
Abstract
Natural products have had an important and decisive role in the development of modern medicinal chemistry and drug design. The development of new bioassay techniques, biotech methods, bio-guided phytochemical studies, automated high-throughput screening, and high performance analytical methods has introduced both new concepts and possibilities for rational drug design and drug discovery. With the development of new spectroscopic techniques, organic chemists have been able to elucidate the complex molecular structures of natural constituents quickly. Secondary metabolites, namely, alkaloids, flavonoids, and terpenoids as anticancer molecules, involving various strategies of treatment, have been discussed with special reference to topoisomerases (Topo), cyclooxygenases (COX), lipoxygenase (LOX), and aromatase as enzymatic targets for various types of cancers. In silico methods or CADD (computer-aided drug design) studies are increasingly being used in both industries and universities. They involve an understanding of the molecular interactions from both qualitative and quantitative points of view. These methods generate and manipulate three-dimensional (3D) molecular structures, calculate descriptors and the dependent molecular properties, model constructions, and employ other tools that encompass computational drug research. Analysis of the molecular structure of a given system allows relevant information to be extracted and to predict the potential of bioactive compounds. Furthermore, in view of the recent advances made in the field of computer-aided drug design, the aim of present chapter is to discuss the use of computational approaches such as ADMET, molecular docking, molecular dynamics simulation, and QSAR to asses and predict the safety, efficacy, potency, and identification of natural potent anticancer molecules.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
9.1 Introduction
The kingdom plantae contributed significantly to the discovery of useful substances that may treat various human diseases. The development of organic chemistry in the nineteenth century occurred in parallel with the study of plants. The plants are the source of a large number of natural products having an undoubtedly unique development and occurrence of complex metabolites (Cragg and Newman 2014). The natural products often resulted from an optimized evolutionary process in which chemicals have been under the selective forces of coevolution, organisms producing substances in the presence of their predators. These products have been utilized by humans since ancient times to treat and cure their diseases. Traditional Chinese medicine is the best example of natural product efficiency, especially in the discovery of new active chemical entities. These compounds are present in fruits and vegetables and are important components of our daily diets. Of the existing plants in the world, most of which are unknown from a scientific point of view, only about 5% of the approximately 250–500 thousand species have been biologically studied and evaluated. Natural products are often phenolic compounds, flavonoids, alkaloids, or terpenes, secondary plant metabolites that may provide several benefits to our health. These benefits include cosmetic action, cardioprotective effects, anti-inflammatory activity, and usefulness in the treatment of cancer and the neglected diseases (Chan et al. 2013; Karioti et al. 2015). The use of natural products has been the single most successful strategy for discovering new medicines, and many medical breakthroughs are based on natural products. Half of the top 20 best-selling drugs are natural chemical compounds, and their total sales amount to US$ 16 billion yearly. These numbers suggest that natural chemicals may well be considered pre-optimized for bioactive potential and therefore possess “drug-like properties” (Kennedy et al. 2009; Wang et al. 2011; Clement 2014). New molecules are continually being reported in the literature, many with relevant pharmacological activity, such as Taxol, forskolin, artemisinin, etc. (Fig. 9.1). It is important to remember that plants have contributed over the years to obtaining various widely used drugs, such as morphine, emetine, vincristine, colchicine, rutin, etc. (Fig. 9.1). In the 1980s, consumers in the USA paid more than 8 billion dollars for prescriptions with active natural products, and about 80% of all people use natural compounds in the treatment of their diseases (Mills et al. 2005; Mishra et al. 2008; Moran et al. 2009).

Structure of some natural products
Several therapeutically active metabolites were isolated from marine organisms, which could be used as effective modulators of biological targets such as phospholipases, adenosine receptors in tumor models (e.g., manoalide, lufarolide, and azidovudine (Fig. 9.2)) inspired by the chemical structure of a substance of marine origin which proved to be active on HIV reverse transcriptase, agent responsible for acquired immunodeficiency syndrome (AIDS), and prostaglandin A2 (Fig. 9.2) (Scotti et al. 2012a; Anjos et al. 2012; Souza et al. 2012; de Araújo et al. 2013; Hatae et al. 2015). New chemical bioactive entity studies done by industrial research laboratories have adopted techniques such as combinatorial chemistry to obtain more compounds. Through this technology, reactions are done in several steps, occurring in parallel or in mixtures, with few reagents. Products are reagent combinations, and therefore large numbers of new compounds can be generated. Thus, the aim of the present chapter is to discuss the use of computational approaches such as ADMET, molecular docking, molecular dynamics simulation, and QSAR to asses and predict the safety, efficacy, potency, and identification of natural potent anticancer molecules.

Structure of some marine natural products
9.2 Medicinal Chemistry and Its Significance
It is dedicated to understanding the molecular mechanism, chemical relationships, and pharmacological activity involved in drug action thru pharmacodynamic and pharmacokinetic factors. The introduction of new technologies has become a prime concept in medicinal chemistry expanding its interdisciplinary character. Most drugs are small bioactive molecules that interact with specific macromolecules or receptors, resulting in their therapeutic effects. Modern computational methods can determine the different qualitative and quantitative contributions of the structural subunits of different drugs. Pharmacokinetic and toxicity factors of new drug candidates can be evaluated virtually using modern computational tools. The computer has become an inseparable ally, allowing computational studies that model the chemistry and molecular dynamics of medicinal chemistry (Tang et al. 2014; Liu et al. 2015; Ding et al. 2015). Using several computational tools, researchers can create new virtual candidate ligands for receptor sites in three dimensions (3D). Pharmaceutical companies report that spending on research and development in 2004 was about 33 billion US dollars, representing real growth year to year in investment; this does not correspond, however, to a proportional increase in discoveries of new active molecular candidates for innovative drugs in the market (Fiorani et al. 1999). Natural products have had an important and decisive role in the development of modern medicinal chemistry and drug design. It has been observed (during the last 200 years) that the complexity, chemical diversity, and biological properties of natural products have all aided in the discovery of important new drugs (von Pawel et al. 2014).
In the past 30 years, the development of new bioassay techniques, biotech methods, bio-guided phytochemical studies, automated high-throughput screening, and high performance analytical methods has introduced both new concepts and possibilities for rational drug design and drug discovery. With the development of new spectroscopic techniques, organic chemists have been able to elucidate the complex molecular structures of natural constituents quickly. Gossypol obtained from the cottonseed oil (Gossypium sp.) is widely used in China as a male contraceptive, and hypericin isolated from Saint John’s wort (Hypericum perforatum) extract is used as an antidepressant (Congur et al. 2015). However, another natural substance of oriental origin is artemisinin isolated from Artemisia annua, a plant known and used in Chinese medicine and whose structural complexity has inspired new, useful drugs for the treatment of malaria. Moreover, the active compounds β-artemether, arte-ether, and sodium artesunate were also obtained without limitations of bioavailability (Fig. 9.2) (Sonderstrup et al. 2015). In silico methods or CADD (computer-aided drug design) studies are increasingly being used in industries or universities which involved an understanding of molecular interactions from both qualitative and quantitative points of views. These methods generate and manipulate three-dimensional (3D) molecular structures, calculate descriptors and the dependent molecular properties, model constructions, and employ other tools that encompass computational drug research. Analysis of the molecular structure of a given system allows relevant information to be extracted and to predict the potential of bioactive compounds (Grunnet et al. 2015). Theoretical studies using in silico methods have aided in the process of drug discovery. Technological advances in the areas of structural characterization, computational science, and molecular biology have contributed to faster planning of new feasible molecules (Yeo et al. 2013). Chemoinformatic studies showed that a large fraction of natural products have structural and physicochemical properties that render them as potential drugs. Some investigators have suggested “natural product-likeness score” as a means to filter large chemical databases and find new entities suitable for testing activity and these products subjected to increase the interest in phytochemistry, biochemistry, and related field of research.
9.3 Docking and Drug Discovery
Drug discovery is a lengthy and expensive process that can take up to 15 years and cost upward of billions of dollars. Most of the drug candidates were failed in clinical trial. Of every 10,000 compounds tested, only 1 or 2 are marketed. Traditionally, new drugs have been discovered through studies of natural or synthetic compounds with biological activity. Today, theoretical methodologies have the advantages of both effectively reducing costs and speeding up drug discovery, which results in earlier drug marketing. Examples are captopril, from Bristol-Myers Squibb used for the treatment of hypertension and heart failure; dorzolamide, an antiglaucoma agent developed by Merck; saquinavir, which is a protease inhibitor used for HIV therapy and produced by Hoffmann-La Roche; and zanamivir, a neuraminidase inhibitor used for influenza treatment (Camacho et al. 2015; Cui et al. 2015).
Computer-aided drug design (CADD) studies used several methodologies of computational chemistry to discover, enhance, and study drugs and their related biologically active molecules. Computational methods help in minimizing not only the number of drug candidates but also their ADME profile and toxicological properties. It is assumed that the biological activity of a drug is generally related to its interaction with a protein or a nucleic acid. The drugs are designed (in silico), based on their interaction (ligand with the macromolecule) observed in three dimensions, using molecular docking, in a structure-based drug design (SBDD) technique. This method is used to investigate and predict how the candidate drug (ligand) interacts at the molecular level, by binding to the target protein or nucleic acid (receptor), and to analyze the energies and interactions involved between them (Campos et al. 2011; Frampton 2013; Brown et al. 2014).
Docking involves molecular biology and computer-assisted drug design. The objective of this method is to predict the predominant binding mode of a ligand to proteins, using complex ligand-protein docking of the known three-dimensional structures. Docking methods can effectively search the high-dimensional spaces which occur and apply a scoring function that correctly ranks candidate dockings. Through docking, it is possible to perform virtual screenings of large libraries of compounds, ranks, results, and their proposed structural hypotheses on of how the ligands might inhibit a target (Echenique and Alonso 2007; da Rocha et al. 2011; Decker 2011). Technological advances, better performing algorithms, and increasing computing power allow timeline molecular docking with thousands of ligands; this is of great importance in the pharmaceutical industry. In recent years, the growing number of publications based on molecular docking demonstrates its importance and effectiveness in drug discovery (Lavecchia and Di Giovanni 2013).
9.4 Treatment of Cancer by Plants and Their Derived Products
Cancer is the set of chronic diseases caused by mutations in protein-coding genes leading to disordered growth and multiplication of abnormal cells to form tumors that destroy tissue and other organs (Charifson and Walters 2014). Being caused by a genetic disorder, the disease development can be simple and fast. It was estimated that 18% of cancer cases reported in 2002 were associated with infections such as hepatitis B and C and papilloma virus (90% of patients with cervical cancer) (Duffy et al. 2012; Chrea et al. 2014). In addition, about 30% of cancers are associated with tobacco smoking and inhalation of pollutants and another 35% to eating habits (Shekhar 2008). Devi et al. (2015) reported that lung, stomach, colorectal, liver, and breast cancers are the major causes of death in the world. Breast cancer is the most common cancer in women, while prostate cancer and lung cancers are common in men (Kothandan and Ganapathy 2014). According to an estimate, 56% of 12.7 million cases and 64% of 7.6 million deaths in 2008 occurred in developed countries, and it is expected to increase up to 11.5 million till 2030 (Kothandan and Ganapathy 2014). At present, the chemotherapy is the most effective method of cancer treatment, but it does not destroy the tumor cells completely or diagnosed in late stage of cancer. Moreover, the disease’s stage and resistance of tumor cells toward drugs and their side effects are main causes behind the failure of the treatment (Talele et al. 2010).
In this regard, the therapeutic potentials of plant and their derived products (alkaloids, flavonoids, and terpenoids) with special reference to topoisomerases (Topo), cyclooxygenases (COX), lipoxygenase (LOX), and aromatase provide an alternative way to target various types of cancers. The computational approaches, namely, ADMET, molecular docking, molecular dynamics simulation, and QSAR, are used in assessing and predicting the safety, efficacy, potency, and identification of these potent anticancer therapeutic molecules. According to the World Health Organization (WHO), 80% of the world’s population utilizes the plants or their derived products to treat the various diseases in developing countries. The plant-derived natural products are of great promise for discovery and development of new pharmaceuticals against diverse human ailments including cancer. At present, out of 121 drugs prescribed for cancer treatment till date, 9 are derived from plants (Talele et al. 2010; Kothandan and Ganapathy 2014). Furthermore, among the FDA-approved anticancer drugs between 1984 and 1994, 60% were isolated from plants (Talele et al. 2010). Among 65 new drugs registered for cancer treatment during the period 1981–2002, 48 were obtained from plants (Chen et al. 2002).
9.4.1 Anticancer Molecules
Tan et al. (2014) investigated the neutral and cationic benzo[c]phenanthridine alkaloids against HCT-116 (colon tumor cells) and HL-60 (promyelocytic leukemia cells) and through computer-aided drug design. The cationic alkaloids (7,8-oxygenated benzo[c]phenanthridine alkaloids, chelerythrine, and NK109) showed better anticancer activity compared to neutral derivatives because these compounds formed shorter bonds between ring A and the substituents influencing the resonance, increasing the inhibition of topoisomerases I and II as evident in the electrostatic potential. Similarly, camptothecin, a monoterpenoid indole alkaloid showing anticancer activity, is isolated from the Camptotheca acuminate (Sagar et al. 2006) and acts as a topoisomerase I inhibitor (Jeong et al. 1999). Researchers in medicinal chemistry developed three semisynthetic syntheses: (1) water-soluble analogue of the plant alkaloid camptothecin, (2) topotecan, and (3) irinotecan, used against ovarian, cervical, colorectal, and lung cancers (Wheat and Currie 2008; Polo and Bravo 2006; Yang and Dou 2010; Thoppil and Bishayee 2011). Similarly, Cui et al. (2015) studied the genes from Catharanthus roseus with strictosidine synthase and geraniol 10-hydroxylase, introduced into Ophiorrhiza pumila hairy roots, generating the transgenic compound. They concluded that there was an increase in the production of camptothecin, which showed antitumor activity against leukemia K562 cell line.
The alkaloid camptothecin was developed as a potent anticancer drug directed against Topo I in the year 1958. Two derivatives of camptothecin, namely, topotecan and irinotecan, are being used as FDA-approved drugs against Topo I (Chalabi et al. 2007; Cheah et al. 2008). Similarly, the first Topo II inhibitor, namely, etoposide, an analogue of alkaloid podophyllotoxin, is also an FDA-approved anticancer drug coming from natural product (Anjos et al. 2012; Souza et al. 2012). Iriodenine has been reported to inhibit both Topo I and II by trapping the cleaved DNA-enzyme intermediate and preventing the release of enzymes (Li et al. 2002). Similarly, the alkaloid eleutherin has been reported to inhibit the action of Topo II by inducing relegation and dissociation of the enzyme from DNA (Vipin et al. 2015). Alkaloids dicentrine (both Topo I and II) and lunacridine (Topo II only) have been reported to inhibit respective topoisomerases by intercalating the DNA helix (Dave and Panchal 2012; Scotti et al. 2012b). However, alkaloids (matrine and tetrandrine) are reported to exert their anticancer activity by induction of cell cycle arrest, apoptosis, as well as inhibition of metastasis and angiogenesis (Morris and Lim-Wilby 2008).
Anonaine is a potential anticancer agent, which inhibits cell proliferation by DNA damage and cell cycle arrest in human lung cancer cell lines H1299 (von Pawel et al. 2014). The anticancer activity of oliveroline has been reported through cytotoxicity in MCF-7 mp53 breast cancer cell lines expressing mutant p53 (Taylor et al. 2002). Furthermore, oliveroline has also been reported to act as an anticancer compound inhibiting proliferation of cells undergoing DNA damage at G2 checkpoint (de Avila and de Azevedo 2014). Sanguinarine has been reported to exert its anticancer activity by induction of cell cycle arrest at different phases or apoptosis in a variety of cancer cell lines (Fiorani et al. 1999). Piperine has been reported to inhibit breast stem cell proliferation without causing toxicity to differentiated (normal) cells (Yuriev and Ramsland 2013). Evodiamine has been reported to exhibit anticancer activities under both in vitro and in vivo conditions by inducing the cell cycle arrest or apoptosis, thereby inhibiting the initiation of proliferation, angiogenesis, invasion, and metastasis in a variety of cancer cell lines (Fiorani et al. 1999; Sonderstrup et al. 2015). Berberine has been reported to inhibit multiple aspects of tumorigenesis and tumor progression under both in vitro and in vivo conditions including the induction of cell cycle arrest at the G1 or G2/M phases and apoptosis (Frampton 2013; Brown et al. 2014; Camacho et al. 2015; Cui et al. 2015).
Tan et al. (2011) reported the antitumor activity of dimers of triphenylethylene-coumarin hybrid containing one amino side chain. The authors attributed the anticancer activity to DNA metabolic enzymes, such as the topoisomerases. Later, Zhu et al. (2015) synthesized monomers and dimers of triphenylethylene-coumarin hybrid containing two amino side chains through condensation of three dicarboxylic acids with the amino monomeric hybrids. These compounds were tested against MCF-7 (human breast cancer), A549 (human lung cancer), K562 (chronic myeloid leukemia), and Hela (cervical carcinoma). The active compounds were evaluated as DNA inhibitors by UV-Vis, fluorescence, and CD spectroscopies and a DNA thermal denaturation experiment to observe interactions, properties, and also conformational changes in DNA morphology. The dimers showed better results than the monomers. The authors supposed that the length of the bond and the basic amino group had an influence on the antiproliferative activity of the coumarins.
In vitro and in vivo studies of a number of plant-derived flavonoids such as isoliquiritigenin, glabridin, protocatechuic acid, apigenin, fisetin, baicalin, daidzein, and gingerol have been reported to exhibit antiangiogenic activity (de Atenção 2013). Various naturally occurring flavonoids and their derivatives have been reported to be active against breast cancer with inhibitory effects on aromatase (Aggarwal et al. 2009; Grivennikov et al. 2011). Out of a total of 282 natural compounds proven with anticancer activity against aromatase, 125 were flavonoids (Ahmedin Jemal et al. 2011). Genistein, a phytosterol belonging to flavonoid family, has been reported to inhibit tyrosine kinase, angiogenesis, arrest of cell cycle in G2/M phase, and induction of apoptosis in human promyelocytic HL-60 cancer cell lines (Tu et al. 2016). Biocalein has been reported to induce apoptosis in cell lines of human hepatocellular carcinoma (HCC) as well as inhibition of Topo II (Wang et al. 2012). Similarly, quercetin has been also reported to induce apoptosis by stimulating release of cytochrome-c to the cytosol and activating caspase 9. A number of phenolic compounds, belonging to flavonoids, extracted from rhizome of ginger, have been reported to exhibit cytotoxic activity in cancer cells. However, resveratrol, a phytoalexin found in grapes, has also been reported to induce apoptosis, through CD95 signaling pathway in breast carcinoma HL60 and T47D cell lines (Qurishi et al. 2010). Similarly, curcumin, an important flavonoid, has been shown to be effective in a variety of cancers including colorectal, pancreatic, gastric, and prostate. It has been reported to be effective on different stages of carcinogenesis, namely, proliferation, angiogenesis, and metastasis. Moreover, curcumin has also been shown to act as a chemo-sensitizer, resulting in the increased activity of other anticancer factors in cases of multidrug-resistant and chemotherapy-resistant cancers (Kumar et al. 2012; Safarzadeh et al. 2014).
Geraniol, an acyclic dietary monoterpenoid, has been shown to inhibit the growth of HepG2 human hepatic carcinoma cell lines by inhibiting 3-hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase, the major rate-limiting enzyme in cholesterol biosynthesis in mammals (Mukherjee et al. 2001). Excisanin A, a diterpenoid, exhibited inhibition of growth of human Hep3B liver cancer cell lines via AKT signaling pathway (Brown et al. 2014). Oridonin (diterpenoid) exhibited antiproliferative activity toward prostate cancer cells. Actein (triterpenoid) was found to inhibit the growth of p53-positive HepG2 cancer cell lines. A triterpenoid, betulinic acid, was reported to inhibit growth of breast cancer cells (Houghton et al. 1991). Ursolic acid, an apentacyclic triterpene, has potent cancer-preventive activity and great therapeutic potential. Breast cancer MCF-7 cells exhibited typical apoptotic features, including chromatin clumps and aggregation and DNA fragmentation after ursolic acid treatment, which was in correlation with the downregulation of Bcl-2 and upregulation of caspase-3 (Kanzawa et al. 2001). Different in vivo studies have shown that xanthorrhizol (sesquiterpenoid) inhibits formation and development of tumors via reducing ornithine decarboxylase, COX-2, and NF-κB signaling activity (Hande 1998). Lycopene (tetraterpenoid) modulated breast cancer gene expression pertaining to various molecular pathways, such as apoptosis, cell communication, mitogen-activated protein kinase (MAPK) pathway, and cell cycle (Fortune and Osheroff 2000). Similarly, it has been reported to trigger G2/M arrest and suppress Bcl-2 expression in breast cancer MCF 7 cell lines (Liu et al. 2009).
9.4.2 Topoisomerase Inhibition by Diterpenes
During the processes of replication and transcription along a stretch in the anterior and posterior region of DNA, strands are separated due to the formation of spirals. Topoisomerases act on the control of spirals, relaxing the DNA, and modifying its tertiary structure without changing the primary structure (Prescott et al. 2007; Brastianos et al. 2007; Konkimalla and Efferth 2010; Chen et al. 2011; Lu et al. 2012). These macromolecules are classified according to the cleavage of DNA strands and the location of the covalent link between the enzyme and the DNA strands. In humans, there are two types of topoisomerases (Fig. 9.3). Enzymes type 1 break only one of the DNA strands, passing it through the intact tape and then reconnecting it. The type 1 topoisomerases (Topo I) can be 1A or 1B. The first, 1A, covalently links the 5′ portion of the DNA and does not need cofactors, while the second, 1B, covalently binds to the phosphate portion 3′ of the DNA using strand and Mg2+ or Zn2+ as cofactor. 1A is formed by topoisomerase type III α and β and type 1B by topoisomerase I and topoisomerase I mitochondrial. Type 2 enzymes (Topo II), performing the process, is similar to type 1; however, instead of a single-stranded DNA, both strands are broken. This group of topoisomerase II α and β and Spo 11 bind to 5′ and are dependent on ATP (Kakarala et al. 2010; Burgeiro et al. 2011; Yang et al. 2012; Millimouno et al. 2014). The findings of previous studies showed that the diterpenes have proven their anticancer activity (Tavares et al. 2006; da Silva et al. 2009; Pita et al. 2012; Scotti et al. 2014; Ishiki et al. 2014).

Classification of topoisomerases
9.4.3 Molecular Modeling and Docking
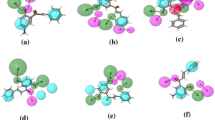
The three-dimensional structures were drawn using HyperChem 8.0.3 software and energy minimized employing the MM+ (Dewar et al. 1985; Cohen 1996) force field without any restrictions. Subsequently, we performed a new geometry optimization based on the semiempirical AM1 method (Leach 2001). The optimized structures were subjected to conformational analysis using a random search method (Staker et al. 2005). Selected dihedral angles were evaluated by rotation in accordance with the standard (default) conditions of the Spartan program (Graille et al. 2008). The diterpene ligands showed two enzymes: topoisomerases I and II (Motohashi et al. 2013). The enzymes were imported from the Protein Data Bank in the Molegro Virtual Docker 6.0 program with a template of complex ligands for the GRID. We selected the MolDock SE algorithm with ten runs for each ligand. The energies (kcal/mol) obtained from the interaction of the ligands and an enzyme are summarized in Table 9.1. We observed that all compounds had the best interaction with Topo I. Compound T1 best interacted in both receptors; on the other hand, A1 showed the highest energy with the two enzymes. We noted that compound T1 forms one hydrogen bond when submitted to docking with Topo I (with the ASP533 residue) and two with residues in Topo II (THR213 and TYR188) (Fig. 9.4). The atisane diterpene forms only steric interactions with ARG364 Topo I and Topo II of TYR188. We concluded that the stability difference observed in the energy of formation can be attributed to hydrogen-bond interactions. Other studies reported the same, as the observations of Laco et al. (2002) which H-bond interactions between the camptothecin and top1/DNA active site are reflected in the values of the energy scores.

Hydrogen bonds between the diterpene T1 and Topo I and II
9.5 Conclusions and Future Prospects
The successful treatment of cancer yet remains a challenge due to the lack of selectivity, toxicity, and development of multidrug-resistant cells to the currently available drugs. The plant-derived products offer a high selectivity, strong activity, low side effects, as well as cancer prevention role by enhancing body immunity. The development of cancer prevention and health-care products from natural products has a broader prospect and greater economic and social benefits when compared with prevalent synthetic anticancer drugs. In this regard, the bioinformatics tools have play a very crucial role in designing and developing new lead therapeutic molecules for the designing of antitumor drugs that use natural products as enzyme inhibitors of human topoisomerases (I and II). These may play an important role in DNA metabolism and inhibition of tumor cells. Some flavonoids and alkaloids like camptothecin and lamellarin D have been extensively examined for the basis for new compounds. In addition, the docking studies, performed with Brazilian diterpenes and both enzymes, showed that the trachylobane diterpene formed a more stable complex due to a hydrogen bond with Top I (with the ASP533 residue) and two hydrogen bonds with residues of Top II (THR213 and TYR188). This study may serve for the basis of compounds which may belong to class II or drug catalytic topoisomerase inhibitors, which interferes with the function of the topoisomerases.
References
Aggarwal BB, Vijayalekshmi RV, Sung B (2009) Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res 15:425–430
Ahmedin Jemal D, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
Anjos JV, Srivastava RM, Costa-Silva JH, Scotti L, Scotti MT, Wanderley AG, Leite ES, Melo SJ, Junior FJ (2012) Comparative computational studies of 3,4-dihydro-2,6-diaryl-4-oxo-pyrimidine-5-carbonitrile derivatives as potential antinociceptive agents. Molecules 17:809–819
Brastianos HC, Sturgeon CM, Roberge M, Andersen RJ (2007) Inhibition of the G2 DNA damage checkpoint by oliveroline isolated from Duguetia odorata. J Nat Prod 70:287–288
Brown DG, Lister T, May-Dracka TL (2014) New natural products as new leads for antibacterial drug discovery. Bioorg Med Chem Lett 24:413–418
Burgeiro A, Gajate C, Dakir EH, Villa-Pulgarín JA, Oliveira PJ, Mollinedo F (2011) Involvement of mitochondrial and B-RAF/ERK signaling pathways in berberine-induced apoptosis in human melanoma cells. Anti-Cancer Drugs 22:507–518
Camacho KM, Kumar S, Menegatti S, Vogus DR, Anselmo AC, Mitragotri S (2015) Synergistic antitumor activity of camptothecin-doxorubicin combinations and their conjugates with hyaluronic acid. J Control Release 210:198–207
Campos HC, da Rocha MD, Viegas FPD, Nicastro PC, Fossaluzza PC, Fraga CA, Barreiro EJ, CJr V (2011) The role of natural products in the discovery of new drug candidates for the treatment of neurodegenerative disorders I: Parkinson’s disease. CNS Neurol Disord-Drug Targets 10:239–250
Chalabi N, Satih S, Delort L, Bignon YJ, Bernard-Gallon DJ (2007) Expression profiling by whole genome microarray hybridization reveals differential gene expression in breast cancer cell lines after lycopene exposure. Biochim Biophys Acta 1769:124–130
Chan JYY, Yuen ACY, Chan RYK, Chan SW (2013) A review of the cardiovascular benefits and antioxidant properties of Allicin. Phytother Res 27:637–646
Charifson PS, Walters WP (2014) Acidic and basic drugs in medicinal chemistry: a perspective. J Med Chem 57:9701–9717
Cheah YH, Nordin FJ, Tee TT, Azimahtol HL, Abdullah NR, Ismail Z (2008) Antiproliferative property and apoptotic effect of xanthorrhizol on MDA-MB-231 breast cancer cells. Anticancer Res 28:3677–3689
Chen YZ, Li ZR, Ung CY (2002) Computational method for drug target search and application in drug discovery. J Theor Comput Chem 1:213–224
Chen BH, Chang HW, Huang HM, Chong IW, Chen JS, Chen CY, Wang HM (2011) (−)-Anonaine induces DNA damage and inhibits growth and migration of human lung carcinoma h1299 cells. J Agric Food Chem 59:2284–2290
Chrea B, O’Connell JA, Silkstone-Carter O, O’Brien J, Walsh JJ (2014) Nature’s antidepressant for mild to moderate depression: isolation and spectral characterization of hyperforin from a standardized extract of St. John’s wort (Hypericum perforatum). J Chem Educ 91:440–442
Clement JA (2014) Recent progress in medicinal natural products drug discovery. Curr Top Med Chem 14:2758
Cohen NC (1996) Guidebook on molecular modeling in drug design. Academic Press, San Diego
Congur G, Erdem A, Mese F (2015) Electrochemical investigation of the interaction between topotecan and DNA at disposable graphite electrodes. Bioelectrochemistry 102:21–28
Cragg G, Newman D (2014) Natural products and drug discovery and development: a history of success and continuing promise for the future. Planta Med 80:750. https://doi.org/10.1055/s-0034-1382292
Cui L, Ni X, Qian J, Teng X, Yang Y, Wu C, Zekria D, Zhang D, Kai G (2015) Co-overexpression of geraniol-10-hydroxylase and strictosidine synthase improves anti-cancer drug camptothecin accumulation in Ophiorrhiza pumila. Sci Rep 5:1–9
da Rocha MD, Viegas FPD, Campos HC, Nicastro PC, Fossaluzza PC, Fraga CA, Barreiro EJ, Viegas C Jr (2011) The role of natural products in the discovery of new drug candidates for the treatment of neurodegenerative disorders II: Alzheimer’s disease. CNS Neurol Disord Drug Targets 10:251–270
da Silva MS, Tavares JF, Queiroga KF, Agra MD, Barbosa JM, Alimeida J, da Silva SAS (2009) Alkaloids and other constituents from Xylopia langsdorffiana (Annonaceae). Quim Nova 32:1566–1570
Dave K, Panchal H (2012) Review on chemogenomics approach: interpreting antagonist activity of secreted frizzled-related protein 1 in glaucoma disease with in silico docking. Curr Top Med Chem 12:1834–1842
de Araújo RS, Guerra FQ, de O Lima E, De Simone CA, Tavares JF, Scotti L, Scotti MT, De Aquino TM, De Moura RO, Mendonça FJ, Barbosa-Filho JM (2013) Synthesis, structure-activity relationships (SAR) and in silico studies of coumarin derivatives with antifungal activity. Int J Mol Sci 14:1293–1309
de Atenção BC (2013) Controle dos cânceres do colo do útero e da mama, 2nd edn. Ministério da Saúde, Brasília
de Avila MB, de Azevedo WF (2014) Data mining of docking results. Application to 3-dehydroquinate dehydratase. Curr Bioinforma 9:361–379
Decker M (2011) Hybrid molecules incorporating natural products: applications in cancer therapy, neurodegenerative disorders and beyond. Curr Med Chem 18:1464–1475
Devi RV, Sathya SS, Selvaraj M (2015) Evolutionary algorithms for de novo drug design – a survey. Appl Soft Comput J 27:543–552
Dewar MJSE, Zoebisch G, Healy EF, Stewart JJPAM (1985) A new general purpose quantum mechanical molecular model. J Am Chem Soc 107:3902–3909
Ding XT, Matsuo K, Xu L, Yang J, Zheng LP (2015) Optimized combinations of bortezomib, camptothecin, and doxorubicin show increased efficacy and reduced toxicity in treating oral cancer. Anti-Cancer Drugs 26:547–554
Duffy R, Wade C, Chang R (2012) Discovery of anticancer drugs from antimalarial natural products: a MEDLINE literature review. Drug Discov Today 17:942–953
Echenique P, Alonso JL (2007) A mathematical and computational review of Hartree-Fock SCF methods in quantum chemistry. Mol Phys 105:3057–3098
Fiorani P, Amatruda JF, Silvestri A, Butler RH, Bjornsti A, Benedetti P (1999) Domain interactions affecting human DNA topoisomerase I catalysis and camptothecin sensitivity. Mol Pharmacol 56:1105–1115
Fortune JM, Osheroff N (2000) Topo II as a target for anticancer drugs: when enzymes stop being nice. Proc Nucleic Acid Res Mol Biol 64:221–253
Frampton CS (2013) An introduction to the special issue on pharmaceuticals, drug discovery and natural products. Acta Crystallogr Sect C 69:1205–1206
Graille M, Cladiere L, Durand D, Lecointe F, Gadelle D, Quevillon-Cheruel S, Vachette P, Forterre P, van Tilbeurgh H (2008) Crystal structure of an intact type II DNA topoisomerase: insights into DNA transfer mechanisms. Structure 16:360–370
Grivennikov SI, Greten FR, Karin M (2011) Immunity, inflammation, and cancer. Cell 140:883–899
Grunnet M, Calatayud D, Schultz NA, Hasselby JP, Mau-Sørensen M, Brünner N, Stenvang J (2015) TOP1 gene copy numbers are increased in cancers of the bile duct and pancreas. Scand J Gastroenterol 50:485–494
Hande KR (1998) Etoposide: four decades of development of a topo II inhibitor. Eur J Cancer 34:1514–1521
Hatae N, Fujita E, Shigenobu S, Shimoyama S, Ishihara Y, Kurata Y, Choshi T, Nishiyama T, Okada C, Hibino S (2015) Antiproliferative activity of O-4-benzo c phenanthridine alkaloids against HCT-116 and HL-60 tumor cells. Bioorg Med Chem Lett 25:2749–2752
Houghton PJ, Cheshire PJ, Myers L, Stewart CF, Synold TW, Houghton JA (1991) Evaluation of 9-dimethylaminomethyl-10 hydroxy-camptothecin (topotecan) against xenografts derived from adult and childhood tumors. Cancer Chemother Pharmacol 31:229–239
Ishiki H, Junior FJ, Santos PF, Tavares JF, Silva MS, Scotti MT (2014) Theoretical research into anticancer activity of diterpenes isolated from the paraiban flora. Nat Prod Commun 9:911–914
Jeong HJ, Shin YG, Kim IH, Pezzuto JM (1999) Inhibition of aromatase activity by flavonoids. Arch Pharm Res 22:309–312
Kakarala M, Brenner DE, Korkaya H, Cheng C, Tazi K, Ginestier C, Liu S, Dontu G, Wicha MS (2010) Targeting breast stem cells with the cancer preventive compounds curcumin and piperine. Breast Cancer Res Treat 122:777–785
Kanzawa F, Koizumi F, Koh Y, Nakamura T, Tatsumi Y, Fukumoto H, Saijo N, Yoshioka T, Nishio K (2001) In vitro synergistic interactions between the cisplatin analogue nedaplatin and the DNA topoisomerase I inhibitor irinotecan and the mechanism of this interaction. Clin Cancer Res 7:202–209
Karioti A, Milosevic-Ifantis T, Pachopos N, Niryiannaki N, Hadjipavlou-Litina D, Skaltsa H (2015) Antioxidant, anti-inflammatory potential and chemical constituents of Origanum dubium Boiss., growing wild in Cyprus. J Enzym Inhib Med Chem 30:38–43
Kennedy DA, Hart J, Seely D (2009) Cost effectiveness of natural health products: a systematic review of randomized clinical trials. Evidence-Based Compl Altern Med 6:297–304
Konkimalla BV, Efferth T (2010) Inhibition of epidermal growth factor receptor over-expressing cancer cells by the aphorphine-type isoquinoline alkaloid, dicentrine. Biochem Pharmacol 79:1092–1099
Kothandan G, Ganapathy J (2014) A short review on the application of combining molecular docking and molecular dynamics simulations in field of drug discovery. J Chosun Nat Sci 7:75–78
Kumar NDR, George CV, Kumar SP, Kumar AR (2012) Cytotoxicity, apoptosis induction and anti-metastatic potential of Oroxylum indicum in human breast cancer cells. Asian Pac J Cancer Prev 13:2729–2734
Laco GS, Collins JR, Luke BT, Kroth H, Sayer JM, Jerina DM, Pommier Y (2002) Human topoisomerase I inhibition: docking camptothecin and derivatives into a structure-based active site model. Biochemistry 41:1428–1435
Lavecchia A, Di Giovanni C (2013) Virtual screening strategies in drug discovery: a critical review. Curr Med Chem 20:2839–2860
Leach AR (2001) Molecular modeling: principles and applications. Prentice Hall, London
Li Z, Wang Y, Mo B (2002) The effects of carotenoids on the proliferation of human breast cancer cell and gene expression of bcl-2. Zhonghua Yu Fang Yi Xue Za Zhi 36:254–257
Liu YC, Chen ZF, Liu LM, Peng Y, Hong X, Yang B, Liu HG, Liang H, Orvig C (2009) Divalent later transition metal complexes of the traditional Chinese medicine (TCM) liriodenine: coordination chemistry, cytotoxicity and DNA binding studies. Dalton Trans 48:10813–10823
Liu YQ, Li WQ, Morris-Natschke SL, Qian KD, Yang L, Zhu GX, Wu XB, Chen AL, Zhang SY, Nan X, Lee KH (2015) Perspectives on biologically active camptothecin derivatives. Med Res Rev 35:753–789
Lu JJ, Bao JL, Chen XP, Huang M, Wang YT (2012) Alkaloids isolated from natural herbs as the anticancer agents. Evidence-Based Compl Altern Med 2012:485042
Millimouno FM, Dong J, Yang L, Li J, Li X (2014) Targeting apoptosis pathways in cancer and perspectives with natural compounds from mother nature. Cancer Prev Res 7:1081–1107
Mills E, Wu P, Johnston BC, Gallicano K, Clarke M, Guyatt G (2005) Natural health product-drug interactions -a systematic review of clinical trials. Ther Drug Monit 27:549–557
Mishra KP, Ganju L, Sairam M, Banerjee PK, Sawhney RC (2008) A review of high throughput technology for the screening of natural products. Biomed Pharmacother 62:94–98
Moran M, Guzman J, Ropars A-L, McDonald A, Jamerson N, Omune B, Ryan S, Wu L (2009) Neglected disease research and development: how much are we really spending. PLoS Med 6:137–146
Morris GM, Lim-Wilby M (2008) Molecular docking. Methods Mol Biol 443:365–382
Motohashi Y, Igarashi M, Okamatsu M, Noshi T, Sakoda Y, Yamamoto N, Ito K, Yoshida R, Kida H (2013) Antiviral activity of stachyflin on influenza: a viruses of different hemagglutinin subtypes. Virol J 10:118. https://doi.org/10.1186/1743-422X-10-118
Mukherjee AK, Basu S, Sarkar N, Ghosh AC (2001) Advances in cancer therapy with plant based natural products. Curr Med Chem 8:1467–1486
Pita JC, Xavier AL, Sousa TK, Mangueira VM, Tavares JF, Júnior RJ, Veras RC, Pessoa HD, Silva MS, Morelli S, Ávila VD (2012) In vitro and in vivo antitumor effect of trachylobane-360, a diterpene from Xylopia langsdorffiana. Molecules 17:9573–9589
Polo MP, Bravo D (2006) Effect of geraniol on fatty-acid and mevalonate metabolism in the human hepatoma cell line Hep G2. Biochem Cell Biol 84:102–111
Prescott TA, Sadler IH, Kiapranis R, Maciver SK (2007) Lunacridine from Lunasia amara is a DNA intercalating topoisomerase II inhibitor. J Ethnopharmacol 109:289–294
Qurishi Y, Hamid A, Zargar MA, Singh SK, Saxena AK (2010) Potential role of natural molecules in health and disease: importance of boswellic acid. J Med Plant Res 4:2778–2785
Safarzadeh E, Shotorbani SS, Baradaran B (2014) Herbal medicine as inducers of apoptosis in cancer treatment. Adv Pharm Bull 4:421–427
Sagar SM, Yance D, Wong RK (2006) Natural health products that inhibit angiogenesis: a potential source for investigational new agents to treat cancer-part 1. Curr Oncol 13:14–26
Scotti L, Scotti MT, Lima ED, da Silva MS, de Lima MDA, Pitta ID, de Moura RO, de Oliveira JGB, da Cruz RMD, Mendonca FJB (2012a) Experimental methodologies and evaluations of computer-aided drug design methodologies applied to a series of 2-aminothiophene derivatives with antifungal activities. Molecules 17:2298–2315
Scotti L, Mendonça Junior FJB, Moreira DRM, Silva MS, da Pitta IR, Scotti MT (2012b) SAR, QSAR and docking of anticancer flavonoids and variants: a review. Curr Top Med Chem 12:2785–2809
Scotti L, Scotti MT, Ishiki H, Junior FJ, Tavares JF, da Silva MS (2014) Prediction of anticancer activity of diterpenes isolated from the Paraiban flora through a Pls model and molecular surfaces. Nat Prod Commun 9:609–612
Shekhar C (2008) In Silico pharmacology: computer-aided methods could transform drug development. Chem Biol 15:413–414
Sonderstrup IM, Nygård SB, Poulsen TS, Linnemann D, Stenvang J, Nielsen HJ, Bartek J, Brünner N, Nørgaard P, Riis L (2015) Topoisomerase-1 and-2A gene copy numbers are elevated in mismatch repair-proficient colorectal cancers. Mol Oncol 9:1207–1217
Souza B, De Oliveira T, Aquino T, de Lima M, Pitta I, Galdino S, Lima E, Gonçalves-Silva T, Militão G, Scotti L, Scotti M (2012) Preliminary antifungal and cytotoxic evaluation of synthetic cycloalkyl b thiophene derivatives with PLS-DA analysis. Acta Pharma 62:221–236
Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB (2005) Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem 48:2336–2345
Talele TT, Khedkar S, Rigby AC (2010) Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr Top Med Chem 10:127–141
Tan W, Lu J, Huang M, Li Y, Chen M, Wu G, Gong J, Zhong Z, Xu Z, Dang Y, Guo J, Chen X, Wang J (2011) Anticancer natural products isolated from chinese medicinal herbs. J Chin Med 6:27. https://doi.org/10.1186/1749-8546-6-27
Tan G, Yao Y, Gu Y, Li S, Lv M, Wang K, Chen H, Li X (2014) Cytotoxicity and DNA binding property of the dimers of triphenylethylene-coumarin hybrid with one amino side chain. Bioorg Med Chem Lett 24:2825–2830
Tang XJ, Han M, Yang B, Shen YQ, He ZG, Xu DH, Gao JQ (2014) Nanocarrier improves the bioavailability, stability and antitumor activity of camptothecin. Int J Pharm 477:536–545
Tavares JF, Queiroga KF, Silva MVB, Diniz M, Barbosa-Filho JM, da Cunha EVL, de Simone CA, de Araujo JX, Melo PS, Haun M, da Silva MS (2006) ent-trachylobane diterpenoids from Xylopia langsdorffiana. J Nat Prod 69:960–962
Taylor RD, Jewsbury PJ, Essex JW (2002) A review of protein-small molecule docking methods. J Comput Aided Mol Des 16:151–166
Thoppil RJ, Bishayee A (2011) Terpenoids as potential chemopreventive and therapeutic agents in liver cancer. World J Hepatol 3:228–249
Tu X, Wang L, Cao Y, Ma Y, Shen H, Zhang M, Zhang Z (2016) Efficient cancer ablation by combined photothermal and enhanced chemotherapy based on carbon nanoparticles/doxorubicin@SiO2 nanocomposites. Carbon 97:35–44
Vipin AM, Baby B, Kumar MS, Ravisankar V, Nazeem PA (2015) Comparative docking studies on the effect of commercial drugs on dipeptidyl peptidase-4 (DPP-4). Int J Pharm Pharm Sci 7:508–510
von Pawel J, Jotte R, Spigel DR, O’Brien ME, Socinski MA, Mezger J, Steins M, Bosquée L, Bubis J, Nackaerts K, Trigo JM (2014) Randomized phase III trial of amrubicin versus topotecan as second-line treatment for patients with small-cell lung cancer. J Clin Oncol 32:4012–4019
Wang BC, Deng J, Gao YM, Zhu LC, He R, Xu YQ (2011) The screening toolbox of bioactive substances from natural products: a review. Fitoterapia 82:1141–1151
Wang CZ, Calway T, Yuan CS (2012) Herbal medicines as adjuvants for cancer therapeutics. Am J Chin Med 40:657
Wheat J, Currie G (2008) Herbal medicine for cancer patients: an evidence based review. Int J Altern Med 5:1–11
Yang H, Dou QP (2010) Targeting apoptosis pathway with natural terpenoids: implications for treatment of breast and prostate cancer. Curr Drug Targets 11:733–744
Yang ZR, Liu M, Peng XL, Lei XF, Zhang JX, Dong WG (2012) Noscapine induces mitochondria-mediated apoptosis in human colon cancer cells in vivo and in vitro. Biochem Biophys Res Commun 421:627–633
Yeo CD, Lee SH, Kim JS, Kim SJ, Kim SC, Kim YK, Kang HH, Yoon HK, Song JS, Moon HS, Kim JW (2013) A multicenter phase II study of belotecan, a new camptothecin analogue, in elderly patients with previously untreated, extensive-stage small cell lung cancer. Cancer Chemother Pharmacol 72:809–814
Yuriev E, Ramsland P (2013) Latest developments in molecular docking: 2010–2011 in review. J Mol Recognit 26:215–239
Zhu M, Zhou L, Yao Y, Li S, Lv M, Wang K, Li X, Chen H (2015) Anticancer activity and DNA binding property of the dimers of triphenylethylene-coumarin hybrid with two amino side chains. Med Chem Res 24:2314–2324
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Scotti, L. et al. (2018). CADD Studies Applied to Secondary Metabolites in the Anticancer Drug Research. In: Akhtar, M., Swamy, M. (eds) Anticancer Plants: Mechanisms and Molecular Interactions. Springer, Singapore. https://doi.org/10.1007/978-981-10-8417-1_9
Download citation
DOI: https://doi.org/10.1007/978-981-10-8417-1_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8416-4
Online ISBN: 978-981-10-8417-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)