
Abstract
Purpose
Klotho has been identified as a tumor suppressor in several human malignancies including hepatocellular carcinoma (HCC). However, the signaling pathways involved in the tumor suppressive role of klotho in HCC have not been reported. Here, we investigated the role of klotho in HCC cell proliferation, apoptosis, autophagy, and invasion, as well as its associated signal transduction pathways.
Methods
Restoration of klotho gene expression was established by delivering a klotho gene expression vector into the human HCC cell lines HepG2 and MHCC-97-H. Cell viability was measured using a cell counting (CCK-8) assay and apoptosis was analyzed through flow cytometry. Autophagy was measured via LC3-I and LC3-II protein expression levels and tumor cell invasion was assessed using a Matrigel invasion chamber assay. Expression and phosphorylation of several apoptosis and survival related proteins were assessed using Western blot assays.
Results
Exogenous klotho gene expression significantly inhibited HCC cell proliferation, induced HCC cell apoptosis, increased LC3-I and LC3-II protein expression in HCC cells, and decreased migration of HCC cells in a Matrigel invasion chamber assay. Exogenous klotho gene expression also down-regulated the phosphorylation levels of the IGF-1 receptor, and the downstream Akt, ERK, and p70S6K proteins. Both apoptosis and autophagy inhibitors decreased klotho-induced apoptosis and autophagy.
Conclusion
Klotho is a tumor suppressor that, through the regulation of IGF-1R phosphorylation and subsequent activation of downstream Akt-p70S6K and ERK signaling, regulates HCC tumor cell proliferation, apoptosis, autophagy and invasion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Primary liver cancer is the fifth most common cancer and the third most frequent cause of cancer-related death worldwide [1, 2]. Hepatocellular carcinoma (HCC) accounts for 4 % of the currently diagnosed cancers, 42 % of which occur in China [3]. The incidence of HCC is increasing in the western world, particularly in the United States, Canada and southern Europe [4]. Despite overall improvements in cancer therapy, the 5 year survival rate of HCC is still approximately 5 %. This is partially due to the fact that HCC is highly aggressive and that the key molecular mechanisms underlying HCC initiation and progression have not been uncovered yet. However, a variety of HCC related signaling pathways has been identified.
Klotho has been shown to function as a tumor suppressor, and our recent work has shown that klotho also acts as a tumor suppressor in HCC and that decreased klotho expression is involved in HCC progression [5]. Klotho has been observed to inhibit insulin/IGF-1 signaling, to induce cell apoptosis and, consequently, to inhibit tumor growth [6, 7]. IGF-1 exerts its roles through the activation of Raf / Mek / Erk (extracellular signal-regulated kinases) and PI3K (phosphoinositide 3-kinase) / Akt / mTOR signaling. A number of studies have revealed that insulin/IGF-1, Raf / Mek / Erk, and PI3K / Akt / mTOR signaling pathways are also involved in hepatocarcinogenesis [8–10]. Therefore, we hypothesized that klotho may inhibit IGF-1 signaling in HCC and, subsequently, induce apoptosis through down-regulation of the Raf/Mek/Erk and PI3K/Akt/mTOR signaling pathways.
Autophagy is a physiological process that eliminates toxic wastes or damaged cellular components in response to stress. As a form of type II programmed cell death, overactive autophagy may cause cell death [11, 12]. A number of studies have reported that autophagy is activated in cancer cells in response to various anticancer therapies, and that autophagy can be considered as a way to kill apoptosis-resistant tumor cells [3]. Autophagy can be activated through multiple signaling pathways. There is increasing evidence supporting the idea that tumor suppressor genes promote autophagy, while oncogenes inhibit autophagy [13]. We, therefore, hypothesized that klotho might also be responsible for regulating autophagy in HCC.
Here, we investigated the involvement of klotho in HCC cell proliferation, apoptosis, autophagy and invasion, as well as its associated signaling pathways, by exogenously expressing the klotho gene in two human HCC cell lines. Our study provides evidence that klotho regulates signaling pathways involved in HCC cell proliferation, apoptosis, autophagy and invasion.
2 Materials and methods
2.1 Cell culture
HepG2 and MHCC-97-H are human hepatocellular cancer-derived cell lines, and L-02 is a normal human liver cell line. HepG2 and MHCC-97-H were chosen for this study because they express low levels of klotho [5]. All cell lines were obtained from the Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai, China). Cells were cultured in either RPMI 1640 or DMEM with 10 % fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37 °C, 5 % CO2.
2.2 Construction of a klotho gene expression vector
The klotho gene was amplified from a cDNA library established from normal liver cells. The open read frame (ORF) klotho cDNA was amplified by a forward primer containing a Bgl II restriction site: ACTCAGATCTGAGCCGGGCGACGGCGCGCAGA and a reverse primer containing a Bam HI restriction site: CGGTGGATCCCCTATTTGTAACTTCTTCTGCC. The amplified klotho ORF was cloned into a pZsGreen1-C1 vector using the Bgl II/Bam HI sites (Clontech, Mountain View, USA). The klotho ORF was fused to the C-terminal end of GFP. A pZsGreen1-C1 vector without insertion was used as a negative control.
2.3 Klotho gene expression analysis by RT-PCR
Cultured cells were homogenized in Trizol reagent (Invitrogen, Carlsbad, CA) using a PowerGen homogenizer (Fisher Scientific, Pittsburgh, PA). Total RNA was isolated following the manufacturer’s instructions. Reverse transcription was performed using a RevertAid™ H Minus First Strand cDNA Synthesis Kit (Thermo Science, Waltham, MA, USA). The Klotho cDNA was amplified using forward primer: 5′-CACGGCAAGGGTGCGTCCAT-3′ and reverse primer: 5′-TCGCGCCCACGAGATGGAGA-′3. The control GAPDH cDNA was amplified using forward primer: 5′-CTCATGACCACAGTCCATGC-3′ and reverse primer: 5′-TTCAGCTCTGGGATGACCTT-3′. RT-PCR products were visualized on a 1.5 % agarose gel containing 0.5 μg/ml of ethidium bromide.
2.4 Cell viability assay
Cell Counting Kit-8 (CCK-8) (Beyotime, Shanghai, China) allows a sensitive colorimetric assessment of cell viability. Briefly, HepG2 and MHCC-97-H cells were seeded into 96-well plates at 1 × 104 cells per well 24 h before transfection. Cells were transfected with a klotho expression vector, an empty vector, or no vector (PBS) using lipofectamine 2,000 according to the user manual (Invitrogen, Grand Island, NY, USA). Cells were then cultured in Opti-MEM (Invitrogen) for 6 h and subsequently cultured in normal growth medium for 72 h. Finally, 10 μl of kit reagent was added to the cells followed by incubation for 1 h. Cell viability was assessed using a microplate reader at 450 nm. All results were normalized to OD values measured from identically conditioned wells with plain medium. Cell proliferation rate (%) = (OD value in treated cells—OD value in background control wells) / (OD value of control cells—OD value in background control wells) × 100 %.
2.5 Cell proliferation and apoptosis assay
HepG2 and MHCC-97-H cells were seeded in 10 cm culture dishes at a density of 2 × 106 cells per dish. After reaching 60 % confluency, cells were transfected with a klotho expression vector, an empty vector, or PBS as described above. Cells were then trypsinized and suspended in 500 μl binding buffer containing 5 μl of Annexin V-FITC and 5 μl Propidium Iodide (Abcam, Cambridge, MA, USA). After incubation in the dark for 1 h, cells were subjected to flow cytometry.
2.6 Treatment with autophagy and apoptosis inhibitors
HepG2 and MHCC-97-H cells at 70 % confluency were transfected with a klotho expression vector, an empty vector, or PBS as described above. Cells transfected with a klotho expression vector or PBS were incubated with 10 mM 3-methyladenine (3-MA) or 20 μM apoptosis inhibitor Z-VAD-FMK for 24 h. Cells were then harvested for Western blot and/or flow cytometry analysis.
2.7 Matrigel invasion assay
The invasive ability of tumor cells was assessed using a Matrigel invasion chamber (BD Biosciences, San Jose, CA) according to the instructions provided by the manufacturer. HepG2 and MHCC-97-H cells (5 × 104/750 μl) were loaded on the transwell chamber (8 μm pore) covered with Matrigel. 10 ng/ml of EGF was placed in the lower chamber. The number of cells that migrated to the lower surface during a 24 h-incubation period was determined by counting the cells in 5 randomly chosen fields [14]. In this assay system, the pore filter was covered with a thin layer of Matrigel. Therefore, the cells have to first digest the Matrigel before they can reach the lower chamber.
2.8 Western blot analysis
Tumor cells were homogenized and Western blotting was performed as previously described [15]. The anti-klotho, anti-Akt, anti-phospho-Akt1, anti-phospho-p70S6K, anti-phospho-IGF-IR, anti-GAPDH, and HRP-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-LC3-I antibody was purchased from Epitomics (Burlingame, CA, USA). The anti-LC3-II, anti-ERK1/2, anti-phospho-ERK, and anti-p70S6K antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Protein concentrations were measured using a BCA Protein Assay kit (Beyotime, Shanghai, China). Twenty μg of total protein was loaded onto a 12 % SDS-PAGE gel and transferred to nitrocellulose membranes. After blocking with 5 % non-fat milk for 1 h, membranes were incubated with a primary antibody overnight at 4 °C and, subsequently, incubated with a HRP-labeled secondary antibody (1:2,000 dilution) for 2 h at room temperature. Reactive proteins were detected using chemiluminescent reagents (Pierce, Rockford, IL, USA). To control for loading efficiency, the blots were stripped and re-probed with an anti-GAPDH antibody. The images were scanned using Adobe photoshop (Adobe, San Jose, CA) and quantified using NIH Image J (see http://rsb.info.nih.gov). All protein expression levels were evaluated relative to α-tubulin.
2.9 Statistical analysis
Data were analyzed using SPSS 13.0 (statistical package for the Social Sciences Version 13.0). Two samples were compared using the χ2 independence test. A p < 0.05 was considered statistically significant.
3 Results
3.1 Klotho expression in normal liver and HCC cells
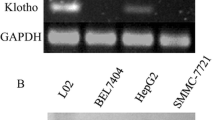
Klotho expression in normal human liver cells and hepatocellular cancer cells was detected using Western blot analyses. As shown in Fig. 1a & b, klotho expression in HepG2 and MHCC-97-H tumor cells was clearly lower than that in L-02 normal cells (p < 0.001). The klotho expression vector (see materials and methods) was effectively transfected in about 70 % of HepG2 and MHCC-97-H cells (Fig. 1c, indicated by GFP expression). Subsequent Western blot analysis showed that the transfection resulted in over-expression of klotho in both HepG2 (Fig. 1d, e) and MHCC-97-H cells (Fig. 1f, g).

Klotho expression in normal liver and tumor cells. a RT-PCR detection of klotho expression in L-02 normal liver cells, and HepG2 and MHCC-97-H HCC cells. b Quantification of klotho mRNA levels in (a). * p < 0.001 vs. L-02 cells. N = 4. c Klotho expression in HepG2 and MHCC-97-H cells transfected with a klotho expression vector. d Western blot analysis of klotho protein expression in HepG2 cells transfected with a klotho expression vector (klotho-V), an empty vector (blank-V), and PBS (non-V). e Quantification of klotho protein levels in (d). * p < 0.001 vs. non-V control. N = 4. f Western blot analysis of klotho protein expression in MHCC-97-H cells. e Quantification of klotho protein levels in f). * p < 0.001 vs. non-V control. N = 4
3.2 Restoration of klotho expression decreases tumor cell proliferation and increases tumor cell apoptosis
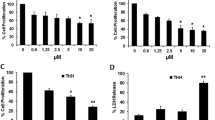
The CCK-8 cell count assay showed that exogenous over-expression of the klotho gene significantly decreased cell viability in both HepG2 (Fig. 2a) and MHCC-97-H (Fig. 2b) cells (p < 0.001). The flow cytometry assay revealed that exogenous over-expression of the klotho gene significantly induced cell apoptosis in both HepG2 (Fig. 2c) and MHCC-97-H (Fig. 2d) cells (p < 0.001). Also, over-expression of the klotho gene significantly increased the amount of G0 cells in both HepG2 (Fig. 2e) and MHCC-97-H cells (Fig. 2f) (p < 0.001), implying cell cycle arrest.

Klotho inhibits cell proliferation and induces apoptosis. a Relative cell viability in HepG2 cells transfected with a klotho expression vector (klotho-V), an empty vector (blank-V), and PBS (non-V). * p < 0.001 vs. non-V. N = 5. b Relative cell viability of MHCC-97-H cells. * p < 0.001 vs. non-V. N = 5. c Flow cytometry of apoptotic HepG2 cells. d Flow cytometry of apoptotic MHCC-97-H cells. e Cell cycle assessment of HepG2 cells. f Cell cycle assessment of MHCC-97-H cells
3.3 Restoration of klotho gene expression induces authophagy and apoptosis
LC3-I and LC3-II are markers for autophagy. Transfection of a klotho expression vector significantly increased LC3-I and LC3-II expression in both HepG2 (Fig. 3a, b, c, p < 0.001) and MHCC-97-H cells (Fig. 3d, e, f, p < 0.001). In HepG2 and MHCC-97-H cells transfected with a klotho expression vector, the autophagy inhibitor 3-MA significantly normalized LC3-I and LC3-II expression levels (p < 0.01). We further tested the effect of an apoptosis inhibitor and found that Z-VAD-FMK significantly decreased klotho-induced LC3-I and LC3-II expression in both HepG2 (p < 0.001) and MHCC-97-H cells (p < 0.001). Next we investigated the role of autophagy and apoptosis inhibitors on apoptosis in HepG2 (Fig. 3g) and MHCC-97-H (Fig. 3h) cells. The flow cytometry assay showed that both autophagy and apoptosis inhibitors significantly inhibited klotho-induced cell apoptosis (p < 0.001).

The effects of apoptosis and autophagy inhibitors. a Western blot analysis of LC3-I and LC3-II expression in HepG2 cells. Cells were transfected with a klotho expression vector (klotho-V), an empty vector (blank-V), and PBS (non-V). LC3-I and LC3-II expression were also measured in HepG2 cells transfected with a klotho expression vector (K-V) and incubated with an autophagy inhibitor (k-V + 3-MA) or an apoptosis inhibitor (k-V + ZVF). ZVF: Z-VAD-FMK. b Quantification of LC3-I levels in HepG2 cells in (a). * p < 0.001 vs. non-V control. # p < 0.01 vs. klotho-V, ∆ p < 0.001 vs. klotho-V. N = 4. c Quantification of LC3-II levels in HepG2 cells in (a). * p < 0.001 vs. non-V control. # p < 0.01 vs. klotho-V, ∆ p < 0.001 vs. klotho-V. N = 4. d Western blot analysis of LC3-I and LC3-II expression in MHCC-97-H cells. e Quantification of LC3-I levels in MHCC-97-H cells in (d). * p < 0.001 vs. non-V control. # p < 0.01 vs. klotho-V, ∆ p < 0.001 vs. klotho-V. N = 4. f Quantification of LC3-II levels in MHCC-97-H cells in (d). * p < 0.001 vs. non-V control. # p < 0.01 vs. klotho-V, ∆ p < 0.001 vs. klotho-V. N = 4. g Percentage of apoptotic cells in HepG2 cell with different treatments. ** p < 0.001 vs. blank-V; * p < 0.01 vs. klotko-V; # p < 0.01 vs. klotho-V. N = 5. h Percentage of apoptotic cells in MHCC-97-H cells. ** p < 0.001 vs. blank-V; * p < 0.05 vs. klotko-V; # p < 0.01 vs. klotho-V. N = 5
3.4 Restoration of klotho expression increases tumor cell invasion
The invasive capacity of tumor cells was assessed using a Matrigel invasion chamber. The number of cells that migrated to the lower chamber was calculated. Transfection of a klotho expression vector significantly decreased the migration of HepG2 and MHCC-97-H cells to the lower chamber (p < 0.001), suggesting that the klotho gene can inhibit hepatocellular cancer cell invasion (Fig. 4).

Klotho increases tumor cell migration. Tumor cell migration was assessed using a Matrigel invasion chamber. The cells that migrated to the lower chamber are shown. Transfection of a klotho expression vector significantly decreased the migration of HepG2 and MHCC-97-H cells to the lower chamber (p < 0.001
3.5 Cellular signaling involved in the inhibitory role of klotho
Next we investigated the expression of signaling molecules involved in cell proliferation and survival. In both HepG2 and MHCC-97-H cells, exogenous over-expression of the klotho gene significantly inhibited phosphorylation of several proliferation- and survival-related molecules. The levels of phospho-IGF-1R, phospho-Akt, phospho-ERK, and phospho-p70S6K were significantly lower in HepG2 (Fig. 5a, c) and MHCC-97-H (Fig. 5b, d) cells transfected with a klotho expression vector than in cells transfected with an empty vector or PBS (p < 0.001). No differences were observed in protein levels of total ERK, Akt, and p70S6K between the 3 treatment groups.

Klotho inhibits phosphorylation of IGF-1, Akt, p70S6K and ERK proteins. a Western blot analysis of protein and phosphorylation levels in HepG2 cells. b Western blot analysis of protein and phosphorylation levels in MHCC-97-H cells. c Quantification of protein levels in HepG2 cells in a). * p < 0.001 vs. non-V control. N = 4. d Quantification of protein levels in MHCC-97-H cells in b). * p < 0.001 vs. non-V control. N = 4. P-IGF-1R phosphor-IGF-1 receptor; p-Akt phosphor-Akt; p-ERK phosphor-ERK; p-p70S6K phospho-p70S6K; klotho-V klotho expression vector; blank-V empty vector; non-V: PBS transfection
4 Discussion
Klotho has been shown to act as a tumor suppressor in several malignancies, including HCC. Our recent work has shown that absence of klotho gene expression is associated with progression of HCC. However, the signaling pathways associated with klotho’s mode of action in HCC survival and growth have thus far remained unknown. Here, the roles played by klotho in cell proliferation, apoptosis and autophagy were investigated through the delivery of a klotho gene expression vector into two HCC cell lines (HepG2 and MHCC-97-H) and one normal liver cell line (L-02). Our results show that restoration of klotho gene expression in HCC cells inhibits tumor cell proliferation, induces apoptosis and authophagy, and reduces tumor cell invasion. In addition, we found that klotho exerts a tumor suppressive role through down-regulation of IGF-1R, Akt, ERK and p70S6K phosphorylation.
These results are similar to those previously reported in lung and colorectal cancers [13, 16]. The signaling pathways involved in the tumor suppressive role of klotho have, as yet, not been fully elucidated. Klotho has been found to inhibit insulin/IGF-1 signaling and, consequently, to induce cell apoptosis and to inhibit tumor growth [17–19]. Klotho regulates insulin/IGF-1 signaling by inhibiting tyrosine phosphorylation of the IGF-1 receptor [20]. Tyrosine phosphorylation of the IGF-1R is also involved in the activation of extracellular signal-regulated kinases (ERK) 1 and ERK2 and PI3K/Akt signaling [21]. In this study, we revealed that restoration of klotho gene expression significantly inhibits IGF-1R, Akt, and ERK phosphorylation in HCC cells. The PI3K-Akt pathway plays a pivotal role in the regulation of apoptosis in many cell types, and the ras-raf-MEK-ERK route has also been proposed to function in cell survival. We, therefore, propose that through inhibiting IGF-1 receptor phosphorylation, klotho inhibits ERK and Akt activities and induces apoptosis in HCC cells. p70S6K is a downstream effector of the PI3K/Akt/mTOR signal transduction pathway. It has been found that inhibition of p70S6K phosphorylation induces cell apoptosis and cell cycle arrest in G1 and G2 in B-cell lymphoma cells [22].
Non-apoptotic cell death is mainly attributed to autophagy, which is considered a viable alternative to kill apoptosis-resistant tumor cells [3]. Autophagy is initiated by the formation of an autophagosome or autophagic vacuole, which then fuses with the lysosomal membrane to deliver its content into the autolysosome for degradation [23]. During autophagosome formation, the microtubule-associated protein light chain 3 I (LC3-I) is conjugated to phosphatidylamine to form LC3-phosphatidylamine, termed LC3-II. Translocation of LC3-II to the autophagosome membrane is an essential step in the formation of the autophagosome [23, 24]. Therefore, LC3-I and LC3-II levels can be used as markers to reflect the activation of autophagy. Here, we detected low LC3-I and LC3-II expression levels in HCC cells and that, after restoration of klotho expression, LC3-I and LC3-II levels significantly increased. Furthermore, the autophagy inhibitor 3-MA inhibited LC3-I and LC3-II expression in cells transfected with a klotho expression vector, suggesting that klotho is an inducer of autophagy in HCC. Thus, klotho is a tumor suppressor capable of inhibiting both apoptosis and autophagy in HCC. However, how klotho inhibits apoptosis and autophagy in tumor cells has so far not been elucidated.
Recent studies have demonstrated that autophagy can be induced by reduced growth factor signaling. Growth factor signaling involved in the insulin/IGF-1-PI3K-Akt- mTORC1 pathway has been shown to regulate cell autophagy through the insulin receptor [25]. In addition, activation of insulin/IGF signaling has been shown to suppress the autophagic-lysosomal pathway [26, 27]. Importantly, the klotho protein functions as a circulating hormone that represses intracellular signals of insulin and IGF-I [20, 28]. In this study, exogenous over-expression of the klotho gene significantly inhibited IGF-1R, Akt, and p70S6K phosphorylation. This suggests that the signaling pathway of klotho-IGF-1R/Akt may be responsible for both apoptosis and autophagy. Unexpectedly, autophagy inhibitors significantly reversed klotho-induced apoptosis, while apoptosis inhibitors significantly inhibited autophagy in HCC cells. This surprising observation suggests that apoptosis and autophagy act through similar pathways. However, the inhibition of apoptosis by an inhibitor to autophagy, and autophagy by an inhibitor to apoptosis, was not 100 % effective, suggesting that apoptosis and autophagy caused by over-expression of klotho may, at least partially, employ different cell death pathways.
In this study, klotho was identified as a tumor suppressor that inhibits tumor cell proliferation, induces cell apoptosis and autophagy as well as inhibits invasion of HCC cells. These tumor suppressive roles of klotho may be associated with a down-regulation of IGF-1 receptor phosphorylation, and a subsequent decreases in Akt/p70S6K and ERK phosphorylation. Our study highlights a central role for klotho in HCC cell survival and suggests that klotho may serve as an ideal target for the development of novel therapeutic agents for HCC.
References
F.X. Bosch, J. Ribes, M. Díaz, R. Cléries, Primary liver cancer: worldwide incidence and trends. Gastroenterology 127, S5–S16 (2004)
M.A. Avila, C. Berasain, B. Sangro, J. Prieto, New therapies for hepatocellular carcinoma. Oncogene 25, 3866–3884 (2006)
H.M. Lin, H.C. Tseng, C.J. Wang, C.C. Chyau, K.K. Liao, P.L. Peng, F.P. Chou, Induction of autophagy and apoptosis by the extract of Solanum nigrum Linn in HepG2 cells. J. Agric. Food Chem. 55, 3620–3628 (2007)
A. Braillon, Hepatocellular carcinoma and evidence-based surgery. World J. Gastroenterol. 15, 5371 (2009)
B. Xie, J. Zhou, L. Yuan, F. Ren, D. Liu, Q. Li, G. Shu, Epigenetic silencing of Klotho expression correlates with poor prognosis of human hepatocellular carcinoma. Human Pathology, in press (2012)
D.E. Arking, D.M. Becker, L.R. Yanek, D. Fallin, D.P. Judge, T.F. Moy, L.C. Becker, H.C. Dietz, KLOTHO allele status and the risk of early-onset occult coronary artery disease. Am. J. Hum. Genet. 72, 1154–1161 (2003)
C.D. Chen, S. Podvin, E. Gillespie, S.E. Leeman, C.R. Abraham, Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. U. S. A. 104, 19796–197801 (2007)
Y. Ohyama, M. Kurabayashi, H. Masuda, T. Nakamura, Y. Aihara, T. Kaname, T. Suga, M. Arai, H. Aizawa, Y. Matsumura, M. Kuro-o, Y. Nabeshima, R. Nagail, Molecular cloning of rat klotho cDNA: markedly decreased expression of klotho by acute inflammatory stress. Biochem. Biophys. Res. Commun. 251, 920–925 (1998)
F. Sahin, R. Kannangai, O. Adegbola, J. Wang, G. Su, M. Torbenson, mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res. 10, 8421–8425 (2004)
E. Schiffer, C. Housset, W. Cacheux, D. Wendum, C. Desbois-Mouthon, C. Rey, F. Clergue, R. Poupon, V. Barbu, O. Rosmorduc, Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 41, 307–314 (2005)
Y. Cheng, F. Qiu, Y.C. Ye, Z.M. Guo, S. Tashiro, S. Onodera, T. Ikejima, Autophagy inhibits reactive oxygen species-mediated apoptosis via activating p38-nuclear factor-kappa B survival pathways in oridonin-treated murine fibrosarcoma L929 cells. FEBS J. 276, 1291–1306 (2009)
B. Levine, J. Yuan, Autophagy in cell death: an innocent convict? J. Clin. Invest. 115, 2679–2688 (2005)
X. Wang, B. Chen, W. Xu, S. Liu, W. Zhao, J. Wu, Combined effects of klotho and soluble CD40 ligand on A549 lung cancer cells. Oncol. Rep. 25, 1465–1472 (2011)
M. Komachi, H. Tomura, E. Malchinkhuu, M. Tobo, C. Mogi, T. Yamada, T. Kimura, A. Kuwabara, H. Ohta, D.S. Im, H. Kurose, I. Takeyoshi, K. Sato, F. Okajima, LPA1 receptors mediate stimulation, whereas LPA2 receptors mediate inhibition, of migration of pancreatic cancer cells in response to lysophosphatidic acid and malignant ascites. Carcinogenesis 30, 457–465 (2009)
X. Zhang, T.H. Lee, C. Davidson, C. Lazarus, W.C. Wetsel, E.H. Ellinwood, Reversal of cocaine-induced behavioral sensitization and associated phosphorylation of the NR2B and GluR1 subunits of the NMDA and AMPA receptors. Neuropsychopharmacology 32, 377–387 (2007)
J. Pan, J. Zhong, L.H. Gan, S.J. Chen, H.C. Jin, X. Wang, L.J. Wang, Klotho, an anti-senescence related gene, is frequently inactivated through promoter hypermethylation in colorectal cancer. Tumour Biol. 32, 729–735 (2011)
L. Lu, D. Katsaros, A. Wiley, I.A. de la Longrais, M. Puopolo, H. Yu, Klotho expression in epithelial ovarian cancer and its association with insulin-like growth factors and disease progression. Canc. Investig. 26, 185–192 (2008)
I. Wolf, S. Levanon-Cohen, S. Bose, H. Ligumsky, B. Sredni, H. Kanety, M. Kuro-o, B. Karlan, B. Kaufman, H.P. Koeffler, T. Rubinek, Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 27, 7094–70105 (2008)
L. Abramovitz, T. Rubinek, H. Ligumsky, S. Bose, I. Barshack, C. Avivi, B. Kaufman, I. Wolf, KL1 internal repeat mediates klotho tumor suppressor activities and inhibits bFGF and IGF-I signaling in pancreatic cancer. Clin. Cancer Res. 17, 4254–4266 (2011)
H. Kurosu, M. Yamamoto, J.D. Clark, J.V. Pastor, A. Nandi, P. Gurnani, O.P. McGuinness, H. Chikuda, M. Yamaguchi, H. Kawaguchi, I. Shimomura, Y. Takayama, J. Herz, C.R. Kahn, K.P. Rosenblatt, M. Kuro-o, Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833 (2005)
R. Kooijman, Regulation of apoptosis by insulin-like growth factor (IGF)-I. Cytokine Growth Factor Rev. 17, 305–323 (2006)
X.F. Zhao, R.B. Gartenhaus, Phospho-p70S6K and cdc2/cdk1 as therapeutic targets for diffuse large B-cell lymphoma. Expert Opin. Ther. Targets. 13, 1085–1093 (2009)
M. Ogata, S. Hino, A. Saito, K. Morikawa, S. Kondo, S. Kanemoto, T. Murakami, M. Taniguchi, I. Tanii, K. Yoshinaga, S. Shiosaka, J.A. Hammarback, F. Urano, K. Imaizumi, Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231 (2006)
H. Chaachouay, P. Ohneseit, M. Toulany, R. Kehlbach, G. Multhoff, H.P. Rodemann, Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother. Oncol. 99, 287–292 (2011)
J.J. Lum, D.E. Bauer, M. Kong, M.H. Harris, C. Li, T. Lindsten, C.B. Thompson, Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248 (2005)
S. Arico, A. Petiot, C. Bauvy, P.F. Dubbelhuis, A.J. Meijer, P. Codogno, E. Ogier-Denis, The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 276, 35243–32546 (2001)
M. Degtyarev, A. De Mazière, C. Orr, J. Lin, B.B. Lee, J.Y. Tien, W.W. Prior, S. van Dijk, H. Wu, D.C. Gray, D.P. Davis, H.M. Stern, L.J. Murray, K.P. Hoeflich, J. Klumperman, L.S. Friedman, K. Lin, Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 183, 101–116 (2008)
C. Mammucari, G. Milan, V. Romanello, E. Masiero, R. Rudolf, P. Del Piccolo, S.J. Burden, R. Di Lisi, C. Sandri, J. Zhao, A.L. Goldberg, S. Schiaffino, M. Sandri, FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471 (2007)
Conflict of interests
All author declared no any conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shu, G., Xie, B., Ren, F. et al. Restoration of klotho expression induces apoptosis and autophagy in hepatocellular carcinoma cells. Cell Oncol. 36, 121–129 (2013). https://doi.org/10.1007/s13402-012-0118-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-012-0118-0