
Abstract
Gene differences predisposing to atherosclerosis disease are important to identify and allow us to understand cellular processes involved in the progression and development of atherosclerotic disease. This protocol describes the experimental procedure for multiplex amplicon sequencing using barcoded primers. The procedures included in this chapter focus on amplicon library preparation (including polymerase chain reaction (PCR) conditions), DNA library quantitation options, next generation pyrosequencing with the 454 GS Junior system and data analysis. The process from DNA to sequencing data can be completed within 5 days.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Recent advances in DNA sequencing technologies have created opportunities for sequencing at an unprecedented depth and breadth and multiplex sequencing has emerged as an important strategy for sequencing of many different samples in parallel. In multiplex sequencing, a unique multiplex identifier (MID), or “barcode” sequence, is added to the DNA that is to be sequenced. After sequencing, reads are sorted into sample libraries via detection of the appropriate barcode.
Atherosclerotic diseases, including coronary artery disease (CAD) and myocardial infarction (MI), are the leading causes of death in the world [1]. The genetic basis of CAD and MI, which are caused by multiple interacting endogenous and exogenous factors, has gained considerable interest in the last years as genome-wide association studies (GWASs) have identified many new susceptibility loci for CAD and MI (for review see [2]). The underlying genes provide new insights into the genetic architecture of these diseases.
This protocol describes the detailed experimental procedure for multiplex amplicon sequencing. The procedure begins with a PCR amplification of a sequence of interest. This is then followed by a second PCR with universal primers containing barcodes and sequences for the 454 sequencing reaction. Optimization will be needed dependent on reagents or instruments for PCR. The amplicons are then quantified prior to sequencing (Fig. 33.1).

Work flow for the sequencing procedure using the 454 GS Junior
2 Materials
2.1 First and Second PCR
Oligonucleotide primers | Life technologies |
Genomic DNA (100 ng) | – |
DNA polymerase and PCR reaction buffer | Bioline reagents |
DNaseZap DNase decontamination | Ambion |
RNase/DNase free water | Ambion |
DNA LoBind 1.5 mL tubes, PCR clean | Eppendorf |
ART self-sealing barrier pipette tips | Sigma-Aldrich |
Mastercycler PCR thermocycler | Eppendorf |
96-well PCR reaction plate | Applied biosystems |
96-well adhesive film | Applied biosystems |
Multipurpose tabletop centrifuge | – |
Refrigerated centrifuge | Sigma-Aldrich |
Vortex | – |
[Optional] Ethidium bromide solution (0.5 μg/ml) | Sigma-Aldrich |
[Optional] Agarose | Sigma-Aldrich |
[Optional] 1X TBE buffer | Thermo scientific |
[Optional] Gel Doc™ XR+ | Bio-rad laboratories |
[Optional] DNA hyperladder I and loading buffer | Bioline reagents |
[Optional] Gel electrophoresis tanks, casts and power unit | Bio-rad laboratories |
2.2 PCR Clean-Up
UltraClean PCR clean-up kit | Mobio |
Multipurpose tabletop centrifuge | – |
ART self-sealing barrier pipette tips | Sigma-Aldrich |
2.3 Amplicon Quantification
2100 Bioanalyzer | Agilent technologies |
DNA 1000 kit | Agilent technologies |
Multipurpose tabletop centrifuge | – |
DNA LoBind 0.5 mL tubes, PCR clean | Eppendorf |
Vortex | – |
2.4 454 Library Preparation, Sequencing and Data Analysis
454 GS Junior sequencer | Roche |
GS Junior Titanium emPCR kit (Lib-A) | Roche |
GS Junior Titanium PicoTiterPlate kit | Roche |
GS Junior Titanium sequencing kit | Roche |
GS Junior Titanium control bead kit | Roche |
3 Detailed Procedure
3.1 First PCR (Target Amplification)
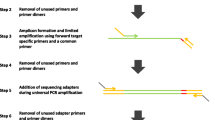
We implemented a two-step PCR procedure [4]. The first reaction involves conventional PCR primers (designed against the region of interest) to amplify the desired sequence form genomic DNA (Fig. 33.2). It is advisable to keep amplified products below 600 bp (see App. A.3). During primer design a ‘universal tail’ is also added to the 5′-end of each primer. The purpose of this modification is to allow the product gained from the first PCR to be used as a template for the second PCR.

Schematic representation of the two PCR reactions. The gene specific sequences for each primer are highlighted in blue. The universal tails which enable secondary primer binding are coloured red. The unique MID codes are highlighted in green and the 454 adaptor sequences are coloured purple
-
1.
On ice prepare the following reaction mixture. Mix the reagents well before use with a vortex. Also allow an additional 10 % volume to account for pipetting inaccuracies:
Reagent | 1 Reaction |
|---|---|
5× PCR reaction buffer (3 mM MgCl2 final concentration) | 5 μl |
10 μM forward primer | 1 μl |
10 μM reverse primer | 1 μl |
dNTPs (10 mM of each) | 2.5 μl |
1.25 U DNA polymerase | 0.25 μl |
Genomic DNA template (100 ng) | As required |
RNase/DNase free water | Up to 25 μl |
Final volume | 25 μl |
-
2.
Pipette the above mixture into a 96 well plate whereby one well contains one DNA individual sample.
-
3.
Attach the 96-well adhesive film tightly to the plate ensuring all of the wells are suitably covered and briefly pulse spin in a refrigerated centrifuge.
-
4.
Place the plate in a thermocycler with the following cycle conditions applied:
Step | Temperature | Time | |
|---|---|---|---|
Initial denaturation | 95 °C | 10 min | |
30 cycles | Denaturation | 95 °C | 1 min |
Annealing | User determined | 45 s | |
Extension | 72 °C | 1 min | |
Final extension | 72 °C | 5 min | |
Hold | 5 °C | Indefinitely | |
-
5.
Once complete the plate can be stored at −20 °C.
It is advisable to run some of the product on an agarose gel along with a DNA ladder to be confident of target amplification and primer specificity e.g. 5 μl of product on a 2 % agarose gel.
-
6.
Before beginning the next PCR reaction all of the samples should be purified to remove any remaining primers and salts (see App. A.1).
3.2 Second PCR (Attachment of Barcodes)
A unique combination of forward and reverse barcode primers (Table 33.1) are then added to each sample in the second PCR (Fig. 33.2). These primers contain the same aforementioned 10 bp universal tails thus allowing attachment to the previously amplified product. Next to the universal tail is a 10 bp MID sequence which is used for sample identification during the sequencing. Adjacent to this is a 25 bp sequence which is able to hybridize to the DNA capture beads during the 454 sample library preparation.
-
1.
On ice prepare the following reaction mixture, remembering to vortex all of the reagents well before use and to add an additional 10 % volume:
Reagent | 1 Reaction |
|---|---|
5× PCR reaction buffer (3 mM MgCl2 final concentration) | 5 μl |
10 μM forward primer | 1 μl |
10 μM reverse primer | 1 μl |
dNTPs (10 mM of each) | 2.5 μl |
1.25 U DNA polymerase | 0.25 μl |
PCR product template | 1 μl |
RNase/DNase free water | 14.25 μl |
Final volume | 25 μl |
-
2.
Pipette the above mixture into a 96 well plate. Each well should contain a unique combination of forward and reverse primers to allow for successful barcoding. This will allow the tracking of each sample when they are later pooled for sequencing.
-
3.
Attach the film lid tightly to the plate ensuring all of the wells are suitably covered and briefly pulse spin in a refrigerated centrifuge.
-
4.
Place the plate in a thermocycler with the following cycle conditions applied:
Step | Temperature | Time | |
|---|---|---|---|
Initial denaturation | 95 °C | 10 min | |
20–30 cycles | Denaturation | 95 °C | 1 min |
Annealing | 60 °C | 45 s | |
Extension | 72 °C | 1 min | |
Final extension | 72 °C | 5 min | |
Hold | 5 °C | Indefinitely | |
-
5.
Once complete the plate can be stored at −20 °C.
It is advisable to run some of the product on an agarose gel along with a DNA ladder to be confident of barcode attachment. If the attachment of the barcodes has been successful then the product size should be 70 bp larger than the product gained from the first PCR.
-
6.
Finally, purify all of the newly gained PCR products (see App. A.1) to prepare for quantification.
3.3 Amplicon Quantification
Precise quantification of the PCR products gained in the previous reaction is essential to allow equal reads when it comes to multiplex sequencing. An inconsistently quantified amplicon library will ultimately lead to difficulties during data normalization. There are numerous methods available to quantify the purified PCR amplicons.
3.3.1 2100 Bioanalyzer
The 2100 Bioanalyzer is able to size and quantify PCR products from just 1 μl of sample via a microfluidic approach. Each sample produces an artificial gel image as well as an electropherogram (Fig. 33.3). In conjunction with the DNA 1,000 kit each chip can simultaneously analyze 12 samples ranging from 25 to 1,000 bp in size. One possible limitation to this assay, however, is the narrow quantitation range and low sensitivity available (Table 33.2).

Left: A gel representation of two samples generated with the 2100 Bioanalyzer on a DNA 1,000 chip. Right: The corresponding electropherogram for sample ‘A1’ denoting the concentration (ng/μl) of the desired amplicon and the two markers
3.3.2 Quant-iT PicoGreen dsDNA Kit
An alternative method for quantifying PCR products prior to sequencing is to use the commercially available Quant-iT PicoGreen dsDNA assay (Invitrogen). The fluorescent stain in the kit specifically binds to double stranded DNA (dsDNA), even in the presence of single stranded nucleic acids and other contaminants, and measured with a standard fluorometer. Quantification is extrapolated from an 8-point standard curve. The assay also supports 96- and 384-well plate formats to increase throughput capabilities.
3.4 454 Library Preparation
Prior to preparing the reagents required for sequencing it is first necessary to dilute and pool the amplicon library. This DNA pool is then processed in an emulsion-based clonal amplification (emPCR) reaction (Fig. 33.4). Finally the DNA capture beads containing the different amplicons are retrieved (approximately 500,000).

Workflow of 454 amplicon library preparation prior to sequencing
All of the above steps including amplicon dilution and emPCR procedures can be found in detail in the Amplicon Library Preparation Manual [5] and the emPCR Amplification Method Manual – Lib-A [6] respectively.
3.5 454 Sequencing
Before a sequencing run can commence a pre-wash of the machine is required to prepare the instrument. The amplicon-enriched beads are injected onto a PicoTiterPlate (PTP) device in a series of layers. Following a final prime of the 454 GS Junior the PTP device is then loaded into the instrument and the sequencing can take place overnight. Raw data generated in a series of images following repeated deoxynucleotide base additions is captured, normalized and collated into runs.
For in-depth methodology on sequencing with the 454 GS Junior refer to the Sequencing Method Manual [7].
3.6 Data Analysis
The benefit of next generation sequencing is having an immense data set containing thousands of reads therefore allowing the confidence to interpret results. However with such a vast supply of results provides difficulties during the analysis stage. Fortunately there is software available to aid in the presentation and interpretation of the experimental data.
A suitable program to analyze amplicon library reads is the GS Amplicon Variant Analyzer (Roche). This software is able to analyze both uni- and bi-directional sequencing reads (see App. A.2). Each amplicon sequenced is compared against a desired reference sequence to highlight any possible variants in the amplicon library (Fig. 33.5). The corresponding MIDs must first be selected on the software for each sample to differentiate between subjects. Flowgrams can also be created which are useful when presenting any novel variant found in a sequencing run (Fig. 33.6).

Using the GS Amplicon Variant Analyzer to highlight single nucleotide polymorphisms. The top line (green) is the reference sequence. The second line is the sample sequence. Nucleotides highlighted (red) are those which differ from the expected sequence. The additional data represents individual reads for that sample

A flowgram demonstrating an A/G substitution. Top: The expected reference sequence. Middle: An amplicon sample sequence. Bottom: The difference between both the amplicon and the reference. Notice a single A removal (−1) and a single G addition (+1)
References
WHO (2013) The top 10 causes of death. [Online]. http://who.int/mediacentre/factsheets/fs310/en/index.html. Accessed 8 Jan 2014
Maouche S, Schunkert H (2012) Strategies beyond genome-wide association studies for atherosclerosis. Arterioscler Thromb Vasc Biol 32(2):170–181. doi:10.1161/ATVBAHA.111.232652
454 Life Sciences Corp. (2013) GS Junior system tables of materials. [PDF] USA. http://454.com/downloads/my454/documentation/gs-junior/system-wide-documents/GSJunior_TablesofMaterials_May2013.pdf. Accessed 7 Jan 2014
Berry D, Ben Mahfoudh K, Wagner MA, Loy A (2011) Barcoded primers used in a multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol 77(21):7846–7849. doi:10.1128/AEM. 05220-11
454 Life Sciences Corp. (2013) Amplicon Library Preparation Manual. [PDF] USA. http://454.com/downloads/my454/documentation/gs-junior/method-manuals/454SeqSys_AmpliconLibraryPrepMethodManual_Jun2013.pdf. Accessed 7 Jan 2014
454 Life Sciences Corp. (2012) emPCR Amplification Method Manual – Lib-A. [PDF] USA. http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJunioremPCRAmplificationMethodManualLib-A_March2012.pdf. Accessed 7 Jan 2014
454 Life Sciences Corp. (2013) Sequencing Method Manual. [PDF] USA. http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJuniorSequencingManual_Jan2013.pdf. Accessed 7 Jan 2014
Roche Diagnostics (2011) GS Junior System. [PDF] Germany. http://454.com/downloads/GSJuniorSystem_Brochure.pdf. Accessed 8 Jan 2014
454 Life Sciences Corp. (2009) Using multiplex identifier (MID) adaptors for the GS FLX titanium chemistry – extended MID set. [PDF] USA. http://454.com/downloads/my454/documentation/technical-bulletins/TCB-09005_UsingMultiplexIdentifierAdaptorsForTheGSFLXTitaniumChemistry-ExtendedMIDSet.pdf. Accessed 7 Jan 2014
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendix
Appendix
1.1 A.1 PCR Clean-Up Approach
There are numerous methods for purifying the newly amplified product following a PCR reaction. The aim is to remove any unused primers therefore reducing the chance of a chimeric product formation between two PCRs. This is of great importance when undertaking this approach to sequencing since the primer sets used from both of the aforementioned PCR reactions share certain sequence homology.
Column based PCR purification kits utilize an adapted silica membrane column to clean-up PCR products in three easy steps: bind, wash and elute. The products recovered are generally greater than 100 bp and depending on the kit used this can be up to 10 kb in size. This rapid approach requires very little laboratory equipment and is relatively cheap.
An alternative to the above post PCR clean-up process is the use of magnetic beads to purify the amplicon of interest. The overall process includes bead addition, separation, wash and elution. This approach is useful for high-throughput requirements. An addition benefit of this technique, in comparison to column based options, is the ability to size select the wanted product via altering the bead:DNA solution ratio. This is particularly useful when more than two products are present.
A third option is to run PCR products on a agarose gel before dissecting the wanted band and ultimately purifying the cut out with a gel purification commercial kit. This method also uses a silica membrane column to capture the amplicons prior to washing. This is mainly used as a last resort when there is unspecific binding present, such as when using bisulphite treated DNA, due to the lower recovery of DNA when compared to the above options.
1.2 A.2 Bidirectional v Unidirectional Sequencing
With the 454 GS Junior there possesses a capability to sequence an amplicon either bidirectionally or unidirectionally. As the name suggests, bidirectional sequencing includes reading the sequence in two directions (forward and reverse) and conversely unidirectional approaches produce reads in only one direction (either forward or reverse). Obviously when wanting to highlight possible variants in amplicons bidirectional sequencing provides a greater deal of confidence and accuracy when analyzing results. Alternatively, unidirectional approaches cannot read in both orientations however these will contain double the number of reads. It is therefore recommended to sequence bidirectionally when investigating rare single nucleotide polymorphisms (SNPs) and unidirectionally when analyzing a wide diversity of amplicons.
1.3 A.3 Amplicon Length Considerations
The average read length for the 454 GS Junior is 400 bp with longest reads being around 600 bp [8] including the two adaptor sequences. It should be noted that shorter than average amplicons (<300 bp) may result in excessive amplification during the emPCR stage when using the suggested conditions. This issue is avoidable by optimizing the emPCR conditions to contemplate for the desired shorter product.
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Bradburn, S. et al. (2015). Protocol for Multiplex Amplicon Sequencing Using Barcoded Primers. In: Slevin, M., McDowell, G. (eds) Handbook of Vascular Biology Techniques. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9716-0_33
Download citation
DOI: https://doi.org/10.1007/978-94-017-9716-0_33
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9715-3
Online ISBN: 978-94-017-9716-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)