
Abstract
Numerous network inference models have been developed for understanding genetic regulatory mechanisms such as gene transcription and protein synthesis. Dynamic Bayesian network effectively represent the causal relationship between genes and gene and protein. Modern approaches employ single multivariate gene expression data set to estimate time varying dynamic Bayesian network. However, evaluating inferred time varying network is infeasible due to the absence of known gold standards. In this paper, the simulation model for time series gene expression level under certain network structure is proposed. The network can be used for assessing inferred data which is estimated based on simulated gene expression data.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
For the past decades, numerous network inference methods have been developed to model underlying genetic regulatory mechanisms such as gene transcription and protein synthesis. The main focus of network inference is on investigating the interactions between genes, and attempt to build descriptive models for understanding complex system. For representing causal relationship dynamic Bayesian network (DBN) is one of well-known probabilistic graphical models. While in static Bayesian network the topology of network is fixed [1,2,3], dynamic Bayesian network is particularly well suited to tackle the stochastic nature of gene regulation and gene expression measurement [4], thus has been widely used for its ability to recover the underlying genetic regulatory network [5]. With development of time series gene experimental expression data estimating time-varying DBN has became feasible. In [4], DBN is inferred based on a penalized likelihood maximization implemented through an extended version of EM algorithm. Also, [6] proposed temporally rewiring networks for capturing the dynamic causal influences between covariates. For estimation, kernel reweighted \( {\text{L}}_{1} \)-regularized auto-regressive procedure is applied.
However, there has been a challenging problem due to the infeasibility to evaluate inferred time-varying Bayesian network. Tranditionally, network inference model has been assessed by comparing predicted genetic regulatory interactions with those known from the biological literature [7]. This approach is controversial due to the absence of known gold standards, which renders the estimation of the sensitivity and specificity, that is, the true and false detection rate, unreliable and difficult.
Rare attempts to generate simulated gene expression data have been developed. In [8], author proposes simulation model for biological system to try on inferred DBN resulted from the simulated gene expression data. [7] develops simulated gene expression data from a realistic biological network involving DNAs, mRNAs, inactive protein monomers and active protein dimers.
Modern approaches such as [6, 9, 10] make an assumption to fully utilize time series dataset: underlying network structure are sparse, vary smoothly across time, and models first-order Markovian. From the assumption, it is derived that temporally adjancent networks are likely to share common edges than temporally distal networks. This assumption makes it possible to reconstruct time varying network with single multivariate time series data. Inituitively, inferred network resulted from time series gene expression data which is generated from underlying network based on the assumption should be maximally equivalent to the underlying network. In other words, time-varying network made up based on the assumption gives upperbound of performance of network inference model in which gene expression data is generated from the underlying network. Therefore, in this paper totally different approach is used for assessing time varying dynamic Bayesian network. First, time varying network is built, and time series dataset is generated from the network. Then the simulated dataset can be used for measuring the performance of methodologies of which their assumption is based on first-order Markovian model.
2 Method
2.1 Preliminaries
Models describing a stochastic temporal processes can be naturally represented as dynamic Bayesian networks [11]. As defined in [6], taking the transcriptional regulation of gene expression as an example, let \( {\mathbf{X}}^{\text{t}} \text{ := }\left( {X_{1}^{t} , \ldots ,X_{p}^{t} } \right)^{T} \in {\mathbb{R}}^{p} \) be a vector representing the expression levels of \( p \) genes at time \( {\text{t}} \), a stochastic dynamic process can be modeled by a “first-order Markovian transition model” \( {\text{p}}({\mathbf{X}}^{\text{t}} |\varvec{X}^{t - 1} ) \), which defines the probabilistic distribution of gene expression at time \( {\text{t}} \) given those at time \( {\text{t}} - 1 \). Under this assumption, likelihood of the observed expression levels of these genes over a time series of \( {\text{T}} \) steps can be expressed as:
where \( \uppi_{\text{i}} \) is the set of genes specifying the gene i, and the transition model \( p({\mathbf{X}}^{\text{t}} |{\mathbf{X}}^{{{\text{t}} - 1}} ) \) factors over individual genes. Each \( p({\text{X}}_{\text{i}}^{\text{t}} |{\text{X}}_{{\uppi_{\text{i}} }}^{\text{t}} ) \) can be viewed as a regulatory gate function that takes multiple covariates and produce a single response. A simple form of the transition model \( {\text{p}}({\mathbf{X}}^{{\mathbf{t}}} |{\mathbf{X}}^{{{\text{t}} - 1}} ) \) in a DBN is a linear dynamic model:
where \( {\mathbf{A}} \) is a matrix of coefficients relating the expressions at time \( {\text{t}} - 1 \) to those of the next time point, and \( \varvec{\upepsilon} \) is a vector of isotropic zero mean Gaussian noise with variance \( \sigma^{2} \).
Our simulator generates time-series gene expression dataset under assumption (2):
where \( x_{\text{i}}^{\text{t}} \) is \( i \)-th gene expression level at time point t, and \( \alpha_{0} \) is the parameter to regulate the influence of the target gene expression level at previous time point on one at time point t. \( \beta_{j} \) is the degree of association that affects gene expression level at target time point. Finally, expression level of each gene at a time point is generated with a noise with 0 mean, and \( \sigma^{2} \) variance.
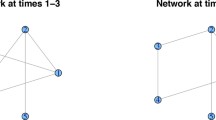
At network building stage, a set of genes is grouped to generate gene expression data based on the group in which a gene is belongs to only one group. Group is made to make it possible to activate associations in the group at the same time. To represent temporal interaction between genes, degree of activation of group is varying over time, and multiple groups are activated at different time point for different time periods. The example of interaction variation is illustrated in Fig. 1.

The examples for variation of interactions possibly appeared in underlying network. \( \varvec{\beta}_{{\varvec{ab}}} \) is the interaction between gene a and b. It is smoothly increased and decreased in activation over time periods. \( \varvec{\beta}_{{\varvec{ad}}} \) repeats to be activated spontaneously.
2.2 Algorithm
The algorithm takes parameters the number of genes n, the number of time points m, target influence parameter \( \alpha_{0} \). And it produces time varying network and time series gene expression data over m time points, and group information of each gene.
At the first stage, time varying Bayesian network is built from line 2 to 5. Then gene expression level is generated based on underlying network structure. At line 2, each node belongs to a group, and their interactions within the group are randomly set at line 3. Finally, activation period of each group is set randomly.
At second stage, time series gene expression data is generated. The expression levels of genes at initial time point are randomly set ranging from 0.3 to 1. \( X^{i} \left[ j \right] \) means gene expression level of j-th gene at time point t, and G[i, j] is group number of interaction between i-th gene and j-th gene. Activation period and degree of activation are contained in the matrix gInfo whose row represents group, and first column for the starting point of activation, and second column for ending point of activation, and third column for degree of activation.

3 Result
This section shows the procedure of parameter optimization to generate gene expression level smoothly varying over time. The parameter \( \alpha_{0} \) is optimized to generate smooth gene expression levels.
First, we attempted to generate small number of genes’ simulated data. As shown in Fig. 2, gene expression level grows up to infinity as time increased because the number of genes having influence on target gene is large. As parameter n is increased, the expression level of target gene is not smoothly varying over time because the target gene affected by its associated gene is changed drastically. It leads us to attempting second experiment with regulation of parameter \( \alpha_{0} \). The configuration of setting target influence parameter to .9 generates gene expression level as shown in Fig. 3.

This is expression level of a gene from 20 genes nodes. The initial expression level is set ranging from 0 to 1

Two examples among 20 genes. The expression level at initial time point is set ranging from 0 to 1. And target influence parameter \( \alpha_{0} \) is set to 0.9
In third experiment, network is built based on group. The associations between genes only appeared in group. Figure 4 illustrates simulation data generated from the group setting. Without setting target influence parameter \( \alpha_{0} \), gene expression level does not look smooth across time.

Gene expression data generated from group setting. The gene expression level at initial time point is set randomly ranging from 0 to 1.
Finally, we investigate how to set \( \alpha_{0} \) to generate smooth time series gene expression data set as the number of nodes increases. The Figs. 5, 6, and 7 illustrates smooth gene expression levels.

Gene expression level resulted from setting \( \alpha_{0} \) to 0.8 and 0.9 for left and right figure respectively. The number of genes is 32.

Gene expression level resulted from setting \( \alpha_{0} \) to 0.9 and 0.95 for left and right figure respectively. The number of genes is 64.

Gene expression level resulted from setting \( \alpha_{0} \) to 0.9 and 0.95 for left and right figure respectively. The number of genes is 128.
4 Conclusion
Traditionally, network inference model has been assessed by comparing inferred network with associations between genes known from the biological literature. This approach is infeasible to measure false detection rate. In this paper, we propose a simulation model for the use of assessing network inference algorithm. The proposed simulator generates time varying Bayesian network, time series gene expression data resulted from the network, and group information of genes. For generating gene expression level smoothly varying across time, target influence parameter has been optimized. The simulated dataset can be used to evaluate network inference algorithms in which smoothness of temporal process is assumed. As future work, simulation model for imitating genetic regulatory system can be developed. Currently, gene expression level is affected only by expression level at previous time point. However, in genetic regulatory system, gene expression level can also be affected by protein. Simulation model that attempts to reflect real regulatory system can be widely used to evaluate network inference model under various network structure.
References
Friedman, N., Linial, M., Nachman, I., Pe’er, D.: Using Bayesian networks to analyze expression data. J. Comput. Biol. 7(3–4), 601–620 (2000)
Werhli, A.V., Husmeier, D.: Reconstructing gene regulatory networks with Bayesian networks by combining expression data with multiple sources of prior knowledge. Stat. Appl. Genet. Mol. Biol. 6(1) (2007)
Needham, C.J., Bradford, J.R., Bulpitt, A.J., Westhead, D.R.: A primer on learning in Bayesian networks for computational biology. PLoS Comput. Biol. 3(8), e129 (2007)
Perrin, B.-E., Ralaivola, L., Mazurie, A., Bottani, S., Mallet, J., d’Alche–Buc, F.: Gene networks inference using dynamic Bayesian networks. Bioinformatics 19(suppl 2), ii138–ii148 (2003)
Yu, J., Smith, V.A., Wang, P.P., Hartemink, A.J., Jarvis, E.D.: Using Bayesian network inference algorithms to recover molecular genetic regulatory networks. In: International Conference on Systems Biology (2002)
Song, L., Kolar, M., Xing, E.P.: Time-varying dynamic bayesian networks. In: Advances in Neural Information Processing Systems, pp. 1732–1740 (2009)
Husmeier, D.: Sensitivity and specificity of inferring genetic regulatory interactions from microarray experiments with dynamic Bayesian networks. Bioinformatics 19(17), 2271–2282 (2003)
Smith, V.A., Jarvis, E.D., Hartemink, A.J.: Evaluating functional network inference using simulations of complex biological systems. Bioinformatics 18(suppl 1), S216–S224 (2002)
Song, L., Kolar, M., Xing, E.P.: KELLER: estimating time-varying interactions between genes. Bioinformatics 25(12), i128–i136 (2009)
Ahmed, A., Xing, E.P.: Recovering time-varying networks of dependencies in social and biological studies. Proc. Nat. Acad. Sci. 106(29), 11878–11883 (2009)
Kanazawa, K., Koller, D., Russell, S.: Stochastic simulation algorithms for dynamic probabilistic networks. In: Proceedings of the Eleventh Conference on Uncertainty in Artificial Intelligence, pp. 346–351. Morgan Kaufmann Publishers Inc. (1995)
Acknowledgement
This research was supported by the MISP (Ministry of Science, ICT & Future Planning), Korea, under the National Program for Excellence in SW supervised by the IITP (Institute for Information & communications Technology Promotion) (R22151610020001002).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this paper
Cite this paper
Lee, G., Lee, H., Sohn, KA. (2017). Generating Time Series Simulation Dataset Derived from Dynamic Time-Varying Bayesian Network. In: Kim, K., Joukov, N. (eds) Information Science and Applications 2017. ICISA 2017. Lecture Notes in Electrical Engineering, vol 424. Springer, Singapore. https://doi.org/10.1007/978-981-10-4154-9_7
Download citation
DOI: https://doi.org/10.1007/978-981-10-4154-9_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4153-2
Online ISBN: 978-981-10-4154-9
eBook Packages: EngineeringEngineering (R0)