenesis
10.5.1 Paradoxical Role of Autophagy in HCC
Autophagy plays a paradoxical role in hepatocarcinogenesis. Immunohistochemical staining has shown that the expression of Beclin-1, a key pro-autophagic protein, was significantly lower in HCC tissues than adjacent tissues and such downregulation was associated with more aggressive clinicopathological phenotypes and poorer overall survival [41]. Numerous tumor suppressors (e.g. XPD, Klotho, Tak1, PTPRO) have also been demonstrated to activate autophagy in HCC cells [42,43,44,45].
The tumor-suppressive function of autophagy was proposed to be mediated through degradation of oncogenic autophagic substrates (e.g. p62/STSTM1, microRNA-224) and maintenance of healthy mitochondria to reduce oxidative stress and DNA damage [46,47,48]. In this connection, gene targeting of p62/SQSTM1, a protein preferentially degraded through the autophagy pathway, has been shown to markedly abrogate the anchorage-independent growth of HCC cells, whereas overexpression of p62/SQSTM1 had opposite effects [48]. To this end, p62/SQSTM1 was reported to take part in the feedforward loop for inducing and sustaining NF-κB activity upon constitutive KRAS activation to promote the development of pancreatic ductal adenocarcinoma [22]. Moreover, knockdown of p62/SQSTM1 inhibits cell migration and invasion in glioblastoma stem cells [49]. Autophagy mediates the degradation of the oncogenic microRNA-224, whose accumulation promotes HCC cell migration and tumor formation through silencing its target gene Smad4 [47]. Autophagy has also reported to mediate the growth-arresting and cytotoxic effects of interferon-γ in HCC cells [50].
Since autophagy is a major catabolic process, autophagy could paradoxically function as a pro-survival mechanism to generate nutrients, especially in time of nutrient deprivation and cellular stress. In this connection, autophagy has been shown to protects cancer cells from the accumulation of damaged organelles or protein aggregates, programmed cell death resulting from detachment from surrounding extracellular matrix (i.e. anoikis), and the toxicity of cancer therapies [3]. In HCC, autophagy inhibition by pharmacological inhibitors or siRNAs has been shown to sensitize cancer cells to the multikinase inhibitor linifanib [51]. MicroRNA-375-mediated inhibition of autophagy also impaired viability of HCC cells in response to hypoxia in vitro and in vivo [52]. Moreover, autophagy could suppress the expression of major tumor suppressors to promote the development of HCC [46].
It is generally believed that an optimal level of autophagy is key to tumor suppression in normal condition whereas this pathway could be subverted by cancer cells for tumor promotion in the later stages of hepatocarcinogenesis [3].
10.5.2 Crosstalk with Cancer-Related Signaling
Autophagy could be induced by Ras-Raf-MEK-ERK, IKK-nuclear factor (NF)-κB, transforming growth factor-β, platelet-derived growth factor, p16/p27/retinoblastoma protein (pRB), p53-DRAM, Ca2+-CaMMKβ, reactive oxygen species (ROS)-ATM-AMPK signaling as well as endoplasmic reticulum stress mediators (e.g. PERK-eIF2α, GRP78/BiP, IRE1-JNK, HDAC6). In contrast, Autophagy is known to be negatively regulated by PI3K-Akt-mTOR signaling, anti-apoptotic members of Bcl-2 family, cytoplasmic p53, FLIP, BRCA1, Jumpy, Naf-1 and rubicon [3].
10.5.3 Emerging Evidence of Involvement of Autophagy in NAFLD-Related HCC
To date, only sporadic studies have directly examined the role of autophagy in NAFLD-related HCC with animal models. Inokuchi-Shimizu and colleagues reported that hepatocyte-specific deletion of the MAP kinase kinase kinase TGF β-activated kinase 1 (TAK1), a positive regulator of AMPK, increased mTOR activity and suppressed autophagy, accompanied by severe hepatosteatosis [44]. The expression of peroxisome proliferator-activated receptor α (PPARα) target genes and β-oxidation, which regulate hepatic lipid degradation, were also repressed. Interestingly, mice with hepatocyte-specific knockout of Tak1 developed spontaneous liver cancer, which expressed high levels of p62/SQSTM1. Inhibition of mTOR activity by rapamycin restored autophagy and prevented HCC development, indicating that induction of autophagy by Tak1 might inhibit fatty liver-associated HCC growth [44].
The tumor-suppressive function of autophagy could also be exemplified by another study reporting that genetic ablation of protein tyrosine phosphatase receptor type O (PTPRO), a known tumor suppressor, produced severe autophagy deficiency, liver injury, insulin resistance, hepatosteatosis and liver tumor formation upon feeding with high-fat diet after diethylnitrosamine (DEN) injection as compared with wild-type littermates [43]. Immunohistochemical staining demonstrated that hepatic PTPRO was reduced while p62/SQSTM1 was increased in NAFLD as compared with normal liver. These findings suggest that low expression of PTPRO in hepatocytes may contribute to the inhibition of autophagy and progression to NASH and NAFLD-related HCC [43].
10.6 Pharmacological Modulation of Autophagy for Treating NAFLD or Preventing NAFLD-Related HCC
Lysosome-dependent degradation of lipid through the autophagic pathway is growingly recognized as a crucial mechanism for lipid utilization whereas dysfunctional autophagy may contribute to NASH and NAFLD-related HCC development. Investigative efforts have thus been put forth to identify pharmacological agents that may restore autophagic functions in NAFLD and thus help to prevent NAFLD-related HCC.
10.6.1 Polyunsaturated Fatty Acids
Polyunsaturated fatty acids are fatty acids (PUFAs) that contain more than one double bond in their backbone. Shen and colleagues demonstrated that dietary PUFAs could increase LC3-II levels and attenuate IL-1β secretion and caspase-1 cleavage in response to lipopolysaccharides in cultured hepatocytes or liver tissues. Autophagy-dependent suppression of nucleotide-binding oligomerization domain leucine-rich repeat-containing receptor protein (NLRP3) inflammasome activation was proposed to mediate the beneficial effect of PUFAs [53].
10.6.2 4-Phenyl Butyric Acid
The chemical chaperone 4-phenyl butyric acid (4-PBA) has been shown to exhibit promising therapeutic effects in a variety of disease models, including metabolic syndrome, inflammatory diseases and cancer. Nissar and colleagues reported that 4-PBA could rectify the accumulation of p62/SQSTM1 and reduce lipid accumulation and apoptosis caused by palmitate in Huh7 hepatoma cells. Atg7 knockdown or pharmacological inhibition of autophagy with 3-methyladenine and chloroquine attenuated the lipid lowering effect of 4-PBA. These findings suggest that 4-PBA could reduce hepatocellular lipid accumulation and lipotoxicity through induction of autophagy [54].
10.6.3 Peretinoin
Peretinoin is an orally available, acyclic retinoid with potential antineoplastic and chemopreventive activities, presumably through activation of nuclear retinoic acid receptors (RAR). In two NASH-HCC mouse models, peretinoin has been shown to significantly improve liver histology and reduce the incidence of liver tumors. Peretinoin increased co-localized expression of LC3B-II and LAMP2, and increased autophagosome formation and autophagy flux in the liver through activating the promoter of Atg16L1, whose expression was reduced in the liver of patients with NASH. Atg16L1 overexpression was found to inhibit palmitate-induced NF-kB activation and IL-6-induced STAT3 activation by inducing the de-phosphorylation of Gp130, a receptor subunit of IL-6 family cytokines [55]. These findings suggest that peretinoin can prevent the development of NASH-HCC through activating autophagy by increasing Atg16L1 expression.
10.6.4 Carbon Monoxide
Carbon monoxide (CO), a reaction product of heme oxygenase activity, has been shown to protect against hepatic steatosis in mice. Subsequent mechanistic investigation demonstrated that carbon monoxide activated the PERK-eIF2α-ATF4 pathway to induce sestrin-2, which contributed to autophagy induction through activation of AMPK and inhibition of mTOR complex 1 [56].
10.6.5 Ginsenoside Rb2
Panax ginseng, a traditional Chinese medicine, has been widely used to treat a variety of metabolic diseases including hyperglycemia, hyperlipidemia, and hepatosteatosis. However, the active ingredient and molecular mechanisms underlying such effects remain largely unknown. Huang and colleagues found that ginsenoside Rb2, a major ginsenoside in Panax ginseng, can restore autophagy and prevent hepatic lipid accumulation in vivo and in vitro via induction of Sirt1 and activation of AMPK [57].
10.6.6 Thyroid Hormones
Iodothyronines are potential pharmacological compounds to treat NAFLD. Two iodothyronines, T2 and T3, both have shown efficacy in reducing the severity of NAFLD in cultured hepatocytes and animal models of NAFLD. Using a targeted metabolomics approach, Iannucci and colleagues found that both T2 and T3 could strongly induce hepatic autophagy and decrease hepatic fat content. However, only T2 was able to rescue the impairment in AKT and MAPK/ERK pathways caused by short-term high-fat diet [58], indicating their differential effects.
10.6.7 Nicotinamide
Nicotinamide, the amide form of nicotinic acid (vitamin B3), could upregulate Sirt1 via the cAMP/PKA/CREB pathway to induce autophagy hepatocytes and thereby attenuating palmitate-induced ER stress and cytotoxicity [59]. These findings suggest that nicotinamide supplementation may represent a therapeutic choice for NAFLD.
10.6.8 Pectic Bee Pollen Polysaccharide
Bee pollen has been used as a nutraceutical against diabetes and obesity. Using high glucose and fatty acid-treated hepatocytes and high fat diet-fed mice, Li and colleagues found that pectic bee pollen polysaccharide from Rosa rugosa could alleviates hepatic steatosis and insulin resistance by promoting autophagy via an AMPK/mTOR-mediated signaling pathway [60], suggesting that this natural compound could be a novel therapeutic agent used for NAFLD.
10.6.9 Caffeine
Caffeine, a psychoactive component in coffee, tea and cola, is the world’s most widely consumed drug. Through genetic, pharmacological, and metabolomic approaches, Sinha and colleagues demonstrated that caffeine could reduce intrahepatic lipid content and stimulate β-oxidation in hepatocytes via concomitantly increasing autophagy and lipid uptake in lysosomes. This beneficial effect was probably mediated through inhibition of mTOR signaling and paralleled with alterations in hepatic amino acids and sphingolipid levels [61].
10.6.10 Epigallocatechin Gallate
Epigallocatechin gallate (EGCG) is a major polyphenol in green tea with anti-inflammatory, anti-cancer, and anti-steatotic properties. EGCG has been shown to reduce hepatosteatosis and concomitantly increase autophagy in mice fed with high-fat diet. In this connection, EGCG increased phosphorylation of AMPK, whose knockdown abrogated autophagy induced by EGCG [62]. These findings suggest that AMPK-dependent induction of hepatic autophagy by EGCG might contribute to its beneficial effects in hepatosteatosis.
10.7 Concluding Remarks and Future Perspectives
NASH has become a dominant cause of HCC and its incidence is on the rise. Autophagy, a self-cannibalistic process, is a major pathway for lipid catabolism. Optimal and timely activation of autophagy also protects hepatocytes from injury and cell death as well as suppresses inflammation. In NAFLD, lipid accumulation, hyper-insulinemia, ER stress and deregulated cytokine expression have been shown to contribute to hepatic autophagy deficiency. Unfortunately, autophagic impairment further promotes these metabolic and molecular abnormalities, thereby creating a detrimental vicious circle. Defective autophagy is causally linked to NAFLD-related HCC, probably through accumulation of p62/SQSTM1, which induces and sustains the oncogenic NF-κB activity, and retention of damaged mitochondria, which produce reactive oxidative species to damage DNA. Nevertheless, it is noteworthy that autophagy could be subverted by HCC cells for opposing tumor suppression or as a pro-survival mechanism in response to therapies in the later stages of cancer development.
Pertinent to clinical practice, several pharmacological agents have been identified for their capacity to restore autophagic flux in NAFLD. These agents might also be promising prophylactics for preventing NALFD-related HCC if hepatocarcinogenesis has not yet been initiated. Nevertheless, the clinical utilization of these agents still awaits further validation in large-cohort human studies. Aside from therapy, recent discovery of circulating p62/SQSTM1 as serological marker [31] may open up a novel avenue for the use of autophagic markers for NAFLD diagnosis.
References
Yu J, Shen J, Sun TT, Zhang X, Wong N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin Cancer Biol. 2013;23(6 Pt B):483–91.
Tian Y, Wong VW, Chan HL, Cheng AS. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin Cancer Biol. 2013;23(6 Pt B):471–82.
Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FK, et al. The autophagic paradox in cancer therapy. Oncogene. 2012;31(8):939–53.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009;2(84):pe51.
Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–62.
Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–5.
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–5.
Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–78.
Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15(6):647–58.
Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284(45):31484–92.
Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58(5):993–9.
Wang Y, Singh R, Xiang Y, Czaja MJ. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology. 2010;52(1):266–77.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–52.
Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1):3–5.
Lim SC, Jeon HJ, Kee KH, Lee MJ, Hong R, Han SI. Involvement of DR4/JNK pathway-mediated autophagy in acquired TRAIL resistance in HepG2 cells. Int J Oncol. 2016;49(5):1983–90.
Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179.
Jones SA, Mills KH, Harris J. Autophagy and inflammatory diseases. Immunol Cell Biol. 2013;91(3):250–8.
Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–5.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30.
Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(1):105–20.
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999;18(11):3044–53.
Lee HM, Shin DM, Yuk JM, Shi G, Choi DK, Lee SH, et al. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J Immunol. 2011;186(2):1248–58.
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–8.
Pan H, Zhang Y, Luo Z, Li P, Liu L, Wang C, et al. Autophagy mediates avian influenza H5N1 pseudotyped particle-induced lung inflammation through NF-kappaB and p38 MAPK signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2014;306(2):L183–95.
Guo L, Stripay JL, Zhang X, Collage RD, Hulver M, Carchman EH, et al. CaMKIalpha regulates AMP kinase-dependent, TORC-1-independent autophagy during lipopolysaccharide-induced acute lung neutrophilic inflammation. J Immunol. 2013;190(7):3620–8.
Luo MX, Wong SH, Chan MT, Yu L, Yu SS, Wu F, et al. Autophagy mediates HBx-induced nuclear factor-kappaB activation and release of IL-6, IL-8, and CXCL2 in hepatocytes. J Cell Physiol. 2015;230(10):2382–9.
Lavallard VJ, Gual P. Autophagy and non-alcoholic fatty liver disease. Biomed Res Int. 2014;2014:120179.
Wu WK, Sakamoto KM, Milani M, Aldana-Masankgay G, Fan D, Wu K, et al. Macroautophagy modulates cellular response to proteasome inhibitors in cancer therapy. Drug Resist Updat. 2010;13(3):87–92.
Wang X, Zhang X, Chu ESH, Chen X, Kang W, Wu F, et al. Defective lysosomal clearance of autophagosomes and its clinical implications in nonalcoholic steatohepatitis. FASEB J. 2018;32(1):37–51.
Fukuo Y, Yamashina S, Sonoue H, Arakawa A, Nakadera E, Aoyama T, et al. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol Res. 2014;44(9):1026–36.
Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol. 2012;18(8):727–35.
Guo R, Nair S, Zhang Y, Ren J. Adiponectin deficiency rescues high-fat diet-induced hepatic injury, apoptosis and autophagy loss despite persistent steatosis. Int J Obes. 2017;41(9):1403–12.
Zhang X, Wu WK, Xu W, Man K, Wang X, Han J, et al. C-X-C motif chemokine 10 impairs autophagy and autolysosome formation in non-alcoholic steatohepatitis. Theranostics. 2017;7(11):2822–36.
Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, et al. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005;54(9):2674–84.
Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87(1):99–109.
Li S, Dou X, Ning H, Song Q, Wei W, Zhang X, et al. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology. 2017;66(3):936–52.
Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24(8):3052–65.
Liu Y, Zhang W, Wu X, Gong J. Foxo3a-dependent Bim transcription protects mice from a high fat diet via inhibition of activation of the NLRP3 inflammasome by facilitating autophagy flux in Kupffer cells. Oncotarget. 2017;8(21):34258–67.
Qiu DM, Wang GL, Chen L, Xu YY, He S, Cao XL, et al. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer. 2014;14:327.
Zheng JF, Li LL, Lu J, Yan K, Guo WH, Zhang JX. XPD functions as a tumor suppressor and dysregulates autophagy in cultured HepG2 cells. Med Sci Monit. 2015;21:1562–8.
Zhang W, Hou J, Wang X, Jiang R, Yin Y, Ji J, et al. PTPRO-mediated autophagy prevents hepatosteatosis and tumorigenesis. Oncotarget. 2015;6(11):9420–33.
Inokuchi-Shimizu S, Park EJ, Roh YS, Yang L, Zhang B, Song J, et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest. 2014;124(8):3566–78.
Shu G, Xie B, Ren F, Liu DC, Zhou J, Li Q, et al. Restoration of klotho expression induces apoptosis and autophagy in hepatocellular carcinoma cells. Cell Oncol (Dordr). 2013;36(2):121–9.
Tian Y, Kuo CF, Sir D, Wang L, Govindarajan S, Petrovic LM, et al. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015;22(6):1025–34.
Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, et al. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology. 2014;59(2):505–17.
Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193(2):275–84.
Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32(6):699–712.
Li P, Du Q, Cao Z, Guo Z, Evankovich J, Yan W, et al. Interferon-gamma induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012;314(2):213–22.
Pan H, Wang Z, Jiang L, Sui X, You L, Shou J, et al. Autophagy inhibition sensitizes hepatocellular carcinoma to the multikinase inhibitor linifanib. Sci Rep. 2014;4:6683.
Chang Y, Yan W, He X, Zhang L, Li C, Huang H, et al. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology. 2012;143(1):177–87. e8
Shen L, Yang Y, Ou T, Key CC, Tong SH, Sequeira RC, et al. Dietary PUFAs attenuate NLRP3 inflammasome activation via enhancing macrophage autophagy. J Lipid Res. 2017;58(9):1808–21.
Nissar AU, Sharma L, Mudasir MA, Nazir LA, Umar SA, Sharma PR, et al. Chemical chaperone 4-phenyl butyric acid (4-PBA) reduces hepatocellular lipid accumulation and lipotoxicity through induction of autophagy. J Lipid Res. 2017;58(9):1855–68.
Okada H, Takabatake R, Honda M, Takegoshi K, Yamashita T, Nakamura M, et al. Peretinoin, an acyclic retinoid, suppresses steatohepatitis and tumorigenesis by activating autophagy in mice fed an atherogenic high-fat diet. Oncotarget. 2017;8(25):39978–93.
Kim HJ, Joe Y, Kim SK, Park SU, Park J, Chen Y, et al. Carbon monoxide protects against hepatic steatosis in mice by inducing sestrin-2 via the PERK-eIF2alpha-ATF4 pathway. Free Radic Biol Med. 2017;110:81–91.
Huang Q, Wang T, Yang L, Wang HY. Ginsenoside Rb2 alleviates hepatic lipid accumulation by restoring autophagy via induction of Sirt1 and activation of AMPK. Int J Mol Sci. 2017;18(5):1063.
Iannucci LF, Cioffi F, Senese R, Goglia F, Lanni A, Yen PM, et al. Metabolomic analysis shows differential hepatic effects of T2 and T3 in rats after short-term feeding with high fat diet. Sci Rep. 2017;7(1):2023.
Shen C, Dou X, Ma Y, Ma W, Li S, Song Z. Nicotinamide protects hepatocytes against palmitate-induced lipotoxicity via SIRT1-dependent autophagy induction. Nutr Res. 2017;40:40–7.
Li X, Gong H, Yang S, Yang L, Fan Y, Zhou Y. Pectic bee pollen polysaccharide from Rosa rugosa alleviates diet-induced hepatic steatosis and insulin resistance via induction of AMPK/mTOR-mediated autophagy. Molecules. 2017;22(5):699.
Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59(4):1366–80.
Zhou J, Farah BL, Sinha RA, Wu Y, Singh BK, Bay BH, et al. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS One. 2014;9(1):e87161.
Acknowledgements
The work was supported by Early Career Scheme (24115815) of the Research Grant Council Hong Kong; Shenzhen Science and Technology Programme (JCYJ20150630165236956, JCYC20140905151710921) of Shenzhen Science and Technology Innovation Commission; and Natural Science Foundation of Guangdong Province (2015A030313886) of Department of Science and Technology of Guangdong Province.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Wu, W.K.K., Zhang, L., Chan, M.T.V. (2018). Autophagy, NAFLD and NAFLD-Related HCC. In: Yu, J. (eds) Obesity, Fatty Liver and Liver Cancer. Advances in Experimental Medicine and Biology, vol 1061. Springer, Singapore. https://doi.org/10.1007/978-981-10-8684-7_10
Download citation
DOI: https://doi.org/10.1007/978-981-10-8684-7_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8683-0
Online ISBN: 978-981-10-8684-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)