
Abstract
Alzheimer’s disease (AD) is a leading cause of progressive dementia and most common neurodegenerative disease worldwide. The prevalence rate of AD is expected to get triple by 2050. Pathological hallmarks of AD are amyloid-β containing plaques and neurofibrillary tangles (NFTs), which are composed of hyperphosphorylated forms of the microtubule-associated protein tau. AD may be classified as early-onset (familial AD) and late-onset (sporadic AD). Familial AD occurs mostly in individuals of age 30–60 years and associated with mutations in amyloid precursor protein (APP) or presenilin (PS1 and PS2) genes, while sporadic AD mainly affects persons after 65 years of age and associated with mutations in apolipoprotein E4 isoform (apoE4) IR dysfunction, etc. Clinical interpretation of AD basically involves progressive deterioration in capabilities of memory, language, calculation, judgment, and behavior. AD is associated with disruption of mitochondrial function, calcium homeostasis, hormonal balance, and increased oxidative stress and neuroinflammation. Animal model has played a major role in defining critical disease-related mechanisms and evaluating novel therapeutic approaches in research. The sporadic form of AD itself probably involves several different etiopathogenic mechanisms. Neuroinflammation, head trauma, brain ischemia, and diabetes have been implicated as risk factors for AD.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Introduction
Alzheimer’s disease (AD) is a leading cause of progressive dementia and most common neurodegenerative disease worldwide. The prevalence rate of AD is expected to get triple by 2050. Pathological hallmarks of AD are amyloid-β containing plaques and neurofibrillary tangles (NFT’s), which are composed of hyperphosphorylated forms of the microtubule-associated protein tau. AD may be classified as early-onset (familial AD) and late-onset (sporadic AD). Familial AD occurs mostly in individuals of age 30–60 years and associated with mutations in amyloid precursor protein (APP) or presenilin (PS1 and PS2) genes, while sporadic AD mainly affects persons after 65 years of age and associated with mutations in apolipoprotein E4 isoform (apoE4) IR dysfunction, etc. Clinical interpretation of AD basically involves progressive deterioration in capabilities of memory, language, calculation, judgment, and behavior. AD is associated with disruption of mitochondrial function, calcium homeostasis, hormonal balance, and increased oxidative stress and neuroinflammation. Animal model has played a major role in defining critical disease-related mechanisms and evaluating novel therapeutic approaches in research. The sporadic form of AD itself probably involves several different etiopathogenic mechanisms. Neuroinflammation, head trauma, brain ischemia, and diabetes have been implicated as risk factors for AD.
Molecular genetic studies have proved the central role of β-amyloid in the pathogenesis of AD. Approximately 5% of the total number of AD cases are because of the mutations in amyloid precursor protein on chromosome 21q21 (APP) and presenilin 1 on chromosome 14q24 (PSEN1), and presenilin 2 on chromosome 1q42 (PSEN2), which are involved in the cleavage of APP. Alzheimer’s disease is associated with two major pathological traits. First, there is an accumulation of β-amyloid (Aβ) peptide. Second, hyperphosphorylation of tau protein occurs, which leads to neurofibrillary tangle formation and loses its microtubule binding and stabilizing role, contributing to neuronal degeneration. Aβ peptides originate from the amyloid precursor protein (APP) by amyloidogenic processing when acted upon by the alternate enzyme b-secretase instead of a-secretase followed by g-secretase. Aβ oligomers are neurotoxic and rapidly blocked long-term potentiation, a classic experimental paradigm for synaptic plasticity, which cause local structural disruption of synapses. Aβ triggers mitochondrial oxidative stress and dysregulation of calcium ion (Ca2+) homeostasis, resulting in impairment of the electron transport chain, increased production of superoxide anion radical (O2), and decreased production of ATP (Fig. 1).

Pathophysiology of Alzheimer’s disease
Moreover, the levels of enzymes involved in energy metabolism (cytochrome c oxidase, pyruvate dehydrogenase complex, and a ketoglutarate dehydrogenase complex) are found to be decreased in brain cells of AD patient. Increased deposition of Aβ results in increased mRNA and protein expression of iNOS and generation of NO. NO can interact rapidly with superoxide anion O2 forming more reactive peroxynitrite (ONOO) and induce lipid peroxidation and functional alterations in proteins and DNA, eventually leading to neuronal death. Aβ deposition activates inflammatory reactions by activating microglia and astrocytes, which leads to release of chemokines such as interleukin 8, interferon-g–inducible protein, macrophage inflammatory protein-1a, macrophage inflammatory protein-1b, and monocyte chemoattractant protein-1 (MCP-1) and cytokines such as IL-1, IL-6, TNF-a, and transforming growth factor-b (TGF-β).
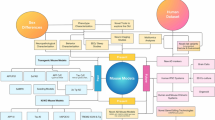
2 Classification of Models AD
Animal models are mainly used to study the mechanisms underlying AD pathogenesis, genetic interactions with genes of interest, and environmental risk factors that cause sporadic AD, as well as to test the therapeutic effects of drugs on neuropathology and cognitive function (Fig. 2).

Classification of animal models of Alzheimer’s disease
2.1 Chemical-Induced AD Models
2.1.1 STZ-Induced AD
The intracerebroventricular (i.c.v.) Streptozotocin (STZ) is a glucosamine–nitrosourea compound that, when metabolized, produces a cytotoxic product that preferentially destroys pancreatic β cells. The alkylating properties of STZ metabolites produce reactive oxygen species and finally resulting in oxidative stress. In moderate-to-low dosage in short-term experiments, STZ systemic administration caused insulin resistance by a decreasing phosphorylation of IRS-1 in rats. IRS-1 plays an important role in transferring signals from insulin receptors to intracellular pathways. Intracerebroventricular STZ (2-deoxy-2-(3-methy1-3-nitrosoureido)) animal model 233 Q19 was developed by Lannert and Hoyer in 1998. It has been reported that there is a decrease in glucose utilization in the brains of AD patients. This leads to the hypothesis that the cognitive dysfunction of AD is related to a reduction in central glucose metabolism. It has been hypothesized that the cholinergic deficit and amyloid accumulation in the brain are caused by a significant decrease in glucose metabolism in the AD brain. Various studies have shown that I.C.V. administration of subdiabetogenic doses of STZ reduce central glucose in rats. STZ administration in non-diabetogenic doses was shown to induce memory impairment, insulin receptor dysfunction in hippocampus and leads to progressive cholinergic impairment, glucose hypo-metabolism, oxidative stress, and neurodegeneration. STZ significantly improves BACE-1, p-p38 MAPK expression, and NF-kB (p65) translocation in astrocytes. Together, these effects significantly result in increase in amyloidogenic processing of APP and eventually cause increased activation of various deleterious pathways leading to neuronal degeneration (Table 1).
Procedure
-
1.
Anesthetize the rats and positioned on digital stereotaxic apparatus.
-
2.
Remove hairs and do sagittal incision on scalp and set the coordinates.
-
3.
Drill the holes of skull which are identified and place cannula into the lateral cerebral ventricles.
-
4.
Animals that take the delivery of either I.C.V. artificial cerebrospinal fluid (ACSF; 10 L/site) or ICV STZ (3 mg/kg bilaterally using a micro syringe). The composition of the ACSF was (in mmol/L): NaCl 147; KCl 2.9; MgCl2 1.6; CaCl2 1.7; dextrose 2.2.
-
5.
To ensure diffusion of the administered drug, the cannula is left in place for a period of 2 min following the injection.
-
6.
The stereotaxic coordinates for ICV injection were 0.8 mm posterior to bregma, 1.8 mm lateral to the sagittal suture, and 3.6 mm beneath the cortical surface.
Advantages
-
ICV application of the STZ has been shown to cause oxidative stress, glucose/energy metabolism alteration, and cholinergic hypo-function accompanied by memory deficit by impairment of the neuronal insulin receptor transduction cascade (including insulin, IR, IRS-1, PI3 K, Akt, and GSK-3).
-
Free radicals generation after ICV STZ injection is an important factor in causing cognitive impairment in rats.
Disadvantages
-
Histological appearance of Aβ and Tau neuropathology takes long time to develop. Technical expertise is required for ICV infusion.
Clinical Relevance
-
It has been demonstrated that AD in postmortem human brains is associated with significantly decreased expression of insulin/IGF trophic factors, their receptors, and IRS proteins, and further enhances the severity with progression of dementia and neurodegeneration (Fig. 3).
Fig. 3 
Amyloid-β-induced neurotoxicity. ROS-reactive oxygen species, NO-nitric oxide, JNK-c-Jun N-terminal kinase, MAPK-mitogen-activated protein kinase, NMDA-, TNF-tumor necrosis factor
-
In postmortem cases of advanced AD, it has been explored and resoluted if the neurodegeneration was correlated with significant abnormalities in the expression of genes encoding insulin, IGF-1 and IGF-2 peptides, their receptors, and downstream signaling mechanisms. In that study, we indicated advanced AD to be associated with remarkably diminished levels of insulin, IGF-1 polypeptide, and receptor genes in the brain.

2.1.2 Amyloid Beta-Induced AD
Aβ oligomers are small assemblies or aggregates of this so-called amyloid beta protein. It was discovered in 1992. Aβ peptide is 39-43 amino acids in length, and these are formed by the cleavage of transmembrane protein amyloid precursor protein (APP) by enzyme secretase. α-secretase is the primary enzyme acts APP under physiological considerations followed by γ-secretase. APP undergoes amyloidogenic processing when acted upon by the alternate enzyme β-secretase instead of α-secretase. The extent of amyloid-β deposits correlates with the degree of neuronal damage, cognitive impairment, and memory loss. The level of cholinergic neurons found to be decreased in amyloid-β-induced animal model of AD. Aβ (1-42) activates microglia as well as astrocytes, initiates a chemotactic inflammatory action, and releases cytokines including superoxide, proinflammatory cytokines such as tumor necrosis factor (TNF) and interleukins (IL-1, IL-6) and excitatory amino acids including glutamate. These inflammatory markers are associated with neurodegenerative processes in AD. The amyloid-β1–42 (Aβ42) peptide rapidly aggregates to form oligomers, protofibrils, and fibrils en route to the deposition of amyloid plaques associated with Alzheimer’s disease. In this model, rats were infused ICV with Aβ (1-42) oligomer (3 nmol/3μl) dissolved in ACSF. It has been demonstrated that oligomeric Aβ alteres the expression of Bcl-2, Bim and Bax and contributes in neuronal apoptosis. On the other hand, Aβ induces NO production either by disrupting calcium homeostasis and subsequent increase in intracellular Ca2+ (nNOS and eNOS-mediated NO release) or by interactions with glial cells (iNOS-mediated NO release). These reactive oxygen species induce a variety of neurotoxic mechanisms, including DNA/ protein alterations, poly ADP-ribose polymerase (PARP) over activation, mitochondrial dysfunction, lipid peroxidation, neuroinflammation, and apoptosis. Also it has been found that there is disruption in the various intracellular signaling pathways by activation of stress-related kinases c-Jun N-terminal kinase (JNK), and p38 is associated with neuronal death in AD models. Amyloid beta itself can generate the free radicals upon interaction with metals, such as iron and aluminum, which cause neuronal cell death and AD (Table 2).
Procedure
-
1.
Anesthetize the rats and positioned on digital stereotaxic apparatus.
-
2.
Remove hairs and do sagittal incision on scalp and set the coordinates.
-
3.
Drill the holes of skull which are identified and place cannula into the lateral cerebral ventricles.
-
4.
Animals receive either I.C.V. artificial cerebrospinal fluid (ACSF; 10 L/site) or ICV STZ (3 mg/kg bilaterally using a microsyringe).
-
5.
Infused I.C.V. in rats with either artificial cerebrospinal fluid (ACSF; in nmol/L: 147 Nacl, 2.9 Kcl, 1.6 Mgcl2, 1.7 CaCl2, and 2.2 dextrose) or amyloid-β1-42 oligomer (3 nmol/3μl) dissolved in ACSF according to the method described by Maurice.
-
6.
Drug solution (3μl) should be injected twice by using Hamilton microsyringe positioned in the injection cannula.
-
7.
The stereotaxic coordinates for ICV injection were 0.8 mm posterior to bregma, 1.8 mm lateral to the sagittal suture, and 3.6 mm beneath the cortical surface.
Advantages
-
The ‘amyloid cascade hypothesis’ emphasizes a central role for Aβ in the pathogenesis of AD. Thus, Aβ has become a major therapeutic target, with various anti-Aβ strategies being pursued.
-
Aβ proteinosis is an important structure of senile plaques and is thought to be the main reason for the loss of neurons and the resulting memory disability.
Disadvantages
-
This model has high mortality rate. Aβ can be infused into animal brain stereotaxically, which requires more care after surgery.
2.1.2.1 Clinical Relevance
-
AD in humans results in significant atrophy of the brain, particularly in the entorhinal cortex, hippocampus, and amygdale.
-
In human, Aβ deposits begin exclusively in the neocortex, then extends into allocortical brain regions, such as the hippocampus. Areas of the diencephalon, where the thalamus and hypothalamus are situated, as well as the striatum and cholinergic nuclei of the basal forebrain are influenced later. Finally, in the last stages of disease, Aβ pathology can be detected in areas of the brain stem and the cerebellum.
2.1.3 Colchicine-Induced AD
Colchicine, an alkaloid extracted from some plants of the lily family, has been used for centuries to treat acute gouty arthritis. It was first isolated in 1820 by the French chemists P.S. Pelletier and J.B. Caventou. In 1883, P.L. Geiger purified an active ingredient, which he named colchicines. Since 1973, it has been recognized as an effective remedy for prophylaxis of attacks of familial Mediterranean fever. Colchicine is a well-known neurotoxin, a cytotoxicant that binds irreversibly to tubulin dimers and induces neurofibrillary degeneration, thereby blocks mitosis and axonal transport, leading to disruption in the microtubule polymerization, death of cerebral granule cells, olfactory bulb neurons, cells of subventricular zone, dentate gyrus cells, and basal forebrain cholinergic neurons, thus causing cognitive impairment. Central administration of colchicine exaggerates free radical generation and oxidative DNA damage that eventually produces marked destruction of hippocampal granule cells and septohippocampal pathways resulting in loss of cholinergic neurons and decreased activities of acetylcholinesterase and choline acetyltransferase, thereby resulting in decreased ability to learn and in loss of memory. Colchicine has been found to be significantly induce free radical generation and deplete antioxidant defense system in rat brains. 15μg/15μl colchicine administered intracerebroventricularly (i.c.v.) in rats to induced memory impairment and oxidative damage. Colchicine is neurotoxic toward various neuronal populations including cerebral granule cells (CGC’s) and causes high molecular weight DNA fragmentation and nucleus condensation and causes caspase-2-induced (induces caspases 3) proteolytic degradation of PARP (poly(ADP-ribose) polymerase, DNA repair enzymes, cytoskeletal proteins, and fodrin (Table 3).
Procedure
-
1.
Anesthetize the rats and positioned on digital stereotaxic apparatus.
-
2.
Remove hairs and do sagittal incision on scalp and set the coordinates.
-
3.
Drill the holes of skull which are identified and place cannula into the lateral cerebral ventricles.
-
4.
Close the scalp with suture. In sham-operated rats, the surgery was identical except for drilling of holes and placement of the cannula.
-
5.
After surgery, gentamicin (5 mg/kg, ip) is given to all animals to prevent sepsis. Intracerebroventricularly (icv) infused with either artificial cerebrospinal fluid (ACSF; in mM: 147 NaCl, 2.9 KCl, 1.6 MgCl2, 1.7 CaCl2, and 2.2 dextrose) or 15 μg colchicine dissolved in ACSF in rats.
-
6.
Solutions were injected using a Hamilton microsyringe positioned in the injection cannula, and the injection cannula was left in place for 2–3 min following infusion.
-
7.
Special care was taken during the postoperative period to provide food and water inside the cage of the rat.
-
8.
The stereotaxic coordinates for ICV injection were 0.8 mm posterior to bregma, 1.8 mm lateral to the sagittal suture, and 3.6 mm beneath the cortical surface.
Advantages
-
Colchicine induces hippocampal lesions resulting in learning and memory impairment, reduction in choline acetyltransferase, suggesting that it could be used as a suitable model for studying Alzheimer’s disease.
Disadvantages
-
The neurotoxic mechanism of colchicine is not fully understood.
-
This model is time-consuming and requires a large number of animals due to high mortality rate.
-
Rats treated with colchicine produce a decrease in appetite and transient diarrhea, adipsia, and aphasia after 7–10 days of its administration. Also there are results in myoclonic twitches, aggressive behavior, decreased body weight, which exhibits acoustic startle behavior and decreased threshold to pain.
Clinical Relevance
-
I.C.V. colchicine model can be further used to understand better SDAT pathogenesis in AMT humans. It has several features that are consonant with SDAT in humans, including an insidious onset, time-dependent changes in behavioral and biochemical patterns.
2.2 Metal-Induced AD
In the AD brain, variation in metal levels may indicate insufficiencies or excesses of specific metalloproteins or defective metal transporters. Indeed, the levels of various Cu and Fe regulatory and storage proteins are changed in AD brain. There is an accumulating indication that interactions between β-amyloid and copper, iron, and zinc are integrated with the pathophysiology of Alzheimer’s disease (AD). Iron and Cu form crucial components of various enzymes required for vital brain functions including energy production, neurotransmitter synthesis, and antioxidant function. Change in metal homeostasis may be a key factor which resulting in AD pathogenesis. It has been found to have role in accumulation of Aβ. Which is mediated by interaction with metals, in particular copper (Cu), zinc (Zn), and iron (Fe). Metal induces oxidative DNA damage by the disruption of calcium homeostasis and intracellular signal transduction pathways. Aβ catalyzes the process of formation of reactive oxygen species (ROS), which in turn contributes to Aβ accumulation by generating modified Aβ species. Due to redox-active nature of Cu and Fe, inappropriate regulation of these metals can lead to reaction with O2 and the formation of ROS, causes cellular toxicity. AD brain indicates marked oxidative damage of proteins, lipids, and nucleic acids. Currently, the copper- and zinc-chelating agent clioquinol depict a potential therapeutic route that may not only obstruct β-amyloid neurotoxicity, but may also reverse the aggregation of neocortical β-amyloid.
2.2.1 Aluminum-Induced AD
The contributions of neurotoxicity of Al in experimental animals were first reported in 1897 by Dollken. It has been also shown that Al salts administered intracerebrally or peripherally in rabbit, cat, mice, rat, and monkey induce the formation of neurofibrillary tangles. Aluminum is the third most common ubiquitous element in the environment. Main sources of aluminum exposure are water, cooking utensils, food additives, grain products, processed cheese, and drugs like antacids and deodorants. Aluminum levels increase in the brain with advancing age. Aluminum (300 mg/kg daily, p.o.) has been shown to abrogate neurotransmission, cognitive behavior. Thus, it has been found to be associated with several neurodegenerative diseases including AD. The neurodegeneration effects of aluminum in experimental animals are dependent on the route of administration, type of aluminum salt, species of animal, dose of treatment, and time of exposure. In most studies, the duration of aluminum exposure ranges from 8 weeks to 6 months. Aluminum has been found to be involved in the formation of paired helical filaments (PHFs) of tau proteins by inhibiting PP2A (protein phosphatase 2A) activity. Moreover, PP2A accounts for approximately 70% of the total tau phosphatase activity in human brain. The mechanism of aluminum also involves accumulation of aluminum in the neurons which leads to cell depolarization and disruption in the Na+/Ca2+ exchange pump. Ultimately cause excessive accumulation of intra-mitochondrial Ca2+ levels leading to opening of the MPT with subsequent release of cytochrome c, activation of caspases and apoptosis. Increase in intra-mitochondrial Ca2+ levels also cause increased production of toxic free radicals. All together, these events cause an increase in amyloid plaques and neurofibrillary tangles in brain. Mechanism of aluminium-induced neurotoxicity is described in Fig. 4 (Table 4).

Mechanism of aluminum-induced AD
2.2.2 Aluminum-, Aβ- and Transforming Growth Factor Beta-1(Tgfβ-1)-Induced AD
The aim was to design an animal model with multifactorial etiologies in animal that mimic the complicated pathological features and biochemical changes of AD. However, most of the classic AD models addressing or targeting single etiologies issues. Therefore, this new animal model may provide a successful multifactorial AD model. Aluminum and Aβ can interact and enhance each other, thereby intensify the neuronal deterioration. Also it has been found that TGF-1 can accelerate the deposition of Aβ in brain and damage capillary vessel to promote the occurrence of AD. Moreover, the impairment of cholinergic system impairment lasts for 3 months in this model. So this could be used for fairly long-term pathogenesis and drug research. It is more close to the process of etiopathology and pathological and biochemical changes of AD than other classic models of AD. IN early-onset familial AD (FAD), highly aggregative Aβ42 is over-produced by mutant presenilin 1/2 genes. Furthermore, Aβ aggregation may be promoted by apolipoprotein E and by environmental factors, such as oxidative stress and diabetes. Oxidative stress-inducing metal ions, such as iron (Fe) Copper (Cu) zinc (Zn) and aluminum (Al), may contribute to the formation of SPs/NFTs and subsequent neuronal damage in the AD brain (Table 5).
Procedure
-
1.
Anesthetize the rats and positioned on digital stereotaxic apparatus.
-
2.
Remove hairs and do sagittal incision on scalp and set the coordinates.
-
3.
Drill the holes of skull which are identified and place cannula into the lateral cerebral ventricles.
-
4.
Coordinates for the injection lateral cerebral ventricle (behind the bregma 1.2 mm, meta 2 mm, deep 4 mm), and the other point was the anterodorsal nucleus of thalamus (at the opposite side of the cerebrum, behind the bregma 2.1 mm, meta 1.4 mm, deep 4.6 mm).
-
5.
The catheter shall be inserted into the lateral cerebral ventricle point, fixed at the cranium with dental base acrylic resin powder and 502 glue mixture, and at the same time, the catheter was blocked by the nylon line core which was a little less than the catheter inner diameter in order to prevent from infection and leakage of cerebrospinal fluid.
2.2.3 AlCl3- and D-Galactose-Induced AD
AlCl3 and D-galactose induce Alzheimer’s disease by alterations in β-amyloid peptide metabolism-related molecules in the early stages of predementia. This model shows alteration in the expression of BACE1 (β–secretase, an enzyme involved in the cleavage of APP), neprilysin (NEP) (Aβ degrading enzymes), insulin degrading enzyme (IDE), and receptor for advanced glycation end products (RAGE), which is strongly involved in Aβ influx back into the brain earlier than Aβ processing. These pathways eventually cause memory impairment and high Aβ levels in the cortex (Co) and hippocampus (Hi), which shows pathological features similar to Alzheimer’s disease. This model showed that the combination of Aland D-gal caused more mouse cognitive impairment as compared to treating with Al and D-gal individually. This model could be employed for the early diagnosis of AD hence helpful in investigating future mechanism and drug screening for AD (Table 6).
Advantages
-
This model could be employed for the early diagnosis of AD hence helpful in investigating future mechanism and drug screening for AD.
-
Most of the classic AD models are mono-factor models, but this model shows multifactor mechanism that mimics the complicated etiology, pathological and biochemical feature of an ideal animal model of AD.
-
The cholinergic system impairment lasts for 3 months in this model.
Disadvantages
-
Aluminum itself is not that sufficient to cause Alzheimer’s disease.
Clinical Relevance
-
However, it was found that in brain tissue from dialysis patients, only 1 case out of 50 exhibited the accumulation of Aβ42 and p-tau. Phosphorylated neurofilaments and cytoplasmic argyrophilic inclusions have been shown to be characteristics of Al-induced pathology in DAE, rather than the paired helical filaments that are consistently observed in AD.
2.2.4 Zinc-Induced AD
Zinc plays an important role in normal growth and development. In physiologic conditions, zinc is also associated in variety of neuronal functions viz neurogenesis, neuronal migration, synaptogenesis, and neurotransmission, thus is highly involved in cognitive functions. A total of 100–200 mg/kg zinc content is required for regulating learning and memory function in rats. Zn is a key modulator for synaptic neural transmission, reaching 150–300 µM during synaptic activity, thus possibly causes Aβ accumulation. It has been indicated that zinc also plays an essential role in cellular homeostasis and various biochemical functions, such as protein synthesis and nucleic acid metabolism. Majority (80–90%) of the zinc present in the brain bounded with proteins, and the remaining percentage is packaged within synaptic vesicles of a large subpopulation of excitatory neurons. The synaptic or vesicular zinc cause activation of several neurotransmitter receptors, including NMDA, AMPA, GABA, and glycine receptors as well as voltage-dependent ion channels in an activity-dependent manner. Zinc is highly enriched in the hippocampus, amygdala, cerebral cortex, thalamus, and olfactory cortex. Recent studies showed that secreted zinc plays vital role in information processing, synaptic plasticity, learning, and memory. In fact, zinc is found to be vital in the hippocampus for the stimulation of long-term potentiation, a form of synaptic information storage thus making it a well-known model for the mechanisms underlying memory formation. In pathologic condition, a significant amount of zinc is released into synapse due to membrane depolarization where it triggers a number of prejudicial signaling processes including those that lead to further ROS generation, marking the start of a positive feedback loop including intracellular zinc release and ROS generation. Synaptic zinc is also connected with neuronal dysfunction by its transport from overactive presynaptic zinc-containing neurons to postsynaptic cells via calcium-permeable channels, including but not restricted to a subclass of AMPA receptors. Zinc has been found to diminish several enzymes, mitochondrial respiration, thus resulting in energy depletion and ROS generation. Excessive glutamate release causes an increase in intracellular Ca2+ levels, which trigger several apoptotic pathways, such as calpain or caspases activation ultimately, resulting in neuronal death. Thus, disruption in zinc homeostasis has been implicated in several neurodegenerative diseases, including Alzheimer’s disease. Both zinc depletion and excess zinc can cause severe damage to neurons.
Both extracellular and intracellular zinc cause the Aβ-induced neurotoxicity during AD. It has been found that plaques themselves are rich in copper and zinc and aggregation of Aβ peptide resulting in increasing the state of oxidative stress during AD through direct formation of oxidants as well as through microglia activation and subsequent production of ONOO− . The ONOO– generation from both neurons and microglia appears to be a lead trigger of zinc-dependent neuronal apoptosis. Moreover, hydrogen peroxide, an oxidant, can cause the release of zinc from MT III, subsequently resulting in accumulation of Aβ. Zinc secreted into the synapse, through zinc-containing glutamatergic vesicles facilitates Aβ aggregation following cleavage of peptide from membrane-bound APP. With age, the concentration of vesicular zinc and the expression of ZnT3 (transporter responsible for packaging zinc into synaptic vesicles) are also found to be diminished in brain. Further, both Aβ aggregates and zinc have been found in mitochondria, lysosomes, and the ER. Similar to Aβ, zinc can also directly bind to tau to facilitate tau hyperphosphorylation, and Aβ can mediate calcium release from the ER, which cause free radical generation in brain.
Advantages
-
Zinc can directly bind to Tau protein and thus contribute in microtubule destabilization, which is one of the major pathologic hallmarks of AD.
Disadvantages
-
The neurotoxic mechanisms of zinc are not clear.
2.3 Lipids-Induced AD
2.3.1 Cholesterol-Induced AD
Cholesterol is important for cell structure, repair and signaling, hormone production, and bile acid synthesis and individuals with enhanced cholesterol levels during midlife have risk to develop AD. Cholesterol may be directly involved in Aβ aggregation: Abnormal oxidative metabolites like cholesterol-derived aldehydes can modify Aβ, which is associated with AD and Aβ, NFT’s pathology. Cholesterol flux is elevated in AD brain, and cholesterol dyshomeostasis is closely associated with Alzheimer’s synaptic loss and cognitive impairment. Individuals with high cholesterol in their early 40s are more likely to develop AD than those with low cholesterol. Alteration in the total cholesterol, HDL and LDL levels, in serum correlate with a disturbed cholesterol metabolism and Aβ load in the AD brain. The cholesterol metabolite 24S-hydroxycholesterol is more soluble than cholesterol and is more easily exported from the brain, and high levels of 24S-hydroxycholesterol have been reported in AD brain. Cholesterol and apoE are involved in fibrillar plaque formation. Lipid rafts are heterogenous, cholesterol- and sphingolipid-rich membrane and responsible for development of nerve axon growth, and they have a major role in APP amyloidogenic processing at γ-secretase level. However, increased cholesterol level (more than 5.8 mmol/L) is correlated to the amyloid plaques associated with AD. Apolipoprotein E (apoE), apoJ, ATP-binding cassette subfamily member 7, and sortilin-related receptor are AD susceptibility genes, which are involved in cholesterol metabolism or transport. ApoE is the major cholesterol carrier in the brain, and it regulates the brain cholesterol metabolism and triglycerides in the body. There are three common alleles of the ApoE genes ε2, ε3, and ε4. ApoE has been demonstrated to play a role in Aβ generation by increasing cellular cholesterol.
2.3.2 Lipopolysaccharides-Induced AD
Lipopolysaccharide (LPS) is a component of the cell wall of gram-negative bacteria, and it has been used experimentally to induce the production of the endogenous IL-1 and b-amyloid precursor protein (β-APP). It is an endotoxin and potent activator of microglia and astrocyte. It plays a substantial role in neuroinflammation and brings on phosphorylation of proteins, protein tyrosine kinase, mitogen-activated protein kinases C and A, G protein, and ceramide-activated protein kinase. LPS are reported to induce various cytokines (interleukin (IL) 1 ß, IL-6, interferon alpha, tumor necrosis factor alpha), cyclooxygenase-2 (COX-2), and amyloid precursor protein mRNA levels within the basal forebrain. These proinflammatory mediators successively activate astrocytes and microglia in hippocampus, pituitary gland, and hypothalamus and produce degeneration of CA3 pyramidal neurons and eventually leading to impairment in spatial memory. Moreover, LPS causes increase in the Ca2+ -independent iNOS activity and protein kinase C activation, which in turn leading NADPH oxidation, free radicals generation and activates protein tyrosine kinase. Further leads to neuronal death and spatial memory impairment. Moreover, intraperitoneal injection of LPS (250 Ìg/kg) has been reported to impair cognition by depressing the social exploration of rats which was thought to be mediated by the central inflammatory cytokine IL-1 (Table 7).
Procedure
-
1.
An (Alzet Palo Alto), CA osmotic minipump (model 2002; 0.5 µl/h) containing LPS (Sigma; E. coli, serotype 055:B5, TCA extraction; 1.0 µg/ml) was implanted into the dorsal abdomen and attached via Tygon tubing (0.060 o.d) to a chronic indwelling cannula (Model 3280P, Osmotic pump connect, 28 gauge, Plastics One, Roanoke, VA) that had been positioned stereotaxically so that the cannula tip extended to these coordinates: 2.5 mm posterior to Lambda, on the midline, and 7 mm ventral to the dura.
-
2.
Controls were infused with artificial cerebrospinal fluid (aCSF): (in mM) 140 NaCl; 3.0 KCl; 2.5 CaCl; 1.0 MgCl; 1.2. 2 2 Na HPO, pH 7.4. The LPS was dissolved in aCSF.
-
3.
The rats were infused with either aCSF or LPS for a total of 4 weeks.
-
4.
A volume overload to the CSF space was discounted because the 0.5 µl/h administered contributed to only about 0.4% of the CSF volume produced by the rat each hour and was only 0.25% of the rat’s total CSF volume.
Advantages
-
LPS can impair cognition by depressing the social exploration of rats which was thought to be mediated by the central inflammatory cytokine IL-1.
Clinical Relevance
-
In AD, brain cholesterol flux is elevated: When compared to controls, higher levels of the more soluble form of cholesterol, 24S-hydroxycholesterol, are found in both CSF and plasma of AD patients, even in early stages of dementia.
-
Levels of total cholesterol and LDL in serum have been found to correlate with Aβ load in the brains of patients with AD. Epidemiological evidence suggests that elevated cholesterol levels during midlife increase the risk of developing AD.
2.4 Aging-Induced AD
2.4.1 D-Galactose-Induced AD
Aging is considered as a major risk factor in AD and mitochondrial dysfunction, and oxidative DNA damage has been demonstrated to have major role in aging. D-gal is a physiological nutrient and a reducing sugar. Generally, galactose is metabolized by D-galactokinase and galactose-1-phosphate uridyltransferase in animals, but its high levels result in abnormality of metabolism. D-galactose is converted into galactitol and started accumulating in the cell, which in turn causes activation of receptor for advanced glycation end products (RAGE), thereby results an increase in oxidative stress and cellular damage. However, high levels of D-galactose cause oxidative metabolism of D-galactose into aldose and hydroperoxide in presence of enzyme galactose oxidase and finally result in excessive ROS and free radical generation in the brain. Mitochondrial dysfunction induces an increased reactive oxygen species and free radical generation and decreased oxidative phosphorylation. It also causes microglia activation and thereby leads to inflammation and causes oxidation of nucleotide acids, especially DNA. Hippocampus plays a major role in formation of memory. Recently, it has been reported that the chronic administration of D-galactose (D-gal) significantly can lead to neuronal damage and symptoms like deterioration of cognitive abilities same as in aging. Also it has been demonstrated that continuous administration of D-gal (s.c.) in mice induced an increase in cell karyopyknosis, apoptosis, and caspase-3 protein levels in hippocampal neurons. Excessive ROS generation coupled with disrupted antioxidant defenses leads to oxidative damage to mitochondria, and chronic administration of D-galactose causes hippocampal dysfunction in rodents, ultimately causes decline in spatial learning and memory function by impaired oxidative defense and mitochondrial complex (I, II, and III) enzyme activities (Table 8).
Advantages
-
It has been reported that the chronic administration of D-galactose (D-gal) can lead to neuronal damage and symptoms like deterioration of cognitive abilities same as in aging.
Clinical Relevance
-
Age-related memory loss starts in the dentate gyrus. A recent gene expression study in the human dentate gyrus postmortem, normalized for gene expression in the entorhinal cortex that is not affected by aging, identified 17 genes showing reliable age-related changes.
3 Conclusion
As Alzheimer’s disease has multiple etiologies, so in this review we focus on different animal models of Alzheimer’s disease that has been identified and discussed the potential of pharmacologically induced rat models of AD, which are more relevant to the sporadic form of AD. Chemical-induced animal models of memory deficits have been more commonly employed for understanding the pathogenesis and for management of dementia and other cognitive deficits. We believe that investigation of currently available animal models for AD will help to clarify the pathogenic mechanism and allow assessment of the effects of new treatment strategies.
Ethical Statement
All institutional guidelines, national guidelines, state and local laws, and regulations with professional standards for the care and use of laboratory animals should be followed. Studies involving animals must state that the institutional animal ethical committee has approved the protocol. For authors using experimental animals, a statement should be made that the animals’ care is in accordance with institutional guidelines and animals used have been treated humanely and with regard for the alleviation of suffering. Researchers should treat animals as sentient and must consider their proper care and use and the avoidance or minimization of discomfort, distress, or pain as imperatives. Animal experiments should be designed only after due consideration of animal health. It should be ensured that all researchers who are using animals have received instruction in research methods and in the care, maintenance and handling of the species being used. All the surgical procedures should be performed under appropriate anesthesia and follow only those procedures which avoid infection and minimize pain during and after surgery.
References
Bensimon G, Chermat (1991) Microtubule disruption and cognitive defects: effect of colchicineon learning behavior in rats. Pharmacol Biochem Behav 38:141–145
Blokland A, Jolles J (1993) Spatial learning deficit and reduced hippocampal ChAT activity in rats after an ICV injection of streptozotocin. Pharmacol Biochem Behav 4:491–494
Chen CF, Lang SY, Zuo PP et al (2006) Effects of D-galactose on the expression of hippocampal peripheral type benzodiazepine receptor and spatial memory performances in rats. Psychoneuroendocrinology 31:805–811
Fang F, Yan N, Feng Z et al (2013) Alzheimer’s disease animal model by aluminum, beta-amyloid and transforming growth factor beta-1. Aging Neurodegeneration 1:15–19
Hauss-Wegrzyniak B, Dobrzanski P, James D et al (1998) Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer’s disease. Brain Res 780:294–303
Hua X (2007) Long-term D-galactose injection combined with ovariectomy serves as a new rodent model for Alzheimer’s disease. Life Sci 80:1897–1905
Kumar MHV, Gupta YK (2003) Effect of centellaasiaticaon cognition and oxidative stress in an intracerebroventricularstreptozotocin model of Alzheimer’s disease in rats. Clin Exp Pharmacol Physiol 30:336–342
Kumar A, Prakash A, Dogra S (2009) Neuroprotective effects of centellaasiatica against intracerebroventricular colchicine-induced cognitive impairment and oxidative stress. Int J Alzheimer’s Dis 1–8
Kumar A, Prakash A, Dogra S (2011) Centellaasiatica attenuates D-galactose-induced cognitive impairment, oxidative and mitochondrial dysfunction in mice. SAGE-Hindawi Access Res Int J Alzheimer’s Dis 1–9
Lu H, Hu J, Jiang Y (2013) Optimal dose of zinc supplementation for preventing aluminum-induced neurotoxicity in rats. Neural Regen 8(29):2754–2762
Luo Y, Niu F, Sun Z (2009) Altered expression of Ab metabolism-associated molecules from D-galactose/AlCl3 induced mouse brain. Mech Ageing Dev 130:248–252
Maurice T, Lockhart BP, Privat A (1996) Amnesia induced in mice by centrally administered beta amyloid peptide involved cholinergic dysfunction. Brain Res 706:181–193
Nitsch R, Hoyer S (1991) Local action of the diabetogenic drug, streptozotocin, on glucose and energy metabolism in rat brain cortex. Neurosci Lett 128:199–202
Nitta A, Itoh A, Hasegawa T, Nabeshima T (1994) β-Amyloid protein-induced Alzheimer’s disease animal model. Neurosci Lett 170:63–66
Patil CS, Singh VP, Satyanarayan PSV et al (2003) Protective effect of flavonoids against aging- and lipopolysaccharide-induced cognitive impairment in mice. Pharmacology 69:59–67
Pitchaimani V, Arumugam S, Thandavarayan RA (2012) Nootropic activity of acetaminophen against colchicine induced cognitive impairment in rats. J Clin Biochem 50:241–244
Salkovic-Petrisic M, Tribl F, Schmidt M (2006) Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the Insulin signalling pathway. J Neurochem 96:1005–1015
Savorya J, Hermanb MM, Ghribi O (2006) Mechanisms of aluminum-induced neurodegeneration in animals. Implications Alzheimer’s Dis 10:135–144
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Shree, S., Bhardwaj, R., Kashish, Deshmukh, R. (2017). Non-transgenic Animal Models of Alzheimer’s Disease. In: Bansal, P., Deshmukh, R. (eds) Animal Models of Neurological Disorders. Springer, Singapore. https://doi.org/10.1007/978-981-10-5981-0_2
Download citation
DOI: https://doi.org/10.1007/978-981-10-5981-0_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-5980-3
Online ISBN: 978-981-10-5981-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)