
Abstract
All the cell types are under strict control of how their genes are transcribed into expressed transcripts by the temporally dynamic orchestration of the transcription factor binding activities. Given a set of known binding sites (BSs) of a given transcription factor (TF), computational TFBS screening technique represents a cost efficient and large scale strategy to complement the experimental ones. There are two major classes of computational TFBS prediction algorithms based on the tertiary and primary structures, respectively. A tertiary structure based algorithm tries to calculate the binding affinity between a query DNA fragment and the tertiary structure of the given TF. Due to the limited number of available TF tertiary structures, primary structure based TFBS prediction algorithm is a necessary complementary technique for large scale TFBS screening. This study proposes a novel evolutionary algorithm to randomly mutate the weights of different positions in the binding motif of a TF, so that the overall TFBS prediction accuracy is optimized. The comparison with the most widely used algorithm, Position Weight Matrix (PWM), suggests that our algorithm performs better or the same level in all the performance measurements, including sensitivity, specificity, accuracy and Matthews correlation coefficient. Our data also suggests that it is necessary to remove the widely used assumption of independence between motif positions. The supplementary material may be found at: http://www.healthinformaticslab.org/supp/ .
Zhao Zhang and Miaomiao Zhao have been contributed equally to this paper.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Transcription of genic regions into RNA molecules is the first step of the biological central dogma, and is dynamically controlled by various transcription factors (TFs) [1]. A TF regulates a gene’s transcription through its dynamic binding to a short (5–20 bps) DNA sequence upstream to the regulated gene. This DNA sequence is the TF’s binding site (TFBS), which is usually highly specific to this TF and is called a motif [2]. Mutations within TFBSs will change the host’s transcription regulatory network, and lead to species specific phenotypes or genetic diseases [3].
There are two major high-throughput strategies to screen the binding sites of a TF in the host genome. Firstly, various high-throughput experimental techniques were developed to screen the TFBSs under the given cell culture conditions, including DNase I footprinting [4], electrophoretic mobility shift assay [5], ChIP-on-chip [6] and ChIP-Seq [7], etc. The dynamic landscape of the transcription regulatory network may be elucidated through these screening techniques. But they are usually costly and labor-intensive, and can only detect the binding sites of one TF under one cell culture condition at a time. Considering the 2,886 transcription factors curated in the human DNA-binding domain (DBD) database [8], and the dynamic nature of transcription regulation, it can be anticipated that the transcription regulatory landscape is significantly under-estimated.
Computational TFBS screening techniques have been used to infer the comprehensive list of TFBSs. The majority of in silico TFBS screening techniques assumes that the binding sites of a given TF have a fixed length, and calculates the similarity score of a query DNA sequence compared with the local oligo-nucleotide frequency patterns in the known TFBSs [9]. The computational techniques include the position weigh matrix (PWM) [10], WebLogo [11], and position specific pairwise score [12], etc. The introduction of TF’s structural information will greatly reduce the false positive rates, as demonstrated by Facelli [13], Saito et al. [14]. But there are only 300 unique human TF structures in the PDB database [ref], and the limited availability of the experimentally detected TF structures restricts the extensive application of these methods [15].
This study hypothesizes that positions contribute differently to the motif scoring based on their nucleotide frequency patterns, and formulates the position contribution as a weight for the position. The vector of weights for different motif positions were randomly mutated by an evolutionary algorithm, with the optimization goal to maximize the overall accuracy. The prediction performance suggests that our algorithm performs similarly or better than the position specific scoring strategies.
2 Materials and Methods
2.1 Data Resources
The proposed algorithm is applied to the following seven transcription factors (TFs), i.e. Ebox, Myc, P53, Q6MAZ, Q601MAZ, V_SREBP_Q3-SREBP (abbreviated as Q3), and V_SREBP2_Q6-SREBP2 (abbreviated as Q6). The known binding sites of these seven transcription factors were manually collected from the database TRANSFAC in August 2012 [16]. Only those binding sites without an “N” letter were kept for further analysis. The target gene sequences and their promoter regions were extracted from the database ENSEMBL [17].
2.2 Motif Screening Problem
The mathematical model of the transcription factor binding site (TFBS) screening problem (sTFBS) is formulated as follows. For a given transcription factor (TF), its known fixed-length binding sites are defined to be the positive dataset P = {M 1, M 2, …, M n }, where |M i | = L. A negative dataset N = {B 1, B 2, …, B m } is randomly extracted from the promoter regions of the genes regulated by the given TF, where |B j | = L, B j has no “N” letters and B j does not overlap with M i . Considering the promoter region is much larger than a TFBS, we set m = 10 × n. A TFBS screening model is denoted as the classification function f(X) ∈ {P, N}, where X ∈ P ∪ N.
Firstly, a similarity score between two fixed-length DNA fragments V = {v 1, v 2, …, v L } and U = {u 1, u 2, …, u L } is defined to be Score(V, U) = (w 1 × S(v 1, u 1) + w 2 × S(v 2, u 2) +···+w L × S(v L , u L )), where the weight vector W = 〈w 1, w 2, …, w L 〉 is the pre-calculated combination pattern, and w i ∈ [0, 1]. The nucleotide similarity score matrix S(v i , u i ) is defined to be 2 if v i = u i , 1 for A versus G or C versus T, and −1 for the other pairs [18]. The combination pattern W = 〈w 1, w 2, …, w L 〉 will be optimized by an evolutionary algorithm, as described in the next section.
This study chose the simple nearest neighbor algorithm as the classification model f(X).

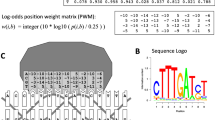
Position Weight Matrix (PWM) algorithm assumes that positions in a fixed-length motif are independent to each other and calculates how a query sequence is similar to the set of known motif occurrences [10, 19]. Firstly, a position conservation factor M i is calculated as \(M_{i} = \sum\nolimits_{{b \in \left\{ {A,T,C,G} \right\}}} {(f_{i} (b)/N - P_{0} (b))^{2} /P_{0} (b)\;,i = 1,2, \ldots ,L,}\) where f i (b) is the observed frequencies of nucleotide b at position i in the set of known motif occurrences, and P 0(b) is the background frequency of nucleotide b. Then the position probability matrix (PPM) is calculated as:
where \(P_{j} (b) = \left\{ {f_{j} (b) + s(b)} \right\}/\left\{ {N + \sum\nolimits_{{b \in \{ A,T,C,G\} }} {s(b)} } \right\}\), and \(s(b) = P_{0} (b)\sqrt N\) is a smoothing factor.
Then the position weight matrix (PWM) is calculated as
where \(w_{i} (b) = \ln \left\{ {P_{i} (b)/P_{0} (b)} \right\}.\)
The standardized similarity score of a query sequence Q is defined to be
where Q i is the i th nucleotide in Q, and b ∈ {A, T, C, G}. For a cutoff S 0, only if S(Q) ≥ S 0, Q is defined as a binding motif of the transcription factor.
2.3 Prediction Performance Measurements and Evaluation
Given the positive dataset P = {M 1, M 2, …, M n }, and the negative dataset N = {B 1, B 2, …, B m }, where |M i | = |B j | = L. M i is a true positive or false negative if SNN(M i ) = P or N, respectively, whereas B j is a true negative or false positive if SNN(B j ) = N or P, respectively. For the classification model SNN(X), the numbers of true positives, false negatives, true negatives and false positives are abbreviated as TP, FN, TN and FP, respectively. The classification performance of the model is measured by sensitivity (Sn), specificity (Sp), accuracy (Ac) and Matthews correlation coefficient (MCC) [20, 21], which are defined as follows. Sn = TP/(TP + FN), Sp = TN/(TN + FP), Ac = (Sn + Sp)/2, and MCC = (TP × TN − FP × FN)/sqrt((TP + FP) × (TP + FN) × (TN + FP) × (TN + FN)), where sqrt(t) is the squared root of t.
A line plot will be generated for the evolutionarily optimized combination pattern W = 〈w 1, w 2, …, w L 〉 for the comparison with the WebLogo plot. TFBS screening algorithms usually use the visual technique WebLogo to demonstrate the DNA compositions at each position in the TFBS, and a higher plotted position suggests a larger information content [11]. An initial weight vector \(W^{0} = \langle w_{1}^{0} ,w_{2}^{0} , \ldots ,w_{L}^{0} \rangle\) is generated from a transcription factor’s WebLogo plot, by scaling the information content at position i to [0, 1] as \(w_{i}^{0}.\)
Two validation strategies are adopted to evaluate the classification algorithm SNN’s prediction performance. Firstly, the algorithm SNN is investigated for its leave-one-out (LOO) cross validation performance, i.e. iteratively choosing one data entry and investigating its prediction by the classification model trained on the rest data sets. The LOO validation strategy has been widely used to measure how a TFBS or other functional element prediction algorithm performs [22, 23]. To further investigate the dataset dependency of the proposed SNN algorithm, this study conducted 3-fold cross validation (3FCV) strategy [24–26]. The basic idea is to randomly split the positive and negative datasets into 3 equal-size subsets {P 1, P 2, P 3} and {N 1, N 2, N 3}, respectively. The prediction results are iteratively investigated for {P i , N i } using the SNN trained on P\P i and N\N i , where i = 1, 2, and 3. A self validation (denoted as Self) is also used to evaluate the self consistency, which is to evaluate how a classification model performs on the training dataset.
2.4 Evolutionary Optimization Algorithm
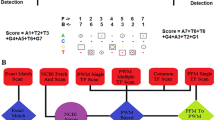
This study proposed an evolutionary optimization algorithm to screen for the weight vector with the best overall accuracy Ac of the algorithm SNN, as shown in Fig. 15.1. The basic idea of an evolutionary optimization algorithm (EOA) is to simulate the natural selection process [27, 28]. Each generation of individuals produce children through the operations of crossing and mutation from a pair of parents. A fitness function is defined to describe how each children fit the natural selection pressure. A better fitness leads to a higher chance to survive into the next generation. The population size is usually fixed to a constant value [11, 29–37]. The initial population W consists of PopSize individual weight vectors, i.e. W i, where i∈{1, 2, …, PopSize}. Each individual W i is an L-dimension vector \(W^{i} = \langle W_{0}^{i} ,W_{1}^{i} , \ldots ,W_{L}^{i} \rangle\), where \(W_{j}^{i}\) is a random value between 0 and 1.

Procedure of the evolutionary optimization algorithm. 5 weight vectors with the best accuracies Ac will be output
MaxGen generations of natural mutation and selection are conducted to find the fittest weight vectors. For a given weight vector W i, an SNN classification model is built, and the overall classification accuracy Ac with the 4-fold cross validation is defined to be the fitness function Ac(W i), as used in step 5. For the population of weight vectors W, Top5(W) consists of 5 weight vectors with the best fitness in the population. The final top 5 weight vectors together with the performance measurements of their classification models are output.
3 Results and Discussion
3.1 Best Parameters for EOA
There are two parameters for the evolutionary algorithm EOA, i.e. the population size PopSize and the generation number MaxGen. Previous studies suggested that PopSize = 100 performs well for the evolutionary optimization problems with individual vector size ~10 [38]. So we firstly fix PopSize = 100, and investigate how the optimization goal, Ac, changes with the increased number of generations, i.e. MaxGen. The parameter MaxGen is set between 0 and 5,000, and the step size is 100. Q6MAZ and Q3 quickly reach the peak Ac value 1.00 after just MaxGen = 200 generations of optimizations, as shown in Fig. 15.2a. The TF genes Ebox, Myc and P53 also reach very high Ac values (>97 %) at just MaxGen = 200. If we choose the Ac value at MaxGen = 5,000 as the final result, all the six investigated TFs reach this peak value at MaxGen = 3,000, as shown in Fig. 15.2a.

Distributions of overall classification accuracy, Ac, for different generation numbers. The population sizes PopSize are fixed to a 100, b 60 and c 140, respectively
We further investigate how the parameter PopSize impacts the optimization performance of EOA, as shown in Fig. 15.2 and Supplementary Figure S1. By choosing PopSize ∈ {20, 40, 60, 80, 100, 120, 140, 160, 200}, the overall accuracy Ac is calculated for generation G∈{0, 100, 200, …, 4,900, 5,000} of EOA on each of the six TFs. Figure 15.2 shows that the TFBS prediction problem of Q6 is the most difficult to be optimized, and reaches the peak values at generations 3,800, 3,000 and 2,600 for PopSize = 60, 100 and 140, respectively. All the other five TFs reach the peak Ac values before the optimization generation 3,000. Similar patterns can be observed for other population sizes PopSize, as in Supplementary Figure S1.
Considering that the running time of the evolutionary algorithm EOA increases linearly with the product PopSize × MaxGen, and the above data, this study will set PopSize = 100 and MaxGen = 3,000 for the following experiments.
3.2 Comparison of PWM and SNN(W0)
We firstly compare the widely used PWM algorithm with the SNN algorithm. WebLogo is also widely used to demonstrate the information content or conservation at each position of a motif [11]. The higher a position is, the larger information content this position has, as shown in Fig. 15.3. And the binding sites of all the seven TFs do show significant patterns in information content of some motif positions. So we hypothesize that the information content from WebLogo plot may represent well the weight of each motif position for the SNN algorithm, and the weight vector is denoted as W 0.

WebLogo plots for the TFs. a Ebox, b Myc, c P53, d Q6MAZ, e Q601MAZ, f Q3 and g Q6. The line plot is for the evolutionarily optimized weight vector by the SNN + EOA algorithms for each TF
Both PWM and SNN score the similarity of a query DNA sequence to the known TFBSs, and this study chooses the cutoff score with Sn ~ Sp for the comparison. In general, the SNN(W 0) algorithm performs similarly well or slightly worse compared with the PWM algorithm, as shown in Table 15.1. Both algorithms produce ~90 % or larger overall accuracy Ac for the TFBS motif screening problem, and the TF Q3 even receives 100 % accurate separation of the positive and negative data entries from both algorithms under the two validation strategies. The biggest difference between the two algorithms is for the TFBS motif screening problem of Myc, where SNN(W 0) performs 5.01 and 5.48 % worse in Ac than PWM using the LOO and 3FCV validations, respectively. So our first hypothesis about the usage of W 0 is reasonable but may need further optimization.
3.3 Comparison of PWM and SNN + EOA
The next hypothesis is that there may exist a weight vector W = 〈w 1, w 2, …, w L 〉 with increased Ac value for the SNN algorithm. Besides the position independent measurements, e.g. PWM or WebLogo, there is no available knowledge about how to optimize the weight vector. So we choose to use the evolutionary optimization algorithm to search for a weight vector with optimal overall accuracy Ac by just random mutations in the weight vectors, as described in Sect. 15.2.4.
After the optimization of MaxGen = 3,000 generations of PopSize = 100 individuals (weight vectors), the motif screening algorithm SNN outperforms the PWM algorithm in any performance measurements for all the seven TFs, as shown in Table 15.2. The PWM algorithm achieves 100 % accuracy for the LOO validation of Q6MAZ and both LOO and 3FCV validations of Q3, and the SNN + EOA algorithm achieves such perfect classification. For the other transcription factors, SNN + EOA outperforms PWM by 0.97–7.83 % in overall accuracy Ac. The measurements MCC ∈ [−1, 1] evaluates how the prediction results match the positive and negative datasets, and a larger MCC means a better prediction. Besides the two TFs Q6MAZ and Q3 that both algorithms perform equally well, SNN + EOA improves the MCC of PWM algorithm by 0.0327–0.2026. The PWM algorithm does not perform well on the dataset of the well-known tumor suppressor P53, as in Table 15.2. It only achieves Sn = 84.78 % and Sp = 96.74 % for the LOO validation of P53, and the overall accuracy is only 90.76 %. SNN + EOA achieves a slightly better specificity (Sp = 97.17 %) and a much better sensitivity (Sn = 100 %). A similar improvement is also achieved by SNN + EOA for the 3FCV validation of P53.
It’s also interesting to observe that the weight vector achieving the best prediction performance does not match the position independent measurement WebLogo, as shown in Fig. 15.3. For the tumor suppressor P53, the optimized weight vector does not agree with WebLogo at positions 4, 5 and 9, as shown in Fig. 15.3c. The information content at position 4 is larger than that at position 5, but their weights in the optimized vector weighs the two positions reversely. And although the information content at position 9 only ranks 8th, position 9 has the second largest weight. Similar discrepancy exists for all the seven investigated TFs, as in Fig. 15.3, and suggests that a concerted weighing of different positions is necessary for motif screening and other similar problems.
References
Crick F (1970) Central dogma of molecular biology. Nature 227(5258):561–563
Ameur A, Rada-Iglesias A, Komorowski J, Wadelius C (2009) Identification of candidate regulatory SNPs by combination of transcription-factor-binding site prediction, SNP genotyping and haploChIP. Nucleic Acids Res 37(12):e85
Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8(3):206–216
Galas DJ, Schmitz A (1978) DNAase footprinting a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res 5(9):3157–3170
Dent C, Latchman D (1993) The DNA mobility shift assay. In: Transcription factors: a practical approach, pp 1–3
Pillai S, Chellappan SP (2009) ChIP on chip assays: genome-wide analysis of transcription factor binding and histone modifications. In: Chromatin protocols. Springer, Berlin, pp 341–366
Johnson DS, Mortazavi A, Myers RM, Wold B (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316(5830):1497–1502
Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA (2008) DBD–taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res 36(Database issue):D88–D92
Stormo GD (2000) DNA binding sites: representation and discovery. Bioinformatics 16(1):16–23
Ben-Gal I, Shani A, Gohr A, Grau J, Arviv S, Shmilovici A, Posch S, Grosse I (2005) Identification of transcription factor binding sites with variable-order Bayesian networks. Bioinformatics 21(11):2657–2666
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14(6):1188–1190
Quader S, Huang CH (2012) Effect of positional dependence and alignment strategy on modeling transcription factor binding sites. BMC Res Notes 5:340
Gorin AA, Zhurkin VB, Wilma K (1995) B-DNA twisting correlates with base-pair morphology. J Mol Biol 247(1):34–48
Oshchepkov DY, Vityaev EE, Grigorovich DA, Ignatieva EV, Khlebodarova TM (2004) SITECON: a tool for detecting conservative conformational and physicochemical properties in transcription factor binding site alignments and for site recognition. Nucleic Acids Res 32(suppl 2):W208–W212
Rose PW, Bi C, Bluhm WF, Christie CH, Dimitropoulos D, Dutta S, Green RK, Goodsell DS, Prlic A, Quesada M et al (2013) The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res 41(Database issue):D475–D482
Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K et al (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34(Database issue):D108–D110
Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S et al (2013) Ensembl 2013. Nucleic Acids Res 41(Database issue):D48–D55
String Alignment using Dynamic Programming.(http://www.biorecipes.com/DynProgBasic/code.html)
Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E (2003) MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 31(13):3576–3579
Xue Y, Zhou F, Zhu M, Ahmed K, Chen G, Yao X (2005) GPS: a comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res 33(Web Server issue):W184–W187
Zhou FF, Xue Y, Chen GL, Yao X (2004) GPS: a novel group-based phosphorylation predicting and scoring method. Biochem Biophys Res Commun 325(4):1443–1448
Sheffield NC, Thurman RE, Song L, Safi A, Stamatoyannopoulos JA, Lenhard B, Crawford GE, Furey TS (2013) Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res 23(5):777–788
Zhou Q, Liu JS (2004) Modeling within-motif dependence for transcription factor binding site predictions. Bioinformatics 20(6):909–916
Cheng C, Ung M, Grant GD, Whitfield ML (2013) Transcription factor binding profiles reveal cyclic expression of human protein-coding genes and non-coding RNAs. PLoS Comput Biol 9(7):e1003132
Zhou F, Xu Y (2010) cBar: a computer program to distinguish plasmid-derived from chromosome-derived sequence fragments in metagenomics data. Bioinformatics 26(16):2051–2052
Qian J, Lin J, Luscombe NM, Yu H, Gerstein M (2003) Prediction of regulatory networks: genome-wide identification of transcription factor targets from gene expression data. Bioinformatics 19(15):1917–1926
Potts JC, Giddens TD, Yadav SB (1994) The development and evaluation of an improved genetic algorithm based on migration and artificial selection. IEEE Trans Syst Man Cybern 24(1):73–86
Tam KY (1992) Genetic algorithms, function optimization, and facility layout design. Eur J Oper Res 63(2):322–346
Anastassopoulos G, Adamopoulos A, Galiatsatos D, Drosos G (2013) Feature extraction of osteoporosis risk factors using artificial neural networks and genetic algorithms. Stud Health Technol Inform 190:186–188
Santiso EE, Musolino N, Trout BL (2013) Design of linear ligands for selective separation using a genetic algorithm applied to molecular architecture. J Chem Inf Model 53(7):1638–1660
Chen JB, Chuang LY, Lin YD, Liou CW, Lin TK, Lee WC, Cheng BC, Chang HW, Yang CH (2013) Genetic algorithm-generated SNP barcodes of the mitochondrial D-loop for chronic dialysis susceptibility. Mitochondrial DNA
Sale M, Sherer EA (2013) A genetic algorithm based global search strategy for population pharmacokinetic/pharmacodynamic model selection. Brit J Clin Pharmacol
Yoon Y, Kim YH (2013) An efficient genetic algorithm for maximum coverage deployment in wireless sensor networks. IEEE Trans Cybern
Azadnia AH, Taheri S, Ghadimi P, Mat Saman MZ, Wong KY (2013) Order batching in warehouses by minimizing total tardiness: a hybrid approach of weighted association rule mining and genetic algorithms. Sci World J 2013:246578
Chuang LY, Cheng YH, Yang CH, Yang CH (2013) Associate PCR-RFLP assay design with SNPs based on genetic algorithm in appropriate parameters estimation. IEEE Trans Nanobiosci 12(2):119–127
Khotanlou H, Afrasiabi M (2012) Feature selection in order to extract multiple sclerosis lesions automatically in 3D brain magnetic resonance images using combination of support vector machine and genetic algorithm. J Med Signals Sens 2(4):211–218
Kou J, Xiong S, Fang Z, Zong X, Chen Z (2013) Multiobjective optimization of evacuation routes in stadium using superposed potential field network based ACO. Comput Intell Neurosci 2013:369016
Deb K, Pratap A, Agarwal S, Meyarivan T (2002) A fast and elitist multiobjective genetic algorithm: NSGA-II. IEEE Trans Evol Comput 6(2):182–197
Acknowledgments
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13040400), Shenzhen Peacock Plan (KQCX20130628112914301), Shenzhen Research Grant (ZDSY20120617113021359), China 973 program (2010CB732606), the MOE Humanities Social Sciences Fund (No.13YJC790105) and Doctoral Research Fund of HBUT (No. BSQD13050). Computing resources were partly provided by the Dawning supercomputing clusters at SIAT CAS.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Shanghai Jiaotong University Press, Shanghai and Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Zhang, Z., Wang, Z., Mai, G., Luo, Y., Zhao, M., Zhou, F. (2015). Evolutionary Optimization of Transcription Factor Binding Motif Detection. In: Wei, D., Xu, Q., Zhao, T., Dai, H. (eds) Advance in Structural Bioinformatics. Advances in Experimental Medicine and Biology, vol 827. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9245-5_15
Download citation
DOI: https://doi.org/10.1007/978-94-017-9245-5_15
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9244-8
Online ISBN: 978-94-017-9245-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)