
Abstract
Vascular cognitive impairment (VCI) describes a spectrum of cognitive changes occurring secondary to damage of the large and small vessels that supply blood to the brain. VCI has been recognized as the second most common cause of dementia and as the most common pathological comorbidity of Alzheimer’s disease. The pathogenesis of VCI appears to be heterogeneous, involving neurodegenerative mechanisms that remain to be fully understood. Stroke and vascular risk factors interfere with many processes subserved by the cerebral vasculature, maintaining cerebral homeostasis (for instance, maintaining and augmenting blood flow, oxygen, glucose supply), providing a structural and chemical barrier between the peripheral circulation and the brain parenchyma, serving intricate immunological functions, and providing a neurogenic niche for brain tissue repair. This chapter discusses the known and theoretical pathophysiological background of VCI, focusing on stroke and disruption of the neurovascular unit (NVU), which contribute to defects in neurotransmitter systems and to disruption of large-scale functionally co-activating networks, which contributes to cognitive deficits and decline.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

7.1 Introduction
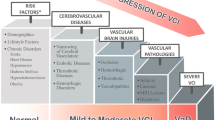
Vascular cognitive impairment (VCI) is defined as a syndrome with evidence of clinical stroke or vascular brain injury associated with cognitive impairment affecting at least one cognitive domain [1]. It includes to a full range of cognitive deficits secondary to cerebrovascular cerebral vascular injury, with vascular dementia being the most severe manifestation. Cerebrovascular pathology has been recognized as the second most common cause of dementia, as well as the commonest pathological comorbidity in Alzheimer’s dementia, highlighting the importance of cerebrovascular factors in cognitive decline from a global perspective. Risk factors for VCI include aging, vascular disorders such as hypertension, diabetes, smoking, and genetic mutations. Historically, VCI was attributed to occlusive large artery disease leading to multiple lacunes or focal territorial, cortical-subcortical infarcts, as suggested by Hachinski’s term “multi-infarct dementia,” introduced in the 1970s to distinguish vascular dementia from the neurodegenerative disorders (Fig. 7.1) [1]. Advances in neuroimaging, and autopsy studies, have revealed new substrates in the pathogenesis of VCI, including subcortical white matter hyperintensities, microinfarcts, microbleeds, and other subtler changes including a decline in the microstructural integrity of brain tissue appearing normal on anatomical brain scans, particularly mild VCI or in early stages. Because the pathogenic mechanisms of VCI are complex and heterogenous, the pathophysiology of VCI remains yet to be fully understood. In this chapter, the known pathophysiological mechanism of VCI will be reviewed. The roles that concepts such as the neurovascular unit, neurotransmitter system, and large-scale neural network may play will be discussed.

A T1-weighted MRI of a 70-year-old woman with multi-infarct dementia who suffered bilateral dorsal medial thalamic infarcts (yellow solid arrows) causing marked short-term memory loss, followed by a right medial frontal infarct (red open arrows) a few years later causing profound apathy and loss of initiation of mental and physical activity
7.2 The Neurovascular Unit (NVU)
The cerebral vasculature provides numerous critical functions. It forms a selectively permeable barrier that orchestrates the crossing or exclusion of molecules to and from the brain parenchyma; it sends and senses signals to accommodate dynamic demands for cerebral oxygenation and glucose; and it fulfills immunological roles to support the defense of the brain against invading microorganisms or harmful materials. To fulfill these roles, the endothelial cells and the vascular smooth muscle cells (VSMC) of the cerebral vasculature work closely with multiple types of cellular components, including pericytes, astrocytes, microglia, and neurons, which collectively form the neurovascular unit (Fig. 7.2). Each of these NVU components plays critical roles in times of neural vascular injury, acting as important determinants of VCI outcomes and progression. The known specific contributions of the NVU components to VCI are reviewed in the following sections.

A cross-sectional schematic representation of the neurovascular unit. The NVU is made of cerebral vasculature (endothelial cells and vascular smooth muscle), pericytes, astrocytes, microglia, and neurons, which work collectively to ensure cerebral homeostasis. The cerebral endothelial cells are conjoined by tight junctions that impede the bidirectional diffusion of polar molecules between blood and the brain. The endothelial cells are surrounded by pericytes embedded in the basal laminal matrix, which are important for maintaining both cerebral blood flow and BBB integrity. Neurons modulate cerebral hemodynamics via astrocytes, star-shaped cells that encase the cerebral blood vessels in their end feet. Microglia are the resident macrophages of the human brain; they work closely with astrocytes to fulfill the immunological roles of NVU. In contrast to the arterioles, pericytes are more plentiful in the capillaries and venules, fulfilling many structural, contractile, immunological, and phagocytic functions in the smaller vessels
7.2.1 The Blood-Brain Barrier (BBB)
Cerebral blood vessels are free of fenestrae—small holes that permit the free diffusion of materials across the vessel walls. Instead, endothelial cells conjoined by occludins and claudins form a physical barrier that impedes the bidirectional diffusion of polar molecules between blood and the brain, which protects the brain from potentially harmful substances from the peripheral circulation. Instead, molecules cross the BBB via active transport due to the presence of multiple transporters, which are critical for the maintenance of cerebral homeostasis, carrying across important substances such as apolipoproteins and pumping out potentially harmful waste products.
BBB integrity is found frequently to be compromised in VCI, manifested as elevated albumin in CSF and detection of fibrinogen and immunoglobulins in injured white matter [1]. The two major causes of BBB leakage appear to be pericyte injury and endothelial dysfunction. Pericytes, the mural cells of cerebral capillaries, have been shown to be critical for the maintenance of BBB integrity [2]. BBB leakage has been reported in multiple rodent models that have pericyte deficiency [2]. Arterial atherosclerosis, stroke, and vascular risk factors like hypertension and diabetes have well-established associations with BBB leakage. In stroke-prone hypertensive rodent models, BBB leakage was found to be associated with ischemic injuries [1], even in the absence of arteriolar occlusion.
One harmful substance in the CNS affected by disruption of BBB integrity is cerebral amyloid, which can be elevated in the CNS due to disruption of active transporter expression. Cerebral amyloid beta (Aβ) is primarily cleared through the perivascular spaces surrounding the penetrating venules and the active transporters like low-density lipoprotein receptor-related protein-1s (LRP-1) across BBB [3]. Impaired Aβ clearance has been linked to LPR-1 deficiency and enlarged perivascular spaces [3]. Thus, BBB defects might result in an elevation in CSF Aβ concentration, which may contribute to the early formation of amyloid plaques on the cerebral vasculature. Amyloid deposition on blood vessel walls (“cerebral amyloid angiopathy”) can further aggravate endothelial dysfunction, resulting in microbleeds, white matter hyperintensities (WMH) visible on T2-weighted MRI, and elevated risk of larger intracerebral hemorrhage (Fig. 7.3). The fibrillation of amyloid beta 42 leads to deposition as amyloid plaques in the neuropil, a well-established feature of AD, are associated with further damage seen as cerebral atrophy and usually cognitive deficits, while the perivascular deposit of amyloid beta 40 is associated with microbleeds. Lobar microbleeds have also been linked to deficits in global cognition and in visuospatial and executive functions [4].

A gradient echo MRI illustrating different manifestations of cerebral amyloid angiopathy in an 81-year-old woman, including large left frontal lobar hematoma cavity surrounded peripherally by hemosiderin (iron-containing macrophages and glial scar; blue arrow). Cerebral microbleed (red open arrow), superficial siderosis (yellow solid arrows), and white matter hyperintensities are also seen (purple)
The presence of BBB defects in both VCI and AD brains suggests a potential interaction between cerebrovascular dysfunction and AD pathology; however, the causal relationships between BBB defects and AD pathology remain to be fully clarified. One hypothesis, proposed by Zlokovic, is that AD is a vascular disorder involving two distinct “hits” as opposed to a pure amyloid pathology (Fig. 7.4) [5]. Zlokovic proposed that the earlier phase of AD could be non-amyloidogenic and purely vascular. The pathogenesis might be initiated by neuronal injuries and white matter infarcts (first hit) secondary to BBB leakage and small vessel hypoperfusion, which might facilitate the development of amyloid accumulation (second hit). This would also be consistent with the observed changes in the transporters that clear Aβ from the brain found in vascular injury, with deficits in perivascular drainage suggested in more recent studies, and with reports that vascular brain changes on MRI can precede amyloid deposition [6]. Under this hypothesis, VCI could be an intermediate phase in the development of some cases of AD, and amyloidosis might contribute to some cases of VCI, placing pure AD and pure VCI on opposite ends of continuous spectrum. Ongoing studies seek to uncover the key determinants of the two-hit pathology model and to clarify the complex interrelationships between cerebral vasculopathy and AD.

The two-hit hypothesis of neurodegeneration due to Alzheimer’s disease and VCI. The two-hit hypothesis [6] postulates cerebral vascular injury as the “first hit” in a two-fold causality of cognitive decline. Cerebral vascular injury caused by vascular risk factors and stroke is considered to damage the blood-brain barrier (BBB), the neurovascular unit, and the surrounding gray and white matter through hypoxia or oxidative stress. The second hit is considered to be amyloid cascade, which damages neurons directly through neuronal toxicity and synaptic dysfunction. These two concomitant insults interact. Amyloid deposited along the arteries gives rise to cerebral amyloid angiopathy (CAA), which compounds vascular injury and impedes amyloid clearance, while BBB deficits further impair the clearance of amyloid, for instance, due to a decrease in LPR1 expression. Under this hypothesis, cerebral vascular pathology and amyloid deposition can lead to cognitive decline due to their reciprocal interaction. Moreover, VCI could be an intermediate phase in the development of some cases of Alzheimer’s disease
7.2.2 Cerebral Vasculature and Neurovascular Coupling
The cerebral vasculature adopts an “outside in” distribution pattern. Cerebral arteries arise from the circle of Willis (located at the base of the brain) and give rise to a rich network of pial vessels along the brain surface. These superficial arteries then divide into long and narrow penetrating smaller arteries and arterioles, without collaterals, to supply blood to the deep subcortical areas. Therefore, any reduction in the blood flow of larger vessels due to occlusion or carotid stenosis would be exaggerated in the smaller vessels, which cannot be compensated for by adjacent vessels. To accommodate this specialized vascular distribution, the high demand for energy consumption, and the lack of local metabolic reserves, the cerebral vasculature has developed a set of well-coordinated signaling mechanisms that facilitate “functional hyperemia,” ensuring sufficient blood flow to distinct cerebral regions. As neural activity changes, vasoactive agents released by endothelial cells and astrocytes act to shape regional hemodynamics. Vascular smooth muscle cells (VSMC) and pericytes (particularly in the smaller vessels) also act to maintain homeostasis by sensing and regulating blood flow directly in the vessels via chemical or physical signals, in a process called autoregulation.
Attenuated cerebral blood flow (CBF) and impaired neurovascular coupling are typical features of VCI. Reduced global resting CBF has been detected in patients with vascular comorbidities or with a heavy burden of white matter injury. Areas surrounding WMH are typically found to have lower vascular reactivity [7]. Interestingly, there is evidence of venous disease in the vicinity of WMH, in addition to arteriolosclerosis. In men with coronary artery disease, CBF was related to poorer global cognitive function and to obesity measures; however, the mechanisms underlying these observations remain to be fully elucidated [8]. Since the deep white matter is particularly vulnerable to CBF deficits, and white matter injury has been linked to executive and psychomotor processing speed, chronic CBF reduction is a likely contributor to cognitive decline. Venous collagenosis further exacerbates alterations in CBF by increasing resistance [9]. Chronic insufficiency in blood supply might also be associated with neuronal loss and brain atrophy, owing to the high demand for energy consumption in neurons.
Different NVU compartments, including VSMCs, endothelial cells, pericytes, and astrocytes, all contribute differently to the impaired CBF and neurovascular coupling. VSMCs, as the sole controller of arterial blood flow, were found to contribute to impaired functional hyperemia [10]. During functional hyperemia, VSMCs hyperpolarize upon the release of nitric oxide released from neurons and endothelial cells, dilating the arteries to increase local blood flow. The VSMC layers also ensure a relatively uniform arterial pressure through autoregulation, preventing the pressure of downstream microvessels from swinging dramatically during cardiac systole and diastole. Therefore, direct injury to the VSMCs affects both resting CBF and functional neurovascular coupling. Stiffness and loss of arterial smooth muscle elasticity secondary to vascular risk factors have been associated with reduced resting white matter CBF and loss of the ability to adapt CBF to metabolic need [1]. Similarly, since the release of NO from endothelial cells directly induces VSMC relaxation, endothelial dysfunction secondary to ischemia or vascular risk factors (e.g., hypertension) can also impair functional hyperemia (Fig. 7.5).

Oxidative stress and neurovascular uncoupling. The synthesis of free radicals in the brain can be initiated by the production of superoxide (O2·) as a product of NADPH oxidase activation. NADPH oxidase can be activated in the presence of inflammatory signals or under conditions of cellular stress. The O2· generated can be deactivated by superoxide dismutase and catalase into water or give rise to a hydroxyl radical (OH·). These reactive oxygen species can bind to adjacent lipids, proteins, or DNA, leading to cellular damage. Superoxide can also directly affect neurovascular coupling through the consumption of nitric oxide (NO), which is the major vasodilatory signal under functional hyperemia. NO is synthesized by eNOS, residing in the endothelial cells, upon activation by factors such as shear stress or muscarinic cholinergic receptor 1 activation during times of neuronal activity. The generated NO will then relax the vascular smooth muscle via cGMP activation, resulting in vasodilation. O2· can react with NO and give rise to another longer-lived free radical species, peroxynitrite (ONOO−), which can also covalently bind to adjacent macromolecules and cause cellular injury. The consumption of NO also impairs the vasodilatory signal, causing defects in neurovascular coupling
Dysfunction of the pericytes and astrocytes can contribute to deficits in neurovascular uncoupling detected in VCI. Pericytes are mural cells embedded in the basement membrane of cerebral capillaries and pre- or postcapillary mirovessels. Pericytes have a direct modulatory effect on capillary blood flow. In rodent models that lack pericyte coverage on part of their capillaries, a delay in stimulus-driven vasodilatory response was observed. Comparing the covered and non-covered capillaries, there was a reduction of capillary blood flow in the non-covered ones [11]. In the same loss of function model, a reduction in global CBF was detected, indicating the critical role of pericytes and capillary vascular coupling in both functional and resting blood flow. Pericyte degeneration is a frequent feature of VCI and brains with evidence of subcortical ischemic vascular disease (SIVD) [11]. Pericyte malfunctioning has also been proposed to contribute to postischemic white matter injury. By adopting a hypercontractile phenotype following ischemic attack, they stall capillary blood supply following stroke and contribute to the secondary microvessel hypoperfusion and white matter injury [12].
Astrocytes are glial cells that serve to intermediate between neurons and cerebral vasculature. They are found to have modulatory effects on both arteriolar and capillary tone, through their interactions with VSMCs and pericytes. The interactions are mediated by astrocytic Ca2+ oscillation and the release of arachidonic acid lipid metabolites, which can result in vasodilation or vasoconstriction through the phospholipase A2 or phospholipase D2 pathways [10]. Presumably, astrogliosis or direct injury to astrocytes would negatively affect neurovascular coupling and CBF. However, the specific mechanisms and the effect size of astrocytic modulation on VSMCs and pericytes remain controversial due to the variable results generated under different experimental settings, which still need to be resolved by future studies.
One of the mechanisms that could injure the NVU directly is oxidative stress. Animal studies have demonstrated a direct connection between cerebral hypoperfusion and oxidative stress [1]. Clinical studies have also reported elevation of isoprostanes, cytokines, and adhesion proteins in both the damaged white matter and the systemic circulation of VCI patient, while peripheral lipid hydroperoxide concentrations were found to be reflective of deep WMH caused by hypertension [13]. At the molecular level, superoxides are generated by nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) in the cerebral endothelial cells in response to hypoxia, inflammation, or hypertensive stimuli, which could react directly with the surrounding macromolecules and lead to endothelial, neuronal, and pericyte dysfunction (Fig. 7.5). The superoxide anion generated can also react with nitric oxide (NO), consuming this critical vasodilator released by both neurons and endothelial cells. Oxidative stress mediators can thus directly reduce CBF and functional hyperemia by inactivating NO (Fig. 7.5). The association between SIVD and oxidative damage to lipid molecules also suggests the potential for oxidative stress to interfere with arachidonic acid lipid-mediated vasodilatory capacity [13]. Perhaps even more importantly, superoxide gives rise to other reactive oxygen species (ROS) or reactive nitrogen species (RNS) like hydrogen peroxide (H2O2), hydroxyl radicals (OH·−), and peroxynitrite (ONOO·−), which initiate a chain of additional redox reactions. It is thought that ROS and RNS generated through these chain reactions propagate injury to multiple components of the cerebral vasculature and NVU and to the adjacent white matter. Accordingly, interventions attempting to reduce free radicals have shown promise in rodent models of cerebral ischemia, and clinical trials are underway.
7.2.3 Glial Cells and Neuroinflammation
In people with vascular disease, inflammation has been associated with cognitive decline and dementia [14]. Upregulation of cerebral cytokines and chemokines was detected in hypertensive mice models and has been directly related to cognitive decline [15]. Even though the mechanism remains unclear, the presence of neuroinflammation has been reported in VCI patients [16], presumably due to both cerebral vascular injury and crosstalk between the periphery and the central nervous system. Neuroinflammation has been proposed to be one of the major mechanisms mediating postischemic secondary cell death and neurovascular dysfunction [17]. In mouse models of vascular dementia, elevated astroglial NF-kB expression has been shown to contribute to white matter damage [18]. Even though the direct link between inflammatory markers and cognitive function in VCI patients remains scarce, animal models have shown the association between elevation of central or peripheral cytokines and cognitive deficits [19], highlighting the potential role of inflammation in VCI.
The specialized foot soldiers of the innate cerebral immune responses are astrocytes, microglial cells, and perivascular macrophages. Astrocytes and microglial cells frequently communicate through the release of inflammatory signals. Just as astrocytes serve as an intermediary between neurons and the cerebral vasculature, they also act as an inflammatory intermediary between multiple cell types. Astrocytes express interleukin receptors, and they are capable of secreting cytokines. Astrocytes can be activated by IL-1β and TNF released from microglia and then release further cytokines that are sensed by the microglia to establish either a feed-forward cycle or a regulatory negative-feedback signal. Constantinescu et al. reported that astrocytes are sensors of hypoxia, becoming activated and releasing pro-inflammatory cytokines like interleukin (IL)-23 [20], which initiates and perpetuates an IL-17-mediated inflammatory response [20]. A similar inflammatory response mediated by interferon-γ (IFN-γ) has also been associated with postischemic injury in animals.
Microglial cells are the macrophages resident in the human brain. They are activated within 2 h post-stroke, releasing IL-1β and TNF and becoming voraciously phagocytotic, enabling them to remove cellular debris [21]. The release of inflammatory factors upon microglial activation, including IL-1β and TNF, IL-17, IL-6, and chemokine ligands (CXCL), can contribute to tissue damage resulting from postischemic neuroinflammation in animal studies, in a cascade ultimately intended to resolve into active post-inflammatory tissue repair [20].
Microglial cells and astrocytes can polarize into different phenotypes depending on the activation signals presented. Secrotome studies have found a shift of astroglial cytokine profiles upon activation by IL-1β and TNF, indicating the potential for astrocytes to change their secretome under inflammatory conditions [22]. Microglia were found to adopt either pro-inflammatory or anti-inflammatory phenotypes following activation [21]. The specific mechanisms driving the polarization of glial cells remain to be fully understood; however, the polarization phenomenon is well preserved among many types of immune cells. One potential mechanism might involve the release of cytokines from populations of pro- and anti-inflammatory peripheral immune cells, which have been shown in animal studies to modulate postischemic brain injury [20]. The polarization phenomenon makes astrocytes and microglial cells potential targets for preventing ischemia-related brain injury and functional recovery.
The mechanisms contributing to the resolution of neuroinflammation following vascular injury also remain under investigation. The presence of both inflammatory and anti-inflammatory T cells has been reported in ischemic tissue. A set of anti-inflammatory regulatory T cells (Treg) can be developed from naïve T helper (Th) cells, in the presence of IL-10 or TGF-β. Experimental depletion of Treg expression in animal models exacerbates functional deficits following ischemia [20]. Further studies are necessary to elucidate the crosstalk between glial cells and Tregs and to harness their potential to reduce postischemic injury mediated by neuroinflammation. Moreover, inflammatory signals which are harmful at one stage may be beneficial at another. For instance, the release of IL-23 or IL-17 may exacerbate postischemic injury but also stimulate the release of growth factors (e.g., brain-derived neurotrophic factor) that participate in angiogenesis and neurogenesis and the repair of the NVU.
7.3 Strategic Pathways of Neurotransmitters
Synapses are considered to be the basic units of neural connectivity. The release of neurotransmitters in synaptic spaces enables the communication between individual neurons and neuronal groups within a certain region and between regions. For instance, glutamatergic and GABAergic neurons that reside universally in the brain will release glutamate/GABA locally, which allows for communication with proximal neurons, whereas cholinergic neurons innervate the entire cortex via long axonal projections emanating from the basal forebrain.
Neurochemical studies and imaging studies in VCI have found abnormities in neurotransmitter systems, even though the underlying mechanisms and the relationship of these defects to neurovascular pathologies remained uncovered. The cholinergic system appeared to be the most commonly impaired system in VCI, manifested as decreases in choline acetyltransferases and cholinergic receptor expression in animal and autopsy studies, respectively [23]. Glutamate, which serves an essential physiological role in long-term potentiation, has also been studied extensively in VCI due to its role in excitotoxicity. Even though memory loss is less pronounced in VCI compared to Alzheimer’s disease, memory deficits are often key features of dementia with vascular contributions.
7.3.1 The Cholinergic System
The cerebral cortex is endowed with a rich system of cholinergic networks, originating from the basal forebrain and the brain stem (Fig. 7.6). Pedunculopontine tegmental (PPT) and laterodorsal tegmental (LDT) nuclei residing in the brain stem mainly project to subcortical regions, which provide cholinergic innervation to the thalamus and the striatum, while basal forebrain cholinergic nucleus, consisting of medial septal nucleus (MS ch1), the diagonal band of Broca nucleus (vDB ch2(v) + 3(h)), and nucleus basalis of Meynert (NBM, ch4) of the substantia innominata (SI), is the major cholinergic input for the cerebral cortex, the hippocampus, and partially the thalamus.

A schematic illustration of cerebral cholinergic projections The cerebral cholinergic network originates from two different nuclei systems, the basal forebrain cholinergic nuclei and the brain stem cholinergic nuclei. Pedunculopontine tegmental (PPT) and laterodorsal tegmental (LDT) nuclei reside in the brain stem. They mainly project to subcortical regions, which provide cholinergic innervation to the thalamus and the striatum. The basal forebrain cholinergic nucleus consists of four different nuclei, the medial septal nucleus (MS ch1), the diagonal band of Broca nucleus (vDB ch2(v) + 3(h)), and the nucleus basalis of Meynert (NBM, ch4) of the substantia innominata (SI). The basal forebrain cholinergic nuclei are the major cholinergic inputs for the cerebral cortex, the hippocampus, and partially the thalamus
Cholinergic neurons play critical roles in cognitive functions, ranging from executive function and memory to emotional processing. Impaired cholinergic cortical innervations have been associated with overall cognitive decline, particularly with reductions in executive function. Clinical trials have shown some potential cognitive benefits of cholinesterase inhibitors in VCI patients [24]. Cholinergic depletion secondary to cerebral vascular injuries is most commonly observed in the lateral projection of basal forebrain cholinergic nuclei, which passes through the external capsule (capsular division) and the claustrum (perisylvian division). The disruption of lateral cholinergic projections is likely to be linked to both white matter lesions (external capsule) and infarctions to the nuclei (claustrum), which in turn reduce cholinergic innervations to the cortex [25]. Direct insult to the NBM due to elongation of the internal carotid artery has also been associated with atrophy and cognitive deficits, indicating a role for large vessel disease [26].
Cholinergic neuronal loss due to cerebral infarction has been reported in several studies; however, the extent of cholinergic loss in VCI appeared to be contradictory [23]. Animal models of VCI utilizing choline acetyltransferase activity as the marker of cholinergic neurons have reported cholinergic depletion in the temporal cortex and the hippocampus; however, a MRI study in patients with multi-infarct vascular dementia found no neuronal changes in the NBM nuclei [27]. It has been hypothesized that this might be because neuronal arborization volume, but not neuronal cell numbers, was impaired in VCI [23]. Another potential mechanism behind the vascular injury-induced cholinergic reduction is proposed to be the decrease in receptor-ligand affinity, supported by the detection of modulated muscarinic expression in VCI [23]. Further studies comparing the volume of cholinergic nuclei between controls and distinct types of VCI are necessary.
Cholinergic tracts disrupted by WMHs (identified through CHIP) have been linked to executive function decline in multiple studies [28]. However, most of the lesions are identified through visually rated white matter hyperintensities that appeared in the cholinergic pathways which does not specify the spatial injuries to the cholinergic microstructure. Cholinergic microstructural changes might also lead to “large-scale network disruption” [29, 30].
A reciprocal correlation between cholinergic system function and cerebral blood flow has been identified, suggesting that changes in either cholinergic signaling or cerebral blood flow could influence the other. Animal studies show that of NBM cholinergic nuclei increased cortical blood flow, either through direct actions of acetylcholine or by activating nitric oxide synthase [23]. Reduction in CBF was suggested to be one of the underlying mechanisms of greater executive function decline in elderly people taking anticholinergic agents [31]. Therefore, the cholinergic system might also represent and important intrinsic link between cerebral blood flow and cognitive function, contributing to cognitive decline induced by vascular injuries.
7.3.2 The Glutamatergic System
Glutamate is the sole excitatory neurotransmitter of the central nervous system. Unlike the cholinergic pathways, which originate from certain nuclei and then divide into multiple sub-projections, glutamate neurons are dispersed throughout the brain, with multiple origins. Glutamate receptors can also be found universally in diverse types of neurons. The loss of glutaminergic synapses, indicated by the reduction of vesicular glutamate transporter 1, has been detected in the temporal cortex of VCI patients, though the glutamate synapse in the frontal cortex seems to be preserved [32]. Since glutamate is also the major player of cerebral excitotoxicity following ischemic or hemorrhagic attacks, research has focused on the utilization of anti-glutamatergic agents in reducing ischemic stroke related neurodegeneration. For instance, memantine was suggested to be potentially beneficial for patients with mild to moderate vascular dementia [33]. The impact of memantine on post-stroke functional recovery is currently under clinical investigation. Moreover, the excitotoxic tryptophan metabolites along the kynurenine pathway amplify glutamatergic signaling and apoptosis, and this has been implicated in post-stroke cognitive impairment [34].
Glutamate is essential for long-term potentiation (LTP), which is the underlying mechanism of learning and memory formation. Both animal studies and clinical studies have confirmed the contribution of glutamate receptors to memory formation, especially the NMDA and AMPA receptors. Both pharmacological and genetic modification of NMDA signaling could affect learning and memory formation in rats [35]. In humans, administration of an AMPA agonist, Ampakine CX516, was shown to have some benefits on short-term memory retention [32].
Even through glutamate has been shown to play an essential role in cognition and ischemia-related neurodegeneration, studies focusing on aspects of glutamatergic pathways in VCI remain scarce. One of the major underlying mechanisms of glutamatergic contribution to VCI might be strategic infarcts disrupting either the frontal parietal, the ventral tegmental, or the temporal glutaminergic projections to other neurotransmitter networks. A study utilizing transcranial magnetic stimulation found that glutamatergic defects might be involved dementia secondary to cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) [36]. Another clinical study has found correlations between temporal and frontal vesicular glutamate transporter (VGLUT1) concentrations and Cambridge Cognition Examination (CAMCOG) scores. The concentration of VGLUT was also reported to be higher in the frontal cortex of post-stroke patients who didn’t develop dementia [32], suggesting the potential correlation between loss of glutaminergic synapses and cognitive impairment in VCI.
7.4 Large-Scale Neural Network Perspectives
Human brains have evolved a set of intricate, interconnected, and functionally crucial networks. Different brain regions may be functionally connected in co-activating networks. The white matter tracts that interconnect these regions are often interrupted in VCI. This is thought to lead to patterns of atrophy, which co-occurs in functionally connected gray matter regions. Each of these types of networks, and their contributions to VCI, will be discussed in the following subsections.
7.4.1 Functional Networks
Functional magnetic resonance imaging (fMRI) allows the partitioning of brain into large-scale functional networks, which is more of a statistical concept built upon the correlations between spatially distinct regions that have common temporal activation patterns. The functional synchronicity between distinct brain regions is most often captured through low-frequency fluctuation signals of cerebral blood oxygen level-dependent (BOLD) fMRI. These functional networks remain relatively stable within a certain state, but they are much more plastic than anatomical networks as we switch between a resting state and various tasks. A single cerebral region could participate in different functional networks and change its connectivity with other regions according to different tasks.
Altered functional connectivity has been detected in multiple neurodegenerative disorders, including AD, multiple sclerosis, and VCI [37]. Alternations usually manifest as decreased or increase in connectivity or simply as changes in regional BOLD signal strength. It is useful to overlap anatomical and fMRI images to study the mechanisms underlying cognitive decline in VCI patients. Cognitive deficits associated with VCI more frequently manifest as decline in executive functions, attention, and psychomotor processing speed, rather than amnesia, particularly in early stages of subcortical VCI. Multiple studies utilizing resting-state fMRI have proposed the possible association between default mode network (DMN) connectivity deterioration and executive defects [38, 39].
DMN is a functional network conjoining frontal cortex, parietal cortex, and subcortical regions. It involves three major hubs, including posterior cingulate cortical (PCC), medial prefrontal cortical, and angular gyrus, as well as several medial temporal subregions of the hippocampus, parietal lobe, and retrosplenial cortex. DMN plays a critical role in externally orientated tasks. In healthy people, DMN is activated when the subject is in a wakeful state but not when focusing on a specific task. It is normally suppressed at times of focused attention [40].
The disruption of DMN resting functional connectivity in SIVD, particularly the connection between PCC and the frontal-subcortical pathways, has been directly linked to slower psychomotor processing speed, poorer executive function on the Stroop test, and decreased memory scores, indicating the role of the DMN in SIVD-induced cognitive impairment. Sun et al. reported disruption of resting functional connectivity in frontoparietal DMN pathways in 16 patients with VCI [38]. The suppression of resting functional connectivity with the PCC was detected in the left middle temporal gyrus, the left anterior cingulate cortex, the left and right middle frontal gyrus, and the left medial frontal gyrus, which overlaps with the DMN. It is thought that the decline in anterior parts of DMN connectivity might be compensated for by enhanced connectivity between PCC and the right inferior temporal cortex, left middle temporal gyrus, and the superior parietal lobule, accounting for these observations of both enhanced and diminished connectivity [38]. These results were confirmed by Yi et al. in 2012 [39], who also reported increased resting-state connectivity between the posterior components of DMN and decreased connectivity between the anterior components of DMN.
The default mode is an example of a large-scale functional network, which has helped to understand how vascular brain changes can interrupt brain circuits, producing some of the most salient cognitive symptoms in VCI. Other circuits, for instance, the attention network that is activated during times of focused mental effort, are activated when the DMN is suppressed, which is also affected in VCI [41].
7.4.2 Network Anatomical Connectivity
The anatomical connections between different cerebral regions can be interrupted by pathology related to VCI. Subcortical WMH burden detected on MRI images has been closely linked to the cognitive decline in both healthy aging population and VCI patients. In cognitively intact elderlies, increasing WMH volumes has been linked to gait disturbance and memory decline [42]; however, depending on the location and the volume of WMHs, the cognitive consequences associated with the WHMs may differ. Strategic WMH disrupting anterior thalamic white matter tracts and forceps were found to inversely correlate to executive function psychomotor processing speed [43], while the WMH load on the cholinergic tracts has also been reported to impair attention in SVD regardless of the overall cognitive status [44].
More subtle changes compromising the microstructure of axon tracts connecting the subcortical structures and the frontal cortex, but not visible on a T2 MRI, might be another underlying cause of functional connectivity defects. Diffusion tensor imaging (DTI) can be used to examine specific bundles of parallel nerve fibers, delineating individual white matter tracts by tracking the diffusivity of water molecules. The two measures most commonly generated from DTI are fractional anisotropy (FA), a measure of water diffusivity in a common orientation, and MD, a measure of the magnitude of water diffusion in any direction. FA is an indicator of microstructural change, which is frequently used to track white matter bundles. A decrease in FA may be an indicator of demyelination or other white matter defects. MD is considered to be more of a measure of cellularity and membrane density, which is inversely related to cell number. Therefore, edema would cause an elevation in MD measurements, while neoplasia would be linked to a decrease in MD values.
In cognitively normal patients with coronary artery disease, Santiago et al. reported significant associations between executive function and normal-appearing white matter microstructural integrity, measured as FA and MD [45]. Psychomotor processing speed and executive function were positively correlated with mean FA in the left parahippocampal, cingulum, and inferior fronto-occipital white matter tracts. Mean MD in the right parahippocampal cingulum, left inferior fronto-occipital, and left superior longitudinal white matter bundles was negatively associated with processing speed and executive function [45]. These indicate a contribution of white matter microstructural changes in early VCI.
The contribution of white matter tract injury at the microstructural level to disrupted functional networks was shown in a study of 20 healthy individuals, combining DTI and fMRI. Microstructural compromise of the cingulate white matter bundle conjoining the PCC and the lateral or medial temporal lobes was reported to correlate with suppressed functional connectivity between those regions [46]. Further studies combining DTI and fMRI in VCI patients would help to elucidate specific roles of white matter microstructural l damage in disrupting the neural circuits involved in executive function.
7.4.3 Structural Covariance Networks
The combination of statistical approaches and structural imaging allows the investigation of structural covariance networks (SCN), which are built based on the correlations between the structural characteristics (cortical thickness, white matter hyperintensity, brain-parenchymal fraction, etc.) of spatially distinct regions of interest (ROI). The SCN approach allows for the isolation of gray matter structural networks and the study of cross tract lesions. By examining covariance, much as in fMRI studies, SCNs overcome the limitation that important structural changes may be related even between regions that are not directly connected by white matter tracts. Yi et al. [39] proposed that gray matter atrophy might contribute partially to the decrease in DMN resting functional connectivity, since accounting for gray matter loss attenuated the differences in functional connectivity between VCI and controls. This finding suggests relationships between the anatomical and functional network changes.
Gray matter networks can be constructed through statistical approaches examining the covariance between the cortical thickness of ROIs. Tuladhar et al. reported an inverse relationship between interhemispheric frontoparietal ROIs’ cortical thickness covariates and white matter burdens [47], describing the contribution of white matter injuries to brain atrophy. Similarly, Nestor et al. showed the connection between subcortical white matter injury and gray matter atrophy in AD patients with SVD. They found that subcortical white matter volume was inversely related to covariance in cortical thickness within the hubs of DMN in AD [48]. Another study by Swardfager et al. reported a significant impact of WMH on verbal recall due to a specific covariance pathway mediated in serial by left temporal lobe atrophy and poorer verbal learning [49]. Further longitudinal studies combining SCN measures and cognitive outcomes are now necessary to confirm relationships with cognitive deficits in VCI.
7.5 Conclusion
VCI, as the second most important cause of dementia, is defined as a full range of heterogenous cognitive disorders attributed to cerebrovascular injuries. Damage to the neurovascular unit, disruption of neurotransmitter systems, and disconnection within large-scale neural networks contribute distinctly to the pathogenesis of VCI. The primary pathogenic mechanisms are considered to be cerebrovascular injuries (endothelial dysfunction, neurovascular uncoupling, etc.) caused by oxidative stress and neuroinflammation secondary to vascular risk factors and stroke, which not only affect BBB integrity and the clearance of Aβ but also impair the resting CBF and functional hyperemia. The interaction between cerebrovascular injuries and Aβ homeostasis reveals a potential link between VCI and AD, suggesting that VCI can contribute to cases of AD dementia. The cerebral hypoperfusion resulting from NVU disruption affects the firing of neurons and interrupts neurotransmitter systems, notably the cholinergic system, and the glutamatergic overstimulation, which has been linked to functional decline and post-stroke cognitive impairment, respectively. The resulting hypoperfusion can also injure the subcortical white matter tracts interconnecting functionally related regions, leading to distinct patterns of atrophy and changes in functional connectivity within neural networks, collectively contributing to psychomotor slowing, executive dysfunction, behavioral inactivation, and also to memory deficits.
References
Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844–66.
Armulik A, Genové G, Mäe M, et al. Pericytes regulate the blood–brain barrier. Nature. 2010;468(7323):557–61.
Ramirez J, Berezuk C, McNeely AA, et al. Imaging the perivascular space as a potential biomarker of neurovascular and neurodegenerative diseases. Cell Mol Neurobiol. 2016;36(2):289–99.
Chung C-P, Chou K-H, Chen W-T, et al. Strictly lobar cerebral microbleeds are associated with cognitive impairment. Stroke. 2016;47(10):2497–502.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–38.
Marnane M, Al-Jawadi OO, Mortazavi S, et al. Periventricular hyperintensities are associated with elevated cerebral amyloid. Neurology. 2016;86(6):535–43.
Makedonov I, Black SE, MacIntosh BJ. Cerebral small vessel disease in aging and Alzheimer’s disease: a comparative study using MRI and SPECT. Eur J Neurol. 2013;20(2):243–50.
MacIntosh BJ, Swardfager W, Robertson AD, et al. Regional cerebral arterial transit time hemodynamics correlate with vascular risk factors and cognitive function in men with coronary artery disease. Am J Neuroradiol. 2015;36(2):295–301.
Keith J, Gao FQ, Noor R, et al. Collagenosis of the deep medullary veins: an underrecognized pathologic correlate of white matter hyperintensities and periventricular infarction? J Neuropathol Exp Neurol. 2017;76(4):299–312.
Kisler K, Nelson AR, Montagne A, et al. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18(7):419–34.
Kisler K, Nelson AR, Rege SV, et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017;20(3):406–16.
Yemisci M, Gursoy-Ozdemir Y, Vural A, et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15(9):1031–7.
Swardfager W, Yu D, Scola G, et al. Peripheral lipid oxidative stress markers are related to vascular risk factors and subcortical small vessel disease. Neurobiol Aging. 2017;59:91–7.
van Exel E, de Craen AJM, Remarque EJ, et al. Interaction of atherosclerosis and inflammation in elderly subjects with poor cognitive function. Neurology. 2003;61(12):1695–701.
Toth P, Tucsek Z, Tarantini S, et al. IGF-1 deficiency impairs cerebral myogenic autoregulation in hypertensive mice. J Cereb Blood Flow Metab. 2014;34(12):1887–97.
Rosenberg GA. Extracellular matrix inflammation in vascular cognitive impairment and dementia. Clin Sci. 2017;131(6):425–37.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808.
Saggu R, Schumacher T, Gerich F, et al. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol Commun. 2016;4(1):76.
Cai M, Lee JH, Yang EJ. Bee venom ameliorates cognitive dysfunction caused by neuroinflammation in an animal model of vascular dementia. Mol Neurobiol. 2017;54(8):5952–60.
Swardfager W, Winer DA, Herrmann N, et al. Interleukin-17 in post-stroke neurodegeneration. Neurosci Biobehav Rev. 2013;37(3):436–47.
Kim J-Y, Kim N, Yenari MA. Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci Ther. 2015;21(4):309–19.
Choi SS, Lee HJ, Lim I, et al. Human astrocytes: secretome profiles of cytokines and chemokines. PLoS One. 2014;9(4):e92325.
Jellinger KA. Pathology and pathogenesis of vascular cognitive impairment: a critical update. Front Aging Neurosci. 2013;5:17.
Muir RT, Lam B, Honjo K, et al. Trail making test elucidates neural substrates of specific poststroke executive dysfunctions. Stroke. 2015;46(10):2755–61.
Liu Q, Zhu Z, Teipel SJ, et al. White matter damage in the cholinergic system contributes to cognitive impairment in subcortical vascular cognitive impairment, no dementia. Front Aging Neurosci. 2017;9:47.
Gao F, Pettersen JA, Bocti C, et al. Is encroachment of the carotid termination into the substantia innominata associated with its atrophy and cognition in Alzheimer’s disease? Neurobiol Aging. 2013;34(7):1807–14.
Kalaria RN. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016;131(5):659–85.
Black S, Roman GC, Geldmacher DS, et al. Efficacy and tolerability of donepezil in vascular dementia: positive results of a 24-week, multicenter, international, randomized, placebo-controlled clinical trial. Stroke. 2003;34(10):2323–30.
Lim J-S, Kim N, Jang MU, et al. Cortical hubs and subcortical cholinergic pathways as neural substrates of poststroke dementia. Stroke. 2014;45(4):1069–76.
McNeely AA, Ramirez J, Nestor SM, et al. Cholinergic subcortical hyperintensities in Alzheimer’s disease patients from the Sunnybrook Dementia Study: relationships with cognitive dysfunction and hippocampal atrophy. J Alzheimers Dis. 2015;43(3):785–96.
Lanctôt KL, O’Regan J, Schwartz Y, et al. Assessing cognitive effects of anticholinergic medications in patients with coronary artery disease. Psychosomatics. 2014;55(1):61–8.
Kirvell SL, Elliott MS, Kalaria RN, et al. Vesicular glutamate transporter and cognition in stroke: a case-control autopsy study. Neurology. 2010;75(20):1803–9.
Mijajlović MD, Pavlović A, Brainin M, et al. Post-stroke dementia—a comprehensive review. BMC Med. 2017;15(1):11.
Gold AB, Herrmann N, Swardfager W, et al. The relationship between indoleamine 2,3-dioxygenase activity and post-stroke cognitive impairment. J Neuroinflammation. 2011;8(1):17.
Rezvani AH. Involvement of the NMDA system in learning and memory. In: Levin ED, Buccafusco JJ, editors. Animal models of cognitive impairment. Boca Raton, FL: CRC Press/Taylor & Francis; 2006.
Palomar FJ, Suarez A, Franco E, et al. Abnormal sensorimotor plasticity in CADASIL correlates with neuropsychological impairment. J Neurol Neurosurg Psychiatry. 2013;84(3):329–36.
Wang L, Zang Y, He Y, et al. Changes in hippocampal connectivity in the early stages of Alzheimer’s disease: evidence from resting state fMRI. NeuroImage. 2006;31(2):496–504.
Sun Y, Qin L, Zhou Y, et al. Abnormal functional connectivity in patients with vascular cognitive impairment, no dementia: a resting-state functional magnetic resonance imaging study. Behav Brain Res. 2011;223(2):388–94.
Yi L, Wang J, Jia L, et al. Structural and functional changes in subcortical vascular mild cognitive impairment: a combined voxel-based morphometry and resting-state fMRI study. PLoS One. 2012;7(9):e44758.
Broyd SJ, Demanuele C, Debener S, et al. Default-mode brain dysfunction in mental disorders: a systematic review. Neurosci Biobehav Rev. 2009;33(3):279–96.
Fernández PJ, Campoy G, García Santos JM, et al. Is there a specific pattern of attention deficit in mild cognitive impairment with subcortical vascular features? Evidence from the attention network test. Dement Geriatr Cogn Disord. 2011;31(4):268–75.
Silbert LC, Nelson C, Howieson DB, et al. Impact of white matter hyperintensity volume progression on rate of cognitive and motor decline. Neurology. 2008;71(2):108–13.
Biesbroek JM, Weaver NA, Hilal S, et al. Impact of strategically located white matter hyperintensities on cognition in memory clinic patients with small vessel disease. PLoS One. 2016;11(11):e0166261.
Ishikawa H, Meguro K, Ishii H, et al. Silent infarction or white matter hyperintensity and impaired attention task scores in a nondemented population: the Osaki-Tajiri project. J Stroke Cerebrovasc Dis. 2012;21(4):275–82.
Santiago C, Herrmann N, Swardfager W, et al. White matter microstructural integrity is associated with executive function and processing speed in older adults with coronary artery disease. Am J Geriatr Psychiatry. 2015;23(7):754–63.
Teipel SJ, Bokde ALW, Meindl T, et al. White matter microstructure underlying default mode network connectivity in the human brain. NeuroImage. 2010;49(3):2021–32.
Tuladhar AM, Reid AT, Shumskaya E, et al. Relationship between white matter hyperintensities, cortical thickness, and cognition. Stroke. 2015;46(2):425–32.
Nestor SM, Mišić B, Ramirez J, et al. Small vessel disease is linked to disrupted structural network covariance in Alzheimer’s disease. Alzheimers Dement. 2017;13(7):749–60.
Swardfager W, Cogo-Moreira H, Masellis M, et al. The effect of white matter hyperintensities on verbal memory; mediation by temporal lobe atrophy. Neurology. 2018;90(8):e673–82.
Acknowledgments
We would like to gratefully acknowledge the support from our colleagues, Sabrina Adamo and Fuqiang Gao from Sunnybrook Research Institute, for their contribution to the processing and labeling of the MR images. W.S. gratefully acknowledges support from the Alzheimer’s Association (US), Brain Canada, The Canadian Partnership for Stroke Recovery, Sunnybrook Health Sciences Centre Department of Psychiatry, Sunnybrook Research Institute Hurvitz Brain Sciences Program, and the University of Toronto Department of Pharmacology and Toxicology. S.E.B. gratefully acknowledges financial and salary support from the Fondation Leducq, Canadian Institutes of Health Research (#125740 & #13129), Heart and Stroke Foundation Canadian Partnership for Stroke Recovery, Hurvitz Brain Sciences Research Program at Sunnybrook Research Institute, and the Linda C. Campbell Foundation. S.E.B. would also like to thank the Sunnybrook Research Institute, Sunnybrook Health Sciences Centre Department of Medicine, and the Brill Chair Neurology, University of Toronto, for financial and salary support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Science+Business Media Singapore
About this chapter

Cite this chapter
Yu, D., Swardfager, W., Black, S.E. (2020). Pathophysiology of Vascular Cognitive Impairment (I): Theoretical Background. In: Lee, SH., Lim, JS. (eds) Stroke Revisited: Vascular Cognitive Impairment. Stroke Revisited. Springer, Singapore. https://doi.org/10.1007/978-981-10-1433-8_7
Download citation
DOI: https://doi.org/10.1007/978-981-10-1433-8_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-1432-1
Online ISBN: 978-981-10-1433-8
eBook Packages: MedicineMedicine (R0)