
Abstract
After the culmination of Human Genome Project in 2003, it was prophesied that the upcoming era in modern biotechnology would pose as a real acid test. While the pre-genomic era was marked by the efforts to sequence the human genome, advancements into the post-genomic era are characterized by the challenge of reaping benefits from these genomic texts. Formidably a large data is generated from high throughput techniques, and it cannot be used efficiently in probing the plant genome and evolution without the aid of bioinformatics. The goal of this field is thus, to provide computational approaches and in silico methodologies for coping with, and interpreting this genomic data to develop new cost-effective, accessible, safe and reliable treatments for diseases such as cancer. A major aspect of cancer research focuses on studying clinically useful plant-derived anticancer agents and promising new plants with anticancer potential. Also, certain agents that have failed in earlier clinical studies are considered for evaluation to obtain novel anticancer drugs using bioinformatics approaches and this field has triggered more interest among researchers in recent years. The aim of this chapter is to merge the sphere of computer-based methods in ‘omics’ technologies with the anticancer analysis of plant sources, and also to cover the sophisticated bioinformatics softwares and tools adopted in the process.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
12.1 Introduction
With an increase in the number of cancer patients across the world, it has become more imperative to reap maximum benefits from anticancer agents. Plants are a crucial source for such compounds and are being researched vigorously for generating lead molecules in the drug development process (Katiyar et al. 2012; Atanasov et al. 2015). However, this process is impaired without the aid of advancing omics technologies that have made non-targeted identification of signaling molecules possible and much more efficient. The understanding of biological processes has improved substantially with the examination of genes and gene products associated with cell growth, apoptosis, necrosis and cellular metabolism. Providing major breakthroughs in the field of oncology, omics has led to an earlier detection and more accurate treatment of cancer (Sallam 2015; Armitage and Southam 2016; Dijkstra et al. 2016; Uzilov et al. 2016).
Genetics and molecular biology experienced a face lift with the emergence of genomics, an interdisciplinary field concerned with the study of genomes, the functions of genes as a unit of inheritance, their interactions with the environment and most importantly, techniques such as DNA sequencing and applications of recombinant DNA. Further, the flow of genetic information through transcriptional and post-transcriptional modifications of DNA gives an entire set of messenger RNA called transcriptome. While microarray analysis is highly prevalent, it is being replaced by RNA-Seq that incorporates next-generation sequencing. In the next step, mature mRNA is decoded by ribosomes, where translation occurs. The resulting proteins are researched through techniques like mass spectrometry and two-dimensional gel electrophoresis under proteomics, a rapidly growing field. Another aspect of post genomics is metabolomics, one of the newest ‘omics’ sciences. Concerned with the study of metabolites through methods like nuclear magnetic resonance spectroscopy, it explores cellular processes and the physiology of cells (Horgan and Kenny 2011). To analyze and interpret biological data from omics, and overcome the obstacle of integration of these technologies, bioinformatics is employed. With a wide range of applications in cancer and plant research, this multifaceted field has become indispensable. Thus, the aim of the chapter is to merge the sphere of computer-based methods in ‘omics’ technologies with the anticancer analysis of plant sources, and also to cover the sophisticated bioinformatics softwares and tools adopted in the process.
12.2 Omics in Anticancer Plants
Coupled with developments in technology, improved sample handling, validated study designs and statistical solutions for data interpretation, omics provide a holistic approach to the interplay between surroundings and human health (Vlaanderen et al. 2010). Since these involve examination of huge numbers of genes, their associated expressions or proteins, the techniques involved are called high throughput. High-throughput screening methods provide effectual assessment of agents or conditions in biological assays (Szymanski et al. 2012) and are pivotal for the discovery of new chemotypes (Macarron et al. 2011). Coalescence of multiple omics data in higher plants is essential to remodel complex networks that typify the phenotypes in the cell. Genomics, transcriptomics, proteomics and metabolomics techniques pave the way for robust and practical plant metabolic engineering (Yonekura-Sakakibara et al. 2013). Exploitation of these technologies has led to the authorization and subscription of many new anticancer plant-derived materials as medicines, due to reduced side effects and efficacious chemoprevention activity (Fridlender et al. 2015).
12.2.1 Genomics in Anticancer Plants
Genome sequences incorporate critical information regarding plant origin, development and epigenomic regulation that serves as the cornerstone of decoding genome diversity and chemodiversity at the microscopic level (Hao and Xiao 2015). High-throughput sequencing of medicinal plants highlights the biosynthetic mechanisms of anticancer compounds, especially secondary metabolites (Boutanaev et al. 2015) and their regulatory pathways.
RAD-Seq (restriction-site associated DNA sequencing) (Rubin et al. 2012), a fractional genome sequencing strategy can construct a RAD library and carry out low-coverage genome sequencing of candidate species. This is an efficient approach for assessing the heterozygosity of the representative genome. Genetic map and physical map are other essential tools for the aggregation of complex plant genomes and research in functional genomics. The results obtained are used in map-based cloning. Other applications include metabolic gene mapping and marker-assisted selection of anticancer traits. This genomic data from the RAD-Seq and genotyping by sequencing (GBS) method can also be utilized to determine the origin and the spatial distribution pattern of existing anticancer plants (Hao and Xiao 2015).
A recent study states that sequencing of the entire genome of Ocimum tenuiflorum has helped to delineate that amino acid mutations present at the loci of genes involved in biosynthesis, confer exceptional pharmaceutical attributes to this herb. As a defense mechanism, this plant generates specialized metabolites like ursolic acid, luteolin, apigenin, taxol, oleanolic acid, sitosterol and eugenol that have anticancer potential. With the genome laid out, the specific genes responsible for curing cancer can be known and can be used to develop targeted drugs (Upadhyay et al. 2015).
Capsicum annuum (Qin et al. 2014), Coffea canephora (Denoeud et al. 2014), Brassica napus (Chalhoub et al. 2014) and Phalaenopsis equestris (Cai et al. 2015) are some representative plant species that have undergone whole-genome sequencing. A comparison between the genome sequences of cultivated pepper Zunla-1 (C. annuum) and its wild progenitor Chiltepin (C. annuum var. glabriusculum) gave an understanding about Capsicum domestication and differentiation. The Capsicum reference genome, along with tomato and potato genomes, provides an insight into the evolution of other Solanaceae species, including the well-known Atropa medicinal plants. This strategy can be applied to probe a number of plant-derived substances exhibiting anticancer activity such as Camptothecin derivatives, Podophyllotoxin derivatives, Taxanes, Vinca alkaloids, etc. (Fridlender et al. 2015).
Microsatellite markers such as Simple Sequence Repeats (SSRs) are present in high frequency in transcribed regions of plants, especially in untranslated regions (UTR) and also possess the ability to associate with many phenotypes. Thus, they have several applications in plant genomics. They aid in plant reproduction, genome regulation, evaluation and organisation. Microsatellite markers have been identified for many anticancer plant families and genera, and these results are useful for gene evolution and protection of genetic resources of these plants. A recent Korean study focussed on Viscum coloratum, a hemiparasitic plant with anticancer properties and 19 novel polymorphic microsatellite loci were developed to aid this plant’s ecological conservation and population genetics studies (Kim et al. 2017). Present in the genome sequences, these markers are crucial for recombination and quantitative genetic variation. Through the current study on evolutionary genomics, there is a great scope of improvement in resolution, making it possible to identify the particular genes responsible for specific innovations. More information on plant evolution may improve the understanding on botanical diversity, including medicinally important traits such as anticancer, antiallergic, anti-inflammatory, etc. properties (Hao and Xiao 2015). Genome sequencing of plant genomes has qualified as a reliable process to delve into a diverse set of concepts of medicinal importance.
12.2.2 Transcriptomics in Anticancer Plants
Whole-genome sequencing is an expensive process. It is also challenging when the genome comprises of a high proportion of repeat sequences, high heterozygosity and non-diploids (Chen et al. 2010). Large-scale comparative transcriptome studies of anticancer plants are more feasible than comparative genomics. Transcriptomics is thus, an effective strategy to retrieve genomic data from several non-model medicinal plants that do not have a reference genome. This information delineates certain relevant traits pertaining to secondary metabolite formation and for studying pharmaceutically important molecular mechanisms (Hao et al. 2015a).
Curcuma longa, commonly known as turmeric, is recognized as an herbal remedy and alternative medicine for cancer. Research has shown that this herbaceous member of the ginger family reduces incidences of gastrointestinal cancers due to the presence of secondary metabolite, Curcumin in its rhizomes. Rhizome transcriptomes of various varieties of Curcuma longa were sequenced via Illumina reversible dye terminator sequencing. This resulted in the availability of transcripts related to terpenoid biosynthesis and biosynthetic pathways of other anticancer phytochemicals like vinblastine, taxol and curcumin. This helped to reinstate the biosynthetic pathways to synthesize various terpenoids in C. longa and contributed to its transcriptomic database(Annadurai et al. 2013).Also, researchers have used the phylotranscriptomic approach to test phylogenetic hypotheses to give information about the evolution of the fundamental anticancer plant traits attributed for their myriad chemodiversity (Hao et al. 2015b). Another medicinal plant species, Chlorophytum borivilianum that derives its medicinal value for its higher saponin content (approximately 17% by dry weight), exhibits antitumor and anticancer properties (Kumar et al. 2010) due to the presence of cytotoxic steroidal glycosides, saponinchloromaloside-A and spirostanol-pentaglycosides embracing beta-D-apiofuranose. High throughput transcriptome sequencing of its leaf RNA was performed through Illumina’s HiSeq 2000 sequencing platform. Bioinformatics tools such as SOAP denovo, Contig Assembly Program (CAP3) assembler and Kegg Orthology-Based Annotation System (KOBAS) were used for the assembly and annotation of the transcriptome. Further, molecular insight into the flavonoid and steroid biosynthesis pathways of this endangered species and bioinformatics analysis showed that its combination with other herbs could be instrumental in oncological treatment (Kalra et al. 2013).
Transcriptomics using DNA microarray has become another effective tool for the study of anticancer plants because of high throughput, sensitivity, precision, specificity, and duplicability. It is implemented widely in Chinese herbal medicine and provides a practical approach that enables researchers to examine the expression of numerous genes concurrently (Lo et al. 2012).Whole transcriptome shotgun sequencing (WTSS), also known as RNA sequencing (RNA-seq), provides whole-transcriptome expression profiles of certain plant extracts as well, hence making integrated analysis of transcriptomics and metabolomics possible in any plant species (Yamazaki et al. 2013). It allows the probing of genes involved in metabolite synthesis of plant specialized products, and the integration of transcriptome data with metabolic profiling data sets to reveal the relationship between genes and metabolites in anticancer plants. Its prime feature is that it makes it possible to obtain gene sequences from plants without a reference genome. A 2013 study used Illumina-derived short read sequences for deep transcriptome analysis of cell suspension cultures and the hairy roots of Ophiorrhiza pumila, a Rubiaceae species that accumulates the anticancer monoterpenoidindole alkaloid, camptothecin. This yielded a 2GB sequence for each sample, and expedited prediction of new and novel genes committed in secondary metabolic pathways. Bioinformatics tools such as the Oases assembler, CAP3 program, Bowtie package and Cufflinks aided this research (Yamazaki et al. 2013).
One of the most medicinally important plants is the Withania somnifera that synthesizes bioactive secondary metabolites known as withanolides. Chemo-profiling of leaf and root tissues of this plant imply dissimilarities in the composition and properties of withanolides in various chemovars. To identify the genes involved in chemotype and tissue-specific withanolide biosynthesis, transcriptomes of leaf and root tissues of discrete chemotypes were established (Gupta et al. 2015). Several medicinal phytometabolites have also been discovered in the buttercup family, Ranunculaceae. Some of them like alkaloids, terpenoids, saponins, and polysaccharides, have expressed antitumorigenic behaviour both in vitro and in vivo. Gene expression profiling and relevant transcriptomics platforms provided an insight into the distinctive effects of plant metabolites on cancer cells with varying physical characteristics (Hao et al. 2017).
12.2.3 Proteomics in Anticancer Plants
Proteomics is a powerful platform to analyse drug-regulated proteins on a large scale and investigate signalling pathway perturbations in cells. It mainly characterizes protein functions, protein-protein interactions in vitro and in vivo, and protein modifications, and has various applications in the research on anticancer plants (Lao et al. 2014). It can also substantiate post-translational protein modifications such as phosphorylation, glycosylation, acetylation, and proteolysis (Zhang et al. 2011). These modifications can occur as cancer progresses or after drug treatment, and can be analysed by proteomic approaches. The mechanism of action of a drug is studied through macro-analysis of protein alterations through proteomic technologies and through the identification of modified proteins as prospective drug targets (Lao et al. 2014). Traditional Chinese medicine (TCM) is an abundant source of anticancer drugs. Bioactive secondary metabolites isolated from TCMs project substantial antitumor effects, although their pathways are still ambiguous. Terpenoids, flavonoids, glycosides and other bioactive TCM products have been studied extensively via proteomics to describe their antitumor activities in various cancers. A significant number of natural agents extracted usually perform tumour-suppression by exclusively targeting mitochondria in malignant cells (Wang et al. 2015).
Analysis of natural flavones such as Baicalein, Tangeretin and Luteolin exhibit anticancer properties but their mechanisms of action are still unclear. However, a proteomic study delineated that Baicalein led to the up-regulation of peroxiredoxin-6, which reduced the generation of reactive oxygen species (ROS) and inhibits colorectal cancer cell proliferation (Huang et al. 2012). Another flavonoid, luteolin demonstrates similar anticancer activity against several forms of cancers, including human hepatic cancers. Proteomics is a multifunctional tool that thus, provides a methodical approach towards understanding the molecular mechanisms of TCM in tumor cells and investigating protein-drug interactions. Stable Isotope Labeling with Amino acids in Cell culture (SILAC) and Isobaric Tag for Relative and Absolute Quantitation (iTRAQ) are chief quantitative approaches for the process (Wang et al. 2015).
Tripterygium wilfordii, a representative TCM has been widely and successfully used to treat numerous diseases such as rheumatoid arthritis and psoriasis. Its anticancer activity and intrinsic action mechanisms have also been investigated intensively and a proteomics study showed that diterpenoid epoxide triptolide, an important bioactive metabolite, has applications in curing colon cancer (Liu et al. 2011). Triptolide treatment induces cell division and the perinuclear translocation of 14–3-3ξ, a key protein pertaining to cell cycle arrest and cell death (Liu et al. 2012). The plant extracts of A. Paniculata have also been found to contain diterpene compounds that encompass medicinally relevant properties against cancer, pathogenic bacteria, virus and hepatitis (Valdiani et al. 2012). There is a lack of comprehensive molecular genetic studies on this medicinal herb from the family Acanthaceae and thus, a huge volume of useful information is obtained from its protein profiling. Proteomic analysis is thus, an efficient methodology to obtain advanced knowledge on the inheritable traits and physiology of anticancer plants (Talei et al. 2014).
Periplocin, sourced from the bark and stems of Periploca graeca, can prevent both lung and colon cancer in vitro and in vivo. It exhibits anticancer effects on the cells via beta-catenin/TCF signalling pathway by inducing apoptosis. According to a study in 2014, quantitative proteomics technologies like tandem mass spectrometry and two-dimensional gel electrophoresis were used to explore the effect of periplocin treatment on human lung cancer cell lines A549. The western blot was used to authenticate the modified proteins and the protein-protein interactions between them were also investigated (Lu et al. 2014). The antioxidant, antineoplastic, antiangiogenic and particularly, anticancer properties of Curcumin and its derivatives have also been thoroughly researched. Several studies have used proteomics to investigate the potential of curcumin against different cancer cell lines. From a study in 2011, proteomic analysis distinguished 12 differentially expressed proteins that boost multiple functional activities in the MCF-7 breast cancer cell line. These functions include DNA transcription, mRNA splicing and translation, amino acid synthesis, protein synthesis, folding and degradation, lipid metabolism, glycolysis, and cell motility (Fang et al. 2011).
Berberine, a natural product obtained from the subterranean part of Coptis chinensis, has also gained interest due to its anti- proliferative properties. Differentially expressed proteins in HepG2 liver cancer cells investigated through proteomic analysis. The results revealed that berberine led to G0 cell cycle arrest and apoptosis (Wang et al. 2016). Gambogic acid, a natural chemical extracted from the brownish or orange resin of Garcinia hanburyi was intensely investigated and found to hinder the growth of various cancer cells via multiple signalling pathways. This xanthonoid has shown promising antitumor activity in clinical trials (Chantarasriwong et al. 2010; Anantachoke et al. 2012; Chen et al. 2012). Research through applied proteomics has shown that stathmin could be a potential target of gambogic acid in hepatocellular carcinoma. Later, more than 80 compounds with anticancer potential were identified from the Garcinia species. Bioassay guided fractionation and systematic proteomic analysis aimed at studying the possible action mechanisms of these active compounds were employed. They showed that 1,3,6,7-tetrahydroxyxanthone, a bioactive metabolite isolated from G. oblongifolia, curtailed cell proliferation by the up-regulation of p16 and 14–3-3σ in hepatocellular carcinoma cells (Fu et al. 2012a). The proteomics data also revealed that 1,3,5-trihydroxy-13,13-dimethyl-2H-pyran [7,6-b] xanthone, which is also derived from G. oblongifolia, can induce cancer cell death by suppressing Heat shock protein 27 (Hsp27) (Fu et al. 2012b). Through 2-DE analysis, it has become evident that Hsp27 plays a crucial role. Tanshione IIA, a phenanthrene quinine extracted from the root of Salvia miltiorrhiza has also been shown to down-regulate Hsp27 expression in cervical cancer cells (Lao et al. 2014). In some cultures, dietary components such as fenugreek seeds are also used for the treatment of cancer. In 2014, the proteomic profiles of these seeds showed that an incidence of primary CNS T cell lymphoma reacted to fenugreek treatment and led to tumor regression. The in vitro effect of fenugreek as a substance that causes cancer cell destruction through cytotoxins, points to the significance of this seed as a treatment for cancer (Alsemari et al. 2014). Proteomics is thus, an essential tool to predict the protein targets of bioactive compounds present in anticancer plants.
12.2.4 Metabolomics in Anticancer Plants
Plants synthesize a vast number of primary and secondary metabolites. Hence, they are being probed extensively to find new chemical entities (NCEs) for drug discovery and development. As chemotherapeutics has many side effects such as fatigue, hair loss, resistance, mouth sores, nausea, blood disorders and nervous system effects, recent attention has shifted to plants that provide a good opportunity for complementary cancer cure (Tecza et al. 2015). More than 50% of anticancer drugs used in therapeutics today are sourced from natural products, whose relevance, however, was undermined due to the arduous method of conventional lead generation (Cragg and Newman 2013; Newman and Cragg 2016). Hence, a dire need of an efficient strategy for the detection of bioactive compounds was felt. Medicinal plant-based metabolomics, a rapidly emerging field, has become a study of prime importance since it has the potential to prevent natural product research from reaching an impasse, aid discovery of anticancer drugs and improve the effectiveness of lead-finding (Kim et al. 2010; Mukherjee et al. 2016; Okada et al. 2010). This approach is being exploited in a wide range of applications including medical science, synthetic biology, Ayurvedic medicine and predictive modelling of plant systems. The working principle of metabolomics basically involves sample preparation, separation of compounds, identification, data processing and finally analysis.
Several therapeutically important secondary metabolites like paclitaxel (taxol), camptothecin (irinotecan, topotecan) and podophyllotoxins (etoposide, teniposide), etc. have been investigatedand have reported to possess anticancer activity. The metabolic profiling and anti-tumorigenic activities of certain widely cultivated plants of the family Compositae were also evaluated. Anticancer potential was studied for human hepatocellular carcinoma (HepG-2) and breast adenocarcinoma (MCF-7) cell lines. Plant species portrayed variable metabolomic profiling. While Artemisia revealed the highest concentration of secondary metabolites, Pulcaria crispa was found to have the most effective in vitro anticancer activity. The latter depicted the maximum inhibition concentration of 50% (IC50) in comparison with the extracts investigated against these cell lines (El-Naggar et al. 2015). With several applications in cancer research such as identification of biomarkers for disease detection, supervision of drug response and analysis of potential cytotoxic effects, metabolomic analysis can carry out extensive studies in a non-invasive way. It disseminates knowledge about cancer metabolites involved in the in situ methodology. It is based on the analysis of the metabolic fingerprint of cancer cells before and after treatments with plant extracts. Nuclear Magnetic Resonance (NMR) spectroscopy based metabolomics is effective for the juxtaposition of metadata of two different measurements to detect any changes. The spectra obtained can be subtracted to reveal the new signals using Heteronuclear Single Quantum Coherence (HSQC) spectroscopy (Kim et al. 2010). According to a study in 2016, NMR spectroscopy and multivariate analysis methods were also used to study the metabolic profile and inhibitory effects of Aloe Vera on Raji cells with an IC50 of 40 μg/ml. The metabolome elaborated on the influence of the surrounding conditions on the genome of an organism. It was reported to exert influence on human hepatocellular carcinoma cells by increasing expression of p53 and Bcl-2 gene. A wonder plant with many beneficial properties, it has significant anticancer and anti-tumorigenic properties (Noorolahi et al. 2016).
Certain secondary metabolites are not detected in plants due to low therapeutic activity and low concentrations. However, because of several intrinsic constituents in herbs and their formulations, synergistic biological activities are produced. In such cases of drug analysis, metabolomics acts as an efficient strategy to comprehend the phytochemical basis of a spectrum of these therapeutically active plant constituents. Owing to the technological boom and high-tech breakthroughs in the isolation and detection of metabolites, the technique of metabolomics is thus, rapidly emerging as a powerful platform for exploitation of anticancer plants, for biomarker-driven drug discovery and development. It offers a promising approach to plant metabolite fingerprinting and such research is urgently needed for better utilization of anticancer plants (Mukherjee et al. 2016).
12.3 Need for Bioinformatics
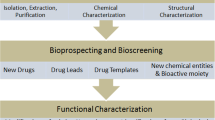
With emerging ground-breaking technologies, current cancer research has plunged into the era of systems biology, which is characterized by massive data generation through omics advances. In the last decade, with developments in modern biological technologies such as pyrosequencing, next generation sequencing and third generation sequencing (Fernandes et al. 2011), scientific discoveries have gained great momentum and sequence generation has become more cost-efficient and speedy. But these high throughput sequencing studies also face drawbacks such as data management, interpretation and quality control (QC) issues. Rapid medical achievements in the laboratory regarding the framework of diseases like cancer are certainly laudable. But, they have not necessarily translated into effective ‘treatment’ breakthroughs. Data sets these days extend from tens to hundreds of gigabytes per run due to which, data storage is another major concern. In other words, biological data are exploding, both in size and complexity. So, next generation analytical tools demand improved robustness, flexibility and cost efficiency. Comprehensive omics investigations, to understand cancer progression and treatment response, integrated with the power of bioinformatics allow exploitation of these achievements for future diagnostic, prognostic and therapeutic purposes. The challenges posed are being tackled effectively by incorporation of bioinformatics, computational biology and wet lab sciences (Al-Haggar et al. 2013) to understand the molecular and genetic bases of diseases like cancer (Fig. 12.1). They also help to enable system-level understanding correlation and dependencies between the molecular components involved.

Integration of omics technologies with bioinformatics in research of anticancer plants
Network analysis has applications in mining of biological knowledge from these data with high-fidelity and performing data-driven biology. Coupled with the newly emerging genome editing advances and exhaustive gene expression using microarrays, it has become a standard tool for studying gene function as well as metabolic pathways that promote cancer cell survival and growth (Yonekura-Sakakibara et al. 2013). Bioinformatics also allows researchers to search biologic databases, compare gene sequences and protein data on a vast scale in order to determine sequences or proteins that vary among cancerous and physiologic cells and tissues or different phenotypes of the same disease. In conjugation with omics, it is used for identification of therapeutic targets and drug design. Relevant bioinformatics technologies have really transformed extant thinking on nuclear genome, transcriptome, proteome and metabolome evolution.
12.4 Bioinformatics Approaches for Applications of Anticancer Plants
There are several bioinformatics tools, programs, web servers, softwares and pipelines that have contributed towards genomics and postgenomicsstudies on anticancer plants or have the potential to do so.
12.4.1 Genomics Applications
This section states the bioinformatics approaches for genomics applications of anticancer plants (Tables 12.1 and 12.2).
12.4.1.1 Genome Assembly
12.4.1.1.1 Phrap
Phrap, a program that assembles shotgun DNA sequence data, can handle large datasets. It permits utilization of the read as a whole and not just the trimmed high quality part. In the presence of repeats, it improves the process of assembly by using a combination of user-supplied and internally computed data quality information. Instead of a consensus, it forms the contig sequence as an assortment of the highest quality parts of the reads. Phrap also provides information regarding assembly to also carry out trouble-shooting of assembly programs successfully (Machado et al. 2011).
12.4.1.1.2 Contig Assembly Program (CAP) and CAP3
The Contig Assembly Program finds the shortest common superstring of a set of fragments. It supports DNA shotgun sequencing with the help of a filter that removes DNA fragment pairs that cannot overlap. For the remaining fragments, computation of maximal-scoring overlapping alignment is done. Then, the fragments are assembled in order of alignment scores. The CAP sequence assembly program has been improved to give the CAP3 program. It uses forward-reverse constraints to rectify assembly errors and connect contigs, and base quality values to compute overlaps between sequence reads, create multiple sequence alignments of reads and give consensus sequences. CAP3 also has the ability to automatically clip 5′ and 3′ poor regions of reads. Compared to Phrap, it is better at constructing scaffolds on low-pass data.
12.4.1.1.3 BAC Card I
Though there are several tools that carry out post-processing of sequence assemblies, there aren’t as many for construction of virtual clone maps from assembly data. These maps are useful for whole genome shotgun assembly validation and integration of different types of genomic data. The BAC Card I tool is used for this purpose. It computes contig scaffolding, looks for misassemblies and carries out intergenome comparison between related strains as well.
12.4.1.1.4 SOAP Denovo and SOAP Denovo 2
SOAP denovo is a method to assemble short-reads, especially from Illumina GA. It can carry out analyses of large plant genomes that have not been explored. However, it needed several improvements to be more efficient. This is solved by its successor, SOAPdenovo2, which has enhanced accuracy, continuity and coverage, especially for repeat regions in contig assembly and scaffold construction (Li et al. 2010).
12.4.1.1.5 Velvet
Velvet is a novel set of de novo assembly methods. It is based on the manipulation of de Brujin graphs for short read sequencing data. It can remove errors, build contigs and simplify repeated regions both in the presence and absence of read pair information. It is particularly useful when an organism without a reference genome needs to be studied (Zerbino 2010).
12.4.1.2 Whole Genome Sequencing Analysis
12.4.1.2.1 SeqMap
SeqMap can be used for mapping millions of short sequences to a genome of billions of nucleotides, using an index-filtering algorithm. It detects the positions in the reference genome where the sequences are located along with several substitutions, insertions and deletions of nucleotide bases. This is extremely important for high-throughput sequencing analysis.
12.4.1.2.2 RMAP
RMAP is aimed at accurately mapping paired-end reads from next-generation sequencing technology. It uses more advanced quality scores that are indicators of error probability. The length of the read must be between 20 and 64bp. The user can fix the maximum number of mismatches between a read and the location to which it maps.
12.4.1.2.3 Zillions of Oligos Mapped (ZOOM)
With next generation sequencing technologies, huge amounts of short reads are generated. However, for the identification of SNPs or unpopular transcripts, there is a need for software that can map these to a reference genome. ZOOM is used for mapping of oligonucleotides back to the genomes through spaced seeds. It is unparalleled in speed and very sensitive. It has applications in personalized medicine for cancer and re-sequencing.
12.4.1.2.4 Next Generation Sequencing Eclipse Program (NGSEP)
A need was felt for this bioinformatics software tool to carry out accurate and efficient analysis of data generated by high throughput sequencing simultaneously with every GBS protocol. NGSEP is useful for genetic mapping of complex traits and other downstream analyses as well. It can play a major role in genetic improvement of anticancer plants and genomic selection through phenotype prediction (Duitama et al. 2014; Perea et al. 2016).
12.4.1.2.5 Mapping and Assembly with Quality (MAQ)
Particularly designed for Illumina-Solexa 1G Genetic Analyzer, Maq is a program for measuring alignment quality of reads generated by next-generation sequencing technologies. It does ungapped alignment at the mapping stage and calls the consensus at assembling stage. There are up to two or three mismatches in the hits in case of single-end reads and around one in case of paired-end reads.
12.4.1.2.6 SAMtools
This is a set of programs for interacting with high-throughput sequencing data. The DNA sequence read alignments can be in SAM (Sequence Alignment/Map), BAM (Binary Alignment/Map) and CRAM formats. They can be viewed, sorted and indexed with the help of the provided tools, and are also inter-convertible. Two key features are that there is a provision to operate compressed BAM files without having to un-compress them and there is also a provision to compress large SAM files for saving space.
12.4.1.3 Nucleotide Sequence Homology Search
12.4.1.3.1 Nucleotide-Nucleotide Basic Local Alignment Search Tool (BLASTN)
The user specifies a nucleotide sequence. BLASTN searches the nucleotide database to carry out comparison with the nucleotide query (Madden 2013).
12.4.1.3.2 Nucleotide 6-Frame Translation-Nucleotide 6-Frame Translation (TBLASTX)
This program is the slowest one in the BLAST family since it incorporates a complex algorithm. The user specifies a nucleotide sequence. TBLASTX translates this query in all six possible reading frames and compares it to all six reading frames of the nucleotide sequence database. This is helpful in detecting distant relationships between nucleotide sequences (Madden 2013).
12.4.1.3.3 Protein-Protein Basic Local Alignment Search Tool (BLASTP)
The user specifies a protein sequence. BLASTP searches the protein sequence database to carry out comparison with the protein query (Madden 2013).
12.4.1.3.4 Protein-Nucleotide 6-Frame Translation (TBLASTN)
The user specifies a protein sequence. TBLASTN compares this query with all six reading frames of the nucleotide sequence database. This is helpful in identification of proteins in uncharacterized genomes (Madden 2013).
12.4.1.4 Comparative Genome Visualisation
12.4.1.4.1 Connectivity Map (cMap) and cMap3D
cMap is a graphical utility for comparing genetic and physical maps within and between related species. It has three components – database cMapDB that can be used for a variety of mapping applications, the user interface and a data retriever. This tool specifies loci, positions and linkage groups. However, it has a limitation of carrying out comparison of only adjacent aligned maps. CMap3D solves this problem by allowing comparsion of several genetic maps in three-dimensional space (Duran et al. 2010; Segal et al. 2012).
12.4.1.4.2 SynBrowse
SynBrowse is a tool for generic sequence comparison that helps in the visualization and analysis of genome alignments within and between species. It is useful for the user to study synteny, homologous genes and other conserved elements between sequences. By carrying out comparison with a reference genome, it also helps in identifying unspecified genes, putative regulatory elements and distinguishable features of a species (Lee et al. 2016).
12.4.1.5 Restriction Site Associated DNA Sequencing Analysis (RAD-Seq)
12.4.1.5.1 pyRAD
pyRAD is a software for the assembly of de novo RAD-seq loci. It has the ability to assemble datasets at fast pace due to its parallel processing and optional hierarchical clustering methods. This pipeline has an advantage over Stacks since there is no disruption of homologous loci clusters by indels in PyRAD (Eaton 2014).
12.4.1.5.2 Radis
Illumina sequencing generates large amounts of data which can be processed for phylogenetic inference using the perl pipeline, RADIS. It makes exploration of RAD-Seq data easier and faster. However, it depends on Stacks for eliminating PCR duplicates, establishing loci and separating data from the multiplex system (Cruaud et al. 2016).
12.4.1.5.3 Stacks
Stacks are a widely used pipeline for converting short-read sequences produced by the Illumina platform to specific loci. Its main function is de novo assembly of RAD sequences for genetic maps. It can be used to examine anticancer plants that have or do not have a reference genome and is hence, flexible software. It can also be used for conducting population genomics to understand adaptation in wild plants with anticancer properties.
12.4.1.6 Protein-Coding Gene Prediction
12.4.1.6.1 Augustus
AUGUSTUS is a web server that predicts genes in eukaryotic genomic sequences by the means of a Generalized Hidden Markov Model (GHMM), a generative probabilistic and statistical model. It helps the server by considering both intrinsic and extrinsic information to give hints about potential protein-coding regions. Finally, the most likely gene structure that is in accord with the constraints specified by the user is the result though there are cases in which no such structure exists. The constraints are significant especially when only a segment of the gene structure is known.
12.4.1.6.2 Exonerate
Exonerate is a genome annotation tool for protein-coding gene prediction. It allows pairwise sequence juxtaposition and gives the user the ability to align sequences using several alignment models that involve either dynamic programming or are a hands-on interaction.
12.4.1.7 Tandem Repeat Prediction
12.4.1.7.1 PolySSR
PolySSR is a novel approach to identify short sequence repeats (SSRs) as well as putatively polymorphic SSRs. The information regarding sequences is derived from public EST databases that include heterozygous individuals and different genotypes. The success rate for potential polymorphic SSR markers is elevated since it considers the presence of SNPs in the flanking regions for PCR-primer design.
12.4.1.7.2 SA-SSR
SA-SSR is accurate and comprehensive software for efficient detection of SSRs in a database with multiple sets of sequences. It is based on suffix and longest common prefix arrays and addresses most of the problems faced by extant tandem repeat prediction algorithms. One of its key features is that it gives a lot of control to the user (Pickett et al. 2016).
12.4.1.7.3 SSR Analysis Tool (SAT)
Microsatellites are one of the most powerful genetic markers. One method for their development is by constructing genomic DNA libraries, followed by DNA sequencing to get SSR markers from the bulk sequence data. But this is a highly intensive and slow process. The SSR Analysis Tool (SAT), a Web application, addresses this challenge effectively. It facilitates the integration, analysis and display of sequence data from these libraries. It can also design PCR primers specifically.
12.4.1.7.4 Poly
There are many programs that detect SSRs tracts but are unable to analyse the results quantitatively. Poly is a tool that carries this out effectively by easily quantifying the frequencies at which SSRs are located relative to other DNA sequence elements, and the tract length as well. It is not a fast program but is technically sounder than its substitutes.
12.4.1.8 Synteny Block Detection
12.4.1.8.1 Synteny Mapping and Analysis Program (SyMAP)
SyMAPv4.0 is a software package that detects, computes, displays, analyses and queries genome syntenic relationships. It has applications only in medium-to-high divergent eukaryotic genomes, and is particularly for studying monocotyledons and eudicotyledons. It has multiple display modes such as circular, side-by-side, dotplot, closeup, etc. (Soderlund et al. 2011).
12.4.1.8.2 SynChro
This tool is created to reconstruct conserved synteny blocks between comparisons of genomes in pairs. It is formulated on a method that simply calculates the Reciprocal Best-Hits (RBH) to build the synteny blocks. It then, finishes off these blocks with non-RBH syntenic homologs automatically. SynChro functions at a fast pace by adjusting a few parameters only and with the help of multiple essential visualization tools (Drillon et al. 2014).
12.4.1.9 Others
12.4.1.9.1 Consed
After sequence assemblies are created with phrap, they need to be viewed. The user should be directed to the variant sites one by one. The incorrect assemblies need to be detected, edited and aligned. All this is done with the help of Consed tool. It picks primers for amplification and templates efficiently. Furthermore, it includes BamScape, which is able to view bam files with unlimited number of reads as well as promote editing of the reference sequence in targeted regions (Gordon and Green Gordon and Green 2013a, b).
12.4.1.9.2 MultiPool
MultiPool is used for mapping of genetic elements from pooled genotyping. When sequencing depth and marker spacing are varied, noise levels become more non-uniform. In such a case, this computational method is used. It allows information sharing across the variant locations as well and uses a dynamic Bayesian network (DBN). This method can be extended to any number of replicates (Gordon and Green Gordon and Green 2013a, b).
12.4.1.9.3 Tassel-GBS
The bioinformatics pipeline, Tassel-GBS was designed to identify and call SNP genotypes by processing next-generation raw genotyping by sequencing data. It can aid GBS in the process of breeding anticancer plants and studying genomic diversity. This method has extremely high utility primarily for two reasons. Firstly, it can be used by those who do not have access to sophisticated computing resources. Secondly, it has the ability for large analyses but it can also be run at smaller scales (Glaubitz et al. 2014).
12.4.1.9.4 Maker
MAKER is a pipeline that allows plants with less complex genomes to annotate their genomes and create databases. It is portable, easily trainable and configurable. In the initial runs, the outputs automatically develop the gene prediction algorithm leading to a better quality gene model in the following runs, and are directly entered into the Generic Model Organism Database (GMOD). The quality indices of the annotations play an instrumental role in the functioning of MAKER (Campbell et al. 2014).
12.4.1.9.5 Bowtie
The Bowtie package is an extremely fast and memory-efficient bioinformatics tool. It aligns short sequencing reads output by next generation sequencing technologies. These alignments can be used to build genome indices and call consensus sequences (Langmead 2010).
12.4.1.9.6 Blast2GO
Blast2GO is an all-inclusive bioinformatics platform for the functional annotation of genomic datasets through high-throughput technologies. Subsequently, data mining is done on the output based on GO vocabulary. With an interactive and user-friendly interface and easy operability, it is a highly suitable software tool for plant genomics research. One of the prime reasons for this is that it can be extended to a vast number of species and be customised accordingly.
12.4.2 Transcriptomics Applications
This section states the bioinformatics approaches for transcriptomics applications of anticancer plants (Table 12.3).
12.4.2.1 Gene Expression Analysis
12.4.2.1.1 Database for Annotation, Visualisation and Integrated Discovery (DAVID)
DAVID is a web-based program that provides functional annotation tools for large lists of genes or proteins. It facilitates the transition from data collection to biological meaning. Its tools summarize the datasets according to Gene Ontology terms, protein functional domains and motifs, and biochemical pathways. They also associate genes with diseases, detect interacting proteins and explore gene names in batch.
12.4.2.1.2 EGAssembler
EGAssembler is a web server which analyses expressed sequence tags and has the ability to process large volumes of data rapidly. It can merge and align genomic fragments generated from shotgun sequencing to give the initial sequence. It has in-built tools as well as tools that can be modified by the user. They are capable of clustering and EST assembly as well.
12.4.2.1.3 KeggOrthology-Based Annotation System (KOBAS)
KOBAS is a web server that has a reservoir of knowledge regarding diseases, gene ontology and enriched pathways, derived from significant databases like KEGG, BioCyc, OMIM, etc. Its improved versions - KOBAS 2.0 and KOBAS 3.0 can be accessed online. It is a popular tool for gene expression analysis that performs statistical tests to give output (Xie et al. 2011).
12.4.2.2 miRNA Prediction
12.4.2.2.1 Ingenuity Pathway Analysis (IPA)
IPA is a powerful web-based software application that has a wide range of features that enable searching, viewing, modelling, understanding, analysing and visualising complex omics’ data. This data can be derived from miRNA and SNP microarrays, RNA-Seq and other omics experiments. It is a popular tool that provides an insight into biological and chemical interactions pivotal to research, and discovers the processes for diseases like cancer, neurological disease, cardiovascular disease, etc. (Henderson-Maclennan et al. 2010).
12.4.2.2.2 psRNATarget
psRNATarget is a novel miRNA target prediction server that features the ability to differentiate between translational and post-translational inhibition. This tool is designed for the analysis of data generated from high-throughput sequencing and transcriptomics technologies. It allows reverse complementary matching between non-coding RNA and the RNA that has already been transcribed, along with the evaluation of the target site (Dai and Zhao 2011).
12.4.2.3 Spliced Read Alignment
12.4.2.3.1 TopHat and TopHat 2
TopHat is a bioinformatic tool for high throughput alignment of shotgun transcriptomiccDNA sequencing reads. It does fast splice junction mapping for RNA-Seq reads by alignment carried out as a two-fold process. Firstly, unspliced reads are aligned with the ultra high-throghput short read aligner Bowtie. Then, the mapping reads are analyzed to discover RNA splice junctions de novo. An improved version of TopHat is TopHat2, which can align reads of various lengths across small indels accurately and produce sensitive alignments even for unfavourable genomes (Kim et al. 2013).
12.4.2.3.2 SpliceMap
SpliceMap is a highly sensitive de novo splice junction discovery tool. It offers support for arbitrarily long RNA-seq read lengths. Though more inclined toward mammalian genomes, it can also be used on anticancer plant genomes (Au et al. 2010).
12.4.2.4 De Novo Transcriptome Assembly
12.4.2.4.1 SOAPdenovo-Trans
RNA-Seq had become a favorable method to tackle the increase in throughputs cost-effectively. But short reads from next generation sequencing make their assembly to recover full transcript sequences a herculean task. SOAPdenovo-Trans, a de novotranscriptome assembler, has been designed specifically to solve this problem. It adjusts with alternative splicing and variable expression levels amid transcripts. This assembler is faster and provides higher contiguity as well as lower obsolescence (Xie et al. 2014).
12.4.2.4.2 Oases
Oases is a de novo transcriptome assembler that performs its functions even when there is no reference genome. It assembles RNA-Seq reads from technologies such as Illumina, SOLiD or 454 to produce transcripts. It uses an array of hash lengths, a dynamic noise filter, a resolution of alternative splicing events and merging of multiple assemblies (Schulz et al. 2012).
12.4.2.5 Others
12.4.2.5.1 Cufflinks
Cufflinks is a great tool for measurement of de novo transcript isoform expression. It carries out assembly of transcripts by assembling the alignments into transcript sets. It is also responsible for estimating their abundances and determining differential expression. Cufflinks regulates RNA-Seq samples by accepting the aligned reads. Then, depending on how many read support each transcript, their relative abundances are estimated (Trapnell et al. 2010).
12.4.2.5.2 miRCat and miRCat2
miRCat can identify mature miRNAs and their precursors from high-throughput plant sRNA datasets. Thus, it does not require a putative precursor sequence since it is predicted by the program itself. A tool depicting high sensitivity and specificity, miRCat searches for sRNA-covered genomic regions after the sequences are mapped to the input genome. After a list of loci has been created, it is probed further for likely miRNA candidates. All reads must have a certain abundance, whose level can be varied using the minimum abundance parameter. Finally, the sRNA read with the most abundance within a locus is identified as the likely miRNA. miRCAt2, an improved version of miRCat, has much lower high false positive and false negative rates, and predicts miRNA loci using a novel entropy-based approach (Paicu et al. 2017).
12.4.3 Proteomics Applications
This section states the bioinformatics approaches for proteomics applications of anticancer plants (Table 12.4).
12.4.3.1 Protein Sequence Analysis
12.4.3.1.1 Cluster Database at High Identity with Tolerance (CD-HIT)
CD-HIT is a popular program that clusters and compares proteins that meet a similarity threshold to reduce redundancy and correct the bias within a dataset. This enhances the analyses of other sequences as well and helps the user in understanding data structures. It can handle large databases and works at a fast pace, hence reducing manual curation (Fu et al. 2012c).
12.4.3.1.2 Conserved Domain Database (CDD)
Conserved Domain Database is a public resource that incorporates domain models derived from databases like Pfam, SMART, etc. It is primarily responsible for proteins’ annotation. One of its key features is that it can identify conserved domains in protein sequences. The NCBI-curated domains use three-dimensional structural data to elucidate on structure relationships (Marchler-Bauer et al. 2013, 2015).
12.4.3.2 Protein-Protein Interaction Prediction
12.4.3.2.1 Search Tool for the Retrieval of Interacting Genes/Proteins (STRING)
STRING is a biological database and web resource that evaluates and integrates physical and functional protein-protein interactions from co-expression data. These associations can be predicted or can be previously known, and are statistically analyzed by the confidence scores generated. STRING incorporates classification systems like GO, Pfam and KEGG as well as sources like experimental data, computational prediction methods and public text collections. It is accessible to all and is updated within regular intervals of time (Szklarczyk et al. 2015).
12.4.3.2.2 Pathway Studio
Pathway Studio is pathway software developed for the visualisation of proteomics data and analysis of protein interaction maps. It has applications in interpretation of gene expression, metabolomics and other high throughput data as well. It is most useful for budding researchers who wish to gain further insight into their independent discoveries and prevents them from carrying out investigations about subjects that have already been deeply explored (Henderson-Maclennan et al. 2010).
12.4.3.3 Protein Database Search
12.4.3.3.1 Position-Specific Iterative BLAST (PSI-BLAST)
PSI-BLAST is tool similar to BLAST with the difference that the former uses position-specific scoring matrices (PSSM) generated during the search. It also searches the protein databases one by one for sequences matching the protein query. It can search the target databases several times using multiple alignments of sequences above a certain score threshold. With every round of searching, a new PSSM is generated. This PSSM is further used to search the database for new matches (Madden 2013).
12.4.3.3.2 Nucleotide 6-Frame Translation-Protein (BLASTX)
The user specifies a nucleotide sequence. BLASTX searches the protein databases to carry out comparison with the translated nucleotide query. It occurs in one step itself (Madden 2013).
12.4.3.4 Mass Spectrometry Based Proteomics
12.4.3.4.1 MassLynx
MassLynx software manages, edits, analyzes and shares mass spectrometry information. It improves the MS system with features like instinctive interface and good instrument control. This software package is extremely versatile and flexible.
12.4.3.4.2 Mascot, and Mascot File Parsing and Quantification (MFPaq)
Mascot software is a benchmark for the identification of proteins from primary sequence databases. This powerful search engine is used for characterization and quantitation of proteins using mass spectrometry data. Unlike its contemporaries, it is capable of integrating all proven search methods like peptide mass fingerprinting, sequence query and identification of fragment ions from uninterpreted MS/MS data (MS/MS Ion Search). The Mascot result files can be easily verified by the web application MFPaQ. It also carries out data quantification using isotopic labeling methods like stable isotope labeling with amino acids in cell culture (SILAC) or isotope-coded affinity tags (ICAT). It can also do so via label-free approaches such as spectral counting or MS signal comparison. MFPaQ also creates and juxtaposes non-redundant protein lists.
12.4.3.4.3 IsobariQ
Isobariq involves isobaric labeling of proteins that plays an integral role in quantitative mass spectrometry. It has applications in data derived from isobaric peptide termini labeling (IPTL), isobaric tags for relative and absolute quantitation (iTRAQ) and tandem mass tags (TMT). The user can study the proteomes extensively and through an interactive graphical user interface. This tool uses variance stabilizing normalization (VSN) algorithms (Arntzen et al. 2011).
12.4.3.5 Others
12.4.3.5.1 HexServer
HexServer is a protein docking server that blazed a trail. Based on the mathematical concept of the Fourier-transform, it was the first one of its kind to be powered by graphics processers. Structures can be inputted from the protein data bank (PDB) to generate a series of docking predictions without the need of any sophisticated computing infrastructure (Macindoe et al. 2010).
12.4.3.5.2 MODELLER
MODELLER is a computer program used for analysis of three-dimensional protein structures and in some cases, quaternary structures as well. The user inputs a sequence alignment that is then, modelled homologically. This software tool is based on satisfaction of spatial restraints, a method that has been derived from the transition of NMR spectroscopy data to three-dimensional structures (Webb and Sali 2014, 2016).
12.4.4 Metabolomics Applications
This section states the bioinformatics approaches for metabolomics applications of anticancer plants (Table 12.5).
12.4.4.1 Metabolite Library
12.4.4.1.1 RIKEN Tandem Mass Spectral Database (ReSpect)
RIKEN tandem mass spectral database is an online database of tandem mass spectroscopy data that has applications in plant metabolomics research. One of its major uses is that it helps in narrowing down complex phytochemical structures to candidate structures. It is constructed on the basis of a fragmentation association rule that helps in assessing confidence levels of annotations obtained by the user, as well as the structural characterization of metabolites (Sawada et al. 2012).
12.4.4.1.2 Metabolite and Tandem MS Database (METLIN)
METLIN is a repository that stores and manages data pertaining to metabolites as well as tandem mass spectrometry. With multiple searching capabilities, it provides MS/MS data at varying collision energies through a positive as well as a negative ionization approach. It helps the user with basic information such as the name, theoretical mass, chemical formula, structure and elemental composition of the metabolite along with its fragment structure. METLIN is also linked with databases like KEGG and HMDB to help the user in compound identification.
12.4.4.2 Metabolomic Network Data and Analysis
12.4.4.2.1 MetaCrop
MetaCrop is a manually-updated database that not just provides high-quality information about metabolic pathways, but also allows its automatic export for building metabolic models. It aids plant metabolomic research by improving yields of crops with potential anticancer properties. It also helps the user with details such as the location of the crop, its transport and related reaction kinetics. Its successor, MetaCrop 2.0 was released in 2011 (Schreiber et al. 2012).
12.4.4.2.2 MetaCore
MetaCore is integrated software suite based on a database of chemical metabolism, molecular interactions and pathways. This database is regularly updated and manually managed. It also provides information regarding toxicity, gene-disease associations, functional analysis of Next Generation Sequencing and many more molecular classes.
12.4.4.3 Mass Spectrometry Based Metabolomics
12.4.4.3.1 MetPa
An integration of statistical enrichment methods with pathway topological characteristics, MetPA is a user-friendly and fully-featured tool. It is responsible for studying relevant metabolic pathways and aids data visualisation through a network system with several features just like Google Maps. It makes data analysis possible by generating a report automatically with the help of statistical procedures (Xia and Wishart 2010).
12.4.4.3.2 MetaboAnalyst
MetaboAnalyst is an online pipeline for analysis and interpretation of data generated from high-throughput metabolomics technologies. Its successors, MetaboAnalyst 2.0 and MetaboAnalyst 3.0 were released in January 2012 and April 2015 respectively. It offers numerous approaches for processing, normalization and annotation of the metabolomic data. It involves advanced statistical analysis methods for carrying out metabolomic studies (Xia et al. 2012, 2015).
12.4.4.4 Nuclear Magnetic Resonance (NMR) Based Metabolomics
12.4.4.4.1 ProMetab
ProMetab is a nuclear magnetic resonance based metabolomics tool that is responsible for data processing. It facilitates the transition from raw NMR spectra to a format for multivariate chemometric and biometric analysis.
12.4.4.4.2 MetaboHunter
MetaboHunter is a user-friendly and freely accessible web server application that enables automatic identification in H-NMR spectra of metabolites. It provides fast-paced metabolic fingerprinting and several effective search methods. Based on compound peak lists and intuitive plotting, it aids in visualisation and is capable of identifying more than 80% of detectable metabolites on an average from spectra of complex mixtures (Tulpan et al. 2011).
12.5 Conclusions and Future Prospects
Recent developments in technologies and instrumentation allow large-scale as well as nano-scale examination of biological samples. Ranging from turmeric (Curcuma longa) to Aloe vera to holy basil (Ocimum tenifluorum) to flowering plants of the coffee and even, asparagus family, plants have multifarious applications in cancer treatment and prevention. The anticancer agents derived from these plants have been classified into vinca alkaloids, epipodophyllotoxins, taxanes and camptothecin derivatives, and have proven to be extremely successful in the discovery of novel medicines. They have untapped potential for novel molecular target discovery and can contribute significantly to the drug development process. Medicinal plants can be probed for compounds with anticancer properties through omics strategies. Since the completion of the first draft of the human genome, an exceptional abundance of biological data has been churned out. Hence, in order to expedite cancer research, it is crucial to integrate systems biology, omics-based technology, bioinformatics and computational biology all together. Interactions and networks between genes and proteins are pivotal for the examination of cancer molecular mechanisms. Weight has shifted from genes to gene products. Hence, it is not only important to strategize and research at the level of the genome, but also at the level of proteome, transcriptome and metabolome. High-throughput sequencing has given an entire facelift to genomics and transcriptomics. Recombinant DNA technology and microarray technology have also become significantly important genomics and transcriptomics techniques respectively. In order to study proteomes, an increasing number of plant biologists are adopting mass spectrometry that aids protein identification, quantitation and structure determination. De novo peptide sequencing is also a popular mass spectrometry technique to determine a peptide sequence without any prior knowledge of the constituent amino acids. It is usually carried out in conjunction with two-dimensional gel electrophoresis that has applications in protein analysis. To study plant metabolism, the omics research technique of nuclear magnetic resonance spectroscopy is also adopted.
In modern biology and medicine, the analysis of huge amount of digital data derived from omics technologies has been overseen by the progressing science of bioinformatics. It allows effortless retrieval, assimilation, prediction and storage of DNA and protein sequence data with the help of efficient software programs and biological databases. With applications in not just genome sequence analysis, but also analysis of gene variation and expression, gene regulation dynamics, protein structure and function, this burgeoning field is being stressed upon globally. It assists molecular biologists in harvesting the fruits put forth by computational biology. This is all the more relevant since disorders are no more investigated by just probing isolated genes. It has been expanded to gene networks, their interactions and roles in diseases like cancer. This has set the ball rolling for a whole new era of personalised medicine. As research in the field of anticancer plants progresses, more and more bioinformatics tools are being developed to make data extraction, processing, management, storage, analysis, interpretation and integration exceedingly efficient. They play a vital role in today’s plant science. The challenge is thus, to employ these technologies effectively and optimally so as to solve oncological problems.
References
Al-Haggar MMS, Khair-Allaha BA, Islam MM, Mohamed ASA (2013) Bioinformatics in high throughput sequencing: application in evolving genetic diseases. J Data Mining Genom Proteom 4:3. https://doi.org/10.4172/2153-0602.1000131
Alsemari A, Alkhodairy F, Aldakan A, Al-Mohanna M, Bahoush E, Shinwari Z, Alaiya A (2014) The selective cytotoxic anticancer properties and proteomic analysis of Trigonella foenum-graecum. BMC Complement Altern Med 14:114. https://doi.org/10.1186/1472-6882-14-114
Anantachoke N, Tuchinda P, Kuhakarn C, Pohmakotr M, Reutrakul V (2012) Prenylated caged xanthones: chemistry and biology. Pharm Biol 50:78–91
Annadurai RS, Neethiraj R, Jayakumar V, Damodaran AC, Rao SN, Katta MA, Gopinathan S, Sarma SP, Senthilkumar V, Niranjan V, Gopinath A (2013) De novo transcriptome assembly (NGS) of Curcuma longa L. rhizome reveals novel transcripts related to anticancer and antimalarial terpenoids. PLoS One 8:e56217. https://doi.org/10.1371/journal.pone.0056217
Armitage EG, Southam AD (2016) Monitoring cancer prognosis, diagnosis and treatment efficacy using metabolomics and lipidomics. Metabolomics 12:146
Arntzen MO, Koehler CJ, Barsnes H, Berven FS, Treumann A, Thiede B (2011) IsobariQ: software for isobaric quantitative proteomics using IPTL, iTRAQ, and TMT. J Proteome Res 10:913–920
Atanasov AG, Waltenberger B, Pferschy-Wenzig EM, Linder T, Wawrosch C, Uhrin P, Temml V, Wang L, Schwaiger S, Heiss EH, Rollinger JM (2015) Discovery and resupply of pharmacologically active plant-derived natural products: a review. Biotechnol Adv 33:1582–1614
Au KF, Jiang H, Lin L, Xing Y, Wong WH (2010) Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res 38:4570–4578
Boutanaev AM, Moses T, Zi J, Nelson DR, Mugford ST, Peters RJ, Osbourn A (2015) Investigation of terpene diversification across multiple sequenced plant genomes. Proc Natl Acad Sci U S A 112:E81–E88
Cai J, Liu X, Vanneste K, Proost S, Tsai WC, Liu KW, Chen LJ, He Y, Xu Q, Bian C, Zheng Z (2015) The genome sequence of the orchid Phalaenopsis equestris. Nat Genet 47:65–72
Campbell MS, Holt C, Moore B, Yandell M (2014) Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinformatics 12:4–11
Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B, Corréa M (2014) Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345:950–953
Chantarasriwong O, Batova A, Chavasiri W, Theodorakis EA (2010) Chemistry and biology of the caged Garcinia xanthones. Chem Eur J 16:9944–9962
Chen S, Sun Y, Xu J, Luo H, Sun C, He L, Cheng X, Zhang B, Xiao P (2010) Strategies of the study on herb genome program. Yao Xue Xue Bao 45:807–812
Chen T, Zhang RH, He SC, Xu QY, Ma L, Wang GC, Qiu N, Peng F, Chen JY, Qiu JX, Peng AH (2012) Synthesis and antiangiogenic activity of novel gambogic acid derivatives. Molecules 17:6249–6268
Cragg GM, Newman DJ (2013) Natural products: a continuing source of novel drug leads. Biochim Biophys Acta 1830:3670–3695
Cruaud A, Gautier M, Rossi JP, Rasplus JY, Gouzy J (2016) RADIS: analysis of RAD-seq data for interspecific phylogeny. Bioinformatics 32:3027–3028
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F, Aprea G, Aury JM (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181–1184
Dijkstra KK, Voabil P, Schumacher TN, Voest EE (2016) Genomics and transcriptomics based patient selection for cancer treatment with immune checkpoint inhibitors: a review. JAMA Oncol 2:1490–1495
Drillon G, Carbone A, Fischer G (2014) SynChro: a fast and easy tool to reconstruct and visualize synteny blocks along eukaryotic chromosomes. PLoS One 9:e92621
Duitama J, Quintero JC, Cruz DF, Quintero C, Hubmann G, Foulquié-Moreno MR, Verstrepen KJ, Thevelein JM, Tohme J (2014) An integrated framework for discovery and genotyping of genomic variants from high-throughput sequencing experiments. Nucleic Acids Res 42:e44. https://doi.org/10.1093/nar/gkt1381
Duran C, Boskovic Z, Imelfort M, Batley J, Hamilton NA, Edwards D (2010) CMap3D: a 3D visualization tool for comparative genetic maps. Bioinformatics 26:273–274
Eaton DA (2014) PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30:1844–1849
Edwards MD, Gifford DK (2012) High-resolution genetic mapping with pooled sequencing. BMC Bioinform 13:S8
El-Naggar SA, Abdel-Farid IB, Elgebaly HA, Germoush MO (2015) Metabolomic profiling, antioxidant capacity and in vitro anticancer activity of some compositae plants growing in Saudi Arabia. Afr J Pharm Pharmacol 9:764–774
Fang HY, Chen SB, Guo DJ, Pan SY, Yu ZL (2011) Proteomic identification of differentially expressed proteins in curcumin-treated MCF-7 cells. Phytomedicine 18:697–703
Fernandes F, da Fonseca PG, Russo LM, Oliveira AL, Freitas AT (2011) Efficient alignment of pyrosequencing reads for re-sequencing applications. BMC Bioinform 12:163
Fridlender M, Kapulnik Y, Koltai H (2015) Plant derived substances with anticancer activity: from folklore to practice. Front Plant Sci 6:799
Fu WM, Zhang JF, Wang H, Tan HS, Wang WM, Chen SC, Zhu X, Chan TM, Tse CM, Leung KS, Lu G (2012a) Apoptosis induced by 1,3,6,7-tetrahydroxy xanthone in Hepatocellular carcinoma and proteomic analysis. Apoptosis 12:842–851
Fu WM, Zhang JF, Wang H, Xi ZC, Wang WM, Zhuang P, Zhu X, Chen SC, Chan TM, Leung KS, Lu G (2012b) Heat shock protein 27 mediates the effect of 1,3,5-trihydroxy-13,13-dimethyl-2H-pyran [7,6-b] xanthone on mitochondrial apoptosis in hepatocellular carcinoma. J Proteome 75:4833–4843
Fu L, Niu B, Zhu Z, Wu S, Li W (2012c) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152
Glaubitz JC, Casstevens TM, Lu F (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9:e90346
Gordon D, Green P (2013a) Consed: a graphical editor for next-generation sequencing. BMC Bioinform 29:2936–2937
Gordon D, Green P (2013b) Consed: a graphical editor for next-generation sequencing. Bioinformatics 29:2936–2937
Gupta P, Goel R, Agarwal AK, Asif MH, Sangwan NS, Sangwan RS, Trivedi PK (2015) Comparative transcriptome analysis of different chemotypes elucidates withanolide biosynthesis pathway from medicinal plant Withania somnifera. Sci Rep 5:18611
Hao DC, Xiao PG (2015) Genomics and evolution in traditional medicinal plants: road to a healthier life. Evol Bioform Online 11:197–212
Hao DC, Chen SL, Osbourn A, Kontogianni VG, Liu LW, Jordán MJ (2015a) Temporal transcriptome changes induced by methyl jasmonate in Salvia sclarea. Gene 558:41–53
Hao DC, Xiao PG, Liu LW, Peng Y, He CN (2015b) Essentials of pharmacophylogeny: knowledge pedigree, epistemology and paradigm shift. China J Chin Mat Med 40:1–8
Hao DC, He CN, Shen J, Xiao PG (2017) Anticancer chemodiversity of Ranunculaceae medicinal plants: molecular mechanisms and functions. Curr Genomics 18:39–59
Henderson-MacLennan NK, Papp JC, Talbot CC, McCabe ER, Presson AP (2010) Pathway analysis software: annotation errors and solutions. Mol Genet Metab 101:134–140
Horgan RP, Kenny LC (2011) Omic technologies: genomics, transcriptomics, proteomics and metabolomics. The Obstet Gynaecol 13:189–195
Huang WS, Kuo YH, Chin CC, Wang JY, Yu HR, Sheen JM, Tung SY, Shen CH, Chen TC, Sung ML, Liang HF (2012) Proteomic analysis of the effects of baicalein on colorectal cancer cells. Proteomics 12:810–819
Kalra S, Puniya BL, Kulshreshtha D, Kumar S, Kaur J, Ramachandran S, Singh K (2013) De novo transcriptome sequencing reveals important molecular networks and metabolic pathways of the plant, Chlorophytum borivilianum. PLoS One 8:e83336
Katiyar C, Gupta A, Kanjilal S, Katiyar S (2012) Drug discovery from plant sources: an integrated approach. Ayu 33:10–19
Kim HK, Wilson EG, Choi YH, Verpoorte R (2010) Metabolomics: a tool for anticancer lead-finding from natural products. Planta Med 76:1094–1102
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36
Kim BY, Park HS, Kim S, Kim YD (2017) Development of microsatellite markers for Viscum coloratum (Santalaceae) and their application to wild populations. Appl Plant Sci 5:1600102
Kumar M, Meena P, Verma S, Kumar M, Kumar A (2010) Anti-tumour, anti-mutagenic and chemomodulatory potential of Chlorophytum borivilianum. A Pac J Cancer Prev 11:327–334
Langmead B (2010) Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics 11:11.7.1–11.7.14. https://doi.org/10.1002/0471250953.bi1107s32
Lao Y, Wang X, Xu N, Zhang H, Xu H (2014) Application of proteomics to determine the mechanism of action of traditional Chinese medicine remedies. J Ethnopharmacol 155:1–8
Lee J, Hong WY, Cho M, Sim M, Lee D, Ko Y, Kim J (2016) Synteny portal: a web-based application portal for synteny block analysis. Nucleic Acids Res 44:W35–W40
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20:265–272
Liu Z, Ma L, Zhou GB (2011) The main anticancer bullets of the Chinese medicinal herb, thunder God vine. Molecules 16:5283–5297
Liu Y, Song F, Wu WK, He M, Zhao L, Sun X, Li H, Jiang Y, Yang Y, Peng K (2012) Triptolide inhibits colon cancer cell proliferation and induces cleavage and translocation of 14-3-3 epsilon. Cell Biochem Funct 30:271–278
Lo HY, Li CC, Huang HC, Lin LJ, Hsiang CY, Ho TY (2012) Application of transcriptomics in Chinese herbal medicine studies. J Tradit Compl Med 2:105–114
Lu Z, Song Q, Yang J, Zhao X, Zhang X, Yang P, Kang J (2014) Comparative proteomic analysis of anti-cancer mechanism by periplocin treatment in lung cancer cells. Cell Physiol Biochem 33:859–868
Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DV, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U (2011) Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov 10:188–195
Machado M, Magalhaes WCS, Sene A et al (2011) Phred-Phrap package to analyses tools: a pipeline to facilitate population genetics re-sequencing studies. Investig Genet 2:3. https://doi.org/10.1186/2041-2223-2-3
Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW (2010) HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res 38:W445–W449
Madden T (2013) The BLAST sequence tool. In: The NCBI handbook, 2nd edn. National Center for Biotechnology Information, Bethesda
Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F (2013) CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41:D348–D352
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43:D222–D226
Mukherjee PK, Harwansh RK, Bahadur S, Biswas S, Kuchibhatla LN, Tetali SD, Raghavendra AS (2016) Metabolomics of medicinal plants-a versatile tool for standardization of herbal products and quality evaluation of Ayurvedic formulations. Curr Sci 111:1624–1630
Newman DJ, Cragg GM (2016) Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 79:629–661
Noorolahi SM, Sadeghi S, Mohammadi M, Azadi M, Rahimi NA, Vahabi F, Arjmand M, Hosseini H, Mosallatpur S, Zamani Z (2016) Metabolomic profiling of cancer cells to Aloe vera extract by 1HNMR spectroscopy. J Metabol 2:1–7. https://doi.org/10.7243/2059-0008-2-1
Okada T, Mochamad Afendi F, Altaf-Ul-Amin M, Takahashi H, Nakamura K, Kanaya S (2010) Metabolomics of medicinal plants: the importance of multivariate analysis of analytical chemistry data. Curr Comput Aided Drug Des 6:179–196
Paicu C, Mohorianu I, Stocks M, Xu P, Coince A, Billmeier M, Dalmay T, Moulton V, Moxon S (2017) miRCat2: accurate prediction of plant and animal microRNAs from next-generation sequencing datasets. Bioinformatics 33:2446–2454
Perea C, De La Hoz JF, Cruz DF, Lobaton JD, Izquierdo P, Quintero JC, Raatz B, Duitama J (2016) Bioinformatic analysis of genotype by sequencing (GBS) data with NGSEP. BMC Genomics 17:S498. https://doi.org/10.1186/s12864-016-2827-7
Pickett BD, Karlinsey SM, Penrod CE, Cormier MJ, Ebbert MT, Shiozawa DK, Whipple CJ, Ridge PG (2016) SA-SSR: a suffix array-based algorithm for exhaustive and efficient SSR discovery in large genetic sequences. Bioinformatics 32:2707–2709
Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, Cheng J, Zhao S, Xu M, Luo Y, Yang Y (2014) Whole-genome sequencing of cultivated and wild peppers provides insights into capsicum domestication and specialization. Proc Natl Acad Sci USA 111:5135–5140
Rubin BE, Ree RH, Moreau CS (2012) Inferring phylogenies from RAD sequence data. PLoS One 7:e33394
Sallam RM (2015) Proteomics in cancer biomarkers discovery: challenges and applications. Dis Markers 2015:321370
Sawada Y, Nakabayashi R, Yamada Y, Suzuki M, Sato M, Sakata A, Akiyama K, Sakurai T, Matsuda F, Aoki T, Hirai MY (2012) RIKEN tandem mass spectral database (ReSpect) for phytochemicals: a plant-specific MS/MS-based data resource and database. Phytochemistry 82:38–45
Schreiber F, Colmsee C, Czauderna T, Grafahrend-Belau E, Hartmann A, Junker A, Junker BH, Klapperstück M, Scholz U, Weise S (2012) MetaCrop 2.0: managing and exploring information about crop plant metabolism. Nucleic Acids Res 40:D1173–D1177
Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28:1086–1092
Segal MR, Xiong H, Bengtsson H, Bourgon R, Gentleman R (2012) Querying genomic databases: refining the connectivity map. Stat Appl Genet Mol Biol 11:1–6. https://doi.org/10.2202/1544-6115.1715
Soderlund C, Bomhoff M, Nelson WM (2011) SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res 39:e68. https://doi.org/10.1093/nar/gkr123
Szklarczyk D, Franceschini A, Wyder S (2015) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43:D447–D452
Szymanski P, Markowicz M, Mikiciuk-Olasik E (2012) Adaptation of high-throughput screening in drug discovery-toxicological screening tests. Int J MolSci 13:427–452
Talei D, Valdiani A, Rafii MY, Maziah M (2014) Proteomic analysis of the salt-responsive leaf and root proteins in the anticancer plant Andrographis paniculata Nees. PLoS One 91:e112907. https://doi.org/10.1371/journal.pone.0112907
Tecza K, Pamula-Pilat J, Lanuszewska J, Grzybowska E (2015) Pharmacogenetics of FAC chemotherapy side effects in breast cancer patients. Heredit Cancer Clin Pract 13:A10. https://doi.org/10.1186/1897-4287-13-s2-a10
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Tulpan D, Léger S, Belliveau L, Culf A, Čuperlović-Culf M (2011) MetaboHunter: an automatic approach for identification of metabolites from 1HNMR spectra of complex mixtures. BMC Bioinform 12:400. https://doi.org/10.1186/1471-2105-12-400
Upadhyay AK, Chacko AR, Gandhimathi A, Ghosh P, Harini K, Joseph AP, Joshi AG, Karpe SD, Kaushik S, Kuravadi N, Lingu CS (2015) Genome sequencing of herb Tulsi (Ocimum tenuiflorum) unravels key genes behind its strong medicinal properties. BMC Plant Biol 15:212. https://doi.org/10.1186/s12870-015-0562-x
Uzilov AV, Ding W, Fink MY, Antipin Y, Brohl AS, Davis C, Lau CY, Pandya C, Shah H, Kasai Y, Powell J (2016) Development and clinical application of an integrative genomic approach to personalized cancer therapy. Genome Med 8:62. https://doi.org/10.1186/s13073-016-0313-0
Valdiani A, Kadir MA, Tan SG, Talei D, Abdullah MP, Nikzad S (2012) Nain-e Havandi (Andrographis paniculata) present yesterday, absent today: a plenary review on underutilized herb of Iran's pharmaceutical plants. Mol Biol Rep 39:5409–5424
Vlaanderen J, Moore LE, Smith MT, Lan Q, Zhang L, Skibola CF, Rothman N, Vermeulen R (2010) Application of omics technologies in occupational and environmental health research; current status and projections. Occup Environ Med 67:136–143
Wang Y, Yu RY, He QY (2015) Proteomic analysis of anticancer TCMs targeted at mitochondria. Evidence-Based Compl Altern Med 2015:539260
Wang N, Wang X, Tan HY, Li S, Tsang CM, Tsao SW, Feng Y (2016) Berberine suppresses cyclin D1 expression through proteasomal degradation in human hepatoma cells. Int J Mol Sci 17:1899. https://doi.org/10.3390/ijms17111899
Webb B, Sali A (2014) Protein structure modeling with MODELLER. Methods Mol Biol 1137:1–15. https://doi.org/10.1007/978-1-4939-0366-5_1
Webb B, Sali A (2016) Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci 86:2.9.1–2.9.37. https://doi.org/10.1002/cpps.20
Xia J, Wishart DS (2010) MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 26:2342–2344
Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS (2012) Metabo analyst 2.0-a comprehensive server for metabolomic data analysis. Nucleic Acids Res 40:W127–W133
Xia J, Sinelnikov IV, Han B, Wishart DS (2015) MetaboAnalyst 3.0-making metabolomics more meaningful. Nucleic Acids Res 43:W251–W257
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316–W322
Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, Zhou X, Lam TW, Li Y, Xu X, Wong GK, Wang J (2014) SOAP de novo-trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30:1660–1666
Yamazaki M, Mochida K, Asano T, Nakabayashi R, Chiba M, Udomson N, Yamazaki Y, Goodenowe DB, Sankawa U, Yoshida T, Toyoda A (2013) Coupling deep transcriptome analysis with untargeted metabolic profiling in Ophiorrhiza pumila to further the understanding of the biosynthesis of the anticancer alkaloid Camptothecin and anthraquinones. Plant Cell Physiol 54:686–696
Yonekura-Sakakibara K, Fukushima A, Saito K (2013) Transcriptome data modeling for targeted plant metabolic engineering. Curr Opin Biotechnol 24:285–290
Zerbino DR (2010) Using the velvet de novo assembler for short read sequencing technologies. Curr Protoc Bioinformatics, Wiley, New York 31:11.5.1–11.5.12. https://doi.org/10.1002/0471250953.bi1105s31
Zhang A, Sun H, Yuan Y, Sun W, Jiao G, Wang X (2011) An in vivo analysis of the therapeutic and synergistic properties of Chinese medicinal formula Yin-Chen-Hao-Tang based on its active constituents. Fitoterapia 82:1160–1168
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mehta, A., Hasija, Y. (2018). Bioinformatics Approaches for Genomics and Post Genomics Applications of Anticancer Plants. In: Akhtar, M., Swamy, M. (eds) Anticancer Plants: Mechanisms and Molecular Interactions. Springer, Singapore. https://doi.org/10.1007/978-981-10-8417-1_12
Download citation
DOI: https://doi.org/10.1007/978-981-10-8417-1_12
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8416-4
Online ISBN: 978-981-10-8417-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)